
Access to 1-indolyltetrahydro-β-carbolines via metal-free cross-dehydrogenative coupling: the total synthesis of eudistomin U, isoeudistomin U and 19-bromoisoeudistomin U†
Ganapathy
Ranjani
 a and
Rajagopal
Nagarajan
*b
a and
Rajagopal
Nagarajan
*b
aSchool of Chemistry, University of Hyderabad, Hyderabad-500046, Telangana, India
bSchool of Chemistry, University of Hyderabad, Hyderabad-500046, Telangana, India. E-mail: nagarajan@uohyd.ac.in
First published on 10th December 2020
Abstract
A highly selective and captivating metal-free cross-dehydrogenative coupling for the cross-coupling of two reactive nucleophiles such as tetrahydro-β-carboline and indoles is developed. A series of 1-indolyltetrahydro-β-carboline derivatives were synthesized in excellent to moderate yields. Temperature, time and concentration control resulted in mono indolylation selectively. Moreover, the total synthesis of eudistomin U and isoeudistomin U and the first total synthesis of 19-bromoisoeudistomin U were accomplished.
Construction of a carbon–carbon bond plays an exemplary role in organic synthesis and serves as the heart of classical and modern synthetic organic chemistry.1 For many years, transition metal-catalysed traditional cross-couplings were mainly used for C–C bond formation.2 Conventional metal-catalysed coupling reactions employ pre-functionalized electrophiles and nucleophiles, and result in stoichiometric amounts of waste during the transmetalation step. Besides, the pre-functionalized electrophiles used in traditional coupling reactions are either directly or indirectly obtained from the corresponding C–H nucleophiles.
To minimize the waste production and address environmental issues, scientists started developing much greener and environmentally benign synthetic reactions.3 If two C–H nucleophiles are coupled directly without any pre-functionalization, then the by-product would be hydrogen, and also the waste production will be minimized. The coupling of two nucleophiles seemed to be highly imaginary until oxidative cross-coupling strategies were developed.3 The future of organic synthesis relies on such oxidative cross-coupling strategies as they have the potential to make synthetic organic chemistry more economical and much eco-friendlier. C–H bonds can generally be classified as reactive and non-reactive. Combinations of transition-metal catalysts, oxidants, and mostly high temperatures are generally needed to activate non-reactive C–H bonds (C–H activation).
Oxidation of the C–H bond becomes easy when it accompanies a hetero-atom (O, N, and S) at the adjacent position (reactive nucleophiles). In oxidative coupling reactions, one of the two nucleophiles involved in the C–C bond formation is in situ converted into an electrophile by means of oxidizing reagents (NCS, NBS, tBuOCl, DDQ, TEMPO salts, and transition metal catalysts). On the other hand, such reactions are highly prone to side reactions (homocoupling, coupling of oxidizing reagents with one of the reactants, over-oxidation, etc.) and poor selectivity. The situation becomes very tough to handle when both the nucleophiles involved in the oxidative cross-coupling are highly reactive. The specific choice of reagents and reaction conditions may solve the issues experienced during the coupling of highly reactive nucleophiles.
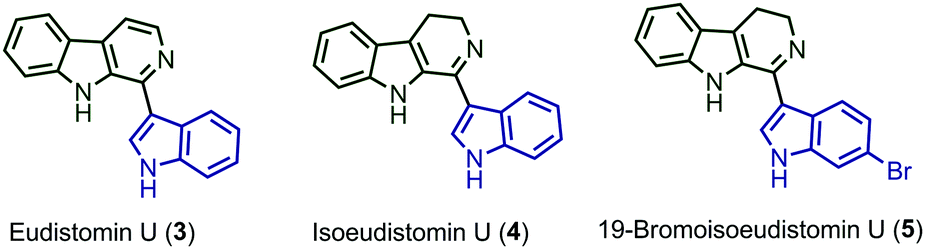
Tetrahydro-β-carboline (THβC) (1) is an annulated indole that is highly reactive and present in many biologically active alkaloids.4a–d Also, many exciting alkaloids are mentioned in the literature with the combination of THβC (1) and indole (2) (Fig. 1). Developing cross-dehydrogenative coupling (CDC) for the coupling of reactive nucleophiles such as indoles and THβC will address the problems associated with the CDC reaction of reactive nucleophiles and also afford biologically important 1-indolyltetrahydro-β-carboline (6) derivatives. Though the oxidation of secondary amine is relatively difficult, the reactivity of indole allows the substrate to undergo further oxidation followed by coupling with one more molecule of indole.5a
 | ||
| Fig. 1 Examples of 1-indolyl-β-carboline alkaloids. | ||
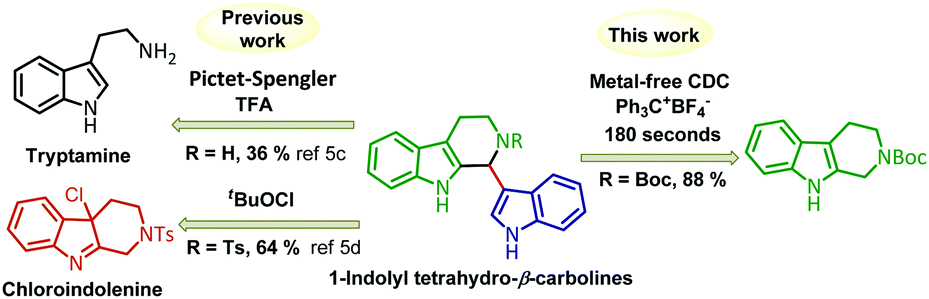
Hence, it will be highly difficult to obtain the mono indolyl products selectively. Since THβC is also reactive, there is a possibility of cleavage of the ring under oxidative conditions,5b due to which no direct CDC methodologies were explored with the combination of THβC and indole. Preparation of 1-indolyl THβC relies on the Pictet–Spengler reaction or less selective chloroindolenine intermediates (Fig. 2).5c,d Hence, we aimed to develop a CDC reaction for the coupling of indole (2) and THβC (1). In the literature, only two reports were found in which CDC is employed for the coupling of tetrahydro-β-carboline with alkenyl/aryl boronates,6 and to the best of our knowledge, no reports were found on the direct CDC of the two reactive nucleophiles such as simple THβC (1) and indole (2) to afford mono indolyltetrahydro-β-carbolines selectively. Keeping the literature gap in mind, we started reaction optimization with different oxidizing reagents and solvents at various temperatures and times (Table 1). N-Chlorosuccinimide (NCS) (five minutes of stirring after the addition of NCS and then indole (2a) was added, followed by 10 minutes of stirring before quenching the reaction) resulted in only 10% of the cross-coupled product 6a (Table 1, entry 1). By decreasing the reaction time to 5 minutes (two minutes of stirring after adding NCS and then indole (2a) was added, followed by 3 minutes of stirring before quenching the reaction), we could get the aimed coupled product selectively without any over oxidation but the conversion was only about 50% (Table 1, entry 2). When tBuOCl, NaOCl and DIB were employed as oxidizing agents, no mono indolyl product 6a formation was observed (Table 1, entries 3–5). With DDQ in DCM (after adding DDQ 1.5 h of stirring and then indole (2a) addition, followed by 30 minutes of stirring before quenching the reaction), 40% of the coupled product was formed (Table 1, entry 6). During the course of optimization, we understood that the time required for the oxidization of THβC 1a is also crucial. We carried out many reactions by increasing the time for the initial oxidation of THβC 1a, and using DDQ, none of our attempts improved the yield of the reaction. Then we prepared a Bobbitt salt and employed it as an oxidizing reagent, and 25% and 50% of coupled product 6a were obtained at 0 °C and −20 °C within 150 seconds, respectively (Table 1, entries 7 and 8). Surprisingly, the yield of the coupled product 6a increased to 82% when the concentration of the reaction decreased to 0.06 M (Table 1, entry 9). When we employed triphenylcarbenium salts as oxidizing agents, fortunately, the yield of the reaction was further improved.
 | ||
| Fig. 2 Previous reports on the synthesis of 1-indolyl THβCs. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| S. noa | Oxidizing reagent | Solvent | T (°C) | Time (min) | Yieldf (%) | ||
| 6a | 7 | 8 | |||||
| where T = temperature.a Reaction conditions: 1a (0.18 mmol, 1 equiv.), 2a (0.2 mmol, 1.1 equiv.), oxidizing reagent (2.2 mmol, 1.2 equiv.) unless otherwise mentioned, solvent (3 mL). T = TEMPO.b Reaction concentration is 0.2 M.c 2 Minutes of stirring after adding the oxidizing reagent and 30 seconds of stirring after adding indole 2a.d Reaction concentration is 0.06 M.e 2.5 Minutes of stirring after adding the oxidizing reagent and 30 seconds of stirring after adding indole 2a, and the reaction concentration is 0.06 M.f Isolated yields. | |||||||
| 1b | NCS (1.0 equiv.) | 1,4-Dioxane | r.t. | 15.0 | 10 | 60 | 20 |
| 2b | NCS (1.0 equiv.) | 1,4-Dioxane | r.t. | 5.0 | 50 | — | — |
| 3b | t BuOCl (1.3 equiv.) | EtOAc–H2O | r.t. | 5.0 | — | 70 | 20 |
| 4b | NaOCl (2.0 equiv.) | DCM–H2O | r.t. | 120 | — | — | — |
| 5b | DIB (1.0 equiv.) | DCM | r.t. | 60 | — | — | — |
| 6b | DDQ (1.0 equiv.) | DCM | r.t. | 120 | 40 | — | — |
| 7bc | T+BF4− | DCM | 0 | 2.5 | 25 | 75 | — |
| 8bc | T+BF4− | DCM | −20 | 2.5 | 50 | 50 | — |
| 9cd | T+BF4− | DCM | −20 | 2.5 | 82 | — | — |
| 10e | Ph3C+PF6− | DCM | −20 | 3.0 | 85 | — | — |
| 11 | Ph 3 C + BF 4 − | DCM | −20 | 3.0 | 88 | — | — |
| 12e | Ph3C+BF4− | DME | −20 | 3.0 | 70 | ||
| 13e | Ph3C+BF4− | THF | −20 | 3.0 | 72 | — | — |
| 14e | Ph3C+BF4− | Toluene | −20 | 3.0 | 60 | — | — |
| 15e | Ph3C+BF4− | EtOAc | −20 | 3.0 | 58 | — | — |
| 16e | Ph3C+BF4− | 1,4-Dioxane | −20 | 3.0 | 24 | — | — |
| 17e | Ph3C+BF4− | CHCl3 | −20 | 3.0 | 45 | — | — |
| 18e | Ph3C+BF4− | DCE | −20 | 3.0 | 54 | — | — |
Hexafluorophosphate salt of the trityl cation afforded 85% of 6a, whereas the tetrafluoroborate salt of the same furnished 88% of 6a in 180 seconds (Table 1, entries 10 and 11). By screening various solvents, we understood that DCM is the best solvent to achieve the targeted compound 6a as a major compound (Table 1, entries 11–18).
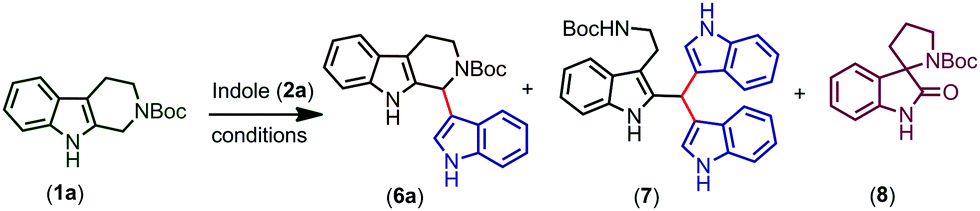
After completing the optimization of the reaction conditions for the synthesis of 6a, we started the investigation of the substrate scope of trityl salt mediated CDC of Boc protected THβC (1a) with different indoles (2a–q) possessing electron donating and electron withdrawing substitutions (Scheme 1). Indoles with EDGs reacted extremely faster and afforded good to excellent yields of the products (6b–f), and the structure of 6c was confirmed using X-ray crystallography (CCDC† number: 1973061). Indoles with EDGs at the second position reacted very fast and also resulted in an excellent yield of the products (6e, 6f). Due to the combined effect of electron donating and electron withdrawing nature of halogens, moderate yields of the product were obtained when the halogens were present on indole (6g–j). As expected, indoles with strong EWGs further resulted in a decreased conversion and therefore gave only a moderate yield of the coupled products (6k–n).
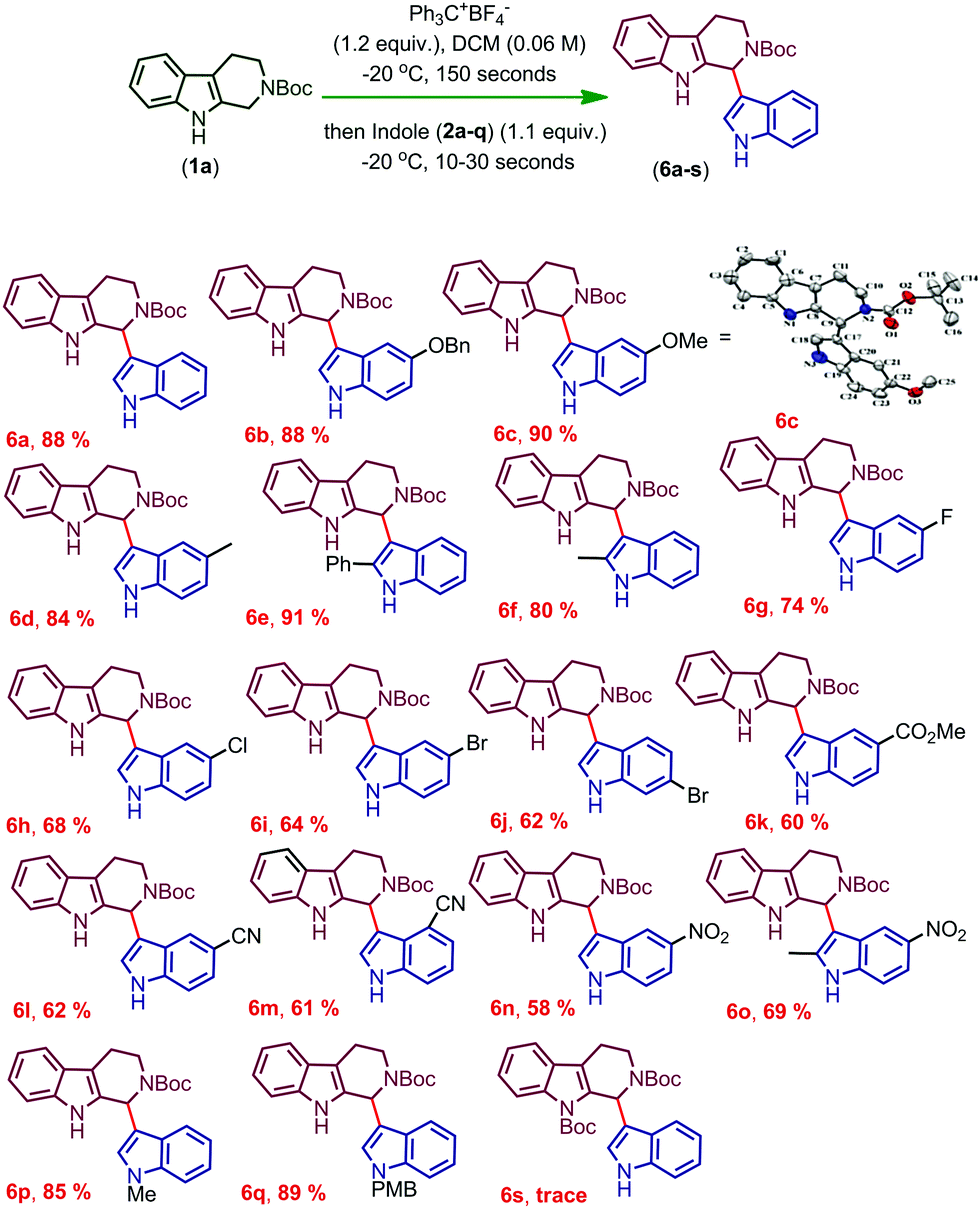 | ||
| Scheme 1 Substrate scope (synthesis of derivatives of 6a).a,b:aReaction conditions: tetrahydro-β-carboline 1a–c (0.185 mmol, 1.0 equiv.), Trityl salt (0.222 mmol, 1.2 equiv.), indoles 2a–q (0.204 mmol, 1.1 equiv.), and DCM (3.0 mL, 0.06 M), 30 seconds. For substrates 2e and 2f, the reaction time is 10 seconds after the addition of indole. bIsolated yields. | ||
Increasing the time of the reaction (to improve the conversion) resulted in a further decrease in the yield of the product in the case of indoles having EWGs (6k–n). Gratifyingly, when an EDG was introduced at the second position of the indole possessing an EWG already on its ring, the conversion and yield of the reaction were further improved (6o). Moreover, the protection of indole N–H with electron releasing groups also resulted in a good yield of the coupled products (6p, q). In the case of N-benzyl protected THβC (1b), no coupled product was observed and when both the N–H groups of tetrahydro-β-carboline were protected with Boc groups (1c), trace amounts of coupled product 6s were obtained (confirmed by TLC). As the nucleophilicity is not sufficient enough to undergo the CDC reaction, no coupled product was observed when 7-azaindole, 3-methylindole, ethyl indole-2-carboxylate, and 5-benzyloxy-ethyl indole-2-carboxylate were used and only oxidative cleavage of tetrahydro-β-carboline (1a) was observed in such cases to afford compound 9 (Scheme 2). Also, a gram scale synthesis of 6a was performed, which resulted in 82% yield (see the ESI†).
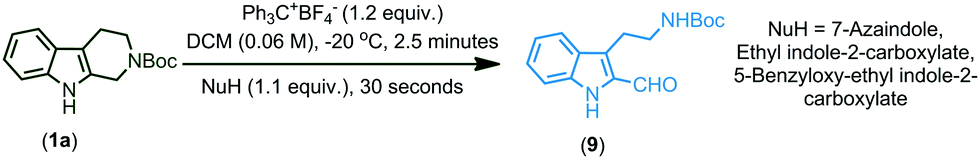 | ||
| Scheme 2 Oxidative cleavage of tetrahydro-β-carboline.a,b aReaction conditions: tetrahydro-β-carboline 1a (0.185 mmol, 1.0 equiv.), Trityl salt (0.222 mmol, 1.2 equiv.), NuH (0.204 mmol, 1.1 equiv.), and DCM (3.0 mL, 0.06 M), bIsolated yields. | ||
In addition, rotamers were found in most of the cases and can be differentiated only in the aliphatic region of the THβC and Boc group of the coupled products. To prove the rotamer formation, we have conducted high temperature NMR by taking 6c as a model substrate, and delightedly, at a higher temperature, multiple peaks that formed at room temperature were merged. Besides, indoles with bulky groups at the 2- and 4-positions did not show rotamer formation (6e, 6f, 6m and 6o). After deprotecting the Boc group of 6a, the rotamers disappeared (10, see the ESI†), which confirms that the rotamers are formed due to the presence of the Boc group.
The proposed mechanism for the trityl salt mediated CDC of tetrahydro-β-carboline 1a with indole 2a and oxidative ring opening by water is described in Fig. 2. The trityl cation oxidizes the compound 1a and affords the intermediate (i) which on nucleophilic attack of indole furnishes the product 6a. When the nucleophilicity of indole is not sufficient enough to couple, attack of water followed by in situ acidic ring opening results in oxidative cleavage of 6a to afford compound 9 (Fig. 3).
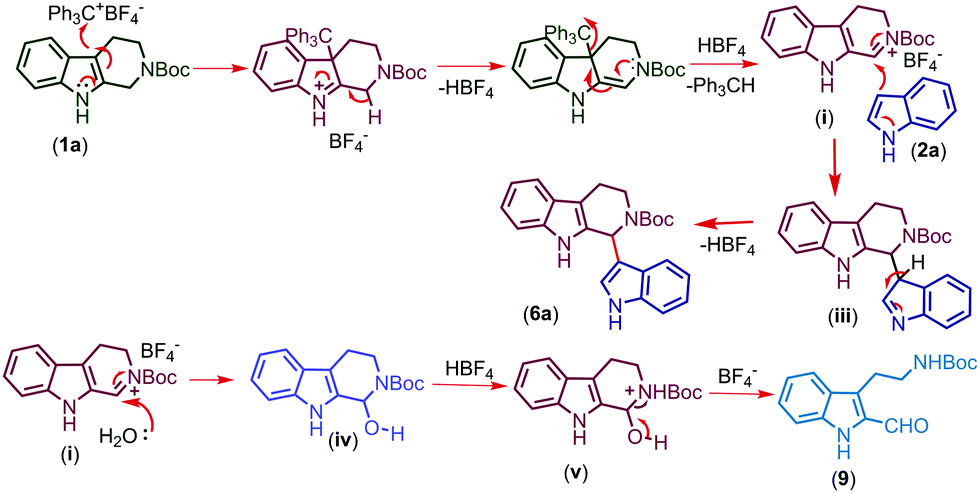 | ||
| Fig. 3 Proposed mechanism of reaction. | ||
After analysing the substrate scope of the reaction, we attempted the total synthesis of alkaloid eudistomin U (3) (Scheme 3). Eudistomin U (3) is a 1-indolyl-β-carboline alkaloid (Fig. 1) and exhibits a wide spectrum of biological properties.7 Eudistomin U (3) and isoeudistomin U (4) were isolated from the marine ascidian Lissoclinum fragile by Francisco et al. in the year 1994.7a 19-Bromoisoeudistomin U (5) was isolated from the marine ascidian Eudistoma along with isoeudistomin U (4) in the year 1996.7b Structural revision of isoeudistomin U (4) was reported by its synthesis in the year 1995,7c whereas the synthesis of alkaloid 5 is not known to date. In 1995, Molina and co-workers successfully completed the first total synthesis of eudistomin U (3).8a Later, a couple of reports were found on the synthesis of 3.5d,8b–m We would like to apply the trityl salt mediated CDC reaction followed by an oxidation/aromatization strategy for the total synthesis of 3, 4 and 5. Compound 6a on Boc deprotection using HCl in 1,4-dioxane afforded compound 10, which on further aromatization using 10% Pd/C in xylene at 140 °C for 6 hours furnished the target alkaloid eudistomin U (3) in 66% yield (over 2 steps). Compound 10 on partial aromatization with IBX afforded the isoeudistomin U in 56% yield (over 2 steps) and compound 6j, on deprotection followed by partial oxidation with IBX, furnished 19-bromoisoeudistomin U in 60% yield (over 2 steps) (Scheme 3, see the ESI,† Table S1).
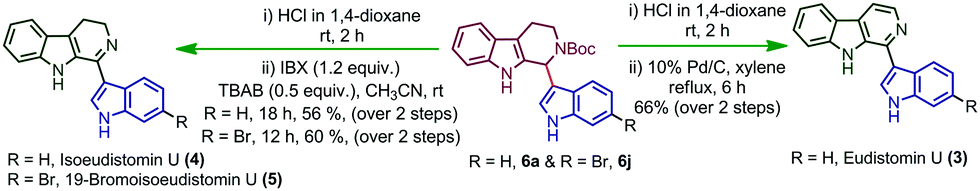 | ||
| Scheme 3 Total synthesis of eudistomin U (3), isoeudistomin U and 19-bromoisoeudistomin U (5). | ||
To conclude, a highly selective and rapid metal-free cross-dehydrogenative coupling strategy for the coupling of highly reactive nucleophiles such as tetrahydro-β-carboline and indoles was demonstrated successfully. Insights into the factors governing the reactivity and reaction's selectivity opened the door to solving the puzzles in the chemistry of highly reactive nucleophiles under CDC conditions. By tuning the solvent concentration, temperature, time and addition mode, we could achieve biologically important mono indolyltetrahydro-β-carboline derivatives 6a–s selectively in excellent to moderate yields. In addition, the total synthesis of alkaloids, eudistomin U (3), and isoeudistomin U (4) and the first total synthesis of 19-bromoisoeudistomin U (5) were successfully completed with overall yields of 58%, 56%, 37%, respectively, in just 3 steps.
We acknowledge SERB, India, for financial support (EEQ/2017/000422) and GR thanks DST, India, for the fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Brahmachari, RSC Adv., 2016, 6, 64676–64725 RSC.
- (a) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS . 111, 1417–1492; (c) Y. Yang, J. Lan and J. You, Chem. Rev., 2014, 114, 9219–9280 CrossRef; (d) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC; (e) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (f) T. L. Mako and J. A. Byers, Inorg. Chem. Front., 2016, 3, 766–790 RSC; (g) K. Zhao, L. Shen, Z.-L. Shen and T.-P. Loh, Chem. Soc. Rev., 2017, 46, 586–602 RSC; (h) P. Y. Choy, S. M. Wong, A. Kapdi and F. Y. Kwong, Org. Chem. Front., 2018, 5, 288–321 RSC; (i) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069–11083 RSC.
- (a) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS; (b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS; (c) C.-Y. Huang, H. Kang, J. Li and C.-J. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS; (d) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082–2086 CrossRef CAS; (e) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS; (f) M. K. Lakshman and P. K. Vuram, Chem. Sci., 2017, 8, 5845–5888 RSC; (g) R. Narayan, K. Matcha and A. P. Antonchick, Chem. – Eur. J., 2015, 21, 14678–14693 CrossRef CAS; (h) P. Caramenti, R. K. Nandi and J. Waser, Chem. – Eur. J., 2018, 24, 10049–10053 CrossRef CAS; (i) Y. Kita, K. Morimoto, M. Ito, C. Ogawa, A. Goto and T. Dohi, J. Am. Chem. Soc., 2009, 131, 1668–1669 CrossRef CAS; (j) J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 10, 4754–4764 CrossRef; (k) R. Samanta, R. Narayan, J. O. Bauer, C. Strohmann, S. Sievers and A. P. Antonchick, Chem. Commun., 2015, 51, 925–928 RSC; (l) J. Dhineshkumar, M. Lamani, K. Alagiri and K. R. Prabhu, Org. Lett., 2013, 15, 1092–1095 CrossRef CAS; (m) L. Lv, D. Zhu and C.-J. Li, Nat. Commun., 2019, 10, 715–722 CrossRef.
- (a) B. F. Bowden, Stud. Nat. Prod. Chem., 2000, 23, 233–283 CAS; (b) F. Liu, L.-Q. Yu, L. Y. Jiang, W.-T. Wud and Q.-D. You, Bioorg. Med. Chem., 2010, 18, 4167–4177 CrossRef CAS; (c) A. A. Kolodina and O. V. Serdyuk, Heterocycles, 2018, 96, 1117–1196 Search PubMed; (d) M. Menna, E. Fattorusso and C. Imperatore, Molecules, 2011, 16, 8694–8732 CrossRef CAS.
- (a) M. Cao, Y. Mao, J. Huang, Y. Ma and L. Liu, Tetrahedron Lett., 2019, 60, 1075–1078 CrossRef CAS; (b) H. Chen, F. Ye, J. Lu and Y. Gao, Org. Lett., 2019, 21, 7475–7477 CrossRef CAS; (c) L. P. P. Liewa, J. M. Fleming, A. Longeon, E. Mouray, I. Florent, M.-L. Bourguet-Kondracki and B. R. Copp, Tetrahedron, 2014, 70, 4910–4920 CrossRef; (d) D. Xu, F. Ye, J. Ye, Y. Gao and H. Chen, Org. Lett., 2019, 21, 6160–6163 CrossRef CAS.
- (a) X. Liu, Z. Meng, C. Li, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 6012–6015 CrossRef CAS; (b) Y. Sun, G. Wang, J. Chen, C. Liu, M. Cai, R. Zhu, H. Huang, W. Li and L. Liu, Org. Biomol. Chem., 2016, 14, 9431–9438 RSC.
- (a) S. A. Adesanya, M. Chobani and M. Pais, J. Nat. Prod., 1992, 55, 525–527 CrossRef CAS; (b) H. Kang and W. Fenical, Nat. Prod. Lett., 1996, 9, 7–12 CrossRef CAS; (c) G. Massiot, S. Nazabadioko and C. Bliard, J. Nat. Prod., 1995, 58, 1636–1639 CrossRef CAS; (d) A. Badre, A. Boulanger, E. Abou-Mansour, B. Banaigs, G. Combaut and C. Francisco, J. Nat. Prod., 1992, 57, 528–533 CrossRef; (e) J. M. Giulietti, P. M. Tate, A. Cai, B. Cho and S. P. Mulcahy, Bioorg. Med. Chem. Lett., 2016, 26, 4705–4708 CrossRef CAS; (f) J. Dai, W. Dan, Y. Zhang and J. Wang, Eur. J. Med. Chem., 2018, 157, 447–467 CrossRef CAS; (g) Y. Xua, C. Afonsoa, Y. Gimbert, F. Fournier, X. Dong, R. RenWen and J.-C. Tabet, Int. J. Mass Spectrom., 2009, 286, 43–52 CrossRef; (h) J. Zheng, Z. Zhang, L. Zhao, X. Sha, X. Dong and R. Wen, Pharm. Biol., 2008, 46, 273–278 CrossRef CAS.
- (a) P. Molina, P. M. Fresneda and S. Garcia-Zafra, Tetrahedron Lett., 1995, 36, 3581–3582 CrossRef CAS; (b) P. Rocca, F. Marsais, A. Godard and G. Quéguiner, Tetrahedron Lett., 1995, 39, 7085–7088 CrossRef; (c) F. Nissen, V. Richard, C. Alayraca and B. Witulski, Chem. Commun., 2011, 47, 6656–6658 RSC; (d) A. D. Yamaguchi, D. Mandal, J. Yamaguchi and K. Itami, Chem. Lett., 2011, 40, 555–557 CrossRef CAS; (e) C. M. Roggero, J. M. Giulietti and S. P. Mulcahy, Bioorg. Med. Chem. Lett., 2014, 24, 3549–3551 CrossRef CAS; (f) D. S. Pakhare and R. S. Kusurkar, Tetrahedron Lett., 2015, 56, 6012–6015 CrossRef CAS; (g) J. D. Panarese and S. P. Waters, Org. Lett., 2010, 12, 4086–4089 CrossRef CAS; (h) K. L. Manasa, Y. Tangella, G. Ramu and B. N. Babu, ChemistrySelect, 2017, 2, 9162–9167 CrossRef CAS; (i) A. Kamal, M. Sathish, A. V. G. Prasanthi, J. Chetna, Y. Tahgella, S. Vunnam, N. Srinivasulu and A. Alarifi, RSC Adv., 2015, 5, 90121–90126 RSC; (j) A. Kamal, M. Sathish, J. Chetna, Y. Tahgella, S. Vunnam, N. Srinivasulu and A. Alarifi, Org. Biomol. Chem., 2015, 13, 8652–8662 RSC; (k) Z. Zhao, Y. Sun, L. Wang, X. Chen, Y. Sun, L. Lin, Y. Tang, F. Li and D. Chen, Tetrahedron Lett., 2019, 60, 800–804 CrossRef CAS; (l) S. Gaikwad, D. Kamble and P. Lokhande, Tetrahedron Lett., 2018, 59, 2387–2392 CrossRef CAS; (m) S. Santhanam, A. Ramu, B. Baburaj and B. K. Kuppusamy, J. Heterocycl. Chem., 2020, 57, 2121–2127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1973061. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06958b |
| This journal is © The Royal Society of Chemistry 2021 |