
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Artificial plant cell walls as multi-catalyst systems for enzymatic cooperative asymmetric catalysis in non-aqueous media†
Luca
Deiana
a,
Abdolrahim A.
Rafi
a,
Veluru Ramesh
Naidu
a,
Cheuk-Wai
Tai
 b,
Jan-E.
Bäckvall
*ac and
Armando
Córdova
*a
b,
Jan-E.
Bäckvall
*ac and
Armando
Córdova
*a
aDepartment of Natural Sciences, Mid Sweden University, Holmgatan 10, 85 179 Sundsvall, Sweden. E-mail: Armando.Cordova@miun.se
bDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, 10 691 Stockholm, Sweden
cDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-10 691 Stockholm, Sweden. E-mail: jeb@organ.su.se
First published on 29th July 2021
Abstract
The assembly of cellulose-based artificial plant cell wall (APCW) structures that contain different types of catalysts is a powerful strategy for the development of cascade reactions. Here we disclose an APCW catalytic system containing a lipase enzyme and nanopalladium particles that transform a racemic amine into the corresponding enantiomerically pure amide in high yield via a dynamic kinetic resolution.
The main structural building block of the plant cell wall is cellulose,1,2 which forms interwoven microfibril networks that are embedded into a polysaccharide and protein matrix to constitute a multilayer architecture (Fig. S1, ESI†).
Advances in biocatalysis are a requirement for addressing the increasing need for sustainable chemistry in the preparation of materials, fine-chemicals and pharmaceutical synthesis.3 The ability of biocatalysts to operate under unnatural conditions (e.g. in non-aqueous media and at high temperatures),4–6 and to catalyze non-natural chemical reactions are important factors for future advancements of the “enzyme universe”.7 To date, the primary strategy for obtaining biocatalyst stability in non-aqueous media is through immobilization within, or on the surface of, solid supports such as silica or fossil-based polymer resins.8 However, often increased stability leads to decrease in biocatalyst activity. Thus, there is a significant incentive for developing new (bio)technologies for the advancement of non-aqueous industrial biocatalysis. The development of homogeneous and heterogeneous multi-catalytic systems for cooperatively advancing chemical transformations by combining different types of catalysts (e.g. metal, enzyme or metal-free)9 is another elegant way of expanding chemical synthesis and biocatalysis.
The construction of protein-derived artificial metalloenzymes for application in chemical synthesis in non-aqueous media is an important area of research that has attracted considerable attention.10–21 Here the state-of-the-art involves directed evolution and/or introducing a transition metal into the enzyme. In the latter approach, the metal of a heme enzyme may be replaced by a transition metal16–18 or a transition metal may be introduced into the active site of the enzyme.10a,19,20 The development of heterogeneous artificial metalloenzyme systems based on dual enzyme/metal nanocatalysis was also recently disclosed.21 However, partial deactivation of the enzyme component led to a limited recycling of the artificial metalloenzyme. This can also be the case for other types of heterogeneous enzyme–metal nanohybrid catalyst systems.22–24
To address this problem, we have focused on a multi-disciplinary approach for the advancement of non-aqueous biocatalysis,8,25 which is based on a synergistically combined platform of cellulose chemistry, nanocatalysis, and heterogeneous catalysis. Based on our research on cooperative heterogeneous catalysis21,22,26–28 and construction/application of functional cellulose-based materials,29,30 we envisioned a novel approach for setting up nature-inspired catalyst systems that are guided by the intriguing assembly of the primary cell wall1,31 of plants. Specifically, self-assembled mixtures of cellulose, polysaccharides, enzymes, surfactants and metals can create an artificial plant cell wall (APCW)-like structure with an embedded multi catalyst system (1), which next could catalyze the stereoselective formation of product D from substrates A + B + C in non-aqueous media (Fig. 1). Herein, we disclose this concept by creation of an assembled PCW mimic, which is employed as a heterogeneous catalyst and a “deracemase” system for converting racemic mixtures of amines 2 to the corresponding chiral amides (R)-3 by dynamic kinetic resolution (DKR).32,33
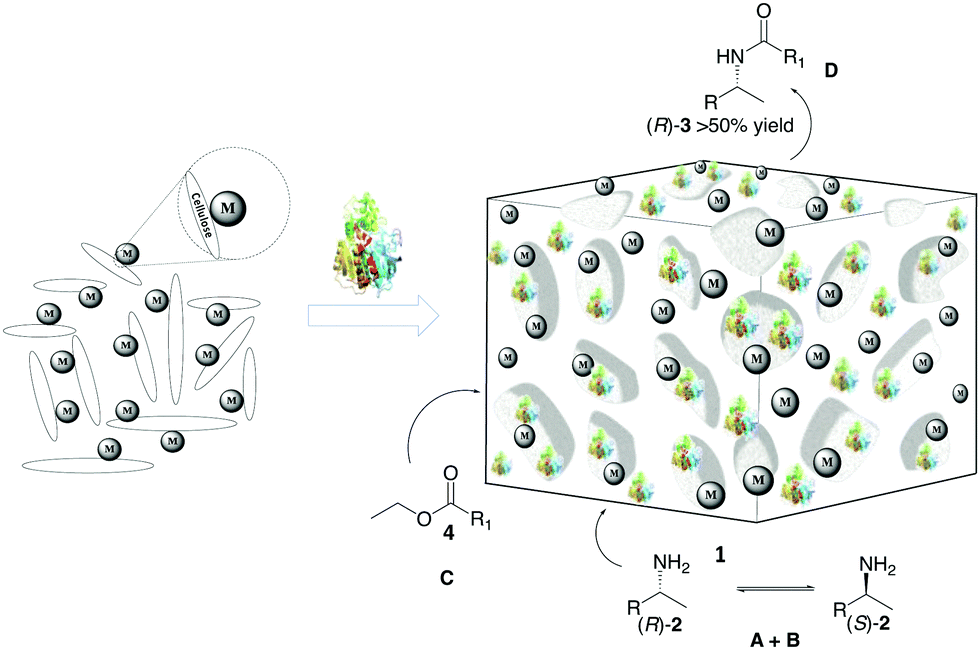 | ||
| Fig. 1 Simplified scheme of self-assembly of components to create an artificial primary cell wall 1, which simultaneously catalyzes racemization and amidation of racemic amines 2. M = metal. | ||
We began our search for catalytically active and synthetically useful APCW catalyst systems by a thorough study of the kinetic resolution of racemic amine 2a using Candida antarctica Lipase B (CALB) as the enzyme of choice (Table S1 and Fig. S2, ESI†). The assembly of various APCWs containing this serine hydrolase revealed a significant enhancement in activity of the enzyme by the different self-assembled polysaccharide-based systems. In fact, CALB is nearly inactive without some type of activation. We also investigated the role of all components used when the APCW contained microcrystalline cellulose (MCC) as the component. The use of cellulose as a support material led to a significant activation as compared to just the use of CALB. Thus, the cellulose immobilization improved the performance of the enzyme in the organic solvent. This is a beneficial effect of immobilization.34 The use of buffer35 within the assembled network was also beneficial but is not necessary. Modifying CALB with a surfactant increased the rate of amidation, but this catalyst system cannot be recycled and used for multiple reactions.36–40 The best self-assembly combination turned out to be non-covalent modification of CALB with surfactant polyethylene glycol hexadecyl ether (Brij) in phosphate buffer together with MCC forming APCW system 3 (APCW3).41 We also successfully created assembled APCWs containing nanofibrillated cellulose (NFC, APCW5), chitosan (APCW6) and nanocrystalline cellulose (CNC, APCW7), respectively. However, in the latter cases (APCW5-7) the E-value (enantiomeric ratio)42 for 2a was lower than that obtained when MCC was used as the polysaccharide component. We next decided to further explore APCWs containing aminopropylsilane modified MCC (MCC-Amp) and Pd(0) nanoparticles (MCC-Amp-Pd(0)-NPs) for the kinetic resolution of rac-2a. Thus, self-assembled APCW8 and APCW9 (Fig. 2a) were evaluated as heterogeneous catalysts. The AmP surface modification as well as the presence of Pd(0)-NPs did not decrease the excellent enantioselectivity of the enzyme (E > 400). We also prepared and evaluated a hybrid catalyst containing CALB/Brij/Pd(0)/buffer salts. This catalyst did not exhibit good selectivity (E = 21). Thus, the presence of the MCC component is essential for reaching a high E-value under the evaluated reaction conditions. With these results in hand, the novel APCW9 structure was chosen and further investigated as the nature-inspired multi-catalyst system. The assembled APCW9 multi-catalyst structure (Fig. 2a) had a narrow distribution of Pd nanoparticles (main distribution of 1.6 to 2.8 nm in size, Fig. 2f) as confirmed by TEM analyses (Fig. 2b and c). The distribution between Pd(0) and Pd(II) in the Pd NPs of APCW9 was difficult to determine due to the presence of the salts of the phosphate buffer; however, it was similar to that of its MCC-AmP-Pd(0) component as determined by XPS (Fig. 2d and e). Thus, we had accomplished the assembly of an APCW structure containing Pd-nanoparticles and enzyme units as well-defined catalysts. Next, the APCW-catalyzed deracemization of amine 2a was investigated (Table 1). We found that APCW9 is an excellent catalyst system, which is able to catalyze the formation of amide 3a under DKR conditions in several solvents (Table 1). The deracemization (DKR process) involves a synergistic interplay between Pd(0)-catalyzed racemization and CALB-catalyzed enantioselective amidation of 2a within the cellulose network of APCW9.43 Another competing catalytic side-reaction that the APCW can catalyze, is deamination of 2a to give the corresponding ethylbenzene (5a). It is noteworthy that the integrated heterogeneous APCW9 catalyst system is more efficient and chemoselective than when using mixed separate Pd-NPs on MCC and CALB on MCC or when using a hybrid-catalyst made of CALB, Brij, Pd(0)-NP and buffer (Table 1, entries 1, 11 and 12).
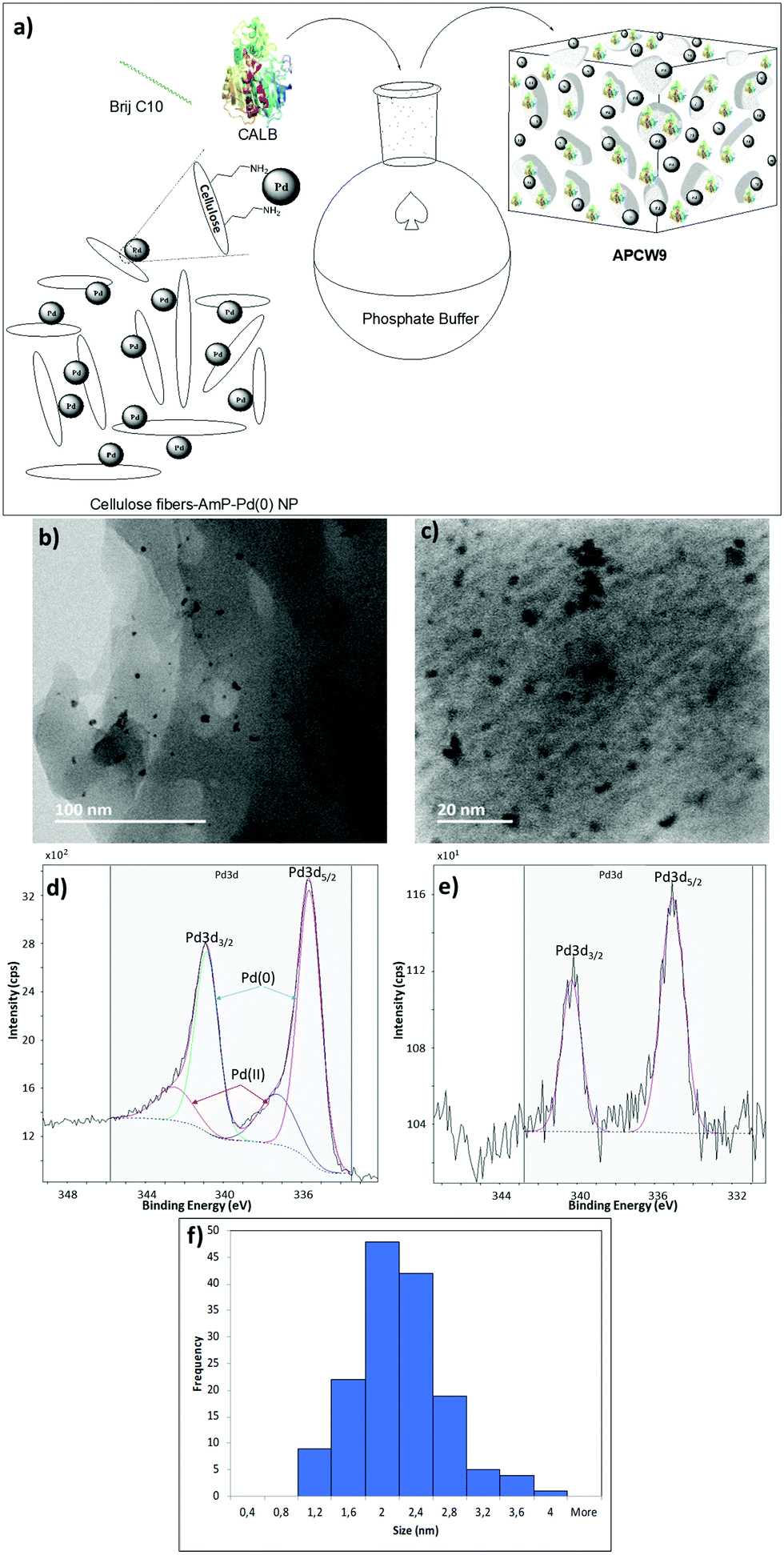 | ||
| Fig. 2 (a) Fabrication of APCW9. STEM images of APCW9, (b) scale bar 100 nm and (c) scale bar 20 nm. XPS spectra of Pd 3d5/2 for (d) MCC-Amp-Pd0 and (e) APCW9 (that had been stirred for 24 h in toluene, at 90 °C, under H2 atmosphere). (f) Size distribution of the Pd nanoparticles of APCW9. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time [h] |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5a:2ab :5a:2ab |
3a Yield [%]c | Ee 3a [%]d |
| a Reaction conditions: 2a (0.25 mmol, 1 equiv.), 4 (2.5 mmol, 10 equiv.), APCW9 (MCC-Amp-Pd0/CALB/Brij/buffer) (9 mg, 1.1 mol% Pd), Na2CO3 (0.75 mmol, 3 equiv.), toluene (1 mL), 90 °C, H2. b Determined by 1H NMR. c Isolated yield. d Determined by chiral HPLC. e Molecular sieves 4 Å (125 mg) were used. f Reaction performed at 70 °C. g APCW9 (18 mg, 2.2 mol% Pd) was used. h APCW10 (CNC-Amp-Pd0/CALB/Brij/buffer) (9 mg, 1.5 mol% Pd). i No catalyst used. ND stands for not determined. j APCW9 (18 mg, 2.2 mol% Pd) was stirred with Na2CO3 under H2 at 90 °C for 23 hours and next 2a and 4 were added. k MCC-Amp-Pd0 (16.5 mg, 12 mol% Pd) and APCW3 (MCC/CALB/Brij/buffer) (9 mg) were used as catalysts. l CALB/Brij/Pd0/buffer (9 mg) was used as a catalyst. m APCW11 (MCC-Amp-PdII/CALB/Brij/buffer) was used as a catalyst. n APCW3 was used as a catalyst. | |||||
| 1 | Toluene | 23 | 87:13:0 |
81 | 99 |
| 2e | Toluene | 23 | 67:33:0 |
67 | 48 |
| 3f | Toluene | 23 | 72:28:0 |
58 | 63 |
| 4 | 1,4-Dioxane | 70 | 70:21:9 |
70 | 99 |
| 5g | 1,4-Dioxane | 24 | 46:54:0 |
44 | 99 |
| 6 | 3-methyl3-pentanol | 70 | 63:23:7 |
62 | 99 |
| 7h | Toluene | 22 | 46:54:0 |
49 | 84 |
| 8h | 1,4-Dioxane | 69 | 32:56:12 |
33 | 99 |
| 9i | Toluene | 23 | 0:0:100 |
ND | ND |
| 10j | Toluene | 44 | 74:26:0 |
70 | 99 |
| 11k | Toluene | 23 | 76:24:0 |
77 | 99 |
| 12l | Toluene | 117 | 65:30:5 |
64 | 99 |
| 13m | Toluene | 22 | 52:48:0 |
54 | 57 |
| 14n | Toluene | 27 | 50:0:50 |
46 | 99 |
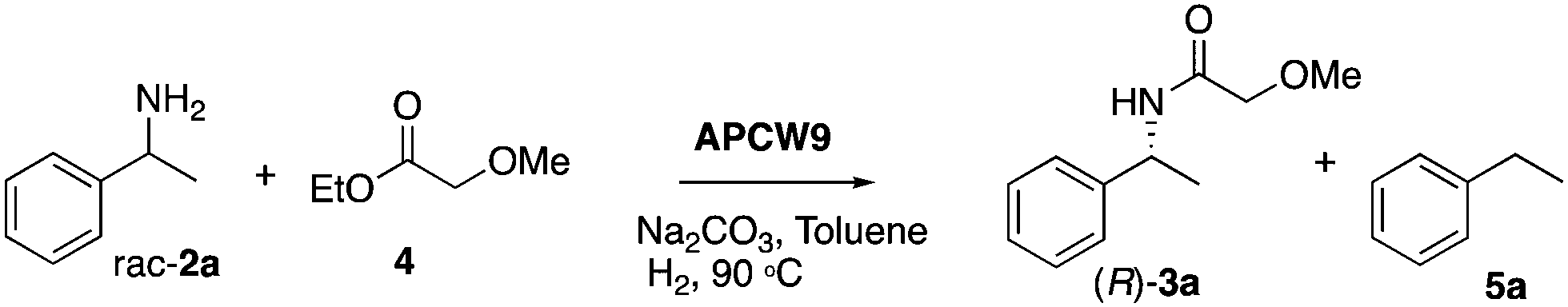
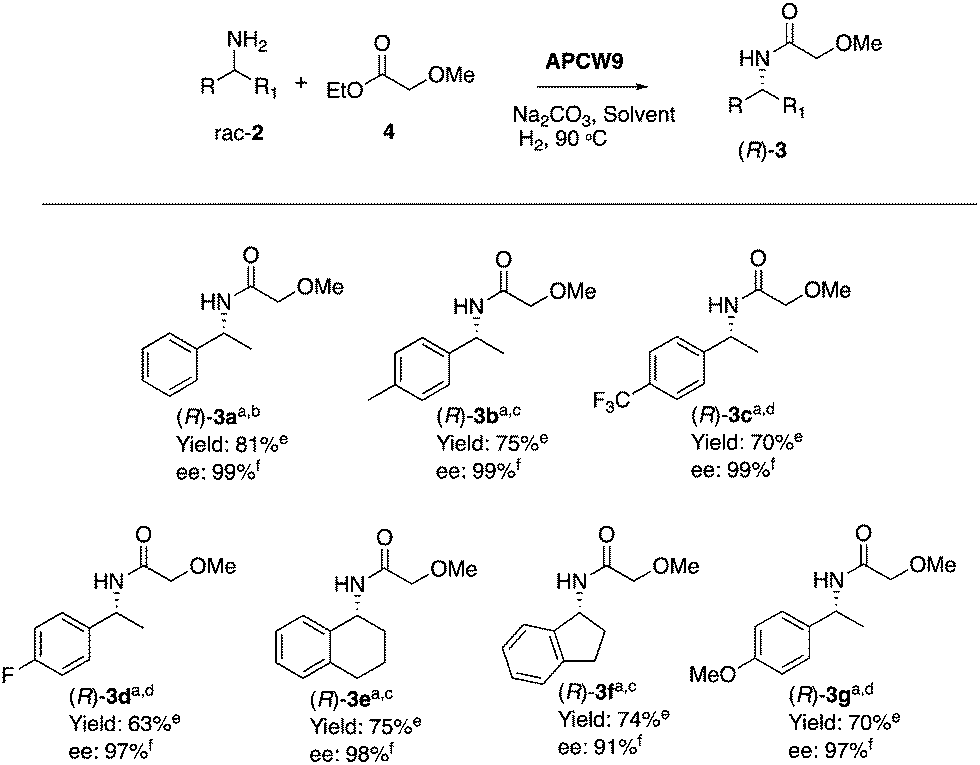
In addition, performing the reaction with APCW11, which contains Pd(II), gave a low ee (entry 13). The reaction with APCW3, which do not contain Pd, led to a kinetic resolution (3a, 46% yield, entry 14). Moreover, the Pd-nanoparticle catalysts are not able to racemize the product (R)-3a just the amine 2a (see ESI†). The heterogeneous APCW9 multi-catalyst system catalyzed the asymmetric synthesis of a variety of amides 3, which were isolated in good to high yields with 91–99% enantiomeric excess (Table 2). We further investigated the sustainable APCW multi-catalyst systems by probing their recyclability. The recycling studies revealed that the APCW catalytic systems, which contained a hydrolytic enzyme can be recycled multiple times when catalyzing the KR of amine 2a maintaining their high enantioselectivity in the organic solvent. For example, the APCW3 system catalyzed the amidation of 2a in high enantioselectivity and the corresponding amide (R)-3a was isolated in 48% yield (50% is the maximum theoretical yield) in >99% ee even after 9 recycles (Fig. S2a, ESI†). It is remarkable that the sustainable APCW9 multi-catalyst system can catalyze the DKR of 2a over several cycles (Fig. S2b, ESI†),44 since most of the previous recycling studies of artificial “deracemases” and hybrid enzyme catalyst systems containing CALB displayed a limited recycling due to significant deactivation of the enzyme catalyst.21 Thus, the disclosed artificial plant-cell wall concept enables several important features of accomplishing effective biocatalysis in non-aqueous media such as maintaining activity and stability in organic solvents, high stereoselectivity, recyclability, asymmetric synthesis, high temperature tolerance (90 °C), and efficient cooperation with chemical catalysts. No leaching of the metal catalyst was observed during the recycling studies.
| a Reaction conditions: 2 (0.25 mmol, 1 equiv.), 4 (2.5 mmol, 10 equiv.), APCW9 (9 mg, 1.1 mol% Pd), Na2CO3 (0.75 mmol, 3 equiv.), solvent (1 mL), 90 °C, H2 balloon. b Reaction performed in toluene for 23 hours. c Reaction performed in 1,4-dioxane for 69 hours. d Reaction performed in 3-methyl 3-pentanol for 69 hours. e Isolated yield. f Determined by chiral HPLC. |
|---|
|
We have demonstrated that sustainable artificial plant cell-wall (APCW) structures, which contain a multi-catalytic system of both enzyme and metal nanoparticle catalysts, can be used to accomplish asymmetric catalysis and cascade synthesis in non-aqueous media. A key feature of the disclosed APCW concept is its intrinsic ability to activate the enzyme protein for catalysis in organic solvents. The heterogeneous polysaccharide-based APCW network having both enzyme and metal nanoparticles in its structure, catalyzed DKR of primary amines by cooperative catalysis. Thus, the assembled sustainable APCW structure acts as a “deracemase”. It is also possible to reuse the bioinspired APCW structures in multiple-reaction cycles. In comparison, previous attempts of constructing artificial “deracemases” using silica-based or cross-linked enzyme aggregate systems had limitations in their recycling due to deactivation of the enzyme component. The concept of introducing catalysts with different functions within an artificial primary cell-wall structure holds great promise in creating other types of APCW catalysts with novel reactivity. Thus, various enzymes and chemical catalysts can be combined within nature-inspired plant cell wall structures to create heterogeneous catalysts for cooperative chemoenzymatic cascade reactions.
The authors thank the European Union, the Swedish Research Council (VR), the Swedish Foundation for Strategic Environmental Research (Mistra: project Mistra SafeChem, project number 2018/11), and the Knut & Alice Wallenberg Foundation (KAW 2016 0072) for financial support. KAW is gratefully acknowledged for an equipment grant for the electron microscopy facilities at Stockholm University.
Conflicts of interest
There are no conflicts to declare.References
- (a) D. J. Cosgrove, Nat. Rev. Mol. Cell Biol., 2005, 6, 850 CrossRef CAS PubMed; (b) B. B. Buchanan, W. Gruissem and R. L. Jones, Biochemistry & Molecular Biology of Plants., American Society of Plant Physiologists, 2000 Search PubMed; (c) E. Sjöström and Wood Chemistry, Fundementals and Applications, Elsevier, 1992 Search PubMed; (d) D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358 CrossRef CAS PubMed.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47 CrossRef CAS PubMed.
- (a) A. J. Ragauskas, et al. , Science, 2006, 311, 484 CrossRef CAS PubMed; (b) R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678 RSC; (c) R. Wohlgemuth, Curr. Opin. Biotechnol., 2010, 21, 713 CrossRef CAS PubMed.
- A. M. Klibanov, Nature, 2001, 409, 241 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, Science, 1984, 224, 1249 CrossRef CAS PubMed.
- P. S. Brogan, L. Bui-Le and J. P. Hallett, Nat. Chem., 2018, 10, 859 CrossRef PubMed.
- H. Renata, H. Z. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223 RSC.
- (a) C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2013, 3, 2856 CrossRef CAS; (b) R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC; (c) O. Pamies and J.-E. Bäckvall, Chem. Rev., 2003, 103, 3247 CrossRef CAS PubMed; (d) F. Rudroff, et al. , Nat. Catal., 2018, 1, 12 CrossRef; (e) R. A. Sheldon and D. Brady, Chem. Commun., 2018, 54, 6088 RSC; (f) O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996 CrossRef CAS PubMed; (g) S. Afewerki and A. Córdova, Chem. Rev., 2016, 116, 13512 CrossRef CAS PubMed; (h) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (i) Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355 CrossRef CAS PubMed.
- (a) M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS; (b) M. Diéguez, J.-E. Bäckvall and O. Pàmies, ed., “Artificial Metalloenzymes and MetalloDNAzymes in Catalysis”, Wiley-VCH, 2018. ISBN: 978-3-527-34178-8 Search PubMed.
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048 CrossRef CAS PubMed.
- S. C. Hammer, G. Kubik, E. Watkins, S. Huang, H. Minges and F. H. Arnold, Science, 2017, 358, 215 CrossRef CAS PubMed.
- J. B. S. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132 CrossRef PubMed.
- K. Chen, X. Huang, S. B. J. Kan, J. Kan, R. Zhang and F. H. Arnold, Science, 2018, 360, 6384 Search PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67 CrossRef CAS PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534 CrossRef CAS PubMed.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102 CrossRef CAS PubMed.
- S. N. Natoli and J. F. Hartwig, Acc. Chem. Res., 2019, 52, 326 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 66 CrossRef PubMed.
- T. Heinisch and T. R. Ward, Acc. Chem. Res., 2016, 49, 1711 CrossRef CAS PubMed.
- K. Engström, E. V. Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C.-W. Tai and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006 CrossRef PubMed.
- (a) T. Görbe, K. P. Gustafson, O. Verho, G. Kervefors, H. Zheng, X. Zou, E. V. Johnston and J.-E. Bäckvall, ACS Catal., 2017, 7, 1601 CrossRef; (b) K. P. J. Gustafson, T. Görbe, G. De Gonzalo, N. Yuan, C. Schreiber, A. Shchukarev, C.-W. Tai, I. Persson, X. Zhou and J.-E. Bäckvall, Chem. – Eur. J., 2019, 25, 9174 CrossRef CAS PubMed.
- (a) M. Filice, M. Marciello, M. del Puerto Morales and J. M. Palomo, Chem. Commun., 2013, 49, 6876 RSC; (b) S. Gao, Y. Liu, L. Wang, P. Liu, J. Gao and Y. Jiang, ACS Catal., 2021, 11, 5544 CrossRef CAS.
- X. Li, Y. Cao, K. Luo, Y. Sun, J. Xiong, L. Wang, Z. Liu, J. Li, J. Ma, J. Ge, H. Xiao and R. N. Zare, Nat. Catal., 2019, 2, 718 CrossRef CAS.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318 CrossRef.
- K. P. J. Gustafson, R. Lihammar, O. Verho, K. Engström and J.-E. Bäckvall, J. Org. Chem., 2014, 79, 3747 CrossRef CAS PubMed.
- L. Deiana, Y. Jiang, C. Palo-Nieto, S. Afewerki, C. A. Incerti-Pradillos, O. Verho, C.-W. Tai, E. V. Johnston and A. Córdova, Angew. Chem., Int. Ed., 2014, 53, 3447 CrossRef CAS PubMed.
- C. Palo-Nieto, S. Afewerki, M. Andersson, C.-W. Tai, P. Berglund and A. Córdova, ACS Catal., 2016, 6, 3932 CrossRef CAS.
- M.-B. Li, Y. Yang, A. A. Rahim, M. Oschmann, E. Svensson Grape, A. K. Inge, A. Córdova and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2020, 59, 10391 CrossRef CAS PubMed.
- A. Córdova, S. Afewerki, R. Alimohammadzadeh, I. Sanhueza, C.-W. Tai, S. H. Osong, P. Engstrand and I. Ibrahem, Pure Appl. Chem., 2019, 91, 865 CrossRef.
- T. Paulraj, S. Wennmalm, D. C. F. Wieland, A. V. Riazanova, A. Dedinaite, T. G. Pomorski, M. Cárdenas and A. J. Svagan, Nat. Commun., 2020, 11, 958 CrossRef CAS PubMed.
- General reviews on DKR: (a) O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996 CrossRef CAS PubMed; (b) R. Marcos and B. Martín-Matute, Isr. J. Chem., 2012, 52, 639 CrossRef CAS Reviews specified on DKR of amines:; (c) Y. Kim, J. Park and M.-J. Kim, ChemCatChem, 2011, 3, 271 CrossRef CAS.
- For representative DKRs of amines see: (a) J. Paetzold and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 17620 CrossRef CAS PubMed; (b) A. Parvulescu, D. De Vos and P. Jacobs, Chem. Commun., 2005, 5307 RSC; (c) M.-J. Kim, W.-H. Kim, K. Han, Y. K. Choi and J. Park, Org. Lett., 2007, 9, 1157 CrossRef CAS PubMed.
- (a) A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185 CrossRef PubMed; (b) S. Datta, L. R. Christena, Y. Rani and S. Rajaram, 3 Biotech., 2013, 3, 1 CrossRef PubMed.
- H. R. Costantiono, K. Griebenow, R. Langer and A. M. Klibanov, Biotechnol. Bioeng., 1997, 53, 345 CrossRef.
- K. Debulis and A. Klibanov, Biotechnol. Bioeng., 1993, 31, 41 Search PubMed.
- K. Griebenow, et al. , J. Am. Chem. Soc., 2001, 123, 5380 CrossRef CAS PubMed.
- F. Bordusa, Chem. Rev., 2002, 102, 4817 CrossRef CAS PubMed.
- M.-J. Kim, Y. Chung, Y. Choi, H. Lee, D. Kim and J. Park, J. Am. Chem. Soc., 2003, 125, 11494 CrossRef CAS PubMed.
- L. Borén, B. Martin-Matute, Y. Xu, A. Córdova and J.-E. Bäckvall, Chem. – Eur. J., 2006, 12, 225 CrossRef PubMed.
- We found that the APCW concept presented here also can activate other hydrolytic enzymes such as subtilisin for use in multiple reaction cycles.
- C.-S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294 CrossRef CAS.
- The success of the process relies on the fact that the Pd under the conditions used only racemizes the amine and not the amide. For racemization of amides by Pd/C under harsh conditions at 130 °C see: G. Valenti, P. Tinnemans, I. Baglai, W. L. Noorduin, B. Kaptein, M. Leeman, J. H. ter Horst and R. M. Kellogg, Angew. Chem., Int. Ed., 2021, 60, 5279 CrossRef CAS PubMed.
- TEM after the 7th catalytic cycle showed that considerable aggregation of Pd had occurred (see Fig. 1Sb in ESI†).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02878b |
| This journal is © The Royal Society of Chemistry 2021 |