
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments and applications of quantitative proteomics strategies for high-throughput biomolecular analyses in cancer research
Hannah N.
Miles†
 a,
Daniel G.
Delafield†
b and
Lingjun
Li
*ab
a,
Daniel G.
Delafield†
b and
Lingjun
Li
*ab
aSchool of Pharmacy, University of Wisconsin-Madison, 777 Highland Avenue, Madison, WI 53705-2222, USA. E-mail: lingjun.li@wisc.edu; Fax: +1-608-262-5345; Tel: +1-608-265-8491
bDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA
First published on 19th May 2021
Abstract
Innovations in medical technology and dedicated focus from the scientific community have inspired numerous treatment strategies for benign and invasive cancers. While these improvements often lend themselves to more positive prognoses and greater patient longevity, means for early detection and severity stratification have failed to keep pace. Detection and validation of cancer-specific biomarkers hinges on the ability to identify subtype-specific phenotypic and proteomic alterations and the systematic screening of diverse patient groups. For this reason, clinical and scientific research settings rely on high throughput and high sensitivity mass spectrometry methods to discover and quantify unique molecular perturbations in cancer patients. Discussed within is an overview of quantitative proteomics strategies and a summary of recent applications that enable revealing potential biomarkers and treatment targets in prostate, ovarian, breast, and pancreatic cancer in a high throughput manner.
 Hannah N. Miles | Hannah N. Miles completed her Bachelor's degree at Carthage College in Kenosha, Wisconsin. She did a year of research within the lab of Dr John L. Nitiss located at the University of Illinois Chicago College of Pharmacy in Rockford. There, her work focused on characterizing novel human topoisomerase II mutants using yeast as a model organism. She became a graduate student at the University of Wisconsin-Madison in 2019 and is currently a member of Dr Lingjun Li's research group within the School of Pharmacy, focusing on a multi-omics approach to characterizing various cellular models of prostate cancer progression in addition to elucidating the role of the post-translational modification glycosylation in various cancer types. |
 Daniel G. Delafield | Daniel G. Delafield received his Bachelor of Science degree from the University of Oklahoma where he began his proteomics research under the guidance of Dr Si Wu. Working in a top-down proteomics research group, he spearheaded glycoform characterization efforts on the way to earning his Master of Science. This training eventually led him to join the Research Group of Professor Lingjun Li in Department of Chemistry and School of Pharmacy at the University of Wisconsin-Madison, where he continues to pursue novel approaches for glycoconjugate separation, identification, and structural characterization. |
 Lingjun Li | Dr Lingjun Li is a Vilas Distinguished Achievement Professor and the Charles Melbourne Johnson Distinguished Chair Professor of Pharmaceutical Sciences and Chemistry at the University of Wisconsin-Madison (UW-Madison). Dr Li received her PhD degree in Analytical Chemistry/Biomolecular Chemistry from the University of Illinois at Urbana-Champaign in 2000. She then did joint postdoctoral research at the Pacific Northwest National Laboratory and Brandeis University before joining the faculty at UW-Madison in December 2002. Dr Li's research interests include the development of novel mass spectrometry (MS)-based tools such as new isotopic and isobaric labeling strategies that enable hyperplexing for quantitative proteomics, peptidomics, and glycomics, and their applications in neuroscience and cancer research. She and her team also develop microscale separations, in vivo microdialysis and imaging MS tools for functional discovery of neuropeptides in model organisms and (glyco)protein biomarkers in neurodegenerative diseases with a strong focus on Alzheimer's disease. Her lab also explores novel use of ion mobility MS to address technical challenges in peptidomic research. Professor Li has established a highly productive research program and published more than 300 peer-reviewed research journal papers and has given more than 200 invited talks. She has successfully trained and graduated 50 PhDs and is currently training 26 PhD graduate students, 4 postdoctoral scientists, and 7 undergraduate students. She has been recognized with numerous awards, including ASMS Research Award, NSF CAREER Award, Sloan Fellowship, PittCon Achievement Award, and ASMS Biemann Medal, and was named one of the Top 50 most influential women in the analytical sciences in 2016 and was recently featured in the 2019 Top 100 Power List by the Analytical Scientist. Dr Li is currently serving as an Associate Editor for the Journal of the American Society for Mass Spectrometry (JASMS) and sitting on the Advisory Board for Analytical and Bioanalytical Chemistry. She is a member of the Board of Directors for the US Human Proteome Organization (US HUPO) and Chair of the Board of Directors for the Chinese American Society for Mass Spectrometry (CASMS). |
Introduction
Mass spectrometry (MS) represents a unique and powerful technological platform in investigative biomolecular research. This high sensitivity regime grants access to the discovery and identification of small molecules,1–3 endogenous peptides,4–7 proteins,8–10 and macromolecular complexes.11–13 The utility of MS is enhanced through the facility of ionizing biomolecule species in solution via electrospray ionization14 (ESI) and matrix-assisted laser desorption/ionization15–17 (MALDI) that provides a means of producing ions from stationary supports and tissue sections. As well, numerous mass analyzers18 have been developed to accommodate high-speed and high-resolution measurements. Realizing the full potential and flexibility of modern MS platforms, as well as their ability to decipher complex biological samples, focus has shifted towards improving instrument efficiency and sample throughput.Gradual improvements in instrument operational speed, the advent of novel dissociation techniques19–22 and implementation of multidimensional ion separation regimes23–25 enable researchers to obtain greater levels of detail from complex mixtures than ever before. However, while shotgun proteomics provides a means for deep proteomic profiling, the typical time course and complexity of a single experiment26 renders repetitive measurements of numerous samples untenable. For this reason, many have turned to multiplexed quantitative proteomics workflows to provide simultaneous deep proteomic profiling of numerous samples while retaining the ability to assign relative and absolute abundance information.
Quantitative proteomics, now comprised of several distinct strategies, operates under the principle that signal response from any given analyte is related to its abundance within the mixture. As such, should an analyte be identified in numerous samples, the relative intensity of the analyte's signal response in each sample can be used to provide a means of relative or absolute quantitation. However, MS reporting signal is divided into numerous channels depending on the quantity and ionization efficiency of all present biomolecules, which indicates the high variability that can arise from even discrete sample changes. In remedy of this ailment—and to remove run-to-run variation—researchers have employed unique chemical modifiers that often incorporate stable isotopes to label biomolecules within solution. These labels result in a unique mass shift for each sample without altering their retention time in liquid-chromatography. These newly tagged analytes may then be combined, measured simultaneously via MS, and then evaluated for the relative abundance of all labeled channels.
These quantitative strategies have provided unique avenues towards the discovery and validation of cancer-specific biomarkers. The ability to analyze numerous samples simultaneously provides researchers not only with a means for high throughput sample profiling, but also a means to uncover what proteomic perturbations are relevant across patients, between control groups, and specific to disease severity and progression. These perturbations and quantitative differences are often discussed in language that is familiar to proteomic researchers but that may create confusion in those coming from adjacent fields of research. Within proteomics, and ubiquitous throughout this review, quantitative differences of proteins, peptides and other biomolecules are described as “up-regulated” or “down-regulated.” These terms are used to describe those species with quantifiable differences against the control, often with statistical significance. Though readers may conjecture that up- or down-regulated protein species are the result of pathway regulation, these hypotheses are often not explored in proteomic literature and may be discussed elsewhere. For this reason, it is important to clarify that differences in regulation are meant only to indicate the quantitative findings presented by the original authors. Regardless of verbiage technicalities, researchers often pursue quantitative proteomics as a facile avenue towards novel biological insight.
Given the significant heterogeneity associated with various cancer subtypes, researchers have sought to employ quantitative proteomics to a litany of biological questions. As seen within, these endeavors have provided seminal insights into the role post-translational modifications play in cancer progression, uncovering up- and down-regulation of biomolecules in disease groups, as well as the efficacy of using protein expression to monitor medical treatment. The true breadth of proteomic cancer research cannot be understated. While quantitative experiments date back several decades, we aim to present a mass spectrometry-centric review. High-throughput quantitative proteomics firmly gained prominence in the early 2000s, providing nearly twenty years of meaningful contributions to cancer detection, identification, and understanding. To provide readers with the most timely and topical review—as well as to provide discussion on future research interests, we have confined our literature review to applications published within the past 5 years. This concise range enables us to provide critical suggestions for researchers seeking to begin or continue their unique cancer research. Here we present a brief introduction to quantitative proteomics methods and recent investigations of prostate, ovarian, breast, and pancreatic cancer.
Quantitative strategies
Quantitative proteomics has experienced substantial growth over the last two decades, due in large part to the invention and development of high-speed, high-resolution mass spectrometry instrumentation. While there are numerous unique and technically driven means to pursue relative and absolute protein quantitation, most applications fall within one of four major categories: metabolic labeling, isotopic labeling, isobaric labeling, and label-free quantitation. Each method has been thoroughly reviewed and in-depth discussion can be found elsewhere. However, in order to provide rationale behind each strategy for use in investigative cancer research, understanding the principles and key considerations of each is imperative.Metabolic labeling
Metabolic labeling is the earliest27,28 and arguably most traditional method of mass spectrometry-based quantitative proteomics. Taking after the classical Meselson-Stahl29 experiment that proved the semiconservative nature of DNA replication, more routine use of mass spectrometry for peptide identification revealed that proteins, too, could be metabolically labeled with stable isotopes to provide ‘heavy’ and ‘light’ isotopologues. Within these experiments, adjacent cell cultures are provided with either unlabeled, naturally occurring amino acids or amino acids that have been labeled with stable isotopes; this also lends itself to the acronym SILAC, Stable Isotopic Labeling with Amino Acids in Cell Culture.27,30 Though SILAC has grown to incorporate numerous stable isotopes, the most traditional SILAC strategy is to grow a control group in the presence of 12C-lysine and 13N-arginine while providing the experimental group with 13C-lysine and 15N-arginine.31 During culture growth, these light or heavy amino acids are incorporated into the protein backbones with no effect on protein function, viability or expression. Digesting these cellular proteins with a proteolytic enzyme (e.g. trypsin) produces peptides that contain a single labeled or unlabeled residue. Peptides are then combined and analyzed via MS, at which point their mass difference can be observed. Evaluating the intensities of the labeled and unlabeled peptide partners allows the relative abundance of peptides and proteins to be determined. Metabolic labeling strategies are of topical interest to groups seeking to reveal how altered growth conditions, drug administration, or environmental perturbations affect protein production and expression. Beyond relative quantitation of proteins and peptides, SILAC-like experiments have been used to probe post-translational modification production and turnover. However, the chief considerations and drawbacks of these methods are (1) the small number of suitable amino acids that may be used for isotope incorporation; (2) poor separation of isotopic envelopes (causing errors in quantitative accuracy); and (3) the inability to incorporate isotopes to biological tissue and biofluid samples. In remedy, researchers may choose to tag proteins and peptides with isotopic labels after extraction and digestion.Isotopic tagging
Isotopic tagging, though similar in nature to metabolic incorporation, comes with a higher level of flexibility and customization.31 Modern research settings have access to a broad array of stable isotopes, the most ubiquitous being 13C, 15N, 2H, and 18O. These isotopes enable researchers to synthesize their own chemical scaffold while varying the incorporation of these isotopes, creating an array of chemical tags with unique masses that may be functionalized and chemically bound to proteolytic peptides to provide them with a mass difference distinguishable via MS.32,33 In this way, the need for metabolic incorporation is completely removed and experimental peptides can be labeled after extraction and digestion. Similar to metabolic labeling, differences in MS1-level signal intensity between labeled species allow for determination of relative quantitation. Furthermore, isotopic labeling can be used as a means of absolute quantitation, whereby internal calibration curves are created and compared to experimental peptides. Overall, isotopic labeling presents highest utility in instances where the sample collection is relatively small because as sample number increases so does spectral complexity, which can create mass overlap between unique peptide species and produce erroneous quantitation estimates. These limitations in mind, the vast improvements in MS operational speeds, resolving power, and scanning depth begged the question as to whether more efficient chemical labels could leverage these instrumental improvements and eliminate the spectral complexity found in complex isotopic tagging experiments.Isobaric labeling
As mass spectrometry technology continued to develop, it became obvious that the spectral complexity associated with high-throughput metabolic labeling and isotopic tagging experiments directly counteracted any instrumental improvements. As such, it became pertinent to find a method for absolute and relative quantitation that alleviate the ailments posed by multiplexed labeling methods while still retaining the facility in quantitative measurements. Remembering that isotopic tags may be constructed to provide a high number of labeling channels, each with a distinct mass difference of >1 Da, isobaric labels correct for this inherent mass difference using a balancing group.34 When implemented, these isobaric labels display virtually indistinguishable masses at the MS1 level, reducing the spectral complexity of high-throughput experiments. Upon selection of a labeled analyte, MS dissociation causes the isobaric tags to fragment and produce ‘reporter ions’ of unique mass. In this way labeled analytes may be selected and fragmented, providing identification and quantitative information in a single step. As a result, the reduced spectral complexity at the MS1 level promotes greater profiling depth of complex samples and provides equivalent quantitative accuracy. The most popular examples of commercial isobaric labels are iTRAQ, Isotopic Tags for Relative and Absolute Quantitation35 and TMT, Tandem Mass Tags.36 However, the broad utility of isobaric labeling has garnered significant attention from the research community, resulting in numerous novel quantitative labeling strategies37,38 that promote quantitative accuracy at significantly reduced cost.Label-free and reaction monitoring
Finally, in instances where sample labeling may not be preferred (i.e., precious samples, low-abundance molecules of interest, or instances where protein targets are known), label-free and reaction monitoring methods provide a suitable alternative.39 Label-free quantitation serves to provide relative quantitation between samples by comparing area-under-curve for detected analytes. This method, though steadily improving with better instrumentation and software tools, is highly susceptible to changes in sample composition, can result in missing values, and is lower throughput than labeled methods. However, label-free quantitation does still represent a meaningful entry point in discovery-based quantitative proteomics, often providing deep sample profiling and elucidating targets for future analyses. In contrast, reaction monitoring workflows (e.g. multiple reaction monitoring, select reaction monitoring, etc.) may be considered one of the most accurate quantitative strategies, being most suited to targeted analyses and instances when internal standards are readily available. Though reaction monitoring strategies are often tailored to fit unique experimental conditions, all workflows bear resemblance to a basic strategy. First, serial dilutions of a purified or synthetic peptide standard are analyzed via targeted MS/MS. In these targeted analyses, the biomolecule(s) of interest are subjected to MS dissociation, with the various fragments observed and recorded. As each biomolecule will provide a unique transition/fragment, the prevalence of these transitions may be used as a proxy for overall biomolecule abundance. In this way, absolute and relative quantitation information can be determined without the need for chemical labeling while eliminating concerns over sample and spectral complexity. Often, it is preferential to incorporate an isotope-encoded standard,40,41 enabling rapid analyses and high quantitative accuracy. Given the variety of quantitative strategies, it is of topical importance to evaluate their efficacy and provide understanding of quantitative accuracy.Diagnostic accuracy
As quantitative proteomics continues to mature, discussions over quantitative accuracy will continue to be a vanguard consideration. Recently, Dowle et al.42 provided an in-depth comparison of multiple quantitative strategies and should evaluated independently by interested parties. Within all quantitative strategies, the primary diagnostic for accuracy and utility are metrics built on specificity and sensitivity. Measures of specificity (i.e. proportion of correctly-identified true positives) and sensitivity (i.e. proportion of correctly-identified true negatives), may be combined into a single metric. This receiver operating characteristic (ROC) is often viewed as a curve with high sensitivity and specificity representing a value close to 1. As demonstrated by Dowel, several commonly used quantitative strategies display high ROC values, providing detailed considerations of the method most appropriate for a range of experiments. This work may serve as a helpful guide when entering or expanding quantitative proteomics experiments.Taken together, metabolic labeling, isotopic tagging, isobaric labeling, and label-free strategies provide a wealth of entry points into quantitative proteomics. This access in mind, the growing needs of the medical community combined with the ever-increasing access to mass spectrometry technology necessitate the utilization and expansion of investigational proteomics to aid in discovering and validating cancer-specific biomarkers.
Prostate cancer
The second leading cancer type in men, prostate cancer is estimated to affect around 12 percent of all men during their lifetime and currently affects over 3 million men within the United States, with the majority of individuals diagnosed being at least 65 and older.43 Androgen deprivation, the first means of therapeutic intervention, can lead to the progression of castration-resistant prostate cancer (CRPC) in some men, a more aggressive stage of cancer resulting in poor prognosis and survival, with the majority of men developing metastases prior to or following diagnosis.44 Further analyses of the literature have characterized these CRPC subtypes and demonstrated the growing emergence of CRPC phenotypes that have either low or negative AR expression for which there are few targeted therapeutics.45 The growing heterogeneity in prostate cancer subtype underscores the urgency to elucidate and discover novel molecular mechanisms underlying pathogenesis for all subtypes. The use of mass spectrometry (MS)-based quantitative proteomics for prostate cancer research in recent years has been a driving force to exploit the factors underlying tumorigenesis and metastasis.Cellular and tissue analyses
Investigations often profile quantitative differences in the proteome via patient-derived tissue samples, cellular models, or genetically engineered mouse models such as the transgenic adenocarcinoma of the mouse prostate (TRAMP) model. One such study by Zhang et al.46 utilized a label-free approach to quantify differences in expression between the prostate glands of TRAMP versus wild-type littermates. Through generation and an in-depth analysis of the quantitative proteomics data, the authors were able to predict and validate the role of platelet-derived growth factor (PDGF)-B overexpression in increased proliferation, thereby highlighting the therapeutic potential of targeting PDGF signaling within prostate cancer. Other label-free approaches have utilized patient-derived tissue samples to profile global differences, including the work of Müller et al.47 using formalin fixed, paraffin embedded tissue from radical prostatectomy, which focused on characterizing differences between nonmetastasizing tumors, metastasizing primary tumors and their distant nodal metastases. Although the analysis had only five biological replicates per tumor type, significant differences in expression were measured that allowed for clear distinction of each and presented several potential proteins whose increased expression in metastatic tumors could be targeted in future therapeutic studies. However, a smaller sample set warrants further investigation into these proteins as potential targets with a larger sample cohort.Methods that incorporate stable isotopes into the peptide backbone, such as SILAC, allow for direct comparison of identical peptides across sample types and is more robust to instrumentational variation compared to unlabeled approaches. Recently, SILAC was used to examine extracellular vesicles (EVs) and the impact that upregulated α(1,6)-fucosyltransferase (FUT8) expression had on biogenesis of these secreted biomolecules.48 This was one of the first reports to map the systematic impact of an overexpressed glycosyltransferase on the EV proteome, specifically of a glycosyltransferase with known oncogenic activity.49,50 FUT8 overexpression showed a decrease in EVs produced compared to wild-type cells and further analysis of intact glycopeptides from LAPC4 EVs showed marked differences in glycosite occupancy between EV populations and revealed a shift in glycoform composition. Miao et al.51 combined the SILAC approach with parallel-reaction-monitoring (PRM) methods to discern differential kinase expression in two bone metastasis-derived prostate lines, PC3 and PC3MLN4.51 Of the kinases that were quantified and found to be differentially expressed, most notably different was mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4), a kinase previously observed to play a role in ovarian cancer.52 One final example using the SILAC strategy by Sbrissa et al.53 investigated the mechanisms of bone metastasis by determining CXCR4-interacting proteins through overexpression and knockdown of CXCR4 in PC3 cell lines. Proteomic analysis found one unexpected protein, phosphatidylinositol 4-kinase III α (PI4KIIIα), to be upregulated and it was found to localize with CXCR4 to lipid rafts and thus promote cancer cell invasion through increasing phosphatidylinositol-4-phosphate production. The discovery of this novel interaction between chemokine receptor and PI4KIIIα and its regulation on tumor cell invasion requires more detailed experiments characterizing the specific molecular details regarding receptor-kinase communication.
Chemical or enzymatic isotopic labeling strategies allow for labeling of more than cell culture models to study prostate cancer. One approach by Lee et al.54 used biotin—both as an isotopic label and for affinity purification—to systematically label cell-surface proteins that could serve to distinguish adenocarcinoma from neuroendocrine prostate cancer. From this proteogenomic investigation, they systematically validated two candidate antigens: FXYD domain containing ion transport regulator 3 (FXYD3) in prostate adenocarcinoma and CEA cell adhesion molecule 5 (CEACAM5) in neuroendocrine prostate cancer. While additional investigation into targeting these antigens is warranted, such a study demonstrates the utility of quantitative proteomics in discovering and validating new therapeutic targets for advanced prostate cancer.
Much of the quantitative research has shifted to the use of isobaric labeling strategies, which allow for increased multiplexing capabilities and decreased instrument variation through reduced overall runs. Zhou et al.55 used 5-plex TMT labeling to perform a large-scale proteomic quantitation of core fucosylated glycopeptides after selective lectin affinity enrichment to differentiate non-aggressive and aggressive prostate cancer cell models (Fig. 1). Over 20 fucosylated proteins were upregulated in the aggressive cell lines and were involved in processes such as cellular signaling, adhesion and extracellular communication. Identification of these fucosylated proteins and their upregulation in aggressive prostate cancer models establishes these proteins as potential targets for further examination into how their upregulation impacts the aggressive phenotype of the associated model. Another advantage to using TMT labeling is that these tags can undergo synchronous precursor selection (SPS)-MS3 quantitation, which allow for more accurate quantitation. Zhou et al.56 utilized a TMT-SPS-MS3 approach on patient-derived tissue samples with varying prostatic phenotypes to determine differential expression of protein complexes. Low-grade prostate cancer samples were found to have upregulation of complexes involved in RNA splicing and downregulation of those associated with cell adhesion, while high-grade prostate tissue samples had increased assembly of antiapoptotic complexes and a similar lower abundance of complexes involving cell adhesion. Such a comprehensive study of individual protein complexes may give way to determining what protein complexes are critical in distinguishing and diagnosing low- and high-grade cancers.
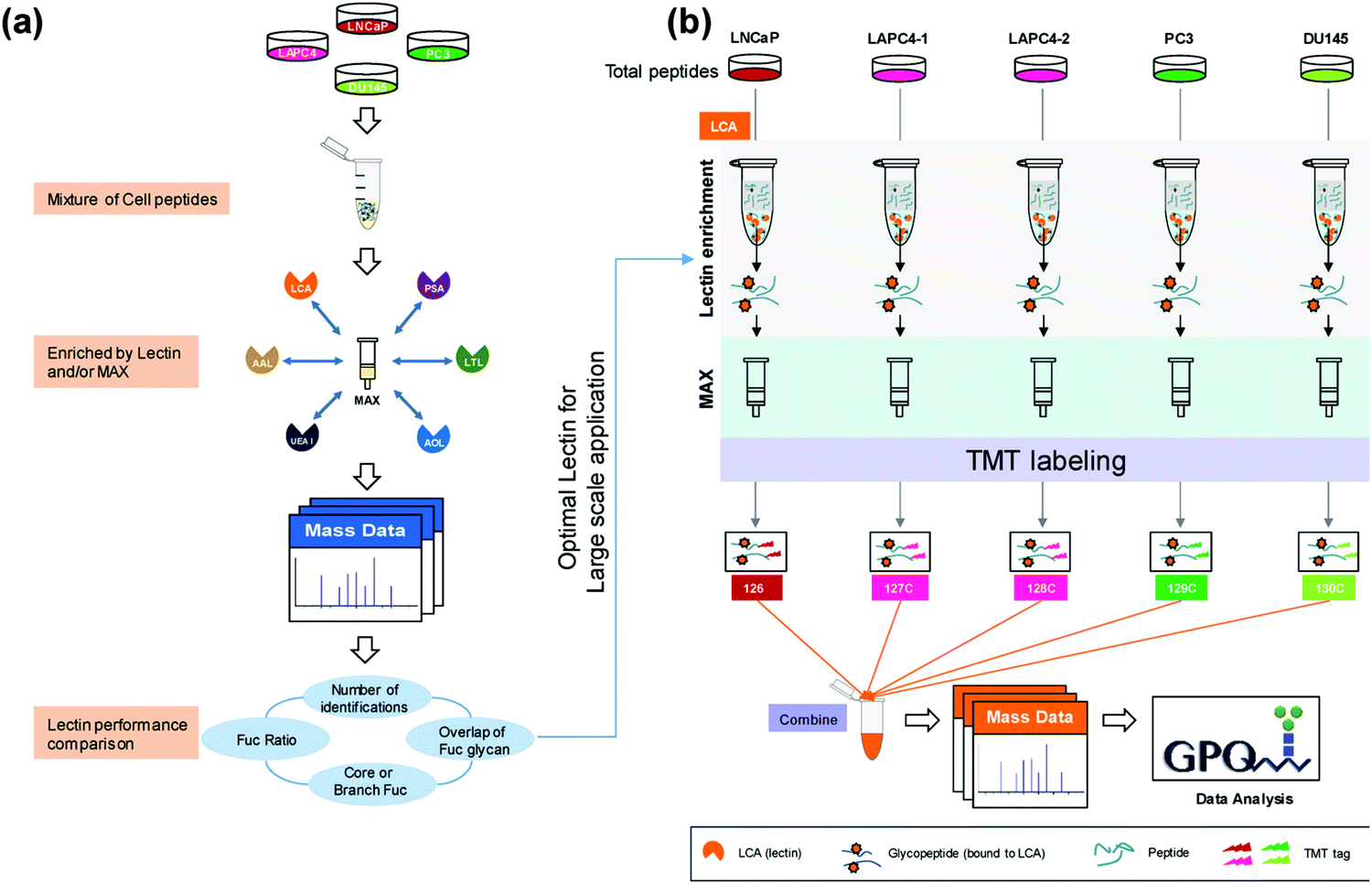 | ||
| Fig. 1 Complete workflow utilized by Zhou et al.55 detailing the quantitative approach to investigate site-specific fucosylation and glycoproteins associated with aggressive prostate cancer phenotypes. The optimized enrichment strategy used to identify glycopeptides contributing to prostate cancer aggressiveness shows promise for application in a variety of cancer glycosylation studies but should also be applied to other prostate cancer models to determine its utility across sample types. Reprinted with permission. | ||
Comparable to TMT labeling, iTRAQ allows for both relative and absolute quantitation of labeled samples. Höti et al.57 set out to examine the mechanisms underlying androgen resistance through a global proteomic approach using iTRAQ, labeling tryptic peptides from two prostate cancer cell models grown in triplicate. One main realization of the data was that androgen resistance cannot be treated with a single therapeutic, as the mechanisms driving resistance involve multiple independent pathways. While unfortunate, these findings did uncover some of the mechanisms driving resistance, including the PI3K/AKT signaling pathway, mitochondrial dysfunction of oxidative phosphorylation complexes and the multicatalytic 26S proteasome. Zhang et al.58 used two sublines of PC-3M to distinguish unique characteristics of highly- and poorly-metastatic potential in prostate cancer. After validation, two proteins were found to potentially contribute to the higher metastatic potential, matrix metallopeptidase 1 (MMP1) and four and a half LIM domains 1 (FHL1). While FHL1 has been extensively studied in a variety of cancer types, the information collected here suggests a unique role of MMP1 for increasing metastatic potential in prostate cancer, presenting the opportunity for future inspection of both MMP1 and other MMPs. Webber et al.59 performed a stromal cell proteomics analysis to differentiate normal from tumor-reactive stromal phenotypes that drive disease progression. One compelling finding was the loss of aldehyde dehydrogenase (ALDH1A1) expression in altered versus normal stromal types, suggesting its potential role as a novel marker of disease-induced changes of the stromal environment. Additional investigations have turned to animal models, as prostate cancers grown in vivo reflect interactions that may otherwise be missed in cell culture models. The Pten-knockout mouse model60 was recently examined by Zhang et al.61 through the combined analysis of iTRAQ proteomics and microarray transcriptomics to identify associated molecular changes in mouse prostate carcinogenesis. Both transcriptomic and proteomic data found that immune and inflammation responses were greatly perturbed, in addition to mediations in central nodal activity through the Akt, NF-κB and P53 signaling pathways.
While tissue-based sampling allows for determination of mechanistic properties of the pathways contributing to tumorigenesis and metastasis, its highly invasive nature is discouraged unless necessary. Even if biopsies are obtained, these analyses are often limited by size constraints, as patient-derived tissues covering all stages of prostate cancer progression can be difficult to obtain in large numbers. Mouse models afford the opportunity to mimic tumor progression and metastasis in vivo, but there are still controversies surrounding prostate-based mouse models due to distinct anatomical differences.62 Cell culture models avoid the translational constraints that other model organisms are bound to, but often omit stromal–epithelial interactions during cell growth, a process that has a great impact on tumor invasiveness and metastatic potential. Additionally, current cell-based models for prostate cancer often either only reflect advanced prostate cancer or require the use of multiple cell lines to cover multiple progression stages, introducing variability that complicates genetic-based analyses. Recent advances in cell-based prostate models have been made that address some of the pitfalls outlined here,63 so future quantitative studies should be selective in the models they choose when profiling.
Biofluid analyses
There is a push to develop biomarker strategies involving the collection of biofluids, a less invasive and more cost-effective method of sample collection. Biofluids—such as blood, tissue-based fluid, saliva, or urine—allow for easier monitoring of patient outcomes, as disease progression and treatment responsiveness can be evaluated with frequent patient sampling. Such biofluid-based monitoring strategies are critical in prostate cancer patients, as a portion of men diagnosed with prostate cancer have tumors that are indolent. One study by Davalieva et al.64 comparatively profiled urine samples using a label-free strategy from patients with prostate cancer, benign prostatic hyperplasia, bladder cancer and renal cancer to determine selective biomarkers for earlier diagnosis of prostate cancer. Of the most promising urinary biomarkers identified by the authors, nine had not yet been associated with prostate cancer, indicating their potential as novel biomarkers and necessitating further research into their associated pathways. Soekmadji et al.65 profiled secretome differences of unlabeled, CD9-positive EVs from cell culture models treated with the hormone dihydrotestosterone (DHT). Their combined analyses determined that DHT treatment increases CD9-positive EV secretion and alters the content of secreted EVs, and in agreement with previous literature highlighting the potential of CD9 EVs as a biomarker for prostate cancer.Reaction monitoring-based strategies are one label-free approach that are typically used after initial discovery for validation and accurate quantitation of biomarkers. Targeted analysis of urinary EVs was completed by Sequeiros et al.66 using SRM to quantify 64 protein candidate biomarkers for prostate cancer. A two-protein combination (ADSV and TGM4) distinguished patients with benign tissue from those with cancer, and a five-protein panel differentiated high-from low-grade prostate cancer (CD63, GLPK5, SPHM, PSA and PAPP), highlighting the advantages of targeted proteomics as a diagnostic tool in the clinic. Kim et al.67 investigated expressed prostatic secretion samples using SRM-based quantitation to determine molecular signatures for extracapsular prostate cancer. From a pool of over 200 potential candidates, these researchers narrowed the candidate list to include 34 peptides representative of 27 unique proteins with promising results as biomarkers. Karasota et al.68 evaluated the analytical performance of multiple SRM- and PRM-based strategies to quantitate kallikrein related peptidase 4 (KLK4) in a variety of biofluid samples. Secreted KLK4 was demonstrated to be present in seminal plasma, and for the first time was investigated as a potential biomarker in both seminal plasma and blood. Taken together, the label-free, targeted proteomics methods used for analysis of biofluids offer a reliable tool for biomarker validation and should thus be considered as useful tools for clinical development.
Fujita et al.69 combined two strategies, initially using iTRAQ for urine samples to profile EVs from patients with a high Gleason score.70 After quantifying 3528 proteins, candidate biomarkers were selected for further quantitation and validation using SRM/MRM. Fatty acid binding protein 5 (FABP5) was highlighted as the most promising biomarker from urinary EVs for the detection and diagnosis of high Gleason score prostate cancer, but further studies would be necessary for confirmation. Yan et al.71 performed an iTRAQ-based analysis on the serum of prostate cancer patients with or without bone metastasis to find potential biomarkers indicative of these metastases. Of the 32 differentially expressed proteins identified, three—CD59, haptoglobin and tetranectin—were selected and validated to be related to prostate cancer bone metastasis, confirming their utility as serum biomarkers. Larkin et al.72 implemented iTRAQ to enhance their proteomic profiling of high-quality serum samples for biomarker discovery. After identification and validation using ELISA, two biomarkers, SAA and TSR1, showed promising results when used in combination with KLK3. However, these results were obtained in a small sample cohort, so further studies with a larger, more diverse sample set are necessary before serious consideration of these proteins as biomarkers. Table 1 summarizes selected prostate cancer biomarkers.
| Proposed biomarker | Source | Findings |
|---|---|---|
| Platelet-derived growth factor (PDGF)-B46 | Prostatic tissue | Overexpressed with increased cancer proliferation |
| α(1,6)-Fucosyltransferase (FUT8)48–50 | LAPC4 and LNCaP cells | Increased FUT8 expression corresponded with decreased extracellular vesicle production |
| Mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4)51,52 | PC3 and PC3MLN4 cells | Differential expression in metastasis-derived cell lines |
| Phosphatidylinositol 4-kinase III α (PI4KIIIα)53 | PC3 cells | Upregulated in PC3 cell lines; promotes cancer cell invasion |
| FXYD domain containing ion transport regulator 3 (FXYD3)54 | PrAd, NEPC cell lines | Biomarker specific to prostate adenocarcinoma |
| CEA cell adhesion molecule 5 (CEACAM5)54 | PrAd, NEPC cell lines | Biomarker specific to neuroendocrine cancer |
| Four and a half LIM domains 1 (FHL1), matrix metallopeptidase 1 (MMP1)58 | PC-3M sublines | Promote higher metastatic potential |
| Aldehyde dehydrogenase (ALDH1A1)59 | Stromal tissue | Loss of expression in altered stromal cell types |
| Actin-depolymerizing factor (ADSV), transglutaminase 4 (TGM4)66 | Urine | Differentiates benign and cancerous tissue |
| CD63 molecule (CD63), glycerol kinase 5 (GLPK5), SPHM sulfohydrolase (SPHM), prostate-specific antigen (PSA) and pappalysin 1 (PAPP)66 | Urine | Distinguishes high- and low-grade cancer |
| Kallikrein related peptidase 4 (KLK4)68 | Seminal fluid | Biomarker available in seminal fluid |
| Fatty acid binding protein 5 (FABP5) | Urine | Utility in detecting, diagnosing high gleason score prostate cancer |
| CD59 molecule (CD59), haptoglobin and tetranectin71 | Serum | Expression correlated to bone metastasis |
The use of quantitative proteomic strategies on patient-derived biofluid samples show promise in the discovery and validation of new biomarkers. Specifically, the KLK family of proteins has been shown in the mentioned literature to have potential in many biofluids and may improve diagnostic accuracy further when combined with others. On the other hand, serum biomarkers in prostate cancer deserve a level of scrutiny as demonstrated by prostate-specific antigen (PSA), a currently approved biomarker whose elevation in serum is also associated with benign prostatic hyperplasia (BPH), resulting in high false positive rates.73 Noting this, prostate cancer biomarkers should be rigorously tested against patients with BPH and other prostatic diseases to ensure accuracy. Such rigorous tests involving larger sample sets can be achieved using the quantitative strategies described above, indicating their potential to advance the knowledge within the field at a rapid pace.
Pancreatic cancer
The seventh leading cause of cancer-related deaths in the world,74 pancreatic cancer has rightfully garnered significant attention from clinical research communities. In-depth proteomic analyses have illuminated the highly dynamic nature of post-translational modifications,75–77 while providing novel insights toward treatment monitoring and severity stratification. The promising results of these profiling experiments in hand, great success has come in the effort to employ quantitative strategies to illuminate dysregulated protein expression, identify treatment pathways, and validate potential biomarkers.Tissue analyses
The prevalence of pancreatic cancer across the world's population has necessitated in-depth proteomic analyses of cancerous tissue and model systems. Model cell lines have enabled researchers to identify pertinent biomolecules specific to pancreatic cancer without the need for invasive, repetitive tissue resection. Naturally, the study of cell lines lends itself to the use of SILAC to perform quantitative investigations. Recently, Liu et al.78 performed secretomic analyses of pancreatic cancer cells (PC-1), revealing 161 proteins with altered expression, including 55 proteins not previously reported. As well, they note a combination panel for cadherin 3 (CDH3), plasmogen activator, urokinase (PLAU), and lunatic fringe (LFNG) proteins that may be useful for improving cancer patient prognoses. Beyond this, Marchand et al.79 employed a three-channel SILAC approach to reveal association of transcription factor EB (TFEB) with nuclear proteins upon inhibition of glycogen synthase kinase-3 (GSK3) and mammalian target of rapamycin (mTOR). Moving beyond SILAC experiments, Shi et al.80 used isotopic dimethyl labeling to examine paracrine communication between pancreatic cancer cells and pancreatic stellate cells (PSCs). This experiment provided the knowledge that leukemia inhibitory factor (LIF) is a key paracrine factor from activated PSCs acting on cancer cells. Employing a novel approach, Roberts et al.81 developed a cysteine-reactive fragment-based ligand library to coordinate novel small molecules that impair pancreatic cancer pathogenicity with druggable hotspots for potential cancer therapy. While numerous SILAC and isotopic tagging workflows exist outside the time frame of this review, the relatively small number of recent applications indicates an area of potential focus for researchers examining pancreatic cancer.Isobaric labeling, however, has seen significant use in the study of pancreatic cancer. Zhang and colleagues82 have provided a meaningful guide for those seeking to perform isobaric labeling experiments using the commercial TMT36 offering from ThermoScientific. Beyond this, Perera et al.83 employed TMT labeling to study pancreatic cancer cell metabolism, revealing the MiT/TFE proteins – MITF, TFE3 and TFEB – are decoupled from regulatory mechanisms, increasing expression levels of lysosomal catabolic function essential for pancreatic ductal adenocarcinoma (PDA) growth. As an alternative to TMT, An et al.84 employed iTRAQ in the analysis of serum exosomes from chemotherapy patients (Fig. 2). Of note, this study indicates patient-derived exosomes play a significant role in cancer metastasis. Furthermore, Li et al.85 demonstrated monumental success in broad protein quantitation while analyzing Peripheral Blood Mononuclear Cells (PBMCs). This study, which employed iTRAQ labeling and 2D-LC-MS quantified 3357 proteins, with 114 being distinguished as dysregulated in the cancer group. These examples of isobaric labeling indicate the broad utility for high throughput analyses of complex pancreatic cancer samples. However, a chief limitation of TMT and iTRAQ is cost, placing their use out-of-reach for many budding research groups. In remedy, Li and colleagues34 developed Dimethyl Leucine (DiLeu) that provides greater multiplexity86–88 than commercial options at a fraction of the cost. DiLeu is available as an isotopic,89 isobaric,34,86,87 and mass-defect90 chemical tag and has even been modified to provide absolute quantitation.89,91 The mass-defect offering, mdDiLeu, has been successfully applied for simultaneous multiomic analysis of pancreatic cancer cells,92 providing uncompromised access to high throughput small molecule and protein quantitation.
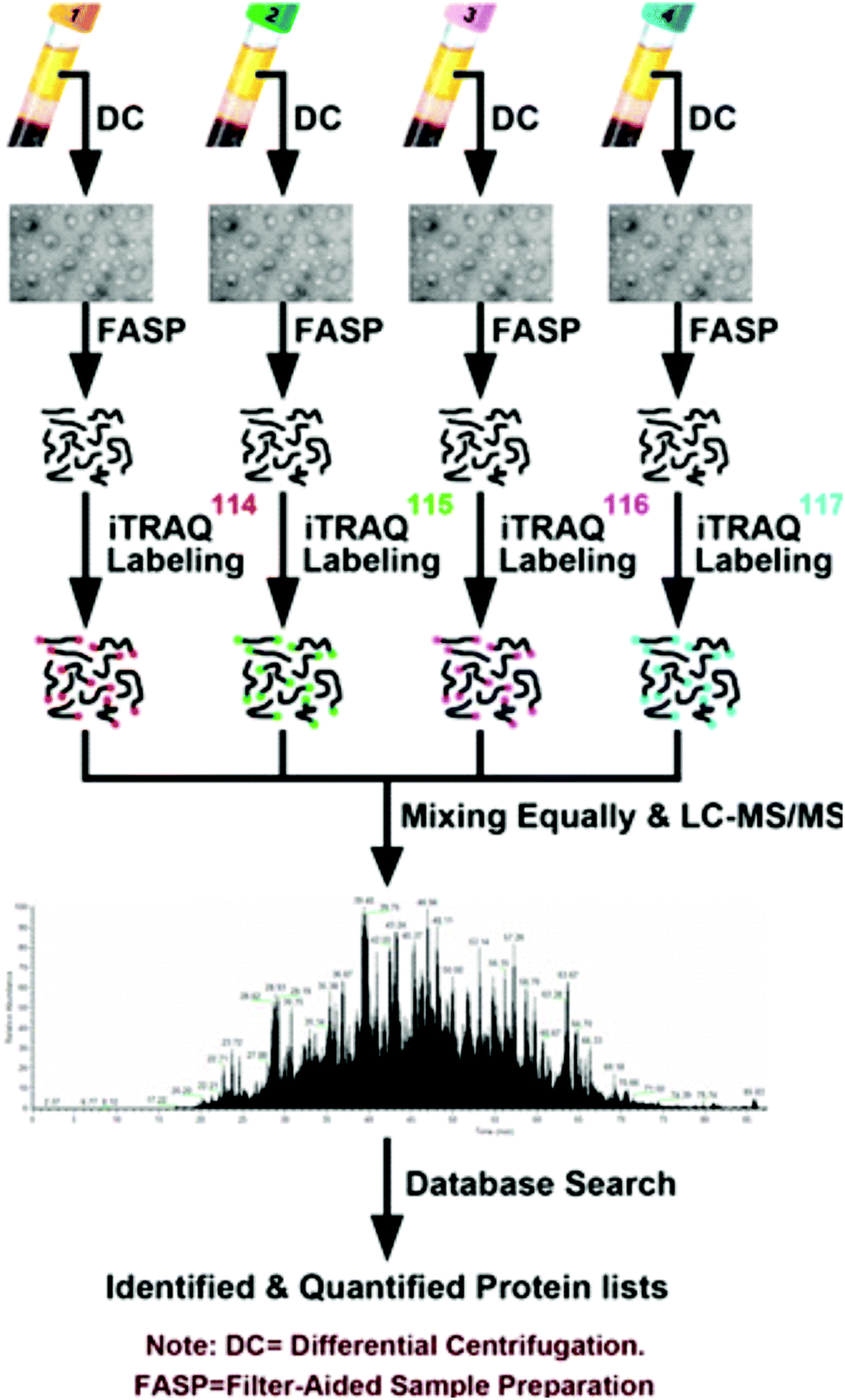 | ||
| Fig. 2 Workflow described by An et al.84 for the quantitative analysis of chemotherapy patient exosomes through iTRAQ labeling and quantitative mass spectrometry. This example of a facile isobaric labeling proteomics experiment provides deep proteomic profiling of multiple complex samples with lower spectral complexity than isotopic labeling methods. Reprinted with permission. | ||
Label-free analyses, too, have seen routine utilization in pancreatic cancer investigations. Wang et al.93 introduced the novel IonStar pipeline for accurate MS1-level protein quantitation. This preliminary example quantified >4000 proteins from 40 biological samples and identified 541 proteins dysregulated groups treated with birinapant and paclitaxel. Later Zhu et al.94 applied the IonStar pipeline to elucidate the relations among relevant signaling pathways during gemcitabine and birinapant treatment. These applications highlight the utility of quantitative proteomics to evaluate treatment efficacy. In a similar vein, Singh et al.95 presented a large-scale, label-free proteomics study to uncover the mechanism by which sanguinarine suppresses cancer proliferation. While quantifying >3100 proteins, 37 biomolecules were identified as differentially expressed, highlighting the pleotropic effects of sanguinarine. Finally, Zhou et al.96 employed parallel reaction monitoring (PRM) to identify 165 potential biomarkers in pancreatic cancer. During validation, brain acid soluble protein 1 (BASP1) was identified as a novel target for pancreatic cancer therapy and is shown to interact with Wilms tumor protein (WT1).
Biofluid analyses
Considering the real-world application of investigational proteomics analyses, a topical concern is the need for invasive patient sampling. This in mind, researchers have long sought to identify cancer-specific analytes from biofluids, which may be sampled repeatedly at lower physical and monetary cost to patients. Though metabolic and isotopic labeling are not well represented in pancreatic cancer research in recent years, Jhaveri et al.97 used a novel serum antibody-based SILAC immunoprecipitation approach, denoted as SASI, to identify specific targets expressed in cancer patients post-vaccine therapy. More popular, however, are applications utilizing isobaric labeling. Sogawa et al.98 employed TMT labeling to ascertain that complement component 4 binding protein alpha (C4BPA) and polymeric immunoglobulin receptor (PIGR) expression was significantly higher in preoperative patients than postoperative. Naba et al.99 identified unique expression levels in 35 proteins as pancreatic cancer islets progressed from hyperplastic to angiogenic to insulomas. Yu et al.100 employed iTRAQ to quantify 4517 proteins in the exosomes of Panc02 and Panc02-H7 cells, notably revealing cancer-derived exosomes promote tumor metastasis. Lin et al.101 and Liu et al.102 further implemented iTRAQ for quantitative evaluations of pancreatic cancer patient serum. An important overlap of these two studies was the identification that apolipoprotein A-1 (APOA1) shows distinct expression in pancreatic cancer patients. Considering this trend was shared between patients expressing carbohydrate antigen (CA) 19-9 and those who are CA19-9-negative, APOA1 presents an area of significant interest moving forward.Similar to the studies presented in pancreatic cancer tissue analyses, label-free quantitation has been routinely employed in quantification of biofluid proteins. Through this quantitative strategy, Ohmine et al.103 successfully validated deoxycytidine kinase (dCK) as a good predictor of progression-free survival and an effective biomarker of gemcitabine sensitivity. Yoneyama et al.104 identified insulin-like growth factor-binding proteins insulin-like growth factor binding protein 2 (IGFBP2) and IGFBP3 as compensatory biomarkers of pancreatic cancer in instances when CA19-9 screening is inconclusive. Park et al.105 performed a large-scale validation of biomarkers, finding that APOA-IV, APOCIII, IGFBP2, and tissue inhibitor of metalloproteinase 1 (TIMP) were significantly altered in pancreatic cancer. Of note, a panel including CA19-9, APOA-IV, and TIMP1 showed improved performance in distinguishing early pancreatic cancer from pancreatitis. Do et al.106 identified 18 biomarker candidates associated with malignancy in intraductal papillary mucinous neoplasms (IPMNs). Finally, Nigjeh et al.107 developed an optimized data-independent acquisition (DIA) workflow to identify and quantify >14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 peptides from ∼2300 plasma proteins (Fig. 3).
000 peptides from ∼2300 plasma proteins (Fig. 3).
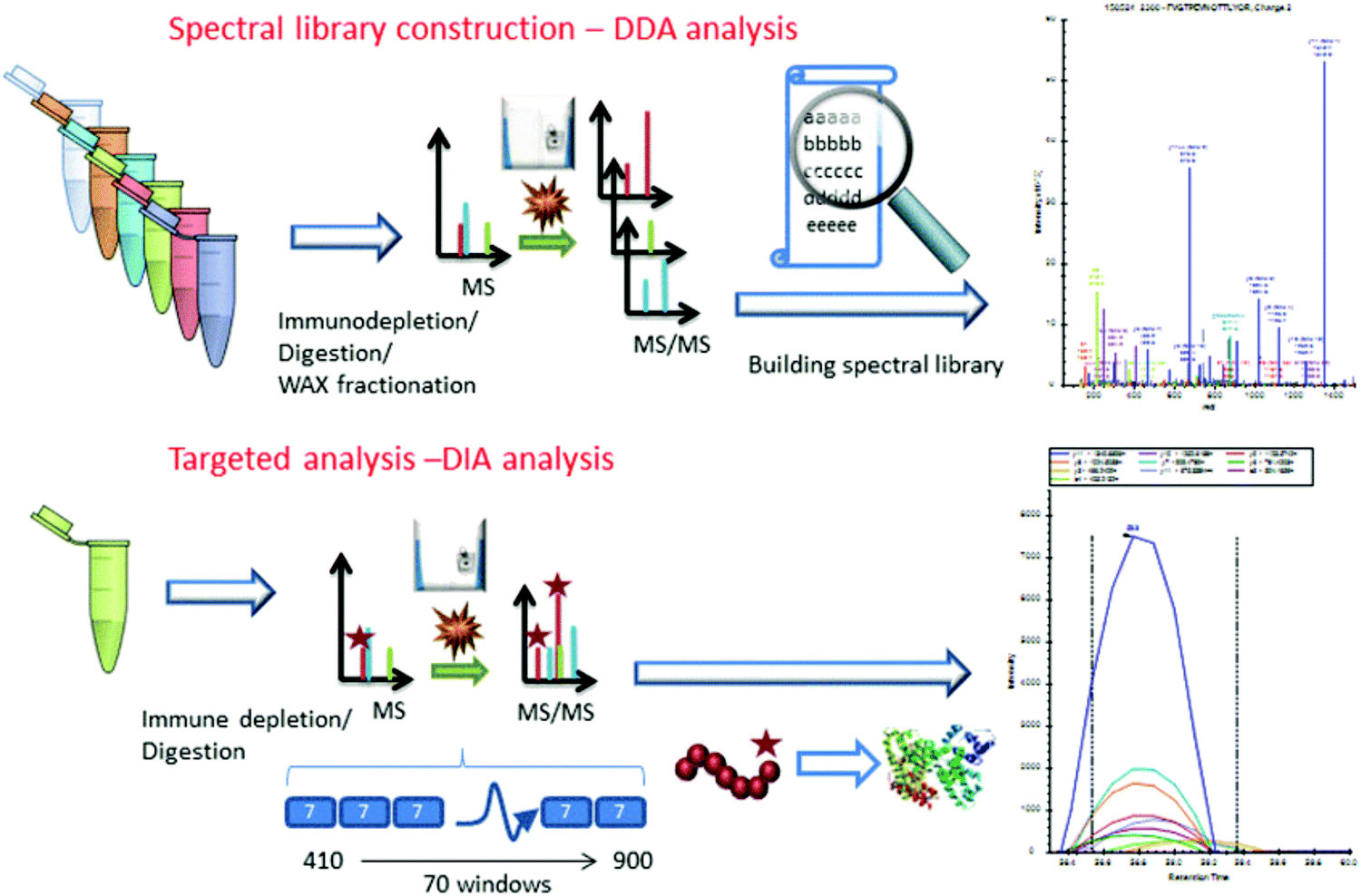 | ||
| Fig. 3 Workflow implemented by Nigjeh et al.107 Quantitative workflows utilizing isobaric labels present the greatest propensity for deep proteome profiling. However, these workflows are limited by their instrument acquisition speed and cycle time required to select and fragment top precursors. For this reason, implementation of DIA strategies presents the ability to sequence a greater number of peptides in the same amount of time. Though the data processing methods are significantly more involved, DIA workflows are sure to be of critical importance to proteome profiling in the coming years. Reprinted with permission. | ||
As seen by the numerous examples of pancreatic cancer tissue and biofluid investigation, quantitative proteomics provides a facile entry point into the field of biomarker identification and validation (Table 2). Considering the agreement across several studies that proteins such as APOA1, APOA4, IGFBP and CA19-9 serve as rigorous biomarkers in pancreatic cancer, future studies should investigate the utility of high throughput label-free, PRM, or MRM screening of these biomolecules. Meaningful evaluation of MS-based protein assays in blind studies may demonstrate potential to accurately identify and diagnose pancreatic cancer at scale. Development of these workflows and associated technology will be vital to understanding the risk factors associated with disease onset and progression, as well as the success of current and novel treatment strategies.
| Proposed biomarker | Source | Findings |
|---|---|---|
| Cadherin 3 (CDH3), plasmogen activator, urokinase (PLAU), lunatic fringe (LFNG)78 | PC-1 cell secretome | Potential for improving cancer patient prognoses |
| Transcription factor EB (TFEB)79 | HEK293, PANC1, MIA PaCa-2 cells | Association with nuclear protein upon inhibition of GSK3 |
| Leukemia inhibitory factor (LIF)80 | Pancreatic stellate cells | Denoted as major paracrine factor |
| Melanocyte inducing transcription factor (MITF), transcription factor binding to IGHM enhancer 3 (TFE3) and transcription factor EB (TFEB)83 | Tissue, PDA cells | Decoupled from regulatory mechanisms, promote catabolic function |
| Brain acid soluble protein 1 (BASP1)96 | Tissue | Novel cancer therapy target |
| Complement component 4 binding protein alpha (C4BPA), polymeric immunoglobulin receptor (PIGR)98 | Serum | Higher expression in preoperative patients than postoperative |
| Apolipoprotein A-1 (APOA1)101,102 | Serum | Distinct expression in both CA19-9 positive and CA19-9-defficient patients |
| Deoxycytidine kinase (dCK)103 | PK9, CFPac-1, PK1, SUIT-2, and AsPC-1 cells | Predictor of progression-free survival, biomarker of gemcitabine sensitivity |
| Insulin like growth factor binding protein 2 (IGFBP2) and IGFBP3104 | Plasma | Compensatory biomarkers when CA19-9 screening is inconclusive |
| Insulin-like growth factor binding protein 2 (IGFBP2) tissue inhibitor of metalloproteinase 1 (TIMP1), apolipoprotein A IV (APOA-IV), apolipoprotein CIII APOCIII105 | Blood | Protein panel highly effective in early detection of pancreatic cancer |
Breast cancer
The high rate of incidence associated with breast cancer, as well as targeted focus drawn from successful advocacy and research fundraising, have shed significant light on the mechanisms of breast cancer. Though this dedicated focus has reduced patient mortality and cancer rates in high income countries, developing nations display the opposite trend.108 Beyond this, breast cancer is of continual interest to the medical community due to the high rate of recurrence and metastasis.109,110 For these reasons, many have turned to quantitative proteomics to aid in stratifying cancer subtypes and identifying potential biomarkers.Tissue and biofluid analyses
Within the timeframe of this review, the majority of quantitative proteomic investigations have been centered on tissue analyses, often employing model cell lines or resected tumor tissue to determine protein expression. Though few applications have employed metabolic labeling for quantitative analyses, Tyanova et al.111 presented a robust investigation that merged quantitative mass spectrometry with traditional RNA- and DNA-based sequencing strategies. Analyzing 40 tumors that were either estrogen receptor positive, Her2 positive, or triple negative, the authors identified an average of >7000 proteins on average, spanning 8 orders of magnitude in protein intensity. Within this study, they combined their quantitative results with microarray analyses and machine learning classification to identify potential subtype-specific therapies.More popular than SILAC-like experiments, isobaric labeling has been extensively employed in breast cancer investigations. Suman et al.112 employed iTRAQ to identify proteins associated with breast cancer subtypes. Notably, this study indicated fibronectin (FN1), alpha-2-macroglobulin (A2M), complement component-4-binding protein-alpha (C4BPA) and complement factor-B (CFB) were critical to subtype differentiation in both plasma and tissue samples. Calderon-Gonzalez et al.113 further employed this technology to identify 306 differentially expressed proteins in breast cancer cell lines. As well, their study indicates large proline-rich protein (BAG6), ATP-dependent RNA helicase (DDX39), annexin A8 (ANXA8) and cytochrome c oxidase subunit 4 (COX4) may serve as useful biomarkers. Gajbhiye et al.114 provided a novel DIA-iTRAQ strategy to uncover proteomic divergence in HER2-enriched cancer cell lines, which allowed for the creation and testing of a 21 protein panel to discriminate cancer and healthy controls. Turning to TMT labeling, Going et al.115 and Clark et al.116 utilized this strategy, illuminating the pathways of action of methoxyclcone in triple negative breast cancer (Fig. 4) and classifying exosomal cargo proteins, respectively. As a cost-effective alternative to these iTRAQ and TMT labeling strategies, DiLeu tagging approach has also successfully been employed in identifying strategies for inhibiting cancer cell proliferation. Within this work, Liu et al.117 revealed that dynamic methylation of pyruvate kinase M2 (PKM2) directly affect the metabolic activity of cancer cells and promotes cell propagation, migration and metastasis. This study, along with those detailed above, serve to indicate the importance of high-throughput quantitative cancer proteomics, outlining potential targets for future treatment strategies.
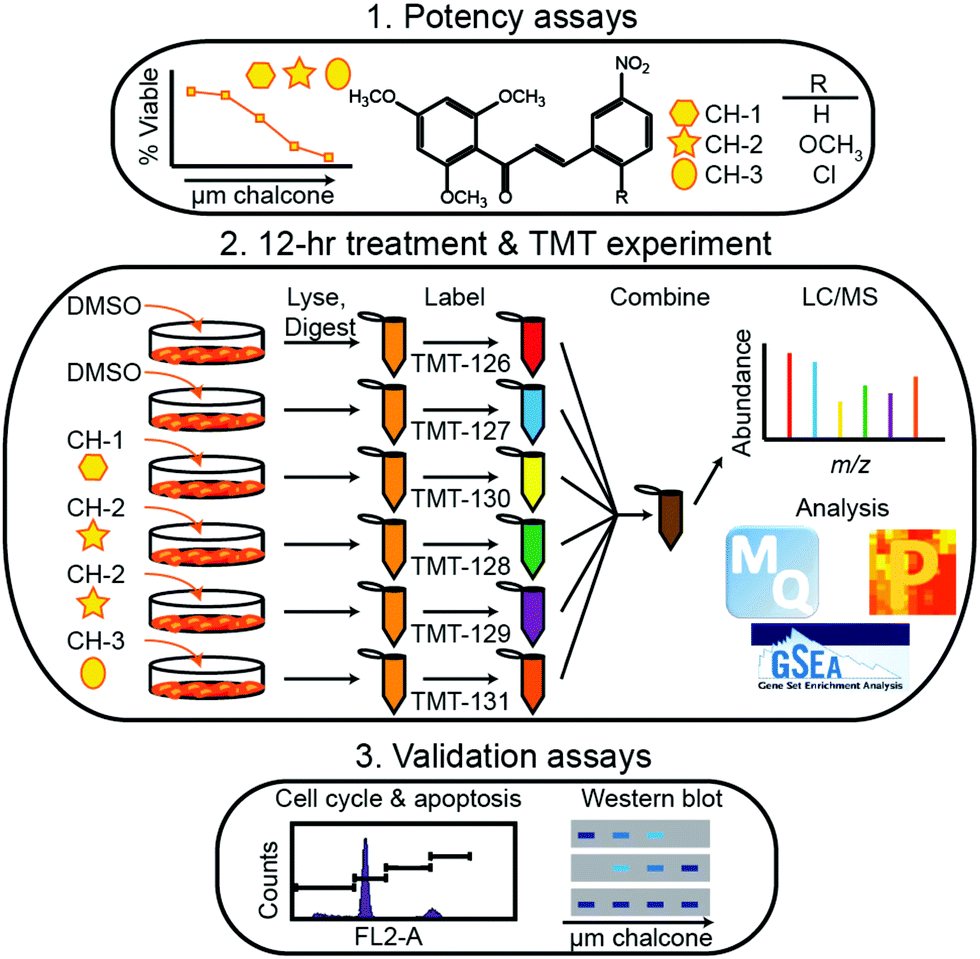 | ||
| Fig. 4 Representative workflow established by Going et al.115 As quantitative proteomics is critical for discovering and validated biomolecules of interest during periods of disease and treatment, this workflow represents an example of how treatment strategies may be controlled and systematically evaluated. While SILAC methods would be useful in situations where cell growth is monitored, isotopic labeling methods may be considered inherently lower throughput due to the increases in spectral complexity they may provide. Reprinted with permission. | ||
A significant entry into quantitative breast cancer proteomics was provided by Johansson et al.118 This study provided in-depth quantitation of 45 breast cancer tumors, spanning each of the 5 PAM50-based molecular classifications. Upon quantitation of 9995 proteins, the authors used these proteome profiles to interpret multiple layers of systems measurements. While each of these studies offered unique insight into uncovering and validating potential biomarkers and investigative strategies, a chief concern among many is the long-term reproducibility of quantitative measurements. Using iTRAQ to quantify proteins from human-in-mouse xenograft tissue, Zhou et al.119 demonstrated that the large majority of quantitative measurements hold consistent over time, but also raised some topical concerns. First, they observed higher variability in quantitation of hydrophilic peptides compared to those of average peptide character, likely due to poor retention of these peptides on column. Second, as researchers have their choice of dissociation methods, this study reveals stepped collision energy offers higher reproducibility between unique measurements. Finally, whereas most commercial software implements a form of scoring to determine the quality of a peptide spectral match (PSM), this study goes further and reveals that a stricter scoring mechanism improves reliability of time-course measurements. This study provides an excellent framework and series of considerations for individuals seeking to begin or improve quantitative mass spectrometry investigations.
Label-free analyses have also been routinely implemented for high throughput biomarker discovery and screening. Among these, Ntai et al.120 compared the quantitation efficiency in bottom-up and top-down analyses of tumor xenografts. Tveitras et al.121 performed comparative analyses of pre-metastatic and metastatic triple negative breast cancer xenograft tissue, uncovering significant changes in expression of haptoglobin, fibrinogen, and thrombospondin-4 and transferrin receptor protein 1 between groups. Wang et al.122 employed a DIA-select reaction monitoring (SRM) approach to reveal distinct proteomic and N-glycoproteomic divergence between normal, precancerous, and cancerous tissues. Gamez-Pozo et al.123 integrated label-free MS quantitation with RT-qPCR to definitively distinguish estrogen receptor positive and triple negative cancer subtypes. Nie et al.124 identified 98 differentially expressed proteins when comparing pure breast cancer stem cells and mature luminal cells. Finally, Warmoes et al.125 elucidated 215 proteins that are significantly enriched in BRCA1-deficient secretome. This study highlights the potential of mass spectrometry to provide sensitive identification of biomarkers in instances when traditional ELISA screening may fall short.
These examples of successful quantitative proteomic analyses in breast cancer applications highlight the flexibility and facility of creating novel workflows to answer an array of biological problems. Knowing there have been a significant number of proteomic measurements made prior to the period in review, these examples of biomarker discovery and validation highlight how rigorous protein MS-based screening assays for the confident identification and stratification of breast cancer may be within reach (Table 3). Assays of this kind, devoid of the need for invasive and repetitive tissue sampling, provide a meaningful conduit towards aiding communities that have limited access to dedicated cancer screening centers and provide direct targets for potential novel therapies.
| Proposed biomarker | Source | Findings |
|---|---|---|
| Fibronectin (FN1), alpha-2-macroglobulin (A2M), complement component-4-binding protein-alpha (C4BPA) and complement factor-B (CFB)112 | Tumor tissue | Critical for subtype differentiation |
| Large proline-rich protein (BAG6), ATP-dependent RNA helicase (DDX39), annexin A8 (ANXA8) and cytochrome c oxidase subunit 4 (COX4)113 | MCF7 and T47D, MDA-MB-231, and SK-BR-3 cells | Putative biomarkers for breast cancer |
| Methylated pyruvate kinase M2 (PKM2)117 | MCF7, MDA-MB-231, HEK293T cells | Promotes cell propagation, migration and metastasis |
| Haptoglobin, fibrinogen, and thrombospondin-4 and transferrin receptor protein 1121 | Pre-/metastatic xenograft tissue | Reveal N-glycoproteomic divergence between normal, precancerous, and cancerous tissues |
Ovarian cancer
Although it has an estimated incidence rate of approximately 2% for 2020, ovarian cancer is the deadliest reproductive cancer in women, with an estimated mortality rate of 5% in women diagnosed with any cancer type and 64% for women diagnosed with ovarian cancer.126 Much emphasis has been placed on the continued research into mechanisms driving ovarian cancer, as late-stage diagnosis of advanced cancer contributes to the high mortality of ovarian cancer. Continued efforts have focused on the identification of critical mechanisms driving disease progression across ovarian cancer subtypes. Quantitative proteomic strategies have continued to increase the depth of knowledge surrounding ovarian cancer and its various subtypes to improve earlier identification strategies and highlight new therapeutic targets.Cellular and tissue analyses
Because the majority of diagnosed ovarian cancer cases have already progressed to a more advanced stage, much quantitative research delves into tissue and cellular proteomic profiling to isolate and exploit dysregulated proteins. While only applicable to cellular-based models, SILAC has been implemented in ovarian cell lines and led to the discovery of critical modulators in ovarian disease progression. Musrap et al.127 cultured the ovarian line OV-90 in adherent and non-adherent conditions using SILAC to compare the impacts of cancer aggregate formation on cellular proteomics. After quantifying 1533 proteins in total, they compared expression with other aggregate-forming lines and saw upregulation of CLCA1, which appeared to affect cancer cell aggregation after further siRNA experimentation. Grassi et al.128 utilized SILAC to quantify epidermal growth factor (EGF)-induced epithelial–mesenchymal transition (EMT) to identify specific mechanisms of this process that may be dysregulated for metastatic purposes. 206 proteins were found to be differentially expressed, some of which included proteins associated with the G1 and G2 checkpoints of the cell cycle, indicating the role of EGF-induced EMT in cell cycle control mechanisms. Another investigation by Ji et al.129 utilized the metabolic strategy to perform an integrated proteomic and N-glycoproteomic analysis of ovarian cancer lines that were either doxorubicin-sensitive or -resistant. They quantified 5509 protein groups and identified 1525 high-confidence N-glycosites corresponding to 740 glycoproteins. Quantifying the protein abundance allowed these researchers to examine glycoprotein abundances and alterations, which provides unique information into the role of N-glycosylation in drug resistance.Applicable to more than just cell culture-based models, isobaric labeling is commonly employed for quantitative experiments applied to ovarian cancer sample sets. Zhang et al.130 used iTRAQ labeling to integrate quantitative proteomics with the transcriptomic profile of ovarian high-grade serous cancer (HGSC) patient biospecimens. Over 3500 proteins were quantified and used in tandem with genomic results to reveal a strong association between specific histone acetylation events and the homologous recombination deficient phenotype seen in patient samples. Hiramatsu et al.131 comparatively profiled HGSC and endometrial carcinoma samples using iTRAQ-based quantitation. Comprehensive analysis revealed 356 quantifiable proteins and identified mitochondrial inner membrane protease subunit 2 (IMP2) and minichromosome maintenance complex component 2 (MCM2) to be modulators of rapid HGSC growth, illustrating the need to examine these two proteins in further ovarian cancer studies.
Alternatively, many other analyses have used the TMT-based isobaric strategy rather than iTRAQ labeling. Recently, Hu et al.132 used an integrated proteomic and glycoproteomic approach with TMT-labeled peptides in their analysis of ovarian HGSC versus non-tumor tissues. These authors combined global proteomics, solid-phase extraction of glycosite-containing peptides (SPEG) and glycan identification via intact glycopeptide analysis to provide a comprehensive view into N-glycoproteomics within ovarian cancer. Their integrated approach yielded promising results, identifying tumor-specific glycosylation and revealing glycosylation enzymes that were correlated with altered glycosylation status. Yoshimura et al.133 treated neighboring peritoneal mesothelial cells with a microRNA shown to be elevated in the serum of ovarian cancer patients to determine its role in cancer invasion and metastasis. The TMT-based proteomics analysis exhibited increased expression of fibronectin and vitronectin, enhancing the ability of the cancer cells to invade the surrounding environment. A straightforward, quantitative comparison of TMT-labeled normal versus cancerous ovarian tissue was performed by Qu et al.134 to find differentially expressed proteins that hold promise in elucidating disease progression. Initial analyses found 498 differentially expressed proteins and highlighted chloride intracellular channel protein 1 (CLIC1), which was examined further and ultimately determined to promote tumorigenesis, making it an attractive therapeutic target. Proteogenomic and phosphoproteomic analysis was performed by McDermott et al.135 to characterize mechanisms driving ovarian HGSC functions down to the post-translational level. Global proteomic analysis led to the identification of 10706 proteins and combined results described a role of histone acetylation as a marker for homologous recombination deficiency, confirming an association earlier proposed by Zhang et al.130 Phosphoproteomics data provided understanding into proliferation-induced replication stress and the impact it has on chromosomal instability in HGSC, implying that mitotic and cyclin-dependent kinases could serve as therapeutic targets after future experimental validation.
Label-free quantitation is frequently employed for ovarian cancer analyses, as the global overview it provides of the proteome allows researchers to identify multiple pathways for further targeted analyses. Chuffa et al.136 used this approach to determine the influence of melatonin treatment on an in vivo model of ovarian cancer. Comparative proteomics analyses showed that downregulation of processes involved in cancer signaling was promoted, underlining molecular targets for therapeutic intervention while indicating the feasibility of melatonin supplementation for ovarian cancer patients. Another comparative analysis by Júnior et al.137 explored the effects of P-MAPA, IL-12 or a combination immunotherapy of the two on the SKOV-3 ovarian cancer cell line. After confirming 532 proteins were identified across all groups, it was noted that combination therapy of P-MAPA and IL-12 was most efficient at regulating proteins involved in metabolic processes that may render cancer cells more vulnerable, suggesting that the use of the two therapies concomitantly is a plausible treatment strategy. Coscia et al.138 used a quantitative, label-free approach in tandem with other quantitative strategies to probe the proteomes of platinum-resistant and -sensitive ovarian HGSC patient-derived tissues (Fig. 5). Multi-level quantitative analyses revealed cancer/testis antigen family 45 (CT45) as a prognostic factor through mediation of chemosensitivity, thereby exposing it as an immunotherapy target.
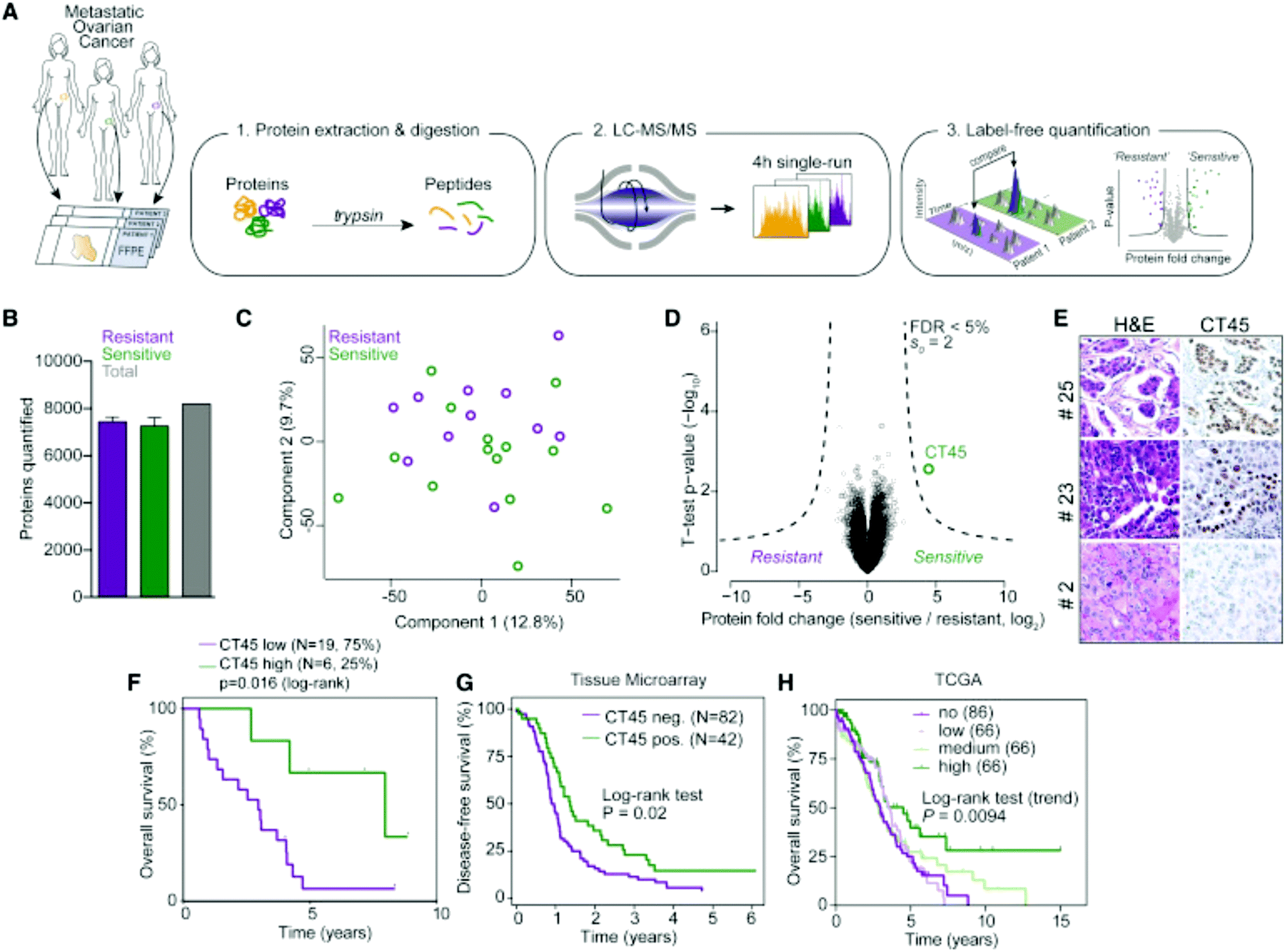 | ||
| Fig. 5 Analysis by Coscia et al.138 to determine proteomic differences in ovarian cancer tissue samples either resistant or sensitive to platinum-based chemotherapeutics. This strategy identified CT45 as a chemosensitivity modulator and demonstrates the ability of quantitative methods to identify factors that play a role in therapeutic resistance. Reprinted with permission. | ||
The quantitative tissue analyses outlined here provide multiple protein targets for the development of new targeted therapies. The role of a defective DNA damage response in ovarian cancer is well established, so the multiple studies highlighting histone acetylation and its role in homologous recombination deficiency is supported by current literature and should be examined in therapeutic development.139 Additional analyses that examine post-translational modifications simultaneously with proteomics should also be explored, as these studies may highlight other processes outside the DNA damage response that promote cancer progression. The experiments above outline the utility that quantitative proteomic approaches hold in advancing the knowledge of the ovarian cancer field.
Biofluid analyses
Quantitative analyses that inspect biofluids of ovarian cancer samples provide valuable information about potential biomarkers that allow for earlier detection and diagnosis, a current area of the ovarian cancer field that is in dire need of new research breakthroughs. Isobaric labeling of ovarian biofluids allow scientists to relatively quantify biomarkers that may otherwise go undetected or are lost during depletion of abundant serum proteins such as albumin. Zhang et al.140 profiled exosomes derived from patient plasma using the TMT tagging strategy. When the 225 proteins identified across all samples were quantitatively compared, proteins associated with the coagulation cascade were found to be differentially expressed and may therefore be promising diagnostic factors for ovarian cancer. Zhang et al.141 went on to further profile circulating exosomes of late-stage cancer patients using iTRAQ. After validation, they determined that apolipoprotein E (ApoE) multiplexed with epithelial cell adhesion molecule (EpCAM), plasminogen (PLG), serpin family C member 1 (serpinC1) and complement component 1q (C1q)were able to accurately diagnose ovarian cancer. It was also noted that activation of coagulation cascades was increased in the ovarian cancer cohort due to increased Factor X levels, demonstrating the impact that tumor-derived extracellular vesicles may have on other biological processes. Swiatly et al.142 examined iTRAQ-labeled serum proteins from healthy control, benign ovarian tumor and ovarian cancer patients. Five proteins were found to be differentially expressed within the ovarian cancer group, and three of these coupled to current biomarkers CA125 and HE4 improved diagnostic discrimination between benign and malignant ovarian tumors. Russell et al.143 used iTRAQ to screen preclinical serum samples for detection of early stage biomarkers and initially identified 90 differentially expressed proteins in ovarian cancer cases. A second targeted analysis of 20 selected candidates revealed vitamin K-dependent protein Z (VKDP), an anticoagulant not previously associated with ovarian cancer, as either a novel independent early detection biomarker or concomitantly with CA125 to increase differential diagnostic capabilities.Although label-free analyses suffer from longer instrument times and potential run-to-run variability, they provide the greatest profiling depth of the multiple quantitative strategies and are vital to finding new ovarian biomarkers. Barnabas et al.144 performed deep proteome profiling of 187 uterine liquid biopsy-derived microvesicles to identify early detection biomarkers. Machine learning algorithms identified a 9-protein signature that correctly identified all Stage I lesions, demonstrating the strength of the panel for future use in early diagnosis. Zhang et al.145 studied the plasma proteins to isolate biomarkers related to chemoresistance of postoperative reoccurrence. These experiments found a total of six dysregulated proteins that could serve as predictive biomarkers for chemoresistance in ovarian cancer patients. The combination of plasma proteomics and metabolomics was utilized by Ahn et al.146 to discover new molecular signatures of ovarian HGSC. Differential expression of 34 metabolites and 197 proteins was found, with three proteins (phosphopantothenoylcysteine synthetase (PPCS), peripheral myelin protein 2 (PMP2) and tubulin beta class I (TUBB)) and two metabolites (L-carnitine and PC-O) related to the carnitine system established as potential markers of cancer plasticity. Hüttenhain et al.147 created a biomarker development strategy for large-scale SRM studies in ovarian cancer plasma samples. After developing a 5-protein signature for ovarian cancer and testing it against the current ELISA-based standard for biomarker tests, it was found that the SRM-based method had sensitivity measurements that exceeded the current ELISA standard, validating its potential for clinical development and use. Rauniyar et al.148 also used a more targeted approach, combining data-independent acquisition methods with PRM to improve identification of ovarian cancer serum biomarkers. They demonstrated that ApoA-IV is a more reliable biomarker than previously determined by immunological assays in addition to the identification of C-reactive protein, transferrin and transthyretin as other available ovarian serum markers. Overall, this study validated the use of quantitative mass spectrometry as a more sensitive and reliable method of quantitation compared to immunological-based procedures.
While the quantitative research mentioned here has progressed ovarian cancer research, continuing studies are still necessary to delve deeper into specific mechanisms of novel markers identified. During the review process, many of the identified studies had a tissue-based proteomics approach and minimal studies focused on biofluid samples (Table 4). More studies focusing on the use of biofluids in ovarian cancer research are critical in the development of novel biomarkers for earlier detection and treatment, and the lack of literature compared to tissue-based studies highlights a current area for further quantitative experimentation in ovarian cancer. In particular, studying the microvesicular proteome for the discovery of novel biomarkers has shown great potential both here and in other quantitative applications. Profiling of extracellular vesicles may prove to be a vital key in the prevention of late-stage diagnosis and increasing the overall survival rate of patients diagnosed with ovarian cancer.
| Proposed biomarker | Source | Findings |
|---|---|---|
| Calcium-activated chloride channel 1 (CLCA1)127 | OV-90 cells | Affects cancer cell regulation |
| Insulin-like growth factor 2 (IMP2) and minichromosome maintenance complex component 2 (MCM2)131 | HGSC and endometrial tissue | Modulators of rapid high-grade serous cancer growth |
| Fibronectin and vitronectin133 | Peritoneal mesothelial cells | Increased expression promotes cancer cell invasion |
| Chloride intracellular channel protein 1 (CLIC1)134 | Tissue | Determined to promote tumorigenesis |
| Histone acetylation130,135 | Tumor tissue | Marker for homologous recombination deficiency |
| Phospholinoleate-palmitoleate anhydride (P-MAPA), interleukin 12 (IL-12)137 | SKOV-3 | Combination immunotherapy is a plausible treatment strategy |
| Cancer/testis antigen family 45 (CT45)138 | Tissue | Found to be a prognostic factor |
| Apolipoprotein E (ApoE), epithelial cell adhesion molecule (EpCAM), plasminogen (PLG), serpin family C member 1 (serpinC1) and complement component 1q (C1q)141 | Circulating exosomes | Diagnostic markers of ovarian cancer |
| Vitamin K-dependent protein Z (VKDP) | Preclinical serum | Novel, early detection biomarker |
| Phosphopantothenoylcysteine synthetase (PPCS), peripheral myelin protein 2 (PMP2) and tubulin beta class I (TUBB)146 | Blood, plasma | Potential markers of cancer plasticity |
| Apolipoprotein IV (ApoA-IV)148 | Serum | More reliable biomarker compared to benchmark proteins |
Conclusions and future directions
The various quantitative strategies outlined here have demonstrated the growing utility of MS-based quantitation methods in cancer diagnosis and research. Quantitative analyses of prostate cancer have been frequently performed within the field due to the growing emergence of resistance to first-line treatments and false diagnoses associated with elevated PSA levels. Multiple members of the KLK family were identified as potential biomarkers and further strengthened when detected in combination with other proteins, suggesting their potential for clinical diagnosis. Targeted validation experiments in a cohort spanning all grades of prostate cancer as well as BPH should be performed before serious consideration is given to using these proteins as biomarkers. Pancreatic studies have been relatively successful in determining sets of robust biomarkers for diagnosis and patient stratification. APOA1, APOA4, IGFBP, and CA19-9 have been indicated in numerous peer-reviewed studies as critical components for pancreatic cancer screening. Future analyses should focus on high throughput reaction monitoring to rapidly screen for these biomarkers. Breast cancer research has seen limited quantitative proteomics studies in recent years, so future efforts of those investigating new biomarkers and determining mechanisms of carcinogenesis should consider quantitative proteomics strategies in their analyses. The small number of studies highlighted here contribute potential protein panels useful for breast cancer screening, but more large-scale studies that confirm the utility of these proteins as biomarkers are necessary. Ovarian research has seen large numbers of tissue- and cellular-based quantitation, but there is a lack of biofluid-based experiments. While tissue-based studies provide large amounts of information that guide knowledge of disease mechanisms, biofluid studies offer important insights that could facilitate the identification and development of protein biomarkers for clinical diagnosis. Due to the lack of biomarkers that detect ovarian cancer at an earlier stage, studies covering biofluids are critical and present an understudied area within the ovarian field.A common drawback of the quantitative studies addressed is that these investigations only determine up- or downregulation of differentially expressed proteins at a single point in time. Time-course evaluations monitoring the differential expression and dynamic changes of these proteins over time could prove to be more useful, as these studies would explain how expression levels change within a single patient over time. In combination with the expression levels across varying disease severity, there is a potential to determine a critical expression level for each stage of cancer progression that determines not only if the patient has cancer, but also the severity of that cancer relative to biomarker concentration levels. Rapid analyses of cancer samples via targeted monitoring strategies offer benefits over current immuno-based assays such as ELISA, demonstrating the advantage of MS-based quantitation for detection and prolonged patient monitoring. Another strategy for improving cancer diagnosis is the integration of additional analyses, such as transcriptomics, metabolomics, or analysis of post-translational modifications and associated crosstalk. Many of the studies outlined here utilized a combined approach to their investigations, leading to the successful identification of a specific protein or process with altered expression in both datasets. These integrated approaches help scientists identify mechanisms driving cancer metastasis and treatment resistance, thus demonstrating their growing utility in future studies. Additional efforts should be made towards understanding communication within the tumor microenvironment, as much remains to be known about the interactions that help a tumor transition from localized to metastatic ability. Finally, studies focusing on single-cell analyses should also be considered for future experiments, as the cellular diversity and heterogeneity provided from such examinations may prove to be critical in understanding specific mechanisms that allow pathogenesis to advance.
Taken together, this review highlights the utility of various quantitative strategies, their associated limitations, and some directions for novel applications in cancer diagnosis and cancer research. As instrumental capabilities continue to grow, it will become necessary for researchers to develop and validate higher throughput labeling strategies that accommodate deeper proteomic profiling. Regardless of the application, quantitative proteomics represents a premier avenue towards cancer biomarker detection, identification, and validation. Continued efforts in the coming years will certainly be centered on the utility of mass spectrometry-based biomarker detection in clinical settings and the development of point-of-care biomolecule screening.
Author contributions
H. N. M., D. G. D., and L. L. wrote, edited and reviewed the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Support for this research was provided in part by the NIH grants U01CA231081, R01 DK071801, RF1 AG052324, and P41GM108538. LL acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.References
- A. A. Aksenov, R. da Silva, R. Knight, N. P. Lopes and P. C. Dorrestein, Global chemical analysis of biology by mass spectrometry, Nat. Rev. Chem., 2017, 1(7), 0054 Search PubMed.
- J.-L. Ren, A.-H. Zhang, L. Kong and X.-J. Wang, Advances in mass spectrometry-based metabolomics for investigation of metabolites, RSC Adv., 2018, 8(40), 22335–22350 Search PubMed.
- G. Paglia and G. Astarita, Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry, Nat. Protoc., 2017, 12(4), 797–813 Search PubMed.
- K. DeLaney, A. R. Buchberger, L. Atkinson, S. Gründer, A. Mousley and L. Li, New techniques, applications and perspectives in neuropeptide research, J. Exp. Biol., 2018, 221(Pt 3), jeb151167 Search PubMed.
- K. DeLaney and L. Li, Data Independent Acquisition Mass Spectrometry Method for Improved Neuropeptidomic Coverage in Crustacean Neural Tissue Extracts, Anal. Chem., 2019, 91(8), 5150–5158 Search PubMed.
- G. Li, D. G. Delafield and L. Li, Improved structural elucidation of peptide isomers and their receptors using advanced ion mobility-mass spectrometry, TrAC, Trends Anal. Chem., 2020, 124, 115546 Search PubMed.
- I. Livnat, H.-C. Tai, E. T. Jansson, L. Bai, E. V. Romanova, T.-T. Chen, K. Yu, S.-A. Chen, Y. Zhang, Z.-Y. Wang, D.-D. Liu, K. R. Weiss, J. Jing and J. V. Sweedler, A d-Amino Acid-Containing Neuropeptide Discovery Funnel, Anal. Chem., 2016, 88(23), 11868–11876 Search PubMed.
- D. J. Ryan, J. M. Spraggins and R. M. Caprioli, Protein identification strategies in MALDI imaging mass spectrometry: a brief review, Curr. Opin. Chem. Biol., 2019, 48, 64–72 Search PubMed.
- K. Srzentić, L. Fornelli, Y. O. Tsybin, J. A. Loo, H. Seckler, J. N. Agar, L. C. Anderson, D. L. Bai, A. Beck, J. S. Brodbelt, Y. E. M. van der Burgt, J. Chamot-Rooke, S. Chatterjee, Y. Chen, D. J. Clarke, P. O. Danis, J. K. Diedrich, R. A. D’Ippolito, M. Dupré, N. Gasilova, Y. Ge, Y. A. Goo, D. R. Goodlett, S. Greer, K. F. Haselmann, L. He, C. L. Hendrickson, J. D. Hinkle, M. V. Holt, S. Hughes, D. F. Hunt, N. L. Kelleher, A. N. Kozhinov, Z. Lin, C. Malosse, A. G. Marshall, L. Menin, R. J. Millikin, K. O. Nagornov, S. Nicolardi, L. Paša-Tolić, S. Pengelley, N. R. Quebbemann, A. Resemann, W. Sandoval, R. Sarin, N. D. Schmitt, J. Shabanowitz, J. B. Shaw, M. R. Shortreed, L. M. Smith, F. Sobott, D. Suckau, T. Toby, C. R. Weisbrod, N. C. Wildburger, J. R. Yates, S. H. Yoon, N. L. Young and M. Zhou, Interlaboratory Study for Characterizing Monoclonal Antibodies by Top-Down and Middle-Down Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2020, 31(9), 1783–1802 Search PubMed.
- K. A. Brown, J. A. Melby, D. S. Roberts and Y. Ge, Top-down Proteomics: Challenges, Innovations, and Applications in Basic and Clinical Research, Expert Rev. Proteomics, 2020, 17(10), 719–733 Search PubMed.
- A. Takemori, D. S. Butcher, V. M. Harman, P. Brownridge, K. Shima, D. Higo, J. Ishizaki, H. Hasegawa, J. Suzuki, M. Yamashita, J. A. Loo, R. R. O. Loo, R. J. Beynon, L. C. Anderson and N. Takemori, PEPPI-MS: Polyacrylamide-Gel-Based Prefractionation for Analysis of Intact Proteoforms and Protein Complexes by Mass Spectrometry, J. Proteome Res., 2020, 19(9), 3779–3791 Search PubMed.
- R. L. Griffiths, E. K. Sisley, A. F. Lopez-Clavijo, A. L. Simmonds, I. B. Styles and H. J. Cooper, Native mass spectrometry imaging of intact proteins and protein complexes in thin tissue sections, Int. J. Mass Spectrom., 2019, 437, 23–29 Search PubMed.
- Z. L. VanAernum, J. D. Gilbert, M. E. Belov, A. A. Makarov, S. R. Horning and V. H. Wysocki, Surface-Induced Dissociation of Noncovalent Protein Complexes in an Extended Mass Range Orbitrap Mass Spectrometer, Anal. Chem., 2019, 91(5), 3611–3618 Search PubMed.
- M. Mann, The ever expanding scope of electrospray mass spectrometry—a 30 year journey, Nat. Commun., 2019, 10(1), 3744 Search PubMed.
- M. Karas, D. Bachmann and F. Hillenkamp, Influence of the wavelength in high-irradiance ultraviolet laser desorption mass spectrometry of organic molecules, Anal. Chem., 1985, 57(14), 2935–2939 Search PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights, Anal. Chem., 2018, 90(1), 240–265 Search PubMed.
- J. Soltwisch, B. Heijs, A. Koch, S. Vens-Cappell, J. Höhndorf and K. Dreisewerd, MALDI-2 on a Trapped Ion Mobility Quadrupole Time-of-Flight Instrument for Rapid Mass Spectrometry Imaging and Ion Mobility Separation of Complex Lipid Profiles, Anal. Chem., 2020, 92(13), 8697–8703 Search PubMed.
- R. Arevalo Jr, Z. Ni and R. M. Danell, Mass spectrometry and planetary exploration: A brief review and future projection, J. Mass Spectrom., 2020, 55(1), e4454–e4454 Search PubMed.
- P. Maitre, D. Scuderi, D. Corinti, B. Chiavarino, M. E. Crestoni and S. Fornarini, Applications of Infrared Multiple Photon Dissociation (IRMPD) to the Detection of Posttranslational Modifications, Chem. Rev., 2020, 120(7), 3261–3295 Search PubMed.
- A. Q. Stiving, Z. L. VanAernum, F. Busch, S. R. Harvey, S. H. Sarni and V. H. Wysocki, Surface-Induced Dissociation: An Effective Method for Characterization of Protein Quaternary Structure, Anal. Chem., 2019, 91(1), 190–209 Search PubMed.
- J. S. Brodbelt, L. J. Morrison and I. Santos, Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules, Chem. Rev., 2020, 120(7), 3328–3380 Search PubMed.
- N. M. Riley, M. S. Westphall, A. S. Hebert and J. J. Coon, Implementation of Activated Ion Electron Transfer Dissociation on a Quadrupole-Orbitrap-Linear Ion Trap Hybrid Mass Spectrometer, Anal. Chem., 2017, 89(12), 6358–6366 Search PubMed.
- P. F. Brandão, A. C. Duarte and R. M. B. O. Duarte, Comprehensive multidimensional liquid chromatography for advancing environmental and natural products research, TrAC, Trends Anal. Chem., 2019, 116, 186–197 Search PubMed.
- L. Ranjbar, J. P. Foley and M. C. Breadmore, Multidimensional liquid-phase separations combining both chromatography and electrophoresis – A review, Anal. Chim. Acta, 2017, 950, 7–31 Search PubMed.
- W. Lv, X. Shi, S. Wang and G. Xu, Multidimensional liquid chromatography-mass spectrometry for metabolomic and lipidomic analyses, TrAC, Trends Anal. Chem., 2019, 120, 115302 Search PubMed.
- A. Michalski, J. Cox and M. Mann, More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent LC-MS/MS, J. Proteome Res., 2011, 10(4), 1785–1793 Search PubMed.
- S.-E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics, Mol. Cell. Proteomics, 2002, 1(5), 376–386 Search PubMed.
- S.-E. Ong and M. Mann, Mass spectrometry–based proteomics turns quantitative, Nat. Chem. Biol., 2005, 1(5), 252–262 Search PubMed.
- M. Meselson and F. W. Stahl, The replication of DNA in Escherichia coli., Proc. Natl. Acad. Sci. U. S. A., 1958, 44(7), 671–682 Search PubMed.
- H. Zhu, T. C. Hunter, S. Pan, P. M. Yau, E. M. Bradbury and X. Chen, Residue-specific Mass Signatures for the Efficient Detection of Protein Modifications by Mass Spectrometry, Anal. Chem., 2002, 74(7), 1687–1694 Search PubMed.
- H.-T. Lau, H. W. Suh, M. Golkowski and S.-E. Ong, Comparing SILAC- and stable isotope dimethyl-labeling approaches for quantitative proteomics, J. Proteome Res., 2014, 13(9), 4164–4174 Search PubMed.
- D. Kovanich, S. Cappadona and R. Raijmakers, Mohammed, S.; Scholten, A.; Heck, A. J. R., Applications of stable isotope dimethyl labeling in quantitative proteomics, Anal. Bioanal. Chem., 2012, 404(4), 991–1009 Search PubMed.
- Y. Wu, F. Wang, Z. Liu, H. Qin, C. Song, J. Huang, Y. Bian, X. Wei, J. Dong and H. Zou, Five-plex isotope dimethyl labeling for quantitative proteomics, Chem. Commun., 2014, 50(14), 1708–1710 Search PubMed.
- F. Xiang, H. Ye, R. Chen, Q. Fu and L. Li, N,N-Dimethyl Leucines as Novel Isobaric Tandem Mass Tags for Quantitative Proteomics and Peptidomics, Anal. Chem., 2010, 82(7), 2817–2825 Search PubMed.
- S. Wiese, K. A. Reidegeld, H. E. Meyer and B. Warscheid, Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research, Proteomics, 2007, 7(3), 340–350 Search PubMed.
- A. Thompson, J. Schäfer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann and C. Hamon, Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS, Anal. Chem., 2003, 75(8), 1895–1904 Search PubMed.
- D. G. Delafield and L. Li, Recent Advances in Analytical Approaches for Glycan and Glycopeptide Quantitation, Mol. Cell. Proteomics, 2021, 20, 100054 Search PubMed.
- N. Rauniyar and J. R. Yates 3rd, Isobaric labeling-based relative quantification in shotgun proteomics, J. Proteome Res., 2014, 13(12), 5293–5309 Search PubMed.
- K. A. Neilson, N. A. Ali, S. Muralidharan, M. Mirzaei, M. Mariani, G. Assadourian, A. Lee, S. C. van Sluyter and P. A. Haynes, Less label, more free: approaches in label-free quantitative mass spectrometry, Proteomics, 2011, 11(4), 535–553 Search PubMed.
- M. A. Kuzyk, D. Smith, J. Yang, T. J. Cross, A. M. Jackson, D. B. Hardie, N. L. Anderson and C. H. Borchers, Multiple Reaction Monitoring-based, Multiplexed, Absolute Quantitation of 45 Proteins in Human Plasma*, Mol. Cell. Proteomics, 2009, 8(8), 1860–1877 Search PubMed.
- T. A. Addona, S. E. Abbatiello, B. Schilling, S. J. Skates, D. R. Mani, D. M. Bunk, C. H. Spiegelman, L. J. Zimmerman, A.-J. L. Ham, H. Keshishian, S. C. Hall, S. Allen, R. K. Blackman, C. H. Borchers, C. Buck, H. L. Cardasis, M. P. Cusack, N. G. Dodder, B. W. Gibson, J. M. Held, T. Hiltke, A. Jackson, E. B. Johansen, C. R. Kinsinger, J. Li, M. Mesri, T. A. Neubert, R. K. Niles, T. C. Pulsipher, D. Ransohoff, H. Rodriguez, P. A. Rudnick, D. Smith, D. L. Tabb, T. J. Tegeler, A. M. Variyath, L. J. Vega-Montoto, Å. Wahlander, S. Waldemarson, M. Wang, J. R. Whiteaker, L. Zhao, N. L. Anderson, S. J. Fisher, D. C. Liebler, A. G. Paulovich, F. E. Regnier, P. Tempst and S. A. Carr, Multi-site assessment of the precision and reproducibility of multiple reaction monitoring–based measurements of proteins in plasma, Nat. Biotechnol., 2009, 27(7), 633–641 Search PubMed.
- A. A. Dowle, J. Wilson and J. R. Thomas, Comparing the Diagnostic Classification Accuracy of iTRAQ, Peak-Area, Spectral-Counting, and emPAI Methods for Relative Quantification in Expression Proteomics, J. Proteome Res., 2016, 15(10), 3550–3562 Search PubMed.
- SEER*Explorer: An interactive website for SEER cancer statistics. Surveillance Research Program. https://seer.cancer.gov/explorer/. (accessed Dec. 4th).
- M. Kirby, C. Hirst and E. D. Crawford, Characterising the castration-resistant prostate cancer population: a systematic review, Int. J. Clin. Pract., 2011, 65(11), 1180–1192 Search PubMed.
- J. E. Vellky and W. A. Ricke, Development and prevalence of castration-resistant prostate cancer subtypes, Neoplasia, 2020, 22(11), 566–575 Search PubMed.
- Y. Zhang, D. Wang, M. Li, X. Wei, S. Liu, M. Zhao, C. Liu, X. Wang, X. Jiang, X. Li, S. Zhang, J. Bergquist, B. Wang, C. Yang, J. Mi and G. Tian, Quantitative Proteomics of TRAMP Mice Combined with Bioinformatics Analysis Reveals That PDGF-B Regulatory Network Plays a Key Role in Prostate Cancer Progression, J. Proteome Res., 2018, 17(7), 2401–2411 Search PubMed.
- A.-K. Müller, M. Föll, B. Heckelmann, S. Kiefer, M. Werner, O. Schilling, M. L. Biniossek, C. A. Jilg and V. Drendel, Proteomic Characterization of Prostate Cancer to Distinguish Nonmetastasizing and Metastasizing Primary Tumors and Lymph Node Metastases, Neoplasia, 2018, 20(2), 140–151 Search PubMed.
- D. J. Clark, M. Schnaubelt, N. Hoti, Y. Hu, Y. Zhou, M. Gooya and H. Zhang, Impact of Increased FUT8 Expression on the Extracellular Vesicle Proteome in Prostate Cancer Cells, J. Proteome Res., 2020, 19(6), 2195–2205 Search PubMed.
- X. Wang, J. Chen, Q. K. Li, S. B. Peskoe, B. Zhang, C. Choi, E. A. Platz and H. Zhang, Overexpression of α (1,6) fucosyltransferase associated with aggressive prostate cancer, Glycobiology, 2014, 24(10), 935–944 Search PubMed.
- N. Höti, T.-S. Lih, J. Pan, Y. Zhou, G. Yang, A. Deng, L. Chen, M. Dong, R.-B. Yang, C.-F. Tu and M. C. Haffner, Kay Li, Q.; Zhang, H., A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance, Cancers, 2020, 12(2), 468 Search PubMed.
- W. Miao, J. Yuan, L. Li and Y. Wang, Parallel-Reaction-Monitoring-Based Proteome-Wide Profiling of Differential Kinase Protein Expression during Prostate Cancer Metastasis in Vitro, Anal. Chem., 2019, 91(15), 9893–9900 Search PubMed.
- C. S. Collins, J. Hong, L. Sapinoso, Y. Zhou, Z. Liu, K. Micklash, P. G. Schultz and G. M. Hampton, A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(10), 3775–3780 Search PubMed.
- D. Sbrissa, L. Semaan, B. Govindarajan, Y. Li, N. J. Caruthers, P. M. Stemmer, M. L. Cher, S. Sethi, U. Vaishampayan, A. Shisheva and S. R. Chinni, A novel cross-talk between CXCR4 and PI4KIIIα in prostate cancer cells, Oncogene, 2019, 38(3), 332–344 Search PubMed.
- J. K. Lee, N. J. Bangayan, T. Chai, B. A. Smith, T. E. Pariva, S. Yun, A. Vashisht, Q. Zhang, J. W. Park, E. Corey, J. Huang, T. G. Graeber, J. Wohlschlegel and O. N. Witte, Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(19), E4473–E4482 Search PubMed.
- J. Zhou, W. Yang, Y. Hu, N. Höti, Y. Liu, P. Shah, S. Sun, D. Clark, S. Thomas and H. Zhang, Site-Specific Fucosylation Analysis Identifying Glycoproteins Associated with Aggressive Prostate Cancer Cell Lines Using Tandem Affinity Enrichments of Intact Glycopeptides Followed by Mass Spectrometry, Anal. Chem., 2017, 89(14), 7623–7630 Search PubMed.
- B. Zhou, Y. Yan, Y. Wang, S. You, M. R. Freeman and W. Yang, Quantitative proteomic analysis of prostate tissue specimens identifies deregulated protein complexes in primary prostate cancer, Clin. Proteomics, 2019, 16(1), 15 Search PubMed.
- N. Höti, P. Shah, Y. Hu, S. Yang and H. Zhang, Proteomics analyses of prostate cancer cells reveal cellular pathways associated with androgen resistance, Proteomics, 2017, 17(6), 1600228 Search PubMed.
- S. Zhang, C. Zheng, S. Yao, Z. Wang, L. Xu, R. Yang, X. Meng, J. Wu, L. Zhou and Z. Sun, Proteomic analysis of human prostate cancer PC-3M-1E8 cells and PC-3M-2B4 cells of same origin but with different metastatic potential, PLoS One, 2018, 13(10), e0206139 Search PubMed.
- J. P. Webber, L. K. Spary, M. D. Mason, Z. Tabi, I. A. Brewis and A. Clayton, Prostate stromal cell proteomics analysis discriminates normal from tumour reactive stromal phenotypes, Oncotarget, 2016, 7(15), 20124–20139 Search PubMed.
- S. Wang, J. Gao, Q. Lei, N. Rozengurt, C. Pritchard, J. Jiao, G. V. Thomas, G. Li, P. Roy-Burman, P. S. Nelson, X. Liu and H. Wu, Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer, Cancer Cell, 2003, 4(3), 209–221 Search PubMed.
- J. Zhang, S. Kim, L. Li, C. J. Kemp, C. Jiang and J. Lü, Proteomic and transcriptomic profiling of Pten gene-knockout mouse model of prostate cancer, Prostate, 2020, 80(7), 588–605 Search PubMed.
- P. J. Hensley and N. Kyprianou, Modeling prostate cancer in mice: limitations and opportunities, J. Androl., 2012, 33(2), 133–144 Search PubMed.
- T. T. Liu, J. A. Ewald, E. A. Ricke, R. Bell, C. Collins and W. A. Ricke, Modeling human prostate cancer progression in vitro, Carcinogenesis, 2019, 40(7), 893–902 Search PubMed.
- K. Davalieva, S. Kiprijanovska, I. M. Kostovska, S. Stavridis, O. Stankov, S. Komina, G. Petrusevska and M. Polenakovic, Comparative proteomics analysis of urine reveals down-regulation of acute phase response signaling and LXR/RXR activation pathways in prostate cancer, Proteomes, 2018, 6(1), 1–25 Search PubMed.
- C. Soekmadji, J. D. Riches, P. J. Russell, J. E. Ruelcke, S. McPherson, C. Wang, C. M. Hovens, N. M. Corcoran, M. M. Hill and C. C. Nelson, Modulation of paracrine signaling by CD9 positive small extracellular vesicles mediates cellular growth of androgen deprived prostate cancer, Oncotarget, 2017, 8(32), 52237–52255 Search PubMed.
- T. Sequeiros, M. Rigau, C. Chiva, M. Montes, I. Garcia-Grau, M. Garcia, S. Diaz, A. Celma, I. Bijnsdorp, A. Campos, P. Di Mauro, S. Borrós, J. Reventós, A. Doll, R. Paciucci, M. Pegtel, I. de Torres, E. Sabidó, J. Morote and M. Olivan, Targeted proteomics in urinary extracellular vesicles identifies biomarkers for diagnosis and prognosis of prostate cancer, Oncotarget, 2017, 8(3), 4960–4976 Search PubMed.
- Y. Kim, J. Jeon, S. Mejia, C. Q. Yao, V. Ignatchenko, J. O. Nyalwidhe, A. O. Gramolini, R. S. Lance, D. A. Troyer, R. R. Drake, P. C. Boutros, O. J. Semmes and T. Kislinger, Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer, Nat. Commun., 2016, 7(1), 11906 Search PubMed.
- T. D. Karakosta, A. Soosaipillai, E. P. Diamandis, I. Batruch and A. P. Drabovich, Quantification of Human Kallikrein-Related Peptidases in Biological Fluids by Multi-Platform Targeted Mass Spectrometry Assays, Mol. Cell. Proteomics, 2016, 15(9), 2863–2876 Search PubMed.
- K. Fujita, H. Kume, K. Matsuzaki, A. Kawashima, T. Ujike, A. Nagahara, M. Uemura, Y. Miyagawa, T. Tomonaga and N. Nonomura, Proteomic analysis of urinary extracellular vesicles from high Gleason score prostate cancer, Sci. Rep., 2017, 7(1), 42961 Search PubMed.
- J. I. Epstein, L. Egevad, M. B. Amin, B. Delahunt, J. R. Srigley and P. A. Humphrey, The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System, Am. J. Surg. Pathol., 2016, 40(2), 244–252 Search PubMed.
- B. Yan, B. Chen, S. Min, Y. Gao, Y. Zhang, P. Xu, C. Li, J. Chen, G. Luo and C. Liu, iTRAQ-based Comparative Serum Proteomic Analysis of Prostate Cancer Patients with or without Bone Metastasis, J. Cancer, 2019, 10(18), 4165–4177 Search PubMed.
- S. E. T. Larkin, H. E. Johnston, T. R. Jackson, D. G. Jamieson, T. I. Roumeliotis, C. I. Mockridge, A. Michael, A. Manousopoulou, E. K. Papachristou, M. D. Brown, N. W. Clarke, H. Pandha, C. L. Aukim-Hastie, M. S. Cragg, S. D. Garbis and P. A. Townsend, Detection of candidate biomarkers of prostate cancer progression in serum: a depletion-free 3D LC/MS quantitative proteomics pilot study, Br. J. Cancer, 2016, 115(9), 1078–1086 Search PubMed.
- S. Loeb, M. A. Bjurlin, J. Nicholson, T. L. Tammela, D. F. Penson, H. B. Carter, P. Carroll and R. Etzioni, Overdiagnosis and overtreatment of prostate cancer, Eur. Urol., 2014, 65(6), 1046–1055 Search PubMed.
- P. Rawla, T. Sunkara and V. Gaduputi, Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors, World J. Oncol., 2019, 10(1), 10–27 Search PubMed.
- S. Rauth, S. Karmakar, A. Shah, R. K. Nimmakayala, R. Bhatia, S. Muniyan, S. Kumar, S. Dutta, K. Datta, S. K. Batra and M. P. Ponnusamy, Abstract 4438: Role of post translational modification of PAF1/PD2 in gemcitabine resistance of pancreatic cancer, Cancer Res., 2019, 79(13 Suppl.), 4438 Search PubMed.
- D. Ansari, W. Torén, Q. Zhou, D. Hu and R. Andersson, Proteomic and genomic profiling of pancreatic cancer, Cell Biol. Toxicol., 2019, 35(4), 333–343 Search PubMed.
- S.-z. Yang, F. Xu, K. Yuan, Y. Sun, T. Zhou, X. Zhao, J. M. McDonald and Y. Chen, Regulation of pancreatic cancer TRAIL resistance by protein O-GlcNAcylation, Lab. Invest., 2020, 100(5), 777–785 Search PubMed.
- P. Liu, Y. Weng, Z. Sui, Y. Wu, X. Meng, M. Wu, H. Jin, X. Tan, L. Zhang and Y. Zhang, Quantitative secretomic analysis of pancreatic cancer cells in serum-containing conditioned medium, Sci. Rep., 2016, 6(1), 37606 Search PubMed.
- B. Marchand, D. Arsenault, A. Raymond-Fleury, F.-M. Boisvert and M.-J. Boucher, Glycogen Synthase Kinase-3 (GSK3) Inhibition Induces Prosurvival Autophagic Signals in Human Pancreatic Cancer Cells, J. Biol. Chem., 2015, 290(9), 5592–5605 Search PubMed.
- Y. Shi, W. Gao, N. K. Lytle, P. Huang, X. Yuan, A. M. Dann, M. Ridinger-Saison, K. E. DelGiorno, C. E. Antal, G. Liang, A. R. Atkins, G. Erikson, H. Sun, J. Meisenhelder, E. Terenziani, G. Woo, L. Fang, T. P. Santisakultarm, U. Manor, R. Xu, C. R. Becerra, E. Borazanci, D. D. Von Hoff, P. M. Grandgenett, M. A. Hollingsworth, M. Leblanc, S. E. Umetsu, E. A. Collisson, M. Scadeng, A. M. Lowy, T. R. Donahue, T. Reya, M. Downes, R. M. Evans, G. M. Wahl, T. Pawson, R. Tian and T. Hunter, Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring, Nature, 2019, 569(7754), 131–135 Search PubMed.
- A. M. Roberts, D. K. Miyamoto, T. R. Huffman, L. A. Bateman, A. N. Ives, D. Akopian, M. J. Heslin, C. M. Contreras, M. Rape, C. F. Skibola and D. K. Nomura, Chemoproteomic Screening of Covalent Ligands Reveals UBA5 As a Novel Pancreatic Cancer Target, ACS Chem. Biol., 2017, 12(4), 899–904 Search PubMed.
- C.-W. Liu and Q. Zhang, Isobaric Labeling-Based LC-MS/MS Strategy for Comprehensive Profiling of Human Pancreatic Tissue Proteome BT, in Tissue Proteomics: Methods and Protocols, ed. M. M. Sarwal, T. K. Sigdel, M. M. Sarwal and T. K. Sigdel, New York, NY, 2018, pp. 215–224 Search PubMed.
- R. M. Perera, S. Stoykova, B. N. Nicolay, K. N. Ross, J. Fitamant, M. Boukhali, J. Lengrand, V. Deshpande, M. K. Selig, C. R. Ferrone, J. Settleman, G. Stephanopoulos, N. J. Dyson, R. Zoncu, S. Ramaswamy, W. Haas and N. Bardeesy, Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism, Nature, 2015, 524(7565), 361–365 Search PubMed.
- M. An, I. Lohse, Z. Tan, J. Zhu, J. Wu, H. Kurapati, M. A. Morgan, T. S. Lawrence, K. C. Cuneo and D. M. Lubman, Quantitative Proteomic Analysis of Serum Exosomes from Patients with Locally Advanced Pancreatic Cancer Undergoing Chemoradiotherapy, J. Proteome Res., 2017, 16(4), 1763–1772 Search PubMed.
- H. Li, Y. Mao, Y. Xiong, H. H. Zhao, F. Shen, X. Gao, P. Yang, X. Liu and D. Fu, A Comprehensive Proteome Analysis of Peripheral Blood Mononuclear Cells (PBMCs) to Identify Candidate Biomarkers of Pancreatic Cancer, Cancer Genomics Proteomics, 2019, 16(1), 81–89 Search PubMed.
- D. C. Frost, T. Greer, F. Xiang, Z. Liang and L. Li, Development and characterization of novel 8-plex DiLeu isobaric labels for quantitative proteomics and peptidomics, Rapid Commun. Mass Spectrom., 2015, 29(12), 1115–1124 Search PubMed.
- D. C. Frost, C. J. Rust, R. A. S. Robinson and L. Li, Increased N,N-Dimethyl Leucine Isobaric Tag Multiplexing by a Combined Precursor Isotopic Labeling and Isobaric Tagging Approach, Anal. Chem., 2018, 90(18), 10664–10669 Search PubMed.
- D. C. Frost, T. Greer and L. Li, High-resolution enabled 12-plex DiLeu isobaric tags for quantitative proteomics, Anal. Chem., 2015, 87(3), 1646–1654 Search PubMed.
- T. Greer, C. B. Lietz, F. Xiang and L. Li, Novel isotopic N,N-dimethyl leucine (iDiLeu) reagents enable absolute quantification of peptides and proteins using a standard curve approach, J. Am. Soc. Mass Spectrom, 2015, 26(1), 107–119 Search PubMed.
- X. Zhong, D. C. Frost and L. Li, High-Resolution Enabled 5-plex Mass Defect-Based N,N-Dimethyl Leucine Tags for Quantitative Proteomics, Anal. Chem., 2019, 91(13), 7991–7995 Search PubMed.
- X. Zhong, Q. Yu, F. Ma, D. C. Frost, L. Lu, Z. Chen, H. Zetterberg, C. M. Carlsson, O. Okonkwo and L. Li, HOTMAQ: a multiplexed absolute quantification method for targeted proteomics, Anal. Chem., 2019, 91(3), 2112–2119 Search PubMed.
- L. Hao, J. Johnson, C. B. Lietz, A. Buchberger, D. Frost, W. J. Kao and L. Li, Mass Defect-Based N,N-Dimethyl Leucine Labels for Quantitative Proteomics and Amine Metabolomics of Pancreatic Cancer Cells, Anal. Chem., 2017, 89(2), 1138–1146 Search PubMed.
- X. Wang, J. Niu, J. Li, X. Shen, S. Shen, R. M. Straubinger and J. Qu, Temporal effects of combined birinapant and paclitaxel on pancreatic cancer cells investigated via large-scale, ion-current-based quantitative proteomics (IonStar), Mol. Cell. Proteomics, 2018, 17(4), 655–671 Search PubMed.
- X. Zhu, X. Shen, J. Qu, R. M. Straubinger and W. J. Jusko, Multi-Scale Network Model Supported by Proteomics for Analysis of Combined Gemcitabine and Birinapant Effects in Pancreatic Cancer Cells, CPT: Pharmacometrics Syst. Pharmacol., 2018, 7(9), 549–561 Search PubMed.
- C. K. Singh, S. Kaur, J. George, M. Nihal, M. C. Pellitteri Hahn, C. O. Scarlett and N. Ahmad, Molecular signatures of sanguinarine in human pancreatic cancer cells: A large scale label-free comparative proteomics approach, Oncotarget, 2015, 6(12), 10335–10348 Search PubMed.
- Q. Zhou, R. Andersson, D. Hu, M. Bauden, T. Kristl, A. Sasor, K. Pawłowski, I. Pla, K. S. Hilmersson, M. Zhou, F. Lu, G. Marko-Varga and D. Ansari, Quantitative proteomics identifies brain acid soluble protein 1 (BASP1) as a prognostic biomarker candidate in pancreatic cancer tissue, EBioMedicine, 2019, 43, 282–294 Search PubMed.
- D. T. Jhaveri, M.-S. Kim, E. D. Thompson, L. Huang, R. Sharma, A. P. Klein, L. Zheng, D. T. Le, D. A. Laheru, A. Pandey, E. M. Jaffee and R. A. Anders, Using Quantitative Seroproteomics to Identify Antibody Biomarkers in Pancreatic Cancer, Cancer Immunol. Res., 2016, 4(3), 225–233 Search PubMed.
- K. Sogawa, S. Takano, F. Iida, M. Satoh, S. Tsuchida, Y. Kawashima, H. Yoshitomi, A. Sanda, Y. Kodera, H. Takizawa, R. Mikata, M. Ohtsuka, H. Shimizu, M. Miyazaki, O. Yokosuka and F. Nomura, Identification of a novel serum biomarker for pancreatic cancer, C4b-binding protein α-chain (C4BPA) by quantitative proteomic analysis using tandem mass tags, Br. J. Cancer, 2016, 115(8), 949–956 Search PubMed.
- A. Naba, K. R. Clauser, D. R. Mani, S. A. Carr and R. O. Hynes, Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression, Sci. Rep., 2017, 7(1), 40495 Search PubMed.
- Z. Yu, S. Zhao, L. Ren, L. Wang, Z. Chen, R. M. Hoffman and J. Zhou, Pancreatic cancer-derived exosomes promote tumor metastasis and liver pre-metastatic niche formation, Oncotarget, 2017, 8(38), 63461–63483 Search PubMed.
- C. Lin, W.-C. Wu, G.-C. Zhao, D.-S. Wang, W.-H. Lou and D.-Y. Jin, ITRAQ-based quantitative proteomics reveals apolipoprotein A-I and transferrin as potential serum markers in CA19-9 negative pancreatic ductal adenocarcinoma, Medicine, 2016, 95(31), e4527–e4527 Search PubMed.
- X. Liu, W. Zheng, W. Wang, H. Shen, L. Liu, W. Lou, X. Wang and P. Yang, A new panel of pancreatic cancer biomarkers discovered using a mass spectrometry-based pipeline, Br. J. Cancer, 2017, 117(12), 1846–1854 Search PubMed.
- K. Ohmine, K. Kawaguchi, S. Ohtsuki, F. Motoi, H. Ohtsuka, J. Kamiie, T. Abe, M. Unno and T. Terasaki, Quantitative Targeted Proteomics of Pancreatic Cancer: Deoxycytidine Kinase Protein Level Correlates to Progression-Free Survival of Patients Receiving Gemcitabine Treatment, Mol. Pharmaceutics, 2015, 12(9), 3282–3291 Search PubMed.
- T. Yoneyama, S. Ohtsuki, K. Honda, M. Kobayashi, M. Iwasaki, Y. Uchida, T. Okusaka, S. Nakamori, M. Shimahara, T. Ueno, A. Tsuchida, N. Sata, T. Ioka, Y. Yasunami, T. Kosuge, T. Kaneda, T. Kato, K. Yagihara, S. Fujita, W. Huang, T. Yamada, M. Tachikawa and T. Terasaki, Identification of IGFBP2 and IGFBP3 As Compensatory Biomarkers for CA19-9 in Early-Stage Pancreatic Cancer Using a Combination of Antibody-Based and LC-MS/MS-Based Proteomics, PLoS One, 2016, 11(8), e0161009 Search PubMed.
- J. Park, E. Lee, K.-J. Park, H.-D. Park, J.-W. Kim, H. I. Woo, K. H. Lee, K.-T. Lee, J. K. Lee, J.-O. Park, Y. S. Park, J. S. Heo, S. H. Choi, D. W. Choi, K.-T. Jang and S.-Y. Lee, Large-scale clinical validation of biomarkers for pancreatic cancer using a mass spectrometry-based proteomics approach, Oncotarget, 2017, 8(26), 42761–42771 Search PubMed.
- M. Do, D. Han, J. I. Wang, H. Kim, W. Kwon, Y. Han, J. Y. Jang and Y. Kim, Quantitative proteomic analysis of pancreatic cyst fluid proteins associated with malignancy in intraductal papillary mucinous neoplasms, Clin. Proteomics, 2018, 15(1), 1–22 Search PubMed.
- E. N. Nigjeh, R. Chen, R. E. Brand, G. M. Petersen, S. T. Chari, P. D. von Haller, J. K. Eng, Z. Feng, Q. Yan, T. A. Brentnall and S. Pan, Quantitative Proteomics Based on Optimized Data-Independent Acquisition in Plasma Analysis, J. Proteome Res., 2017, 16(2), 665–676 Search PubMed.
- O. M. Ginsburg and R. R. Love, Breast cancer: a neglected disease for the majority of affected women worldwide, Breast J., 2011, 17(3), 289–295 Search PubMed.
- S. Miah, C. A. Banks, M. K. Adams, L. Florens, K. E. Lukong and M. P. Washburn, Advancement of mass spectrometry-based proteomics technologies to explore triple negative breast cancer, Mol. BioSyst., 2016, 13(1), 42–55 Search PubMed.
- R. M. Sallam, Proteomics in cancer biomarkers discovery: challenges and applications, Dis. Markers, 2015, 2015, 321370 Search PubMed.
- S. Tyanova, R. Albrechtsen, P. Kronqvist, J. Cox, M. Mann and T. Geiger, Proteomic maps of breast cancer subtypes, Nat. Commun., 2016, 7(1), 10259 Search PubMed.
- S. Suman, T. Basak, P. Gupta, S. Mishra, V. Kumar, S. Sengupta and Y. Shukla, Quantitative proteomics revealed novel proteins associated with molecular subtypes of breast cancer, J. Proteomics, 2016, 148, 183–193 Search PubMed.
- K. G. Calderón-González, M. L. Valero Rustarazo, M. L. Labra-Barrios, C. I. Bazán-Méndez, A. Tavera-Tapia, M. E. Herrera-Aguirre, M. M. Sánchez del Pino, J. L. Gallegos-Pérez, H. González-Márquez, J. M. Hernández-Hernández, G. León-Ávila, S. Rodríguez-Cuevas, F. Guisa-Hohenstein and J. P. Luna-Arias, Determination of the protein expression profiles of breast cancer cell lines by quantitative proteomics using iTRAQ labelling and tandem mass spectrometry, J. Proteomics, 2015, 124, 50–78 Search PubMed.
- A. Gajbhiye, R. Dabhi, K. Taunk, G. Vannuruswamy, S. RoyChoudhury, R. Adhav, S. Seal, A. Mane, S. Bayatigeri, M. K. Santra, K. Chaudhury and S. Rapole, Urinary proteome alterations in HER2 enriched breast cancer revealed by multipronged quantitative proteomics, Proteomics, 2016, 16(17), 2403–2418 Search PubMed.
- C. C. Going, D. Tailor, V. Kumar, A. M. Birk, M. Pandrala, M. A. Rice, T. Stoyanova, S. Malhotra and S. J. Pitteri, Quantitative Proteomic Profiling Reveals Key Pathways in the Anticancer Action of Methoxychalcone Derivatives in Triple Negative Breast Cancer, J. Proteome Res., 2018, 17(10), 3574–3585 Search PubMed.
- D. J. Clark, W. E. Fondrie, Z. Liao, P. I. Hanson, A. Fulton, L. Mao and A. J. Yang, Redefining the Breast Cancer Exosome Proteome by Tandem Mass Tag Quantitative Proteomics and Multivariate Cluster Analysis, Anal. Chem., 2015, 87(20), 10462–10469 Search PubMed.
- F. Liu, F. Ma, Y. Wang, L. Hao, H. Zeng, C. Jia, Y. Wang, P. Liu, I. M. Ong, B. Li, G. Chen, J. Jiang, S. Gong, L. Li and W. Xu, PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis, Nat. Cell Biol., 2017, 19(11), 1358–1370 Search PubMed.
- H. J. Johansson, F. Socciarelli, N. M. Vacanti, M. H. Haugen, Y. Zhu, I. Siavelis, A. Fernandez-Woodbridge, M. R. Aure, B. Sennblad, M. Vesterlund, R. M. Branca, L. M. Orre, M. Huss, E. Fredlund, E. Beraki, Ø. Garred, J. Boekel, T. Sauer, W. Zhao, S. Nord, E. K. Höglander, D. C. Jans, H. Brismar, T. H. Haukaas, T. F. Bathen, E. Schlichting, B. Naume, J. Geisler, S. Hofvind, O. Engebråten, G. A. Geitvik, A. Langerød, R. Kåresen, G. M. Mælandsmo, T. Sørlie, H. K. Skjerven, D. Park, O.-J. Hartman-Johnsen, T. Luders, E. Borgen, V. N. Kristensen, H. G. Russnes, O. C. Lingjærde, G. B. Mills, K. K. Sahlberg, A.-L. Børresen-Dale and J. Lehtiö, Consortia Oslo Breast Cancer Research, C., Breast cancer quantitative proteome and proteogenomic landscape, Nat. Commun., 2019, 10(1), 1600 Search PubMed.
- J.-Y. Zhou, L. Chen, B. Zhang, Y. Tian, T. Liu, S. N. Thomas, L. Chen, M. Schnaubelt, E. Boja, T. Hiltke, C. R. Kinsinger, H. Rodriguez, S. R. Davies, S. Li, J. E. Snider, P. Erdmann-Gilmore, D. L. Tabb, R. R. Townsend, M. J. Ellis, K. D. Rodland, R. D. Smith, S. A. Carr, Z. Zhang, D. W. Chan and H. Zhang, Quality Assessments of Long-Term Quantitative Proteomic Analysis of Breast Cancer Xenograft Tissues, J. Proteome Res., 2017, 16(12), 4523–4530 Search PubMed.
- I. Ntai, R. D. LeDuc, R. T. Fellers, P. Erdmann-Gilmore, S. R. Davies, J. Rumsey, B. P. Early, P. M. Thomas, S. Li, P. D. Compton, M. J. C. Ellis, V. K. Ruggles, D. Fenyö, E. S. Boja, H. Rodriguez, R. R. Townsend and N. L. Kelleher, Integrated bottom-up and top-down proteomics of patient-derived breast tumor xenografts, Mol. Cell. Proteomics, 2016, 15(1), 45–56 Search PubMed.
- M. K. Tveitarås, F. Selheim, K. Sortland, R. K. Reed and L. Stuhr, Protein expression profiling of plasma and lungs at different stages of metastatic development in a human triple negative breast cancer xenograft model, PLoS One, 2019, 14(5), 1–16 Search PubMed.
- Z. Wang, H. Liu, Y. Yan, X. Yang, Y. Zhang and L. Wu, Integrated Proteomic and N-Glycoproteomic Analyses of Human Breast Cancer, J. Proteome Res., 2020, 19, 3499–3509 Search PubMed.
- A. Gámez-Pozo, J. Berges-Soria, J. M. Arevalillo, P. Nanni, R. López-Vacas, H. Navarro, J. Grossmann, C. A. Castaneda, P. Main, M. Díaz-Almirón, E. Espinosa and E. Ciruelos, Fresno Vara, J. Á., Combined Label-Free Quantitative Proteomics and microRNA Expression Analysis of Breast Cancer Unravel Molecular Differences with Clinical Implications, Cancer Res., 2015, 75(11), 2243–2253 Search PubMed.
- S. Nie, S. P. McDermott, Y. Deol, Z. Tan, M. S. Wicha and D. M. Lubman, A quantitative proteomics analysis of MCF7 breast cancer stem and progenitor cell populations, Proteomics, 2015, 15(22), 3772–3783 Search PubMed.
- M. Warmoes, S. W. Lam, P. van der Groep, J. E. Jaspers, Y. H. C. M. Smolders, D. L. Boer, V. T. Pham, S. R. Piersma, S. Rottenberg, E. Boven, J. Jonkers, P. J. van Diest and C. R. Jimenez, Secretome proteomics reveals candidate non-invasive biomarkers of BRCA1 deficiency in breast cancer, Oncotarget, 2016, 7(39), 63537–63548 Search PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2020, Ca-Cancer J. Clin., 2020, 70(1), 7–30 Search PubMed.
- N. Musrap, A. Tuccitto, G. S. Karagiannis, P. Saraon, I. Batruch and E. P. Diamandis, Comparative proteomics of ovarian cancer aggregate formation reveals an increased expression of calcium-activated chloride channel regulator 1 (CLCA1), J. Biol. Chem., 2015, 290(28), 17218–17227 Search PubMed.
- M. L. Grassi, C. d. S. Palma, C. H. Thomé, G. P. Lanfredi, A. Poersch and V. M. Faça, Proteomic analysis of ovarian cancer cells during epithelial-mesenchymal transition (EMT) induced by epidermal growth factor (EGF) reveals mechanisms of cell cycle control, J. Proteomics, 2017, 151, 2–11 Search PubMed.
- Y. Ji, S. Wei, J. Hou, C. Zhang, P. Xue, J. Wang, X. Chen, X. Guo and F. Yang, Integrated proteomic and N-glycoproteomic analyses of doxorubicin sensitive and resistant ovarian cancer cells reveal glycoprotein alteration in protein abundance and glycosylation, Oncotarget, 2017, 8(8), 13413–13427 Search PubMed.
- H. Zhang, T. Liu, Z. Zhang, S. H. Payne, B. Zhang, J. E. McDermott, J. Y. Zhou, V. A. Petyuk, L. Chen, D. Ray, S. Sun, F. Yang, L. Chen, J. Wang, P. Shah, S. W. Cha, P. Aiyetan, S. Woo, Y. Tian, M. A. Gritsenko, T. R. Clauss, C. Choi, M. E. Monroe, S. Thomas, S. Nie, C. Wu, R. J. Moore, K. H. Yu, D. L. Tabb, D. Fenyö, V. Vineet, Y. Wang, H. Rodriguez, E. S. Boja, T. Hiltke, R. C. Rivers, L. Sokoll, H. Zhu, I. M. Shih, L. Cope, A. Pandey, B. Zhang, M. P. Snyder, D. A. Levine, R. D. Smith, D. W. Chan, K. D. Rodland, S. A. Carr, M. A. Gillette, K. R. Klauser, E. Kuhn, D. R. Mani, P. Mertins, K. A. Ketchum, R. Thangudu, S. Cai, M. Oberti, A. G. Paulovich, J. R. Whiteaker, N. J. Edwards, P. B. McGarvey, S. Madhavan, P. Wang, G. A. Whiteley, S. J. Skates, F. M. White, C. R. Kinsinger, M. Mesri, K. M. Shaw, S. E. Stein, D. Fenyo, P. Rudnick, M. Snyder, Y. Zhao, X. Chen, D. F. Ransohoff, A. N. Hoofnagle, D. C. Liebler, M. E. Sanders, Z. Shi, R. J. C. Slebos, L. J. Zimmerman, S. R. Davies, L. Ding, M. J. C. Ellis and R. R. Townsend, Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer, Cell, 2016, 166(3), 755–765 Search PubMed.
- K. Hiramatsu, K. Yoshino, S. Serada, K. Yoshihara, Y. Hori, M. Fujimoto, S. Matsuzaki, T. Egawa-Takata, E. Kobayashi, Y. Ueda, E. Morii, T. Enomoto, T. Naka and T. Kimura, Similar protein expression profiles of ovarian and endometrial high-grade serous carcinomas, Br. J. Cancer, 2016, 114(5), 554–561 Search PubMed.
- Y. Hu, J. Pan, P. Shah, M. Ao, S. N. Thomas, Y. Liu, L. Chen, M. Schnaubelt, D. J. Clark, H. Rodriguez, E. S. Boja, T. Hiltke, C. R. Kinsinger, K. D. Rodland, Q. K. Li, J. Qian, Z. Zhang, D. W. Chan, H. Zhang, A. Pandey, A. Paulovich, A. Hoofnagle, B. Zhang, D. R. Mani, D. C. Liebler, D. F. Ransohoff, D. Fenyo, D. L. Tabb, D. A. Levine, E. Kuhn, F. M. White, G. A. Whiteley, H. Zhu, I.-M. Shih, J. Bavarva, J. E. McDermott, J. Whiteaker, K. A. Ketchum, K. R. Clauser, K. Ruggles, K. Elburn, L. Ding, L. Hannick, L. J. Zimmerman, M. Watson, M. Thiagarajan, M. J. C. Ellis, M. Oberti, M. Mesri, M. E. Sanders, M. Borucki, M. A. Gillette, M. Snyder, N. J. Edwards, N. Vatanian, P. A. Rudnick, P. B. McGarvey, P. Mertins, R. R. Townsend, R. R. Thangudu, R. D. Smith, R. C. Rivers, R. J. C. Slebos, S. H. Payne, S. R. Davies, S. Cai, S. E. Stein, S. A. Carr, S. J. Skates, S. Madhavan, T. Liu, X. Chen, Y. Zhao, Y. Wang and Z. Shi, Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma, Cell Rep., 2020, 33(3), 108276 Search PubMed.
- A. Yoshimura, K. Sawada, K. Nakamura, Y. Kinose, E. Nakatsuka, M. Kobayashi, M. Miyamoto, K. Ishida, Y. Matsumoto, M. Kodama, K. Hashimoto, S. Mabuchi and T. Kimura, Exosomal miR-99a-5p is elevated in sera of ovarian cancer patients and promotes cancer cell invasion by increasing fibronectin and vitronectin expression in neighboring peritoneal mesothelial cells, BMC Cancer, 2018, 18(1), 1–13 Search PubMed.
- H. Qu, Y. Chen, G. Cao, C. Liu, J. Xu, H. Deng and Z. Zhang, Identification and validation of differentially expressed proteins in epithelial ovarian cancers using quantitative proteomics, Oncotarget, 2016, 7(50), 83187–83199 Search PubMed.
- J. E. McDermott, O. A. Arshad, V. A. Petyuk, Y. Fu, M. A. Gritsenko, T. R. Clauss, R. J. Moore, A. A. Schepmoes, R. Zhao, M. E. Monroe, M. Schnaubelt, C.-F. Tsai, S. H. Payne, C. Huang, L.-B. Wang, S. Foltz, M. Wyczalkowski, Y. Wu, E. Song, M. A. Brewer, M. Thiagarajan, C. R. Kinsinger, A. I. Robles, E. S. Boja, H. Rodriguez, D. W. Chan, B. Zhang, Z. Zhang, L. Ding, R. D. Smith, T. Liu and K. D. Rodland, Consortium, C. T. A., Proteogenomic Characterization of Ovarian HGSC Implicates Mitotic Kinases, Replication Stress in Observed Chromosomal Instability, Cell Rep. Med., 2020, 1(1), 100004 Search PubMed.
- L. G. A. Chuffa, L. A. Lupi Júnior, F. R. F. Seiva, M. Martinez, R. F. Domeniconi, P. F. F. Pinheiro, L. D. dos Santos and F. E. Martinez, Quantitative Proteomic Profiling Reveals That Diverse Metabolic Pathways Are Influenced by Melatonin in an in Vivo Model of Ovarian Carcinoma, J. Proteome Res., 2016, 15(10), 3872–3882 Search PubMed.
- L. A. L. Júnior, M. S. Cucielo, R. F. Domeniconi, L. D. Dos Santos, H. S. Silveira, I. Da Silva Nunes, M. Martinez, F. E. Martinez, W. J. Fávaro and L. G. D. A. Chuffa, P-MAPA and IL-12 Differentially Regulate Proteins Associated with Ovarian Cancer Progression: A Proteomic Study, ACS Omega, 2019, 4(26), 21761–21777 Search PubMed.
- F. Coscia, E. Lengyel, J. Duraiswamy, B. Ashcroft, M. Bassani-Sternberg, M. Wierer, A. Johnson, K. Wroblewski, A. Montag, S. D. Yamada, B. López-Méndez, J. Nilsson, A. Mund, M. Mann and M. Curtis, Multi-level Proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy Target in Ovarian Cancer, Cell, 2018, 175(1), 159–170 Search PubMed.
- M. E. Gee, Z. Faraahi, A. McCormick and R. J. Edmondson, DNA damage repair in ovarian cancer: unlocking the heterogeneity, J. Ovarian Res., 2018, 11(1), 50 Search PubMed.
- W. Zhang, X. Ou and X. Wu, Proteomics profiling of plasma exosomes in epithelial ovarian cancer: A potential role in the coagulation cascade, diagnosis and prognosis, Int. J. Oncol., 2019, 54(5), 1719–1733 Search PubMed.
- W. Zhang, P. Peng, X. Ou, K. Shen and X. Wu, Ovarian cancer circulating extracelluar vesicles promote coagulation and have a potential in diagnosis: an iTRAQ based proteomic analysis, BMC Cancer, 2019, 19(1), 1095 Search PubMed.
- A. Swiatly, A. Horala, J. Matysiak, J. Hajduk, E. Nowak-Markwitz and Z. J. Kokot, Understanding ovarian cancer: iTRAQ-based proteomics for biomarker discovery, Int. J. Mol. Sci., 2018, 19(8), 2240 Search PubMed.
- M. R. Russell, M. J. Walker, A. J. K. Williamson, A. Gentry-Maharaj, A. Ryan, J. Kalsi, S. Skates, A. D'Amato, C. Dive, M. Pernemalm, P. C. Humphryes, E. O. Fourkala, A. D. Whetton, U. Menon, I. Jacobs and R. L. J. Graham, Protein Z: A putative novel biomarker for early detection of ovarian cancer, Int. J. Cancer, 2016, 138(12), 2984–2992 Search PubMed.
- G. D. Barnabas, K. Bahar-Shany, S. Sapoznik, L. Helpman, Y. Kadan, M. Beiner, O. Weitzner, N. Arbib, J. Korach, T. Perri, G. Katz, A. Blecher, B. Brandt, E. Friedman, D. Stockheim, A. Jakobson-Setton, R. Eitan, S. Armon, H. Brand, O. Zadok, S. Aviel-Ronen, M. Harel, T. Geiger and K. Levanon, Microvesicle proteomic profiling of uterine liquid biopsy for ovarian cancer early detection, Mol. Cell. Proteomics, 2019, 18(5), 865–875 Search PubMed.
- Z. Zhang, K. Qin, W. Zhang, B. Yang, C. Zhao, X. Zhang, F. Zhang, L. Zhao and B. Shan, Postoperative recurrence of epithelial ovarian cancer patients and chemoresistance related protein analyses, J. Ovarian Res., 2019, 12(1), 1–8 Search PubMed.
- H.-S. Ahn, J. Yeom, J. Yu, Y.-I. Kwon, J.-H. Kim and K. Kim, Convergence of Plasma Metabolomics and Proteomics Analysis to Discover Signatures of High-Grade Serous Ovarian Cancer, Cancers, 2020, 12(11), 3447 Search PubMed.
- R. Hüttenhain, M. Choi, L. Martin de la Fuente, K. Oehl, C.-Y. Chang, A.-K. Zimmermann, S. Malander, H. Olsson, S. Surinova, T. Clough, V. Heinzelmann-Schwarz, P. J. Wild, D. M. Dinulescu, E. Niméus, O. Vitek and R. Aebersold, A Targeted Mass Spectrometry Strategy for Developing Proteomic Biomarkers: A Case Study of Epithelial Ovarian Cancer, Mol. Cell. Proteomics, 2019, 18(9), 1836–1850 Search PubMed.
- N. Rauniyar, G. Peng, T. K. T. Lam, H. Zhao, G. Mor and K. R. Williams, Data-Independent Acquisition and Parallel Reaction Monitoring Mass Spectrometry Identification of Serum Biomarkers for Ovarian Cancer, Biomarker Insights, 2017, 12, 1–12 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |