
Clickable decellularized extracellular matrix as a new tool for building hybrid-hydrogels to model chronic fibrotic diseases in vitro†
Cassandra L.
Petrou
ab,
Tyler J.
D’Ovidio
 b,
Deniz A.
Bölükbas
cd,
Sinem
Tas
cd,
R. Dale
Brown
e,
Ayed
Allawzi
e,
Sandra
Lindstedt
dfg,
Eva
Nozik-Grayck
e,
Kurt R.
Stenmark
e,
Darcy E.
Wagner
cd and
Chelsea M.
Magin
*ab
b,
Deniz A.
Bölükbas
cd,
Sinem
Tas
cd,
R. Dale
Brown
e,
Ayed
Allawzi
e,
Sandra
Lindstedt
dfg,
Eva
Nozik-Grayck
e,
Kurt R.
Stenmark
e,
Darcy E.
Wagner
cd and
Chelsea M.
Magin
*ab
aDepartment of Bioengineering, University of Colorado, Anschutz Medical Campus, 12700 E 19th Ave MS C272 Aurora, Denver, CO 80045, USA
bDivision of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado, Anschutz Medical Campus, 12700 E 19th Ave MS C272 Aurora, Denver, CO 80045, USA. E-mail: chelsea.magin@cuanschutz.edu
cDepartment of Experimental Medical Sciences, Lund Stem Cell Center, and Wallenberg Molecular Medicine Center, Lund University, Sweden
dWallenberg Center for Molecular Medicine, Lund University, Sweden
eDepartment of Pediatrics and Medicine, Cardiovascular Pulmonary Research Laboratory, University of Colorado, Anschutz Medical Campus, USA
fCardiothoracic Surgery and Transplantation, Lund University Hospital, Sweden
gDepartment of Clinical Sciences, Lund University, Sweden
First published on 17th April 2020
Abstract
Fibrotic disorders account for over one third of mortalities worldwide. Despite great efforts to study the cellular and molecular processes underlying fibrosis, there are currently few effective therapies. Dual-stage polymerization reactions are an innovative tool for recreating heterogeneous increases in extracellular matrix (ECM) modulus, a hallmark of fibrotic diseases in vivo. Here, we present a clickable decellularized ECM (dECM) crosslinker incorporated into a dynamically responsive poly(ethylene glycol)-α-methacrylate (PEGαMA) hybrid-hydrogel to recreate ECM remodeling in vitro. An off-stoichiometry thiol–ene Michael addition between PEGαMA (8-arm, 10 kg mol−1) and the clickable dECM resulted in hydrogels with an elastic modulus of E = 3.6 ± 0.24 kPa, approximating healthy lung tissue (1–5 kPa). Next, residual αMA groups were reacted via a photo-initiated homopolymerization to increase modulus values to fibrotic levels (E = 13.4 ± 0.82 kPa) in situ. Hydrogels with increased elastic moduli, mimicking fibrotic ECM, induced a significant increase in the expression of myofibroblast transgenes. The proportion of primary fibroblasts from dual-reporter mouse lungs expressing collagen 1a1 and alpha-smooth muscle actin increased by approximately 60% when cultured on stiff and dynamically stiffened hybrid-hydrogels compared to soft. Likewise, fibroblasts expressed significantly increased levels of the collagen 1a1 transgene on stiff regions of spatially patterned hybrid-hydrogels compared to the soft areas. Collectively, these results indicate that hybrid-hydrogels are a new tool that can be implemented to spatiotemporally induce a phenotypic transition in primary murine fibroblasts in vitro.
1. Introduction
Pathologic tissue remodeling is a hallmark of chronic fibrotic diseases.1–4 It is characterized by persistent and excessive production of biochemically abnormal extracellular matrix (ECM), resulting in spatially heterogeneous increases in tissue stiffness.5–8 Emerging evidence indicates that cell–matrix interactions drive the progression of fibrosis, yet it is not clear whether changes in ECM composition, or the subsequent alterations in mechanical properties of the tissues, are the more potent driver of fibrosis.9–16 Recently, Parker et al. proposed a model of pulmonary fibrosis in which an initial insult creates a lesion in the lung that is repaired with abnormal ECM. It is suggested that this pathologic ECM locally corrupts nearby fibroblasts, further remodeling the surrounding area and hence spreading fibrosis.11 Herrera et al. suggest that these findings reveal mechanisms of fibrotic progression that can be self-perpetuating.14 Thus, using pulmonary fibrosis as an archetype of chronic fibrotic disease, we present a novel hybrid-hydrogel that allows us to decouple fibrotic tissue composition from subsequent changes in mechanical properties to study the dynamic biological processes occurring in fibrosis.Fibroblasts are a heterogeneous and versatile cell population with remarkable plasticity.17 In healthy tissues these cells can differentiate into activated myofibroblasts marked by expression of Collagen 1a1 (Col 1a1) and alpha-smooth muscle actin (αSMA) to promote wound healing. After healing, the majority of these activated cells undergo apoptosis to restore homeostasis.18In vitro studies of myofibroblast activation in response to modulus changes in biomaterials have revealed that this differentiation is reversible.19–21 However, a subpopulation of persistently activated, apoptosis-resistant fibroblasts have been identified in fibrotic tissues.22,23 Currently the mechanisms underlying this progression, and the potential to reverse the persistently activated phenotype by therapeutic treatment, remain largely undefined.
Advances in lung decellularization techniques have fueled a growing interest in building biomaterials from decellularized ECM (dECM).24–26 Hilster et al., for example, reported a protocol for fabricating hydrogels using dECM derived from control and fibrotic human lungs. While these materials comprise the complex biochemical cues that cells encounter in vivo,27 they are limited by poor mechanical properties that do not fully recapitulate diseased lung tissue. The elastic modulus of human lung tissue ranges from 1 to 5 kPa (healthy) to greater than 10 kPa (fibrotic).28,29 Hydrogels comprised of dECM from healthy and fibrotic tissues demonstrated a similar trend, 1.5 kPa and 6.8 kPa, respectively, but ultimately could not match the increased stiffness of fibrotic lung.29 To overcome this limitation, Sava et al. coated polyacrylamide-based hydrogels of modulus values ranging from 1.8 kPa to 23.7 kPa with healthy and fibrotic human-lung dECM. The results of these studies demonstrated that changes in αSMA expression and organization were mechanosensitive regardless of composition.30 Although this approach enabled researchers to decouple dECM composition from mechanical properties, the coating procedure limited experiments to 2D.30 In another study, dECM was methacrylated and covalently crosslinked with gelatin methacrylamide to form 3D hydrogels from two naturally derived polymers with elastic modulus values ranging from approximately 12 kPa to 66 kPa.31 While these experiments were certainly a breakthrough in modeling fibrotic disease in vitro, these static systems do not reproduce the spatiotemporal microenvironmental changes that occur during fibrotic disease progression and have been implicated as a major driver of cellular activation and disease progression.11,15,16,32,33
Dual-stage polymerization systems are one innovative way to recreate heterogeneous and localized ECM stiffening in vitro.34–37 These dual-stage polymerization systems often include a “click” reaction that proceeds under mild conditions and is highly reactive in a fast, specific, and efficient manner.38 The specificity of click crosslinking allows the user to exploit sequential reactions to control hydrogel mechanical properties. Dynamic responsiveness has been initiated in a variety of hydrogels by user-controlled mechanisms, such as light, to probe how cells interact with and receive information from the extracellular microenvironment.39 While these systems are capable of recapitulating the spatiotemporal heterogeneity of the biophysical changes that occur in the ECM during disease progression, two key limitations remain: (1) these biomaterials do not recapitulate the dynamic alterations in ECM composition that characterize fibrotic disease, and (2) existing material systems that mimic the water content of tissues are susceptible to hydrolysis over the long culture periods required to emulate chronic disease.
Here, we present a strategy for synthesizing clickable dECM and combining it with a hydrolytically stable, phototunable poly(ethylene glycol) (PEG) backbone in a dual-stage polymerization system that allows us to decouple fibrotic tissue composition from subsequent changes in mechanical properties to dynamically study the biological processes occurring in fibrosis. The novel hybrid-hydrogel system provides predictable control of initial matrix mechanical properties over a large range of moduli (E = 3.63 ± 0.24 to 13.35 ± 0.83 kPa) and facilitates spatiotemporal control over precise increases in elastic modulus in situ. Primary platelet-derived growth factor receptor alpha-positive (PDGFRα+) fibroblasts from the alveolar niche40 were isolated from adult dual-transgenic reporter mice. These mice express green or red fluorescent protein in response to Col1a1 or αSMA or transgene expression,41 respectively, and are used to monitor cellular responses to this new biomaterial. PDGFRα+ fibroblasts from the alveolar niche were selected for these assays because this is the proposed site of initial injury and remodeling in pulmonary fibrosis.42–44 Fibroblast activation was characterized by measuring expression of these transgenes in response to initial substrate moduli, dynamic stiffening ranging from healthy (E = 1–5 kPa) to diseased levels (E > 10 kPa), and patterns of alternating soft and stiff areas to mimic the effect of heterogenous mechanical properties observed in fibrosis.45 We demonstrate the utility of this hybrid-hydrogel system for dynamically probing cell–matrix interactions with spatial control, highlighting a new approach for understanding the biochemical and biophysical contributions to fibrotic disease progression.
2. Materials and methods
2.1. Small molecule and macromer synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2).
CH2)–), 5.90 (s, 1H, –C = CH2), 6.31 (s, 1H, –CCH2).
CH2).
CH2)–), 5.90 (s, 1H, –C = CH2), 6.31 (s, 1H, –CCH2).
2.2. dECM derivation and thiolation
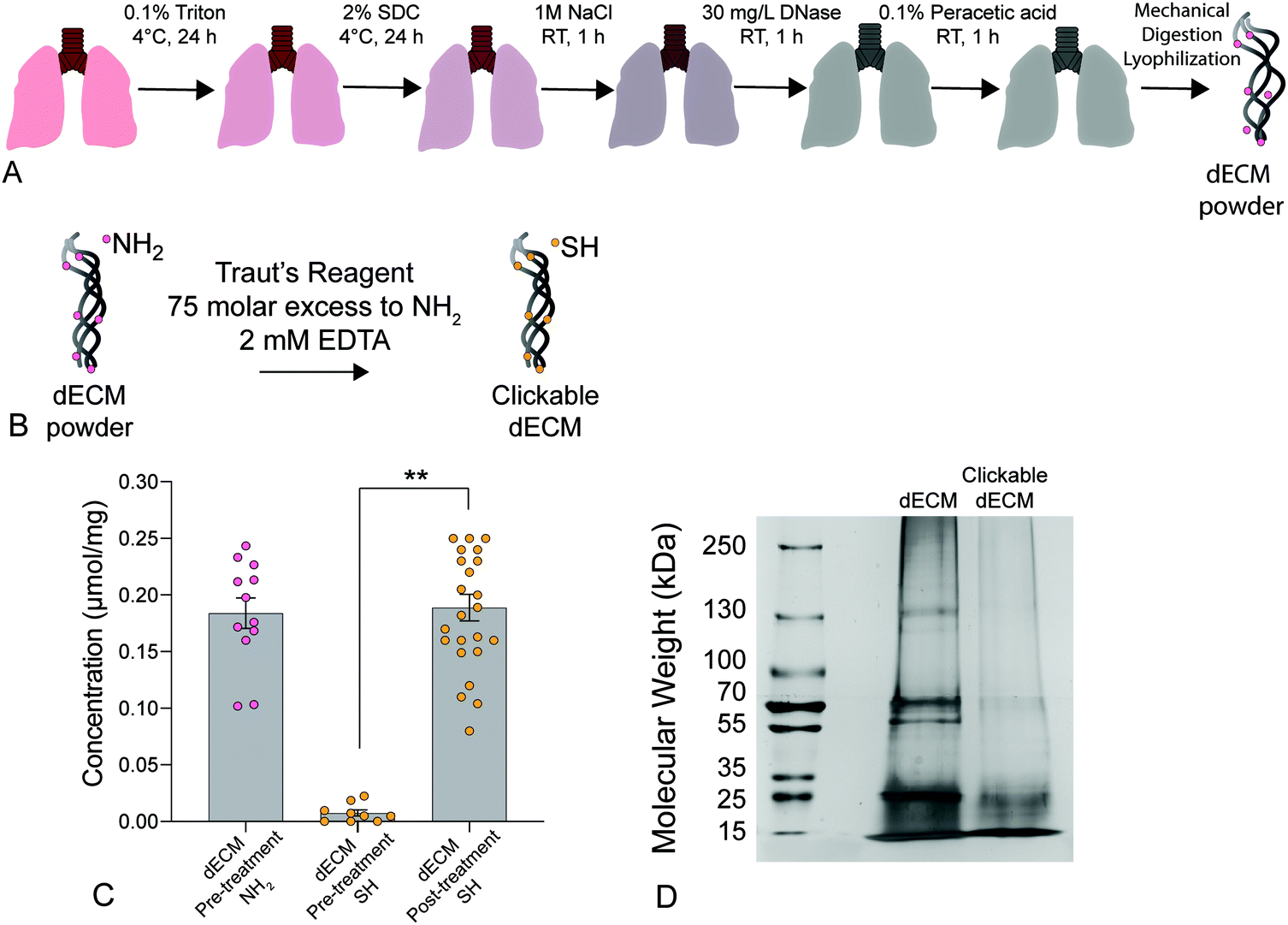 | ||
| Fig. 1 (A) Schematic depicting the lung decellularization process. Briefly, native lungs were sequentially perfused with Triton X-100, sodium deoxycholate (SDC) solution, DNAse solution, and peracetic acid to remove all cellular components before being mechanically digested and lyophilized to form a powder. (B) Decellularized porcine ECM was treated with Traut's reagent at a 75-molar excess to primary amines (NH2) and 2 mM EDTA to convert free primary amines to thiols creating a clickable dECM crosslinker. (C) A significant increase in thiol concentration was measured post-treatment using an Ellman's assay (n = 6, **p < 0.0001, ANOVA, Tukey test). (D) Silver-stained SDS-PAGE results showed a loss of high molecular weight proteins (≥250 kDa) and an increase in proteins below 15 kDa following thiolation, indicating that that the treatment likely cleaved a portion of the dECM proteins into clickable dECM peptides. | ||
A Pierce™ Silver Stain Kit (ThermoFisher) was used to qualitatively analyze protein size distribution in dECM compared to thiolated-dECM. Lyophilized dECM and thiolated-dECM were lysed in RIPA buffer for 1 h on ice and loaded into sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. After resolving the protein by size, the gels were silver stained according to the manufacturer's protocol to visualize the size (via molecular weight) of dECM proteins, peptides, and fragments before and after thiolation.54
2.3. Hydrogel formation
The thiol-functionalized dECM (clickable dECM) and 1,4-dithiothreitol (DTT; Acros Organics) crosslinkers were reacted with PEGαMA in a base-catalyzed Michael addition reaction off-stoichiometry at a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 thiol to αMA ratio. The hydrogel formulation was optimized by varying the percentage of DTT to clickable dECM to achieve a desired elastic modulus. The clickable dECM was dissolved in 15 mM solution of tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Fisher Scientific) for 1 h at a 20× molar ratio to the thiol concentration as determined by the Ellman's assay. Stock solutions of PEGαMA (0.4 mg μL−1), DTT (500 mM), and a peptide sequence that mimics adhesive ligands (0.2 mM; CGRGDS; GL Biochem) were prepared in 0.3 M, pH 8 4-(2-hydroxyethyl)-1piperazineethanesulfonic acid buffering agent (HEPES; Life Technologies). A precursor solution was prepared by combining the clickable dECM, DTT, CGRGDS, and then adding the PEGαMA. Hydrogels were polymerized by placing 40 μL drops of the precursor solution between two hydrophobic glass slides treated with SigmaCote (Sigma). The reaction was allowed to proceed for 1 h at 37 °C. Hydrogels were equilibrated in PBS at 4 °C, with or without 2.2 mM LAP photo-initiator for 24 h. Hydrogels swollen in LAP were exposed to light (365 nm light,10 mW cm−2) for 5 min using an OmniCure Series 2000 UV lamp (Lumen Dynamics) to create stiff hybrid-hydrogel samples. For cell experiments, the hydrogel-forming stock solutions were dissolved in sterile 0.3 M, pH 8 HEPES. The precursor solution was made from the resulting stocks under sterile conditions. Glass coverslips (18 mm; Fisher Scientific) were silanated with (3-aminopropyl)trimethoxysilane (Sigma) using a liquid deposition technique.55 Hydrogel precursors were deposited in 90 μL drops between hydrophobic glass slides and silanated cover slips for 1 h at 37 °C. Hydrogels were then swollen in complete medium (DMEM/F12; Gibco) supplemented with 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 2.5 μg mL−1 amphotericin B (Life Technologies), and 10% fetal bovine serum (FBS; ThermoFisher) for 24 h at 37 °C. The medium was formulated with or without 2.2 mM LAP for stiffening or use as soft hybrid-hydrogel samples in experiments, respectively.
:8 thiol to αMA ratio. The hydrogel formulation was optimized by varying the percentage of DTT to clickable dECM to achieve a desired elastic modulus. The clickable dECM was dissolved in 15 mM solution of tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Fisher Scientific) for 1 h at a 20× molar ratio to the thiol concentration as determined by the Ellman's assay. Stock solutions of PEGαMA (0.4 mg μL−1), DTT (500 mM), and a peptide sequence that mimics adhesive ligands (0.2 mM; CGRGDS; GL Biochem) were prepared in 0.3 M, pH 8 4-(2-hydroxyethyl)-1piperazineethanesulfonic acid buffering agent (HEPES; Life Technologies). A precursor solution was prepared by combining the clickable dECM, DTT, CGRGDS, and then adding the PEGαMA. Hydrogels were polymerized by placing 40 μL drops of the precursor solution between two hydrophobic glass slides treated with SigmaCote (Sigma). The reaction was allowed to proceed for 1 h at 37 °C. Hydrogels were equilibrated in PBS at 4 °C, with or without 2.2 mM LAP photo-initiator for 24 h. Hydrogels swollen in LAP were exposed to light (365 nm light,10 mW cm−2) for 5 min using an OmniCure Series 2000 UV lamp (Lumen Dynamics) to create stiff hybrid-hydrogel samples. For cell experiments, the hydrogel-forming stock solutions were dissolved in sterile 0.3 M, pH 8 HEPES. The precursor solution was made from the resulting stocks under sterile conditions. Glass coverslips (18 mm; Fisher Scientific) were silanated with (3-aminopropyl)trimethoxysilane (Sigma) using a liquid deposition technique.55 Hydrogel precursors were deposited in 90 μL drops between hydrophobic glass slides and silanated cover slips for 1 h at 37 °C. Hydrogels were then swollen in complete medium (DMEM/F12; Gibco) supplemented with 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 2.5 μg mL−1 amphotericin B (Life Technologies), and 10% fetal bovine serum (FBS; ThermoFisher) for 24 h at 37 °C. The medium was formulated with or without 2.2 mM LAP for stiffening or use as soft hybrid-hydrogel samples in experiments, respectively.
2.4. Hybrid-Hydrogel characterization
Rheology was used to assess the mechanical properties of the hydrogels following gelation. Hydrogel samples (height = 300 μm; diameter = 8 mm) were loaded onto a Discovery HR2 rheometer (TA Instruments) with an 8 mm parallel plate geometry and the Peltier plate set at 37 °C. The geometry was lowered until the instrument read 0.03 N axial force and the gap distance was noted. The gap distance between the plate and the geometry was adjusted until the storage modulus measurement (G′) plateaued and a percent compression of the specific hydrogel was defined and used thereafter.56 The samples were subjected to frequency oscillatory strain with a frequency range of 0.1 to 100 rad s−1 at 1% strain. The elastic modulus (E) was calculated using rubber elastic theory, assuming a Poisson's ratio of 0.5 for bulk measurements of elastic hydrogel polymer networks.21Hybrid-hydrogel morphology was visualized by scanning electron microscopy (SEM). Briefly, soft and stiffened hybrid-hydrogels were frozen in liquid nitrogen and then lyophilized at −80 °C for 24 h (Freezone 4.5, Labconco, US). Samples were subsequently sputter-coated with 2 nm platinum/palladium (80/20) in a Quorum Q150T ES turbo pumped sputter coater and examined with the secondary electron detector at 1.5 kV in a Jeol JSM-7800F FEG-SEM.
Distribution of the clickable dECM and the PEG backbone components within hybrid-hydrogels was visualized via confocal microscopy. Clickable dECM crosslinker was treated with TCEP for 1 h as described above. AlexaFluor™ 647 C2 Maleimide (ThermoFisher) at 0.8 mM was added to this solution and allowed to react for 2 h to conjugate the dye to the thiols on the crosslinker. Hybrid-hydrogels were polymerized by placing a 40 μL drop of the precursor solution containing the labeled dECM crosslinker on a silanated glass slide and allowing the reaction to proceed for 1 h at 37 °C. The PEG component of these hybrid-hydrogel samples was visualized through immunostaining. Briefly, samples were blocked with 5% bovine serum albumin (BSA; ThermoFisher) for 1 h. Recombinant anti-PEG antibody produced in rabbit (ab170969; abcam) was diluted 1:10 in an immunofluorescence (IF) solution containing 3% v/v BSA with 0.1% v/v Tween 20 (Sigma) in PBS. Samples were incubated with the primary antibody solution overnight at 4 °C. After washing three times with IF solution, the hybrid-hydrogels were incubated with goat-anti-rabbit IgG Alexa Fluor-488 secondary antibody (1:200 in IF solution, ThermoFisher) overnight at 4 °C. Samples were rinsed with PBS three times and imaged on a Zeiss LSM780 confocal microscope.
2.5. Hydrolytic stability
Hybrid-hydrogels and fully synthetic hydrogels (17 wt% poly(ethylene glycol)-methacrylate (PEGMA; 10 kg mol−1) crosslinked with 100% DTT) were fabricated, swollen in 2.2 mM LAP, stiffened by exposure to light (365 nm light,10 mW cm−2) for 5 min (OmniCure Series 2000; Lumen Dynamics), and two assays were performed on each condition every 10 days for up to 60 days to examine the hydrolytic stability. Rheology was completed as described above on each sample before the sample was placed in DI water, lyophilized, and weighed to record the dry polymer mass.2.6. Spatial patterning of hybrid-hydrogel modulus
Hydrogels were fabricated as described above for cell experiments and swollen in DMEM/F12 supplemented with 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 2.5 μg mL−1 amphotericin B, 1% FBS, 2 mM LAP, and 10 μM methacryloxyethyl thiocarbamoyl rhodamine B (Polysciences Inc.). Hybrid-hydrogels were exposed to 365 nm light at 10 mW cm−2 through a chrome-on-quartz photomask to spatially pattern defined regions of increased elastic modulus. Two line patterns were produced with either 50- or 100-micron width and spacing.2.7. Primary cell isolation
All animal procedures were performed in an AAALAC-accredited facility in accordance with the Guide for the Care and Use of Laboratory Animals57 and approved by the University of Colorado Denver Institutional Animal Care and Use Committee. Male and female, 8–12 week-old, dual-transgenic reporter C57BL/6J mice were bred for this study. Fibroblasts from this GFP-Col1a1 × RFP-αSMA strain express green fluorescent protein (GFP) or red fluorescent protein (RFP) transgenes upon the expression of Col1a1 and αSMA promoters, respectively.41 Wildtype littermates, C57BL/6J mice, 8–12 weeks old, (GFP−, RFP−) which resulted from the breeding protocol were used for cell viability experiments.Cells isolated from enzymatically dispersed whole lung were sorted using magnetic microbeads conjugated with specific monoclonal antibodies to purify a PDGFRα-positive fibroblast population, as follows. At the time of animal sacrifice, the heart–lung block was collected. The lungs were filled with room temperature dispase solution (5 U mL−1; Life Technologies) and allowed to collapse. Next, lungs were inflated through the trachea with 1% low melt melting point agarose (LMP Ultrapure; Life Technologies) and placed in PBS on ice until the agarose solidified. The lungs were transferred to fresh dispase solution (5 U mL−1) in a 50 mL conical tube and incubated for 45 min at room temperature. Then, the lungs were transferred to GentleMACS C tubes (Miltenyi Biotec, Inc.) containing 3 mL of digestion solution: complete DMEM with high glucose (Life Technologies) supplemented with 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 2.5 μg mL−1 amphotericin B, and 10% FBS with DNAse solution (0.33 U mL−1; Life Technologies). The lungs were sequentially digested using a GentleMACS Dissociator (Miltenyi Biotec, Inc.) at 275 rpm for 37 seconds and 3300 rpm for 38 seconds. Digest solution was filtered using a 40 μm cell strainer, centrifuged at 1500 rpm for 10 min to remove the supernatant, and the cells were resuspended in complete medium for counting.
After counting, cells were resuspended in a buffer consisting of 0.5% bovine serum albumin (BSA) and 2 mM EDTA in PBS (PEB buffer). 10 μL of magnetic microbeads conjugated to monoclonal anti-mouse CD31 and to monoclonal anti-mouse CD45 antibodies (Miltenyi Biotec, Inc.) were added to the solution for every 107 cells counted to magnetically label the mature endothelial cells and leukocytes respectively. The solution was triturated twice to mix and incubated at 4 °C for 15 min. The cells were then washed with PEB buffer, centrifuged, and resuspended in 500 μL PEB buffer. Next, magnetically labeled cells in PEB buffer were applied to sorting columns (LS; Miltenyi Biotec, Inc.) and placed in the magnetic field of a Quadromacs Separator (Miltenyi Biotec, Inc.) and allowed to flow through at room temperature until the column reservoir was empty and the columns were rinsed with 9 mL of PEB buffer. Cells that passed through the columns in this step were the CD45−/CD31− fraction, and the CD45+/CD31+ cell fraction was discarded. The CD45−/CD31− fraction was counted, centrifuged to remove the supernatant, and resuspended in 90 μL PEB buffer for every 107 cells counted. 10 μL of magnetic microbeads conjugated to monoclonal anti-mouse CD140a antibodies (Miltenyi Biotec, Inc.) were added to the solution for every 107 cells counted to magnetically label PDGFRα+ fibroblasts. The solution was triturated twice to mix and incubated at 4 °C for 15 min. This cell suspension was loaded into a new sorting column, placed into the magnetic field, and rinsed to remove unlabeled cells. The column was removed from the magnetic separator and immediately flushed with 5 mL PEB buffer into a 50 mL conical tube for collection. This resulting solution contained the desired cell fraction (CD45−, CD31−, and CD140a+).58
2.8. Cellular viability
Sorted PDGFRα+ fibroblasts from wildtype C57BL/6J mice, 8–12 weeks old, (GFP−, RFP−) were seeded onto soft and stiff hybrid-hydrogels (n = 6) at a density of 10000 cells per cm2 and cultured in complete medium (DMEM/F12 supplemented with 100 U mL−1 penicillin, 100 μ mL−1 streptomycin, and 2.5 μg mL−1 amphotericin B and 10% FBS). Cell-seeded hydrogels were incubated with 10% v/v PrestoBlue™ Cell Viability Reagent (ThermoFisher) in culture medium for 3 h in a humidified incubator (37 °C, 5% CO2) on days 1, 3, 5, 7, and 9. Three aliquots of the media containing viability reagent were then transferred to a 96-well plate and read on a plate reader (540 nm excitation, 600 nm emission; Synergy H1 Hybrid Multi Mode Reader; BioTek). Average fluorescence intensity values for all conditions at each time point were normalized to the respective readings acquired on day 1 to calculate normalized metabolic activity.
Sorted PDGFRα+ fibroblasts from wildtype C57BL/6J mice, 8–12 weeks old, (GFP−, RFP−) were seeded onto soft and stiff hydrogels at a cell density of 10000 cells per cm2 and separate cell-seeded hydrogels were stained on days 1 and 7 with 1 mM calcein-AM (ThermoFisher) diluted 1:3000 in media to visualize live cells and 2 μg mL−1 molecular probe Hoechst 33342, Trihydrochloride, Trihydrate (Tocris) to visualize cell nuclei and incubated for 30 min at 37 °C. The cells were then rinsed with PBS and imaged. Sorted PDGFRα+ fibroblasts from wildtype C57BL/6J mice, 8–12 weeks old, (GFP−, RFP−) were also seeded onto soft hydrogels for stiffening experiments. Photo-initiator (LAP) was added to the media on day 6. Hydrogels were stiffened on day 7 by exposure to light (365 nm light, 10 mW cm−2) for 5 min and then stained and imaged on day 9 as described above. Cells stained by both calcein-AM and Hoechst were determined to be alive, while cells only stained by Hoechst were determined to be dead.59
2.9. Cellular activation
PDGFRα+ fibroblasts from dual-reporter (GFP-Col1a1 × RFP-αSMA) mice (8–12 weeks old) were seeded onto soft or stiff hydrogels at a density of 10000 cells per cm2 and cultured in medium (DMEM/F12 supplemented with 100 U mL−1 penicillin, 100 μ mL−1 streptomycin, and 2.5 μg mL−1 amphotericin B, and 1% FBS). Cells were intentionally seeded at a relatively low density to enable single cell analysis without cells reaching confluence.60 All cell-seeded hydrogels were incubated in a humidified incubator (37 °C, 5% CO2). On day 6, the cell culture medium was replaced with complete medium containing 2.2 mM LAP photo-initiator on half of the soft hydrogels. The following day (day 7) soft and stiff hydrogels (n = 4) were collected for analysis and the soft hydrogels treated with LAP (n = 4) were stiffened by exposure to 365 nm light at 10 mW cm−2 for 5 min, rinsed three times to remove any residual LAP, and incubated (37 °C, 5% CO2) for two more days before being collected for analysis on day 9 (Fig. 5a). This process was repeated for a total of three biological replicates. All samples were rinsed with PBS, fixed with 4% v/v paraformaldehyde (Electron Microscopy Sciences) in PBS for 30 min at room temperature, and quenched with 100 mM glycine (Sigma) in PBS for 15 min at room temperature. Following fixation, cells were rinsed with PBS, permeabilized with 0.2% Triton X-100 for 10 min at room temperature, and then stained with DAPI (1:10000, Sigma) for 15 min at room temperature. Finally, samples were washed with PBS and mounted using Prolong Gold Antifade reagent (ThermoFisher) to preserve for imaging.
2.10. Spatial control over cellular activation
Sorted PDGFRα+ dual-reporter fibroblasts were seeded onto spatially patterned (i.e. stiffened) hydrogels (n = 6) at a cell density of 15000 cells per cm2 and cultured in medium supplemented with 1% FBS. Samples were incubated in a humidified incubator (37 °C, 5% CO2) for seven days, then collected and prepared for activation analysis as described above.
2.11. Fluorescence microscopy and image analysis
All microscopy was performed using an upright, epifluorescent microscope (BX-63, Olympus). Ten fields of view were randomly selected and imaged on each sample at 10× magnification. Image analysis for activation experiments was performed using ImageJ software to count cells positive for GFP-Col1a1 and/or RFP-αSMA. These cell counts were divided by the total cell number acquired by counting DAPI-positive nuclei to calculate the proportion of GFP-Col1a1-positive and RFP-αSMA-positive cells on each sample.2.12. Statistical analysis
All quantitative hydrogel characterization was performed with a minimum of n = 3 technical replicates. All in vitro experimental studies involving cell culture for the evaluation of activation were performed with n = 4 technical replicates with 3 biological replicates. Data were presented as mean ± standard error of the mean (SEM) or 95% confidence interval as described in each figure caption. GraphPad Prism 8 Software was used to perform all statistical analyses. One-way analysis of variance (ANOVA) with Tukey's post hoc multiple comparisons tests was done on each experimental measure with multiple groups for pairwise comparisons among conditions with a 95% confidence interval. A 2-tailed Student's t-test was used when comparing fewer than three groups. P-Values of <0.05 were considered significant and designated on plots as *<0.05 or **<0.0001. Linear regression analysis with a 95% confidence interval was completed to compare trends over time.3. Results and discussion
3.1. Hybrid-hydrogel characterization
We present a method for synthesizing a clickable dECM crosslinker that facilitates a dual-stage polymerization reaction providing dynamic control over matrix mechanical properties in real time that is hydrolytically stable and can be implemented in 3D. First, porcine lungs were decellularized52 and mechanically digested into small fragments (Fig. 1a). Next, clickable dECM was generated by converting the naturally occurring primary amines on native dECM molecules to thiol moieties using Traut's reagent (Fig. 1b). The average primary amine concentration of porcine dECM was measured to be 0.184 ± 0.0135 μmol mg−1 by a Ninhydrin assay. The average thiol concentration, measured by Ellman's assay before treatment with Traut's reagent, was negligible (0.00753 ± 0.0273 μmol mg−1). Following treatment with Traut's reagent, however, this thiol concentration increased significantly to 0.189 ± 0.0117 μmol mg−1, indicating successful addition of thiols to dECM (P < 0.05, Tukey Test) (Fig. 1c). Traut's reagent has been used extensively to thiolate natural polymers and growth factors,61 but the impact of this reaction on dECM is not yet well understood. Therefore, we sought to investigate whether the thiolation process induced degradation of dECM molecules. As many conventional protein measurement techniques rely on the detection of amines, we performed silver staining of dECM pre and post-thiolation on SDS-PAGE gels. Silver staining is a commonly used protein detection technique with high sensitivity that relies on the binding of silver ions to the negative side chains of proteins54 and thus avoids potential interference due to thiolation of amine groups. Silver staining of dECM before thiolation revealed a wide distribution of proteins from >15 kDa to some above 250 kDa. In contrast, a loss of high molecular weight proteins (≥250 kDa) and an increase in proteins <15 kDa was observed following thiolation, indicating that that the treatment likely cleaved a portion of the dECM proteins into clickable dECM peptides (Fig. 1d).To demonstrate the utility of this clickable dECM crosslinker, it was incorporated into a PEGαMA-based stiffening hybrid-hydrogel system. First, clickable dECM was reacted off-stoichiometry with DTT and a peptide sequence mimicking a binding region on the basement membrane protein fibronectin (CGRGDS) through a thiol–ene Michael addition reaction. This thiol–ene polymerization was preceded by a “click” orthogonal step-growth mechanism where one thiol of the clickable dECM, DTT, or CGRGDS, reacted with one αMA, leading to a homogeneous distribution in crosslinks.39 The hybrid-hydrogel was then dynamically stiffened by sequentially reacting the residual αMA moieties in the presence of LAP photoinitiator via a light-initiated homopolymerization (Fig. 2a). Rheological measurements were performed to quantify the shear elastic modulus (G′) of hybrid-hydrogels containing various amounts of clickable dECM and converted to elastic modulus (E′) using rubber elasticity theory assuming a Poisson's ratio of 0.5 for bulk measurements of elastic hydrogel polymer networks.21 This assumption has been used extensively to determine the bulk modulus of PEG-based hydrogels19,62,63 and has been previously confirmed using atomic force microscopy.64 The elastic modulus scaled directly with total molar percent of clickable dECM as expected (Fig. 2b). The final formulation for the hybrid-hydrogels consisted of 15 wt% PEGαMA and a molar thiol ratio of 75% DTT to 25% clickable dECM with 1 mM CGRGDS pendant peptide. The soft hybrid-hydrogel exhibited an elastic modulus of 3.63 ± 0.24 kPa within the range of healthy lung tissue (1 to 5 kPa)28,65 (Fig. 2c). Following sequential crosslinking, stiff hybrid-hydrogels were dynamically stiffened to an elastic modulus of 13.35 ± 0.83 kPa, replicating fibrotic stiffness (>10 kPa)28,65 and demonstrating temporal, user-defined control over in situ stiffening (Fig. 2c). The storage modulus and the equilibrium volumetric swelling ratio of hydrogels are proportional to the density of crosslinks within the polymer network.66 The equilibrium volumetric swelling ratio of the soft hybrid-hydrogels was measured to be approximately twice that of the stiffened hybrid-hydrogels, indicating that crosslinking density increased following the stiffening reaction (Fig. S5, ESI†).
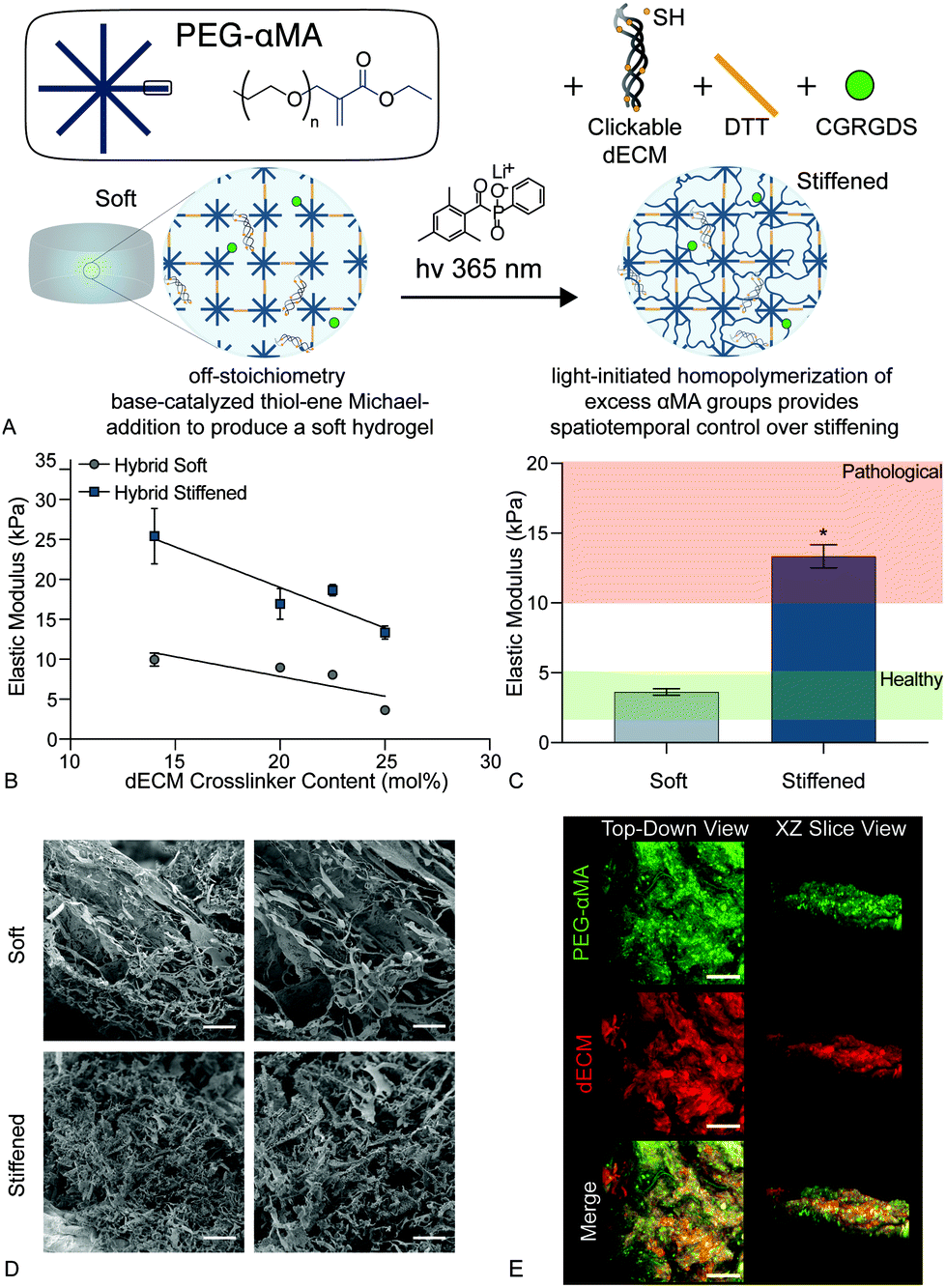 | ||
| Fig. 2 (A) Schematic of the dual-stage polymerization reaction that combined PEGαMA and the clickable dECM crosslinker with DTT and CGRGDS to enable spatiotemporal control over stiffening. (B) The elastic modulus of soft and stiffened hybrid hydrogels decreased with increasing dECM content ranging from 14 mol% to 25 mol% dECM (n = 4, mean ± SEM). (C) Hybrid hydrogel formulations were adjusted so that soft and stiffened samples resulted in elastic modulus values within healthy and pathologic ranges, respectively (n = 4, mean ± SEM, *p < 0.05, ANOVA, Tukey test). (D) Scanning electron micrographs of soft and stiffened hybrid hydrogels. Scale bars, 25 μm. (E) Representative confocal images of hybrid hydrogels stained for PEGαMA (green) and dECM (red) show uniform mixing of the two components throughout the samples. Scale bars, 50 μm. | ||
Likewise, scanning electron micrographs showed a loosely organized morphology within the soft hybrid-hydrogels that became more highly interconnected upon stiffening (Fig. 2d). The initial thiol-Michael addition polymerization preceded a step-growth mechanism where one thiol reacted with one αMA. This mechanism produced a homogeneous distribution of PEGαMA and clickable dECM throughout hybrid-hydrogels as visualized by confocal microscopy (Fig. 2e).
3.2. Hydrolytic stability
Synthetic PEG-based hydrogels have been widely employed to study the cell–matrix interactions associated with the initiation of fibrotic disease.19,37,63,67In vitro studies of fibroblast activation in response to modulus changes in PEG-based biomaterials have revealed that this differentiation is reversible when high modulus hydrogels (>15 kPa) are softened (<7 kPa).19–21 Hydrolysis in these traditional Michael-addition, thiol–ene biomaterials occurs preferentially at ester linkages between the polymer backbone (e.g., PEG) and the acrylate or methacrylate (MA) functional end groups that facilitate polymerization,68 and this leads to the breakdown of the crosslinking within step or chain growth networks (Fig. 3a). The presence of an ester bond between the PEG macromers and functional groups in many of these materials has resulted in irreversible hydrolytic cleavage that degrades hydrogel samples completely within 21 days.69 This hydrolysis limits our ability to model chronic diseases, such as fibrosis, that develop over long time periods. Therefore, the hybrid-hydrogel system presented here was designed to withstand hydrolysis over the long culture periods required to emulate chronic disease by conjugating the MA to the PEG backbone on the opposite side of the carbonyl as a typical MA group. This unique placement of the ester allowed hydrolysis to occur without affecting the crosslinked polymer network (Fig. 3b).48 Additionally, the presence of the carbonyl group imparted high reactivity during chain-growth homopolymerization that is lacking for typical vinyl monomers. Hydrolytic stability of stiffened PEGαMA hybrid-hydrogels was monitored by measuring bulk mechanical properties and mass loss over 60 days in culture. These results were compared to stiffened, fully synthetic PEGMA. The elastic modulus of stiffened PEGαMA hybrid-hydrogels remained stable over 60 days, with a slope that was not significantly different from zero (m = 0.009, p < 0.05, linear regression) (Fig. 3c). The stiffened PEGMA hydrogel, however, began to degrade after just 10 days in PBS, and the elastic modulus of this material decreased below a level recapitulating fibrotic tissue between day 10 and 20, and decreased linearly over the course of 60 days (m = −0.265, p < 0.0001) (Fig. 3a). PEGMA hydrogel mass also decreased at a faster rate (m = −0.576, p = 0.086, linear regression) than the stiffened PEGαMA hybrid-hydrogels (m = −0.399, p = 0.421, linear regression), although these trends are not statistically significant (Fig. 3d).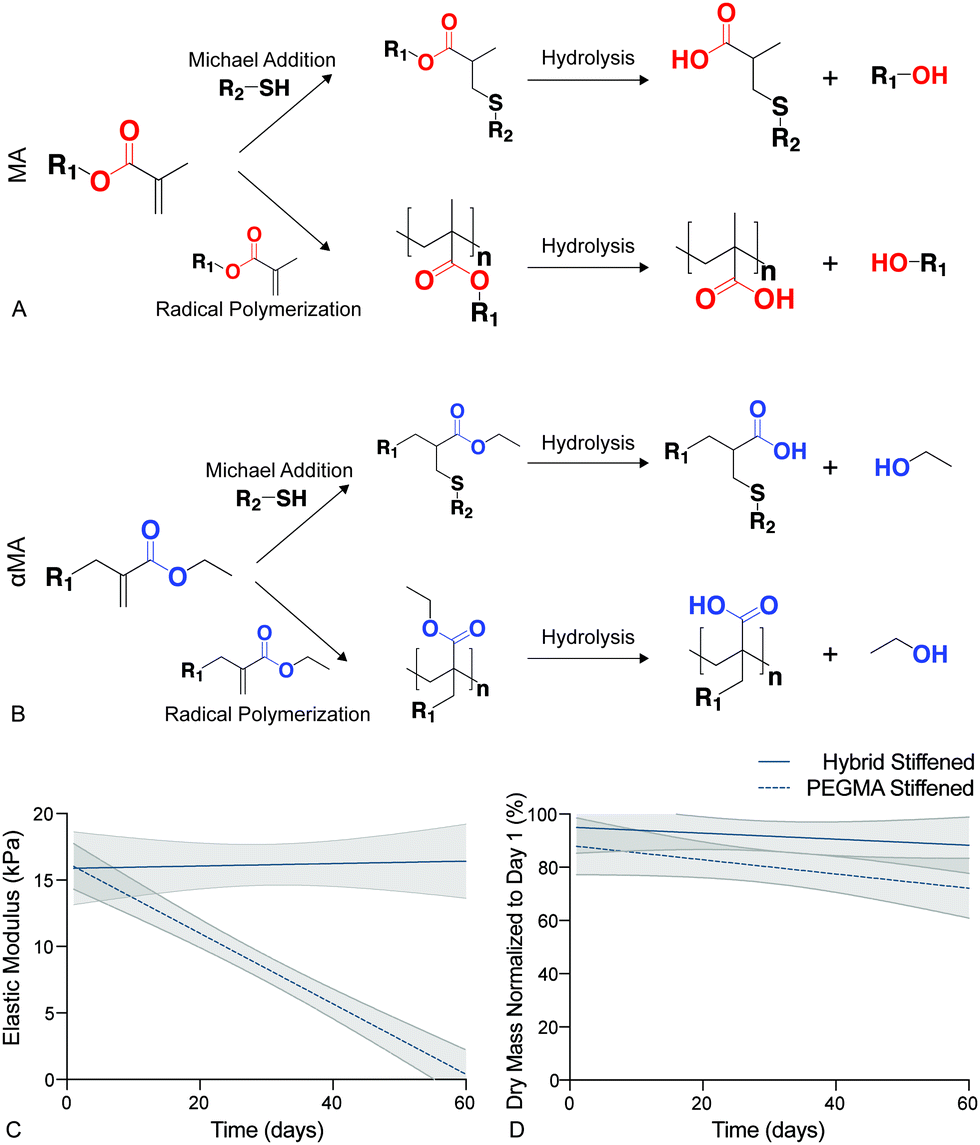 | ||
| Fig. 3 (A) Hydrolysis in traditional PEGMA biomaterials occured preferentially at ester linkages between PEG and the MA functional end groups leading to cleavage of the polymer network and a decrease in elastic modulus. (B) The hybrid-hydrogel system was designed to withstand hydrolysis by conjugating the MA to the PEG backbone on the opposite side of the carbonyl as a typical MA group. This αMA functional group allowed hydrolysis to occur by releasing an ethanol molecule and not degrading the crosslinked polymer network. (C) Linear regression analysis of the elastic modulus for stiffened PEGαMA hybrid-hydrogels and PEGMA synthetic hydrogels showed that the elastic modulus of the hybrid-hydrogel did not significantly decrease over 60 days (m = 0.009, p = 0.81), while the elastic modulus of the PEGMA hydrogels significantly decreased (m = −0.265, p < 0.0001). PEGMA hydrogel modulus values fell below the range considered pathological (>10 kPa) by Day 20 (n = 4, shaded areas represent 95% confidence intervals). (D) Linear regression analysis of stiffened PEGαMA hybrid and PEGMA synthetic hydrogels dry mass measurements over 60 days revealed that the PEGMA hydrogels may be losing mass at a faster rate than the hybrid-hydrogels (m = −0.576, p = 0.086 versus m = −0.399, p = 0.421, respectively), however these trends are not statistically significant. | ||
3.3. Cell viability
To confirm that this new hybrid-hydrogel system was cytocompatible, wildtype PDGFRα+ fibroblasts were seeded onto soft or stiff samples and metabolic activity was measured over time using a resazurin-based PrestoBlue™ Cell Viability assay. Metabolic activity significantly increased over nine days on both soft and stiff hybrid-hydrogels compared to day 1 (Fig. 4a). This increase in metabolic activity can be attributed to cellular proliferation over time. Representative images of cells stained for Calcein-AM (green) and Hoechst (blue) confirmed fibroblast viability on soft and stiff hybrid-hydrogels on days 1 and 7. All cells cultured on soft hybrid-hydrogels were collected for analysis on day 7, while soft hybrid-hydrogels that were stiffened on day 7 were collected for viability analysis on day 9. Results confirmed fibroblast viability through the dynamic stiffening process. Cells positive for green and blue were considered live, while cells stained for blue only were considered dead (Fig. 4b).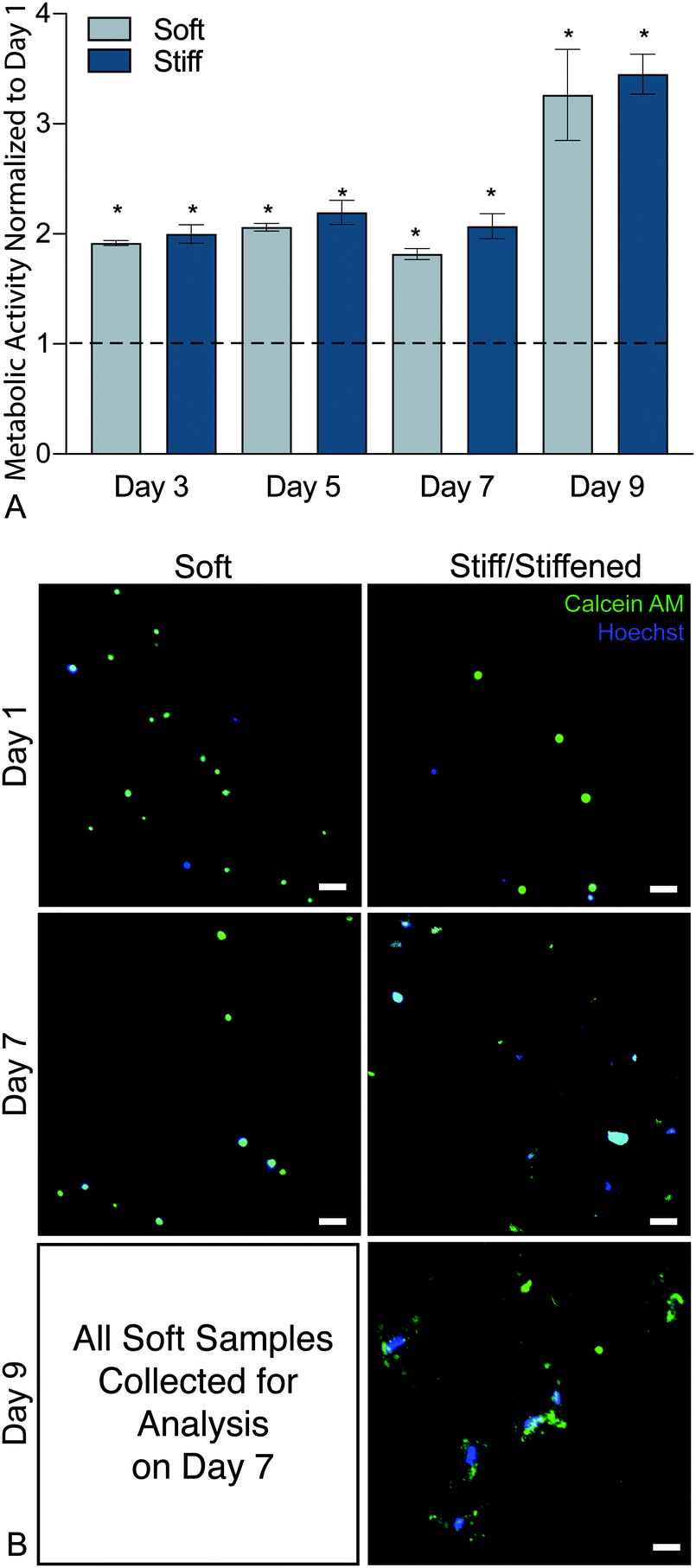 | ||
| Fig. 4 (A) Metabolic activity results from days 3, 5, 7, and 9 were normalized to initial readings at day 1 and indicated that both soft and stiff hybrid-hydrogel substrates supported a significantly increased levels of cellular viability through day 9. (N = 6, mean ± SEM, *p < 0.05, ANOVA, Tukey Test). (B) Representative images of cells stained for Calcein-AM (green) and Hoechst (blue). Cells positive for green and blue were considered live, while cells stained for blue only were considered dead. Scale bars, 25 μm. | ||
3.4. Cell activation
It is well established that both composition and mechanical properties of ECM are significantly altered during the progression of fibrosis and that these alterations influence cellular function.11,12,33,70 Deciphering whether composition or mechanical properties are the major drivers of disease has remained challenging due to a limited number of experimental techniques that allow for precise spatiotemporal control over these parameters. Booth et al. cultured primary human lung fibroblasts on acellular normal and fibrotic human lung slices that had significantly different moduli (1.6 ± 0.08 kPa and 7.3 ± 0.6 kPa, respectively) and observed a significant increase in the production of αSMA in the cells seeded on the fibrotic sections compared to cells on normal lung slices.33 While the use of acellular normal and fibrotic human lung mimics the in vivo scenario, this system is not readily amenable for studying the relative contribution of ECM composition and stiffness. Recently, to overcome this limitation, polyacrylamide hydrogels of distinct moduli were functionalized with solubilized dECM from control and fibrotic human lungs to decouple mechanical properties from substrate stiffness. This study found that substrate stiffness was the dominant factor initiating activation of fibroblasts, and pericytes cultured on medium (4.4 ± 0.5 kPa) and high (23.7 ± 2.3 kPa) modulus substrates replicated transitioning and fibrotic human lung, respectively.30 These cells expressed significantly increased levels of αSMA when compared to cells cultured on soft hydrogels (1.8 ± 0.5 kPa) replicating healthy lung tissue.30 These results demonstrated that changes in αSMA expression and organization were mechanosensitive regardless of composition. However, this culture system does not allow for temporally changing mechanical properties over time. These systems enabled researchers to elucidate certain aspects regarding the influence of lung composition and stiffness on fibroblast activation in a static microenvironment, but the remaining limitation was that these systems could not be altered over time to recapitulate disease.The hybrid-hydrogel system engineered and presented in the current article offers an in vitro system to allow delineation of the contributions of composition versus mechanical environment during disease initiation and progression in vitro. The ability of the hybrid-hydrogel system to provide temporal control over substrate modulus in the presence of cells permits the evaluation of the effect of dynamic modulus variation on primary murine PDGFRα+ dual-reporter fibroblasts. Here, we used PDGFRα+ dual-reporter fibroblasts to allow real-time analysis of fibroblast activation (i.e., col1a1 and αSMA transgene expression). PDGFRα+ dual-reporter fibroblasts were seeded onto soft hybrid-hydrogels, photoinitiator (LAP) was added to culture media on day 6, and 365 nm UV light at 10 mW cm−2 (hν) was applied for 5 min at day 7 to stiffen these substrates. Fibroblast activation on these stiffened samples was compared to cells cultured on soft or stiff hybrid-hydrogel controls (Fig. 5a). There was a significant increase in the expression of myofibroblast transgenes Col1a1 and αSMA, respectively, when PDGFRα+ dual-reporter fibroblasts were cultured on stiff (87.2%, 90.3%) and dynamically stiffened hydrogels (88.6%, 88.9%) compared to soft hydrogels (36.7%, 37.2%) (Fig. 5b, p < 0.0001, ANOVA, Tukey Test). The higher levels of myofibroblast transgene expression on the stiffened hydrogels were comparable to those measured in cells cultured only on stiff hydrogels, and demonstrates that the fibroblasts were activated in response to in situ stiffening. Representative images also showed a morphological difference between PDGFRα+ dual-reporter fibroblasts cultured on the soft substrates (small and rounded) and those grown on the stiff and stiffened substrates (larger and more spread out; Fig. 5c). This change in cellular morphology and the presence of αSMA stress fibers are hallmarks of the myofibroblast phenotype that have been attributed to increases in substrate moduli.19,21,37,71 Similar responses were observed in primary dual-reporter fibroblasts cultured on fully synthetic PEGαMA hydrogel controls in response to the in situ stiffening (Fig. S6, ESI†).
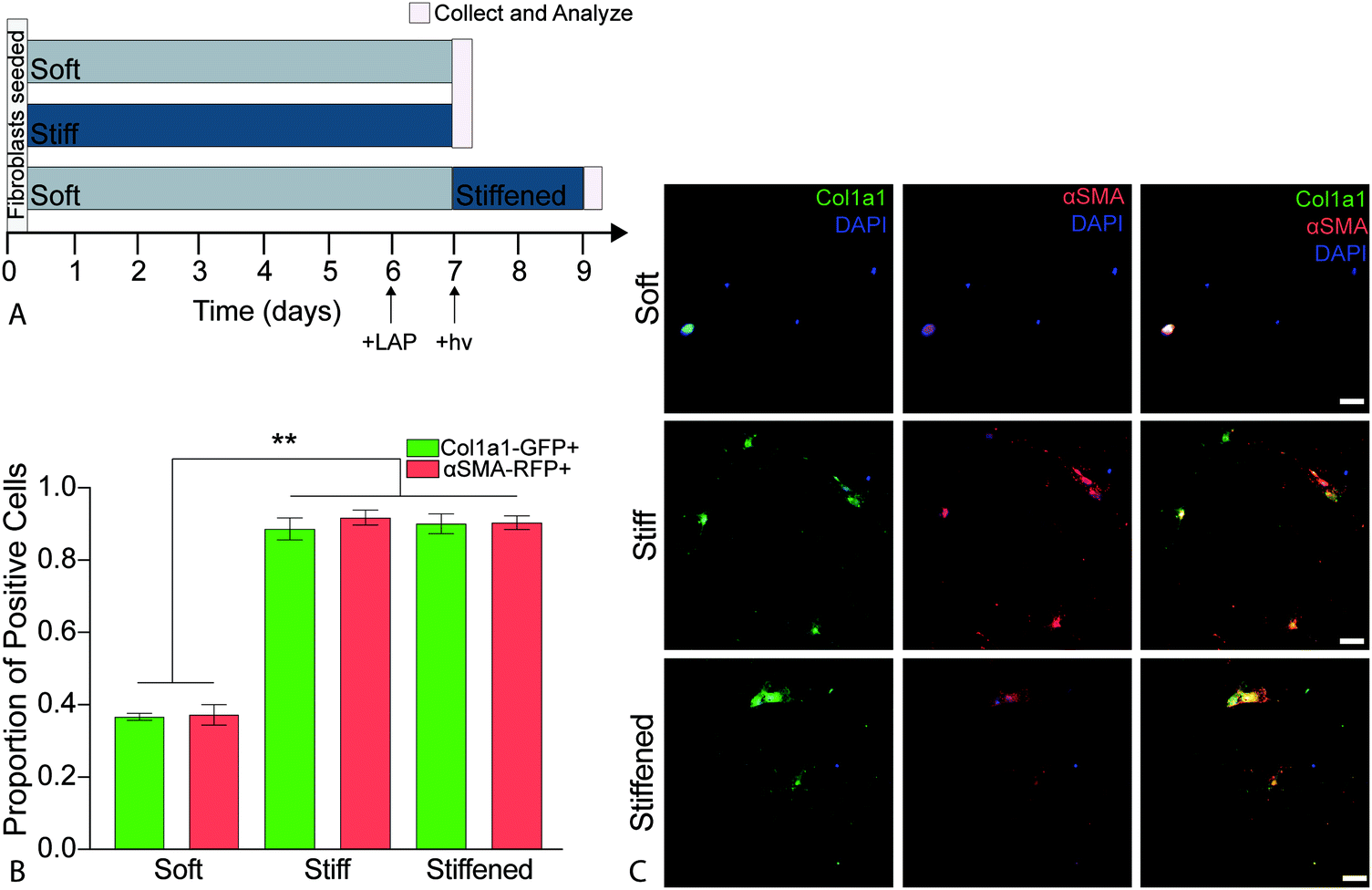 | ||
| Fig. 5 (A) Schematic depicting the timeline for temporal stiffening during activation experiments. Gray and dark blue bars indicated the culturing time of dual-reporter fibroblasts on soft and stiff substrates, respectively. Cells were cultured in 1% FBS media for all conditions. The photoinitiator (LAP) was added to culture media on day 6 for hydrogels to be stiffened, and 365 nm UV light at 10 mW cm−2 (hν) was applied for 5 min at day 7. Pink lines represent when samples were collected and analyzed. (B) Average proportion of dual-reporter fibroblasts that positively expressed Col1a1-GFP (green) and αSMA-RFP (red) for soft, stiff and stiffened conditions (n = 6, mean ± SEM). Significantly more cells cultured on stiff and stiffened substrates expressed Col1a1-GFP and αSMA-RFP than those cultured on soft substrates. (ANOVA, Tukey Test, **p < 0.0001). (C) Representative images of dual-reporter fibroblasts on soft and stiff hybrid-hydrogels on day 7 and stiffened hybrid hydrogels on day 9 showed expression of Col1a1-GFP and αSMA-RFP. Dual-reporter fibroblasts also demonstrated increased spreading and more spindle-like morphology on the stiff and stiffened hybrid-hydrogels compared to fibroblasts cultured on soft hybrid-hydrogels. Scale bars, 25 μm. | ||
Spatial heterogeneity is another hallmark of fibrotic disease that is important to replicate in vitro. Gradient stiffness polyacrylamide hydrogel substrates with modulus values ranging from 0.1 to 50 kPa that mimicked the increasing stiffness of crosslinked fibrotic lesions observed in murine bleomycin models showed notable transitions in fibroblast morphology compared to spindle-shaped cells typical of activated myofibroblasts observed in vivo at higher stiffness levels.16 Additionally, human lung fibroblasts seeded onto these materials expressed gradual increases in procollagen I and αSMA along the stiffness gradient, indicating that the matrix stiffness progressively activated fibroblasts. Another group investigated the influence of pattern size on hepatic stellate cells using UV-induced secondary crosslinking restricted with a photomask to spatially control mechanical properties with a modulus range of 2.5 ± 0.6 kPa outside the patterns to 15.3 ± 5.7 kPa within the patterns.16 There was an expression of high levels of αSMA and type I collagen on stiffer substrates, and the cells responded based on the local stiffness within the patterns. However, they remained quiescent on stiff substrates if the feature size was not sufficient to allow cell spreading.34
To investigate the influence of the spatial distribution of increases in matrix stiffness on PDGFRα+ dual-reporter fibroblasts over 7 days, patterned hybrid-hydrogels were fabricated by exposing soft substrates to light through a chrome-on-quartz photomask comprised of either 50- or 100-micron wide lines (Fig. 6a). Fibroblasts expressed significantly higher levels of the col1a1 transgene on both patterns within the stiff regions compared to the soft regions (Fig. 6b). Trends towards greater differences in expression were observed for both transgenes between the soft and stiff regions on the 100 micron pattern, demonstrating that tuning spatial patterning might impact the degree of cellular activation (Fig. 6c). Collectively, these studies have revealed that the phenotype of PDGFRα+ dual-reporter fibroblasts is highly dependent on substrate mechanical properties, and spatiotemporally stiffening can recreate the heterogeneous mechanical cues that cells encounter in vivo during fibrotic disease progression.
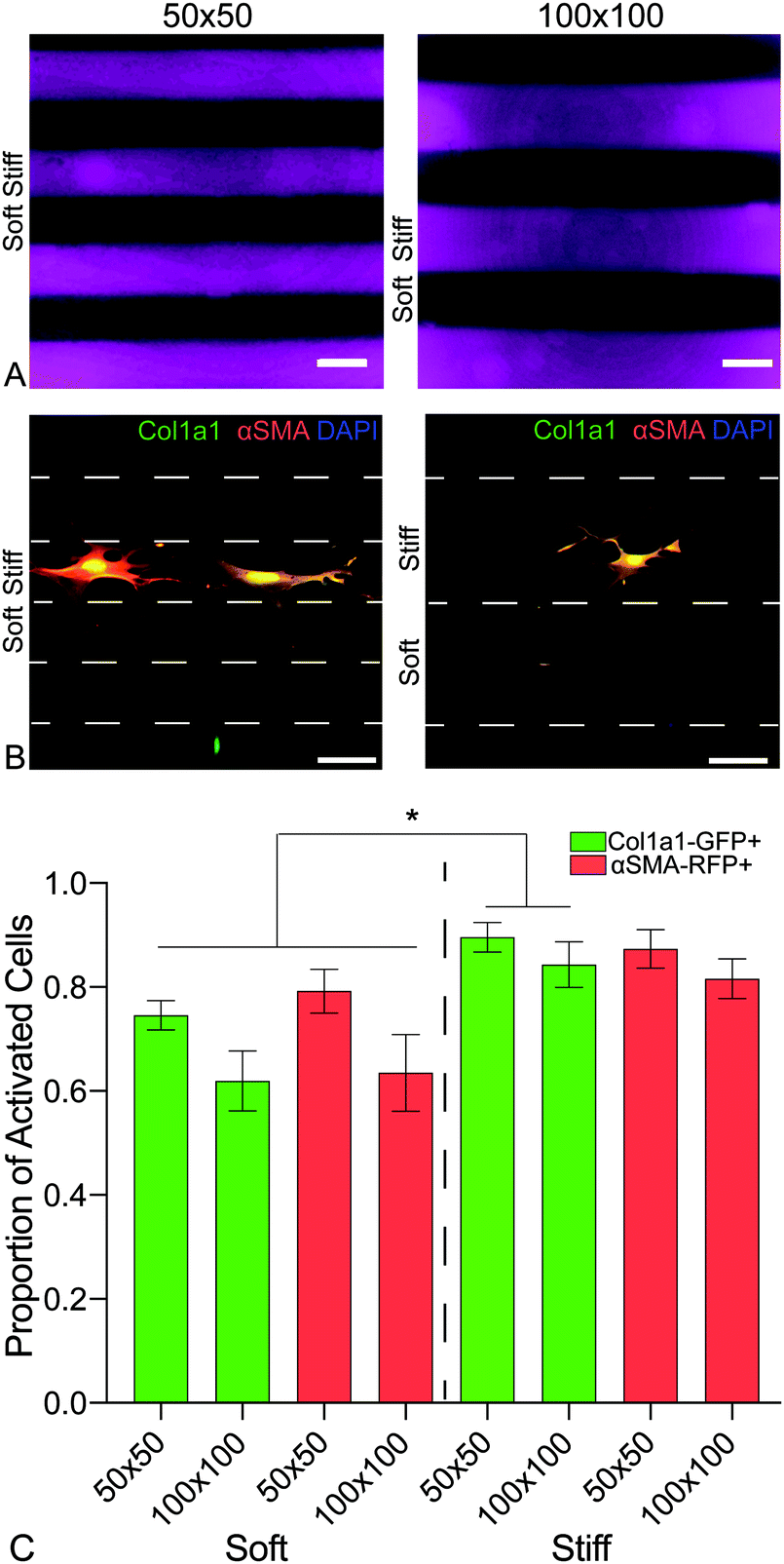 | ||
| Fig. 6 (A) A chrome on quartz photomask with two line patterns of either 50- or 100-micron width and spacing was placed in close contact with the hybrid hydrogel surfaces, which were exposed to 365 nm, 10 mW cm−2 at for 5 min, to spatially pattern defined regions of increased elastic modulus. (B) Representative images of PDGFRα+ dual reporter cells on both patterns. (C) Cells expressed significantly higher levels of col1a1 on both sizes within the stiff regions when compared to cells within the soft regions. There was an emerging trend of a bigger difference of expression with the larger spacing. This data is evidence of ability to spatially activate cells on the hybrid-hydrogel system. | ||
4. Conclusion
Here, a hydrolytically stable hybrid-hydrogel stiffening system with clickable dECM and a phototunable PEG backbone was synthesized and characterized. These hybrid-hydrogels integrated complex biologically relevant compositions into biomaterials that facilitated spatiotemporal control over mechanical properties to generate a platform for studying the dynamic molecular and cellular mechanisms underlying fibrosis. The dual-stage polymerization mechanism provided control over initial elastic modulus and supported spatiotemporal control over precise increases in local mechanical properties in situ, recreating the heterogeneous ECM stiffening that cells encounter in vivo. Using pulmonary fibrosis as a model of chronic fibrotic disease, we employed this in vitro system to investigate the response of PDGFRα+ fibroblasts from dual-transgenic reporter mice to local matrix stiffening. Experimental results indicated that fibroblasts cultured on stiff and temporally stiffened substrates with moduli replicating diseased tissue exhibited increased activation through the measurement of Col1a1 and αSMA transgene expression compared to those grown on soft substrates replicating healthy tissue. A phenotypic transition from quiescent to activated fibroblasts was initiated by exploiting a sequential crosslinking reaction scheme in these novel hybrid-hydrogels. In the present article, clickable dECM provided the complex compositional properties of healthy lung ECM. However, future experiments could include clickable dECM derived from fibrotic dECM to enable the decoupling of fibrotic tissue composition from mechanics for fundamental studies to probe how fibroblasts interact with and receive information from the extracellular microenvironment. This versatile system could also enable the encapsulation of healthy or fibrotic PDGFRα+ fibroblasts within 3D hybrid-hydrogels to investigate cellular responses to dynamic biophysical changes in the extracellular environment in a more physiologically relevant way. Harnessing independent and dynamic control over the presentation of biochemical and biophysical cues to cells cultured within 3D hybrid-hydrogels could allow future experiments with more control over experimental parameters that will improve our ability to study the cellular and molecular mechanisms underlying fibrotic disease initiation and progression.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Carol Mirita and Radu Moldovan, PhD from the Advanced Light Microscopy Core Facility at the University of Colorado Anschutz Medical Campus for assistance obtaining confocal images. The authors are also grateful to Dillon Jarrell, University of Colorado, Anschutz Medical Campus for assisting with photomask design as well as Cary Kuliasha, PhD and the Nanoscale Research Facility at the University of Florida for photomask fabrication. This work was supported by funding from the National Science Foundation Division of Materials Research (NSF CAREER 1941401 to CMM), the National Institutes of Health (NHLBI P01 HL14985 to KRS), and the Department of Defense (PR 192068 to CMM and PR 181125 to KRS). The Knut and Alice Wallenberg foundation is acknowledged for generous support (DEW). This project has received funding from a European Research Council (ERC) Starting Grant under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 805361) to DEW. ST is supported by a RESPIRE3 Postdoctoral Fellowship supported by the European Respiratory Society and the European Union's H2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement (Grant No. 713406). The authors also thank the Lund University Bioimaging Center (LBIC) for providing infrastructure support for SEM measurements.References
- M. E. Smithmyer, L. A. Sawicki and A. M. Kloxin, Biomater. Sci., 2014, 2, 634–650 RSC.
- A. Desmouliere, I. A. Darby and G. Gabbiani, Lab. Invest., 2003, 83, 1689–1707 CrossRef PubMed.
- S. Meran and R. Steadman, Int. J. Exp. Pathol., 2011, 92, 158–167 CrossRef CAS PubMed.
- J. H. W. Distler, A.-H. Györfi, M. Ramanujam, M. L. Whitfield, M. Königshoff and R. Lafyatis, Nat. Rev. Rheumatol., 2019, 15, 705–730 CrossRef CAS PubMed.
- M. Selman and A. Pardo, Respir. Res., 2002, 3, 3 CrossRef PubMed.
- M. Selman and A. Pardo, Proc. Am. Thorac. Soc., 2006, 3, 364–372 CrossRef CAS PubMed.
- M. Korfei, S. Schmitt, C. Ruppert, I. Henneke, P. Markart, B. Loeh, P. Mahavadi, M. Wygrecka, W. Klepetko, L. Fink, P. Bonniaud, K. T. Preissner, G. Lochnit, L. Schaefer, W. Seeger and A. Guenther, J. Proteome Res., 2011, 10, 2185–2205 CrossRef CAS PubMed.
- M. G. Jones, A. Fabre, P. Schneider, F. Cinetto, G. Sgalla, M. Mavrogordato, S. Jogai, A. Alzetani, B. G. Marshall, K. M. A. O’Reilly, J. A. Warner, P. M. Lackie, D. E. Davies, D. M. Hansell, A. G. Nicholson, I. Sinclair, K. K. Brown and L. Richeldi, JCI Insight, 2016, 1, e86375 Search PubMed.
- T. A. Wynn and T. R. Ramalingam, Nat. Med., 2012, 18, 1028–1040 CrossRef CAS PubMed.
- T. N. Wight and S. Potter-Perigo, Am. J. Physiol.: Gastrointest. Liver Physiol., 2011, 301, G950–G955 CrossRef CAS PubMed.
- M. W. Parker, D. Rossi, M. Peterson, K. Smith, K. Sikström, E. S. White, J. E. Connett, C. A. Henke, O. Larsson and P. B. Bitterman, J. Clin. Invest., 2014, 124, 1622–1635 CrossRef CAS PubMed.
- J. K. Burgess, T. Mauad, G. Tjin, J. C. Karlsson and G. Westergren-Thorsson, J. Pathol., 2016, 240, 397–409 CrossRef CAS PubMed.
- Y. Zhou, J. C. Horowitz, A. Naba, N. Ambalavanan, K. Atabai, J. Balestrini, P. B. Bitterman, R. A. Corley, B. S. Ding, A. J. Engler, K. C. Hansen, J. S. Hagood, F. Kheradmand, Q. S. Lin, E. Neptune, L. Niklason, L. A. Ortiz, W. C. Parks, D. J. Tschumperlin, E. S. White, H. A. Chapman and V. J. Thannickal, Matrix Biol., 2018, 73, 77–104 CrossRef CAS PubMed.
- J. Herrera, C. A. Henke and P. B. Bitterman, J. Clin. Invest., 2018, 128, 45–53 CrossRef PubMed.
- A. J. Haak, Q. Tan and D. J. Tschumperlin, Matrix Biol., 2018, 73, 64–76 CrossRef CAS PubMed.
- F. Liu, J. D. Mih, B. S. Shea, A. T. Kho, A. S. Sharif, A. M. Tager and D. J. Tschumperlin, J. Cell Biol., 2010, 190, 693–706 CrossRef CAS PubMed.
- Z. Borok, J. A. Whitsett, P. B. Bitterman, V. J. Thannickal, D. N. Kotton, S. D. Reynolds, M. A. Krasnow, D. W. Bianchi, E. E. Morrisey, B. L. Hogan, J. M. Kurie, D. C. Walker, D. C. Radisky, S. L. Nishimura, S. M. Violette, P. W. Noble, S. D. Shapiro, C. J. Blaisdell and H. A. Chapman, Proc. Am. Thorac. Soc., 2011, 8, 215–222 CrossRef PubMed.
- S. H. Phan, Proc. Am. Thorac. Soc., 2008, 5, 334–337 CrossRef PubMed.
- K. M. Mabry, S. Z. Payne and K. S. Anseth, Biomaterials, 2016, 74, 31–41 CrossRef CAS PubMed.
- H. Wang, L. A. Leinwand and K. S. Anseth, Nat. Rev. Cardiol., 2014, 11, 715 CrossRef PubMed.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS PubMed.
- D. M. Habiel and C. Hogaboam, Front. Pharmacol., 2014, 5, 2 Search PubMed.
- C.-J. Hu, H. Zhang, A. Laux, S. S. Pullamsetti and K. R. Stenmark, J. Physiol., 2019, 597, 1103–1119 CrossRef CAS PubMed.
- L. T. Saldin, M. C. Cramer, S. S. Velankar, L. J. White and S. F. Badylak, Acta Biomater., 2017, 49, 1–15 CrossRef CAS PubMed.
- B. M. Young, K. Shankar, C. K. Tho, A. R. Pellegrino and R. L. Heise, Acta Biomater., 2019, 100, 223–234 CrossRef CAS PubMed.
- R. A. Pouliot, P. A. Link, N. S. Mikhaiel, M. B. Schneck, M. S. Valentine, F. J. Kamga Gninzeko, J. A. Herbert, M. Sakagami and R. L. Heise, J. Biomed. Mater. Res., Part A, 2016, 104, 1922–1935 CrossRef CAS PubMed.
- M. M. De Santis, D. A. Bölükbas, S. Lindstedt and D. E. Wagner, Eur. Respir. J., 2018, 52, 1601355 CrossRef PubMed.
- F. Liu and D. J. Tschumperlin, J. Visualized Exp., 2011, 2911, DOI:10.3791/2911.
- R. H. J. de Hilster, P. K. Sharma, M. R. Jonker, E. S. White, E. A. Gercama, M. Roobeek, W. Timens, M. C. Harmsen, M. N. Hylkema and J. K. Burgess, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2020, 318, L698–L704 CrossRef CAS PubMed.
- P. Sava, A. Ramanathan, A. Dobronyi, X. Peng, H. Sun, A. Ledesma-Mendoza, E. L. Herzog and A. L. Gonzalez, JCI Insight, 2017, 2(24), 96352 CrossRef PubMed.
- J. Visser, P. A. Levett, N. C. te Moller, J. Besems, K. W. Boere, M. H. van Rijen, J. C. de Grauw, W. J. Dhert, P. R. van Weeren and J. Malda, Tissue Eng., Part A, 2015, 21, 1195–1206 CrossRef CAS PubMed.
- A. Marinković, F. Liu and D. J. Tschumperlin, Am. J. Respir. Cell Mol. Biol., 2013, 48, 422–430 CrossRef PubMed.
- A. J. Booth, R. Hadley, A. M. Cornett, A. A. Dreffs, S. A. Matthes, J. L. Tsui, K. Weiss, J. C. Horowitz, V. F. Fiore, T. H. Barker, B. B. Moore, F. J. Martinez, L. E. Niklason and E. S. White, Am. J. Respir. Crit. Care Med., 2012, 186, 866–876 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- A. M. Rosales, C. B. Rodell, M. H. Chen, M. G. Morrow, K. S. Anseth and J. A. Burdick, Bioconjugate Chem., 2018, 29, 905–913 CrossRef CAS PubMed.
- A. M. Rosales, S. L. Vega, F. W. DelRio, J. A. Burdick and K. S. Anseth, Angew. Chem., Int. Ed., 2017, 56, 12132–12136 CrossRef CAS PubMed.
- K. M. Mabry, R. L. Lawrence and K. S. Anseth, Biomaterials, 2015, 49, 47–56 CrossRef CAS PubMed.
- C. M. Magin, D. L. Alge and K. S. Anseth, Biomed. Mater., 2016, 11, 022001 CrossRef PubMed.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- J. A. Zepp, W. J. Zacharias, D. B. Frank, C. A. Cavanaugh, S. Zhou, M. P. Morley and E. E. Morrisey, Cell, 2017, 170, 1134–1148.e1110 CrossRef CAS PubMed.
- S. T. Magness, R. Bataller, L. Yang and D. A. Brenner, Hepatology, 2004, 40, 1151–1159 CrossRef CAS PubMed.
- P. A. Reyfman, J. M. Walter, N. Joshi, K. R. Anekalla, A. C. McQuattie-Pimentel, S. Chiu, R. Fernandez, M. Akbarpour, C.-I. Chen, Z. Ren, R. Verma, H. Abdala-Valencia, K. Nam, M. Chi, S. Han, F. J. Gonzalez-Gonzalez, S. Soberanes, S. Watanabe, K. J. N. Williams, A. S. Flozak, T. T. Nicholson, V. K. Morgan, D. R. Winter, M. Hinchcliff, C. L. Hrusch, R. D. Guzy, C. A. Bonham, A. I. Sperling, R. Bag, R. B. Hamanaka, G. M. Mutlu, A. V. Yeldandi, S. A. Marshall, A. Shilatifard, L. A. N. Amaral, H. Perlman, J. I. Sznajder, A. C. Argento, C. T. Gillespie, J. Dematte, M. Jain, B. D. Singer, K. M. Ridge, A. P. Lam, A. Bharat, S. M. Bhorade, C. J. Gottardi, G. R. S. Budinger and A. V. Misharin, Am. J. Respir. Crit. Care Med., 2018, 199, 1517–1536 CrossRef PubMed.
- R. C. Chambers and P. F. Mercer, Ann. Am. Thorac. Soc., 2015, 12, S16–S20 CrossRef PubMed.
- A. Camelo, R. Dunmore, M. A. Sleeman and D. L. Clarke, Front. Pharmacol., 2013, 4, 173 Search PubMed.
- F. Liu, D. Lagares, K. M. Choi, L. Stopfer, A. Marinković, V. Vrbanac, C. K. Probst, S. E. Hiemer, T. H. Sisson, J. C. Horowitz, I. O. Rosas, L. E. Fredenburgh, C. Feghali-Bostwick, X. Varelas, A. M. Tager and D. J. Tschumperlin, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2014, 308, L344–L357 CrossRef PubMed.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- D. Davis-Hall, V. Nguyen, T. J. D'Ovidio, E. Tsai, G. Bilousova and C. M. Magin, Adv. Biosyst., 2019, 3, 1900022 CrossRef.
- X. Tong, J. Lai, B.-H. Guo and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1513–1516 CrossRef CAS.
- M. S. Hahn, L. J. Taite, J. J. Moon, M. C. Rowland, K. A. Ruffino and J. L. West, Biomaterials, 2006, 27, 2519–2524 CrossRef CAS PubMed.
- S. Lin-Gibson, S. Bencherif, J. A. Cooper, S. J. Wetzel, J. M. Antonucci, B. M. Vogel, F. Horkay and N. R. Washburn, Biomacromolecules, 2004, 5, 1280–1287 CrossRef CAS PubMed.
- D. E. Wagner, N. R. Bonenfant, D. Sokocevic, M. J. DeSarno, Z. D. Borg, C. S. Parsons, E. M. Brooks, J. J. Platz, Z. I. Khalpey, D. M. Hoganson, B. Deng, Y. W. Lam, R. A. Oldinski, T. Ashikaga and D. J. Weiss, Biomaterials, 2014, 35, 2664–2679 CrossRef CAS PubMed.
- F. E. Uhl, D. E. Wagner and D. J. Weiss, Methods Mol. Biol., 2017, 1627, 253–283 CrossRef CAS PubMed.
- J. C. Grim, I. A. Marozas and K. S. Anseth, J. Controlled Release, 2015, 219, 95–106 CrossRef CAS PubMed.
- K. R. Chirco, K. S. Worthington, M. J. Flamme-Wiese, M. J. Riker, J. D. Andrade, B. M. Ueberheide, E. M. Stone, B. A. Tucker and R. F. Mullins, Acta Biomater., 2017, 57, 293–303 CrossRef CAS PubMed.
- C. M. Kirschner and K. S. Anseth, Small, 2013, 9, 578–584 CrossRef CAS PubMed.
- T. K. L. Meyvis, S. C. De Smedt, J. Demeester and W. E. Hennink, J. Rheol., 1999, 43, 933–950 CrossRef CAS.
- I. f. L. A. Research, Journal, 2011.
- J. Green, M. Endale, H. Auer and A.-K. T. Perl, Am. J. Respir. Cell Mol. Biol., 2016, 54, 532–545 CrossRef CAS PubMed.
- P. E. Farahani, S. M. Adelmund, J. A. Shadish and C. A. DeForest, J. Mater. Chem. B, 2017, 5, 4435–4442 RSC.
- H. Ma, A. R. Killaars, F. W. DelRio, C. Yang and K. S. Anseth, Biomaterials, 2017, 131, 131–144 CrossRef CAS PubMed.
- M. Gajendiran, J.-S. Rhee and K. Kim, Tissue Eng., Part B, 2017, 24, 66–74 CrossRef PubMed.
- S. T. Gould, N. J. Darling and K. S. Anseth, Acta Biomater., 2012, 8, 3201–3209 CrossRef CAS PubMed.
- C. M. Kirschner, D. L. Alge, S. T. Gould and K. S. Anseth, Adv. Healthcare Mater., 2014, 3, 649–657 CrossRef CAS PubMed.
- K. E. Bailey, C. Pino, M. L. Lennon, A. Lyons, J. G. Jacot, S. R. Lammers, M. Konigshoff and C. M. Magin, Am. J. Respir. Cell Mol. Biol., 2020, 62, 14–22 CrossRef CAS PubMed.
- F. Liu, C. M. Haeger, P. B. Dieffenbach, D. Sicard, I. Chrobak, A. M. F. Coronata, M. M. S. Velandia, S. Vitali, R. A. Colas, P. C. Norris, A. Marinković, X. Liu, J. Ma, C. D. Rose, S.-J. Lee, S. A. A. Comhair, S. C. Erzurum, J. D. McDonald, C. N. Serhan, S. R. Walsh, D. J. Tschumperlin and L. E. Fredenburgh, JCI Insight, 2016, 1(8), e86987 Search PubMed.
- C. M. Kirschner and K. S. Anseth, Acta Mater., 2013, 61, 931–944 CrossRef CAS PubMed.
- H. Wang, S. M. Haeger, A. M. Kloxin, L. A. Leinwand and K. S. Anseth, PLoS One, 2012, 7, e39969 CrossRef CAS PubMed.
- A. Metters and J. Hubbell, Biomacromolecules, 2004, 6, 290–301 CrossRef PubMed.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- H. B. Schiller, I. E. Fernandez, G. Burgstaller, C. Schaab, R. A. Scheltema, T. Schwarzmayr, T. M. Strom, O. Eickelberg and M. Mann, Mol. Syst. Biol., 2015, 11, 819 CrossRef PubMed.
- S. R. Caliari, M. Perepelyuk, B. D. Cosgrove, S. J. Tsai, G. Y. Lee, R. L. Mauck, R. G. Wells and J. A. Burdick, Sci. Rep., 2016, 6, 21387 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00613k |
| This journal is © The Royal Society of Chemistry 2020 |