
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe relationship between oxide-ion conductivity and cation vacancy order in the hybrid hexagonal perovskite Ba3VWO8.5†
Asma
Gilane
ab,
Sacha
Fop
 a,
Falak
Sher
b,
Ronald I.
Smith
c and
Abbie C.
Mclaughlin
*a
a,
Falak
Sher
b,
Ronald I.
Smith
c and
Abbie C.
Mclaughlin
*a
aThe Chemistry Department, University of Aberdeen, Meston Walk, Aberdeen AB24 3UE, UK. E-mail: a.c.mclaughlin@abdn.ac.uk
bDepartment of Chemistry and Chemical Engineering, SBA School of Science and Engineering, Lahore University of Management Sciences (LUMS), Lahore, Pakistan
cISIS Facility, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
First published on 22nd July 2020
Abstract
Significant oxide ionic conductivity has recently been reported in cation-deficient hexagonal perovskite Ba3M′M′′O8.5 derivatives (M′ = Nb; M′′ = Mo, W), with disordered hybrid 9R-palmierite average structures. Here, we present a study of the crystal structure and electrical properties of the related compound Ba3VWO8.5. Electrical characterization demonstrates that Ba3VWO8.5 is also an oxide ion conductor with a bulk conductivity of 2.0 × 10−3 S cm−1 in air at 900 °C, thus revealing that it is possible to obtain oxide ion conducting Ba3M′M′′O8.5 materials with a variety of different M′M′′ combinations. Whilst Ba3NbMoO8.5 and Ba3NbWO8.5 present a random distribution of cationic vacancies, X-ray and neutron diffraction experiments demonstrate that the cationic vacancies are ordered on the M2 sites in Ba3VWO8.5, resulting in a structure where M1Ox palmierite-like layers are separated by empty octahedral cavities. Bond-valence site energy (BVSE) analysis on the different phases reveals that ordering of the cationic vacancies hinders long-range oxygen diffusivity parallel to the c-axis in Ba3VWO8.5 explaining the reduced ionic conductivity of this compound. These results suggest that, together with the dominant 2-dimensional conduction pathway along the palmierite-like layers, additional diffusion routes parallel to the c-axis provide a relevant contribution to the conductivity of these Ba3M′M′′O8.5 systems by creation of a complex 3-dimensional ionic percolation network, the topology of which depends on the particular arrangement of cation and anion vacancies.
Introduction
Solid oxide fuel cells (SOFCs) are electrochemical devices able to generate electricity from renewable energy sources, with high efficiency and low emissions of pollutants.1–3 However, oxide ion transport in most commercially available solid electrolytes is enabled only at high working temperatures (>700 °C), thus making the commercialization of SOFCs particularly challenging.2,4,5 The high operating temperature reduces the components' durability, results in slow start up times and the necessitation of expensive materials for seals and interconnects.1,4 In order to overcome these issues, it is desirable to develop novel electrolyte materials with significant oxide ion conductivity at intermediate temperatures (300–600 °C). Ionic conduction in solid oxides is strongly related to the crystal structure, and as a result the discovery and characterization of novel oxide ion conductors in various structural families has attained significant attention from researchers.6 Several fast oxide ion conductors have been reported in systems such as fluorite-type structures,7–9 silicon and germanium apatites,10 rare earth molybdates (La2Mo2O9, LAMOX materials),11 Bi4V2O11-derived oxides (BIMEVOX),12 and compounds containing GaO4 tetrahedral moieties.13,14 High oxide ion conductivity has also been observed in different cubic and layered perovskite systems.15–18 The perovskite ABO3 structure is generally composed by a framework of corner-sharing BO6 octahedral units, in which the A cations occupy the interstitial voids. When there is a large difference in size between the constituent cations, some of the B octahedra share faces to release the strain caused by the size mismatch between the cations, resulting in the formation of hexagonal polytypes.19 Mixed combinations of corner-sharing and face-sharing octahedra can give rise to a variety of hexagonal perovskite derivatives able to accommodate cationic and anionic vacancies.19,20 Recently, we have reported the first hexagonal perovskite derivative with significant oxide ion conductivity, Ba3NbMoO8.5.21 Ba3NbMoO8.5 exhibits bulk oxide ion conductivity of 2.2 × 10−3 S cm−1 at 600 °C, competitive with other electrolyte materials, and the conductivity is predominantly ionic in a wide pO2 range. Ba3NbMoO8.5 is a cation-deficient hexagonal perovskite derivative with an average structure composed of a hybrid of the 9R perovskite and palmierite units.21 Variable coordination M1Ox polyhedral units are generated within the palmierite-like layers due to considerable positional oxygen disorder. Average octahedral and tetrahedral units arise from partial occupation of two crystallographic oxygen positions (O2 and O3),21,22 with the existence of 5-fold geometries on the local-scale.23,24 The random distribution of intrinsic oxygen vacancies and available oxygen sites results in a continuous 2-dimensional ionic diffusion pathway for oxide migration along the palmierite-like layers.25 The cationic vacancies are disordered on two mutually exclusive metal sites (M1 and M2), leading to the formation of complex disordered stacking configurations of M1Ox and M2O6 polyhedral units.23,26 Substantial oxide ion conductivity has been discovered in other compounds of the Ba3M′M′′O8.5 (M′ = Nb; M′′ = Mo, W) family.26–30 In particular, members of the Ba3Nb1−xVxMoO8.5 series present enhanced ionic conductivity, with the x = 0.1 composition showing a bulk conductivity of ∼0.01 S cm−1 at 600 °C, one order of magnitude higher than the conductivity of Ba3NbMoO8.5.26 Furthermore, Ba3NbWO8.5 exhibits similar conductivity to Ba3NbMoO8.5 at 600 °C and greater stability in a reducing 5% H2/N2 atmosphere.27 High oxide ion and proton conductivity has also been reported in Ba7Nb4MoO20, a hexagonal derivative with an average crystal structure composed of a disordered arrangement of 12R hexagonal perovskite and palmierite-like layers, similar to Ba3NbMoO8.5.31 The structures of these cation-deficient hexagonal perovskite derivatives are versatile and able to accommodate various transition metal cations, potentially enabling ionic conduction in different disordered layered systems.In this study, we present the crystal structure and electrical properties of the hexagonal perovskite derivative Ba3VWO8.5. This phase has been synthesized previously, but the conductivity has not been measured, nor the structure characterized by neutron diffraction.32 Impedance spectroscopy measurements reveal that Ba3VWO8.5 is also an oxide ion conductor at high and intermediate oxygen partial pressures, with a bulk conductivity of 2.0 × 10−3 S cm−1 at 900 °C. Hence, it is possible to synthesise hexagonal perovskite oxide ion conductors without the elements of Mo and Nb, opening up further opportunities for discovering novel oxide and/or proton conductors in the hexagonal perovskite family. While all the Ba3M′M′′O8.5 phases reported so far are isostructural with Ba3NbMoO8.5, in Ba3VWO8.5 the cationic vacancies are ordered on the M2 sites, thus resulting in a structure where M1Ox palmierite-like layers are separated by empty octahedral cavities. Bond-valence site energy (BVSE) analysis on the average structure confirms the presence of diffusion pathways along the palmierite-like layers but demonstrates the absence of long-range oxygen mobility parallel to the c-axis. The relationship between the electrical properties and BVSE calculations on Ba3NbMoO8.5, Ba3NbWO8.5 and Ba3VWO8.5 suggests that additional pathways parallel to the c-axis may provide a relevant contribution to the conductivity of these systems, highlighting the importance of both cation disorder and the chemistry of the M′M′′ cations in defining the ionic conducting properties of Ba3M′M′′O8.5 derivatives.
Experimental
Ba3VWO8.5 was prepared by the solid-state reaction of stoichiometric quantities of Ba(NO3)2 (99.999%, Aldrich), WO3 (99.9%, Aldrich) and V2O5 (≥99.6%, Aldrich). The starting materials were ground, pressed into a pellet and then calcined in air at 900 °C for 10 hours. The pellet was subsequently ground, re-pelletized and sintered at 1060 °C for 24 hours in air with a heating rate of 5 °C min−1 and then cooled down to room temperature with a cooling rate of 5 °C min−1. The last sintering step was repeated multiple times until a phase pure sample was obtained. The synthesis of Ba3VWO8.5 is very sensitive to sintering temperature and a phase pure material is only obtained over a very narrow temperature range (1060 °C ± 10 °C). This could be a result of Ba volatilisation. Impurities of BaWO4 form above this temperature.Powder X-ray diffraction (XRD) data were collected with a PANalytical Empyrean Powder diffractometer with Cu Kα1 radiation at room temperature. Data were acquired in the range 10° ≤ 2θ ≤ 110°, with a step size of 0.013°. Time-of-flight neutron powder diffraction experiments were performed at room temperature on the Polaris diffractometer33 at the ISIS pulsed neutron source (Rutherford Appleton Laboratory). A sample of 5 g of Ba3VWO8.5 was loaded into an 8 mm diameter cylindrical vanadium can and data were recorded for a total of 350 μA h proton charge to the ISIS target, corresponding to ∼2 hours exposure in the neutron beam. Rietveld analysis was performed using the GSAS/EXPGUI package.34 The background was fitted by the Chebyschev polynomial function and the peak shapes were modelled using profile function 3, which is a convolution of a pseudo-Voigt function with two back-to-back exponentials.
For the impedance spectroscopy measurements, a pellet of ∼10 mm diameter and ∼1.5 mm thickness was prepared using a sample of Ba3VWO8.5 sintered at 1060 °C for 48 hours to achieve 90% of the theoretical density. Platinum electrodes were painted on both sides of the pellet using Pt paste (Metalor 6082). Impedance measurements were acquired in the frequency range 0.1 Hz to 1.0 MHz with a Solartron 1260 impedance analyser with an applied alternating voltage of 0.1 V. Data were recorded upon cooling from 900 °C to 350 °C in a sealed tube furnace under a flow of dry air, oxygen, nitrogen and 5% H2/N2 gas mixture. Dry gaseous atmospheres were obtained by flowing the employed gas through a column of a commercial desiccant (Drierite) (pH2O < 10−4 atm). Impedance data were measured every ∼15 °C allowing 2 hours equilibration time at each temperature. The recorded impedance data were corrected using the geometrical factor and analysed with the ZView software (Scribner Associates, Inc.)
Bond-valence site energy (BVSE) calculations were performed with the softBV program,35,36 using the structural models from Rietveld refinement as input. Details about the softBV approach and the type of force-field employed for the BVS analysis can be found elsewhere.35,36 Bond-valence site energy landscapes for the interaction of a test O2− ion were calculated for a dense grid of points with a resolution of 0.1 Å. Diffusion pathways were identified with regions of low bond-valence site energy by direct visualization of the connectivity of the isosurfaces and by examination of the calculated pathway segments. BVSE models of migration barriers were plotted from the calculated energy profiles of the pathway segments.
Analysis of minimum bonding ellipsoids using the PIEFACE software was employed to evaluate the effect of doping on the relaxation of the average metal coordination polyhedra.37 The polyhedral distortion was quantified by the standard deviation, σ(R), of the three principal ellipsoids' radii (Rx, Ry, Rz).
Results and discussion
The X-ray diffraction pattern of Ba3VWO8.5 (Fig. 1) shows that there are no impurities in the as prepared sample and that the peaks could be indexed in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m H, as previously reported for Ba3NbMoO8.5.21
m H, as previously reported for Ba3NbMoO8.5.21
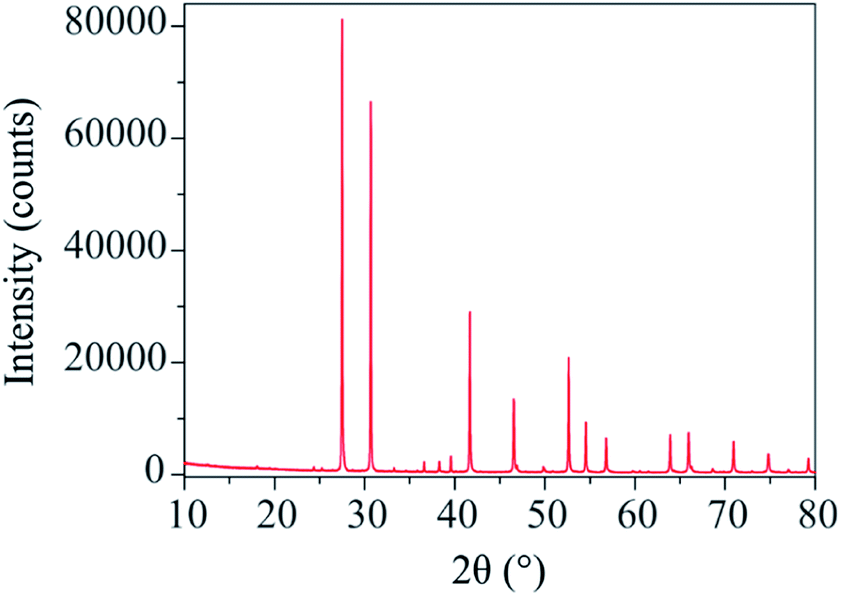 | ||
| Fig. 1 X-ray diffraction pattern of Ba3VWO8.5 which can be indexed by the Rm H space group. | ||
The phase stability of Ba3VWO8.5 was investigated by annealing the sample in air at selected temperatures between 550 °C and 900 °C for 24 hours. There is no evidence of Bragg reflections from impurity phases after annealing, as shown in the X-ray diffraction data in Fig. 2a. This indicates that Ba3VWO8.5 is stable in air between 500 to 900 °C. This contrasts with Ba3NbMoO8.5, which is not stable between 650–1000 °C, with the formation of BaMoO4 and Ba7Nb4MoO20 impurities above 650 °C.21 The Ba3VWO8.5 sample was also annealed at 900 °C for 24 hours under the flow of a range of different gases (O2, N2, 5% H2/N2). The XRD data demonstrate that the Ba3VWO8.5 phase is stable in both oxidizing and reducing environments (Fig. 2b).
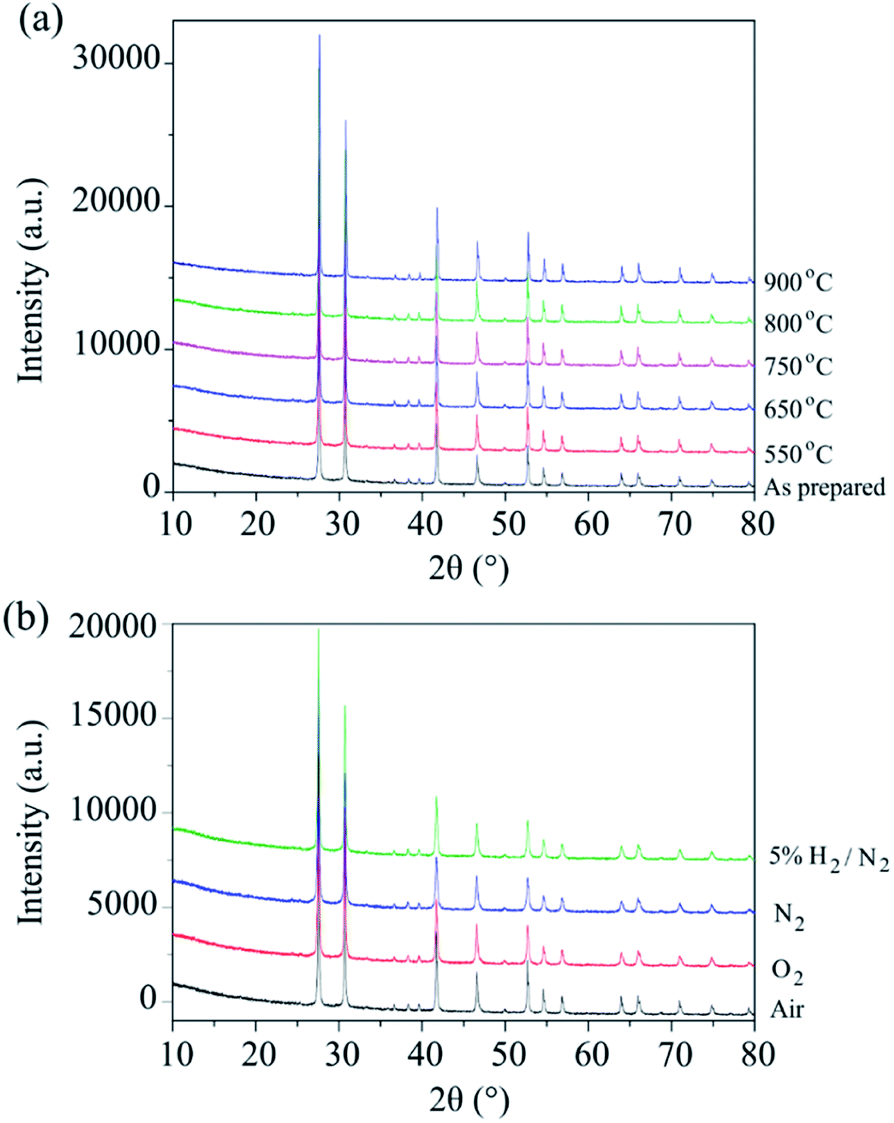 | ||
| Fig. 2 (a) XRD patterns of Ba3VWO8.5 recorded after a 24 hour anneal in air at selected temperatures. (b) XRD patterns of Ba3VWO8.5 recorded after 24 hours annealing at 900 °C under different flowing gases (air, O2, N2, 5% H2/N2). | ||
AC impedance spectroscopy was used to investigate the ionic conductivity of Ba3VWO8.5. Typical complex impedance plane (Z*) plots are displayed in Fig. 3a and b. The bulk (C ∼7–12 pF cm−1) and grain boundary responses (C ∼30–40 pF cm−1) have very similar time constants and overlap in the high-intermediate frequency regime (Fig. 3a), resulting in a broad and depressed grain arc.
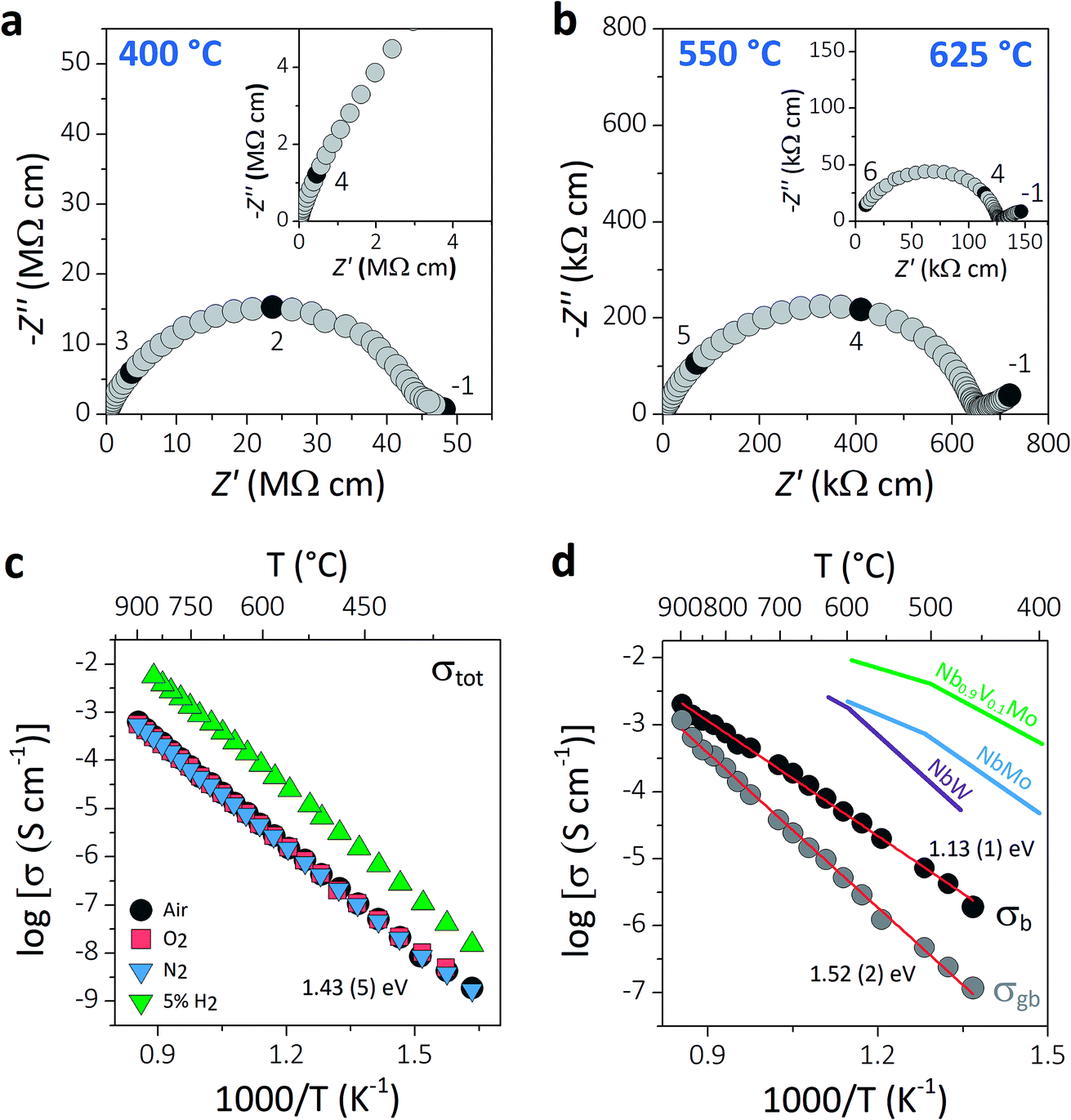 | ||
| Fig. 3 (a and b) Typical complex impedance plots recorded in oxygen at 400 °C, 550 °C, and 625 °C. The inset in (a) shows a magnification of the high frequency region. The numbers and corresponding filled symbols indicate selected frequency decades. (c) Arrhenius plot of the total conductivity of Ba3VWO8.5 recorded in a range of different dry atmospheres (air, O2, N2, 5% H2/N2), with the reported activation energy in dry air. (d) Arrhenius plots of the bulk and grain boundary conductivity of Ba3VWO8.5; red lines show the linear fit to the data with the corresponding activation energies. The bulk conductivities of Ba3NbMoO8.5 (NbMo, ref. 21), Ba3NbWO8.5 (NbW, ref. 27) and Ba3Nb0.9V0.1MoO8.5 (Nb0.9V0.1Mo, ref. 26) are reported for comparison. | ||
A low-slope electrode signal is observed at low frequencies above 450 °C (Fig. 3b). This feature indicates finite length diffusion typical of a Warburg electrode response and is evidence of ionic conduction in a material with partially blocking electrodes.38 The electrode response becomes less resistive upon increasing the oxygen partial pressure, consistent with oxygen ion diffusion. Total resistivity (Rb + Rgb) values were obtained from the high frequency intercept of the grain boundary semicircle on the real axis at all temperatures. The total conductivity values of Ba3VWO8.5 recorded in a range of different atmospheres are presented in the Arrhenius plot in Fig. 3c. Ba3VWO8.5 exhibits a total conductivity of 6.2 × 10−4 S cm−1 in air at 900 °C, with an activation energy of 1.43 (5) eV. There is no change in the conductivity values recorded in air, O2 and N2, revealing that the total conductivity of Ba3VWO8.5 is independent of oxygen partial pressure at high pO2 (see ESI Fig. S1†), so that pure oxide ion conduction is observed at high pO2. There is an increase in the conductivity in 5% H2/N2, which reveals that Ba3VWO8.5 exhibits an electronic component in the more reducing atmosphere. This is analogous to Ba3NbMoO8.5, where concentration cell experiments revealed a minor electronic component,21 but is in stark contrast to Ba3WNbO8.5 and Ba3Nb0.9V0.1MoO8.5, for which impedance spectroscopy measurements under 5% H2/N2 revealed a negligible electronic conductivity contribution.26,27
Equivalent circuit analysis was used to extract the individual bulk (Rb) and grain boundary resistivity values (Rgb). An equivalent circuit constituted by a series combination of a resistor (R) in parallel with a constant phase element (CPE) was employed to model the bulk and grain boundary impedance responses of the data collected in dry air (see ESI Fig. S2†). Representative equivalent circuit fits of the complex impedance data at 500 °C and 700 °C are shown in ESI Fig. S3.† The Arrhenius plot in Fig. 3d shows the bulk conductivity of Ba3VWO8.5 compared with the bulk conductivities of Ba3NbMoO8.5, Ba3NbWO8.5 and B3Nb0.9V0.1MoO8.5. The bulk conductivity of Ba3VWO8.5 is 5.0 × 10−5 S cm−1 at 600 °C, two orders of magnitude lower than the bulk conductivity of Ba3NbMoO8.5 (2.2 × 10−3 S cm−1) and Ba3NbWO8.5 (1.8 × 10−3 S cm−1) and three orders of magnitude lower than the conductivity of the V-doped B3Nb0.9V0.1MoO8.5 composition (1.0 × 10−2 S cm−1). The grain boundaries of Ba3VWO8.5 present reduced conductivity (5.3 × 10−6 S cm−1 at 600 °C) and considerably higher activation energy (1.52 (2) eV) than for ionic transport in the bulk domain (1.13 (1) eV). The grain boundaries clearly constitute the most resistive part, dominating the total resistivity of the material, as reported also for other Ba3M′M′′O8.5 derivatives.21,27 Materials containing barium that are sintered at high temperature can have a different chemical composition of the grain boundaries resulting in a higher grain boundary resistivity due to the presence of blocking effects, as in the case of Ba3Nb0.8W1.2O8.6.28 This is generally caused by barium volatilisation and/or segregation of secondary phases in the grain boundary region and is a common problem in various Ba-containing oxide ion and proton conductors.39–42 Further sintering studies will be required in order to reduce the grain boundary resistivity of Ba3M′M′′O8.5 derivatives and further exploit their high bulk conductivity.
Time-of-flight (TOF) neutron powder diffraction data43 were recorded on the Polaris diffractometer in order to determine the crystal structure of Ba3VWO8.5 and to obtain accurate oxygen occupancies. Ba3VWO8.5 was previously reported to crystallize with a 9R hexagonal polytype structure with stacking sequence (hhc)3, in which trimers of face-sharing (V/W)O6 octahedra are connected via corner sharing.32 This model, which contains purely octahedral coordination of the metals, resulted in a poor Rietveld fit. The hybrid 9R perovskite – palmierite average structure previously reported for Ba3M′M′′O8.5 (ref. 20, 21 and 27) was therefore employed as a starting model for Ba3VWO8.5. In this model, the oxygen atoms occupy three different sites. The oxygen atom at Wyckoff position 18h (O1) is common to both the 9R and palmierite structures. The 9R perovskite structure also has an oxygen atom at Wyckoff position 9e (O2), whereas the palmierite structure has an oxygen atom at 6c (O3). The hybrid average structural model contains oxygen on all three sites. The intrinsic oxygen vacancies are randomly distributed along the pseudo-cubic BaO2.5 layers, resulting in partial occupation of the average O2 and O3 crystallographic sites, and in the formation of mixed average tetrahedral and octahedral metal coordination units within the palmierite-like layers. Large positional oxygen disorder along the O2–O3 layers leads to local disruption of the crystallographic oxygen site configurations and generates 4-, 5-, and 6-fold coordination geometries on the local scale.23,24 The barium atoms occupy the 6c and 3a Wyckoff positions (Ba1 and Ba2 respectively), while the transition metal cations are at Wyckoff position 6c (M1) and at Wyckoff position 3b (M2).21,27 Refinement of the fractional occupancy of the transition metals in Ba3VWO8.5 from neutron diffraction data is difficult because vanadium has a low scattering cross section for neutrons.44 For this reason, we first performed Rietveld refinement of the structural model from high-resolution X-ray diffraction data in order to determine the fractional occupancies of tungsten and vanadium within the crystal structure of Ba3VWO8.5.
In the reported hybrid structural model of Ba3M′M′′O8.5, the transition metals and the cationic vacancies are distributed in a disordered pattern over the M1 and M2 sites.21,27 In contrast, unconstrained refinement of the W and V site occupancies in Ba3VWO8.5 reveals that the M1 site is fully occupied, while the M2 site is empty. Examination of X-ray difference Fourier maps shows no evidence of significant missing scattering density on or in proximity of the (0,0,0.5) M2 site (VW in Fig. 4). For comparison, an X-ray difference Fourier map calculated on Ba3NbMoO8.5 by setting the occupancy of M2 to zero clearly reveals un-modelled electron density located at 6c (0,0,∼0.52) (NbMo in Fig. 4), above and below the nominal 3b position, in agreement with previous reports of splitting of the M2 site.23,26
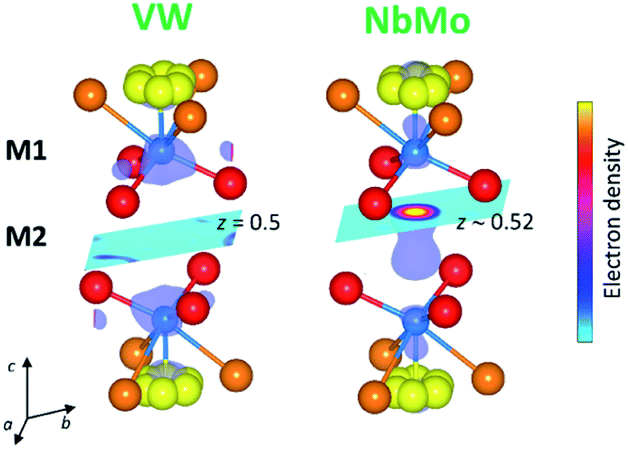 | ||
| Fig. 4 Volumetric data obtained from the X-ray difference Fourier analysis on the refined structures of Ba3VWO8.5 (VW) and Ba3NbMoO8.5 (NbMo). The difference Fourier map for Ba3VWO8.5 shows no evidence of electron density on or in proximity of the 3b (0,0,0.5) M2 site. In contrast the map for Ba3NbMoO8.5, calculated by setting the occupancy of M2 to zero, clearly indicates missing electron density located for the most part at the 6c (0,0,∼0.52) site. Sections represent slices of the volumetric Fourier data seen along the [001] direction at z = 0.5 (VW) and at z ∼0.52 (NbMo). The metal cations are represented by the blue spheres, while the oxygen atoms are in red (O1), orange (O2) and yellow (O3). | ||
Adopting the vacant M2 model for Ba3VWO8.5 resulted in a significant improvement of the goodness of fit parameters, which were reduced from χ2 = 5.34, Rwp = 8.87% to χ2 = 2.93, Rwp = 5.55%, and in a good match between the observed and calculated X-ray diffraction histograms (see ESI Fig. S4 and Table S1†). The V and W occupancies obtained from the high-resolution X-ray diffraction data were then fixed in the refinement of the Ba3VWO8.5 structure from neutron diffraction data. Refining the oxygen site occupancies from the collected neutron diffraction data gave an overall oxygen stoichiometry of 8.5; O1 is fully occupied, while the O2 and O3 crystallographic positions are partially occupied. The fractional occupancies of the Ba atoms refined to within ±1% of full occupancy and were fixed at 1.0. Refinement of the atomic displacement parameters, U, for all atoms was performed anisotropically. The oxygen atom O3 exhibits large anisotropic thermal displacement parameters U11 = U22 = 0.0706 (6) Å2, giving evidence of disorder at the O3 apical site of the M1O4 pseudo-tetrahedron, on the ab plane. Therefore, the general position 36i was assigned to O3 instead of the ideal 6c position in the palmierite structure, in order to account for the disorder of the oxygen atoms within the M1Ox polyhedra on the 3-fold axis. The splitting of this site has previously been reported also for Ba3NbMoO8.5.21 The O3 atom was subsequently modelled by using a single isotropic Uiso factor. An excellent Rietveld fit to this model was obtained (space group Rm H; a = b = 5.82254 (6) Å, c = 21.1327 (2) Å, V = 620.45 (2) Å3; χ2 = 4.397, Rp = 2.12% and Rwp = 1.53%; Fig. 5, Table 1).
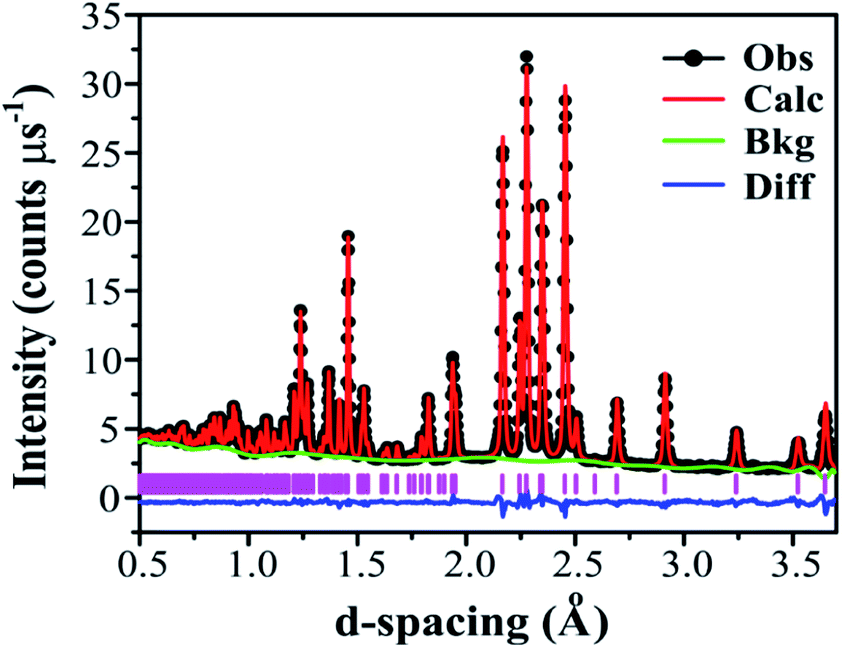 | ||
| Fig. 5 Fitted neutron diffraction histogram for Ba3VWO8.5 at ambient temperature. The TOF neutron diffraction data from the 90° bank detector of the Polaris diffractometer is shown. Black dots indicate the observed data, the red line the Rietveld fit, the blue line indicates the difference between the observed and the calculated patterns, the green line the background function, and the pink vertical bars the reflection positions. | ||
m H model from powder neutron diffraction dataa
| Atom | Site | x | y | z | Fraction | U 11 = U22 | U 33 | U 12 |
|---|---|---|---|---|---|---|---|---|
|
a Data were refined in space group Rm H with χ2 = 4.397, Rp = 2.12%, Rwp = 1.53%; refined unit cell parameters; a = b = 5.82254(6) Å, c = 21.1327(2) Å, V = 620.45 (2) Å3. Uij (in Å2) represent atomic displacement parameters; U13 and U23 are zero. The M1 site is composed of 50% V and 50% W.
|
||||||||
| Ba1 | 3a | 0 | 0 | 0 | 1 | 0.0140 (2) | 0.0064 (3) | 0.00703 (8) |
| Ba2 | 6c | 0 | 0 | 0.20693 (4) | 1 | 0.0111 (2) | 0.0148 (3) | 0.00559 (8) |
| M1 | 6c | 0 | 0 | 0.39415 (7) | 1 | 0.0097 (4) | 0.0137 (7) | 0.00480 (2) |
| O1 | 18h | 0.17269 (4) | 0.82730 (4) | 0.10188 (2) | 1 | 0.0149 (1) | 0.0136 (2) | 0.0115 (2) |
| O2 | 9e | 0.5 | 0 | 0 | 0.361 (1) | 0.0446 (6) | 0.0305 (9) | 0.0279 (3) |
| O3 | 36i | 0.0526 (4) | 0.0103 (9) | 0.32659 (6) | 0.118 (1) | 0.0212 (6) | — | — |
Significant differences exist between the average structures of Ba3VWO8.5 and Ba3M′M′′O8.5. The average structure of Ba3VWO8.5 is composed of M1Ox polyhedral units spaced by empty octahedral cavities (Fig. 6a, VW), as a consequence of ordering of the cationic vacancies on the M2 sites. In contrast the average structure of Ba3NbMoO8.5 is formed by hybrid trimer stacks of face-sharing polyhedral units (Fig. 6a, NbMo) in which the cationic vacancies are disordered on two metal sites M1 and M2 which, due to the short separation between the positions, are mutually exclusive and cannot be occupied simultaneously. The random distribution of cation vacancies leads to the formation of local complex disordered stacking M1-vac-M1, M1-M2-vac, or vac-M2-M1 configurations, resulting in a random arrangement of face-sharing or separated M1Ox and M2O6 polyhedral units (Fig. 6b, bottom).23,26 Instead, the ordered distribution of cationic vacancies in Ba3VWO8.5 results in an ordered stacking of palmierite-like layers separated by empty octahedral sites (Fig. 6b, top). The average structure of Ba3VWO8.5 exhibits oxygen disorder similar to Ba3M′M′′O8.5, with competitive occupation of the two average crystallographic O2/O3 sites leading to the formation of average M1(O13O3) tetrahedral units and average M1(O13O23) octahedral units. It is expected that the occurrence of neighbouring tetrahedral and octahedral units, in conjunction with the large positional oxygen disorder, results in disruption of the local oxygen configuration and in the formation of local intermediate 5-fold geometries as reported for Ba3NbMoO8.5.23,24
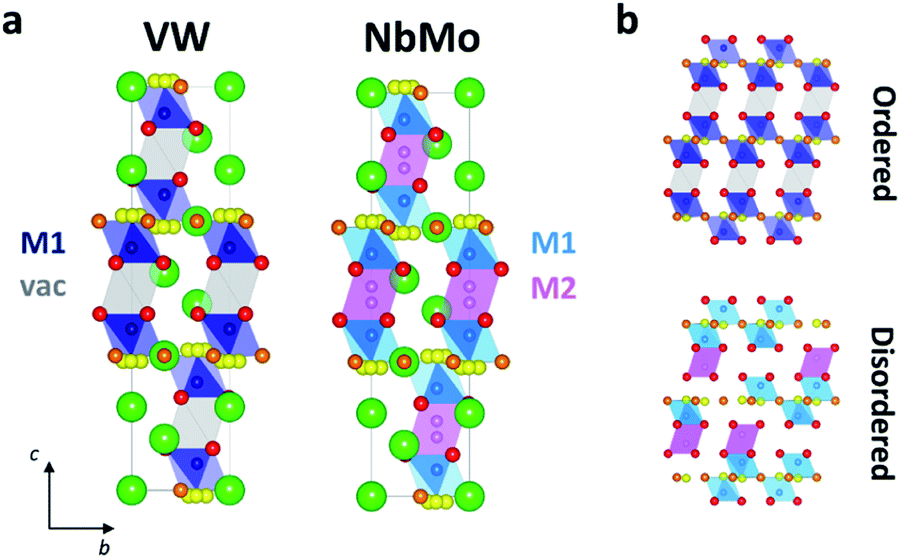 | ||
| Fig. 6 (a) Average crystal structure of Ba3VWO8.5 (VW) and Ba3NbMoO8.5 (NbMo). The green spheres are the Ba atoms, while the oxygen atoms are represented by red (O1), orange (O2) and yellow (O3) spheres. Blue and light blue polyhedra represent the average M1Ox units created by partial occupation of O2 and O3. The grey polyhedra in VW represents the empty octahedral cavities. M2O6 octahedra are represented in magenta in NbMo. (b) Representation of the ordered stacking of palmierite-like layers and cation vacancies in Ba3VWO8.5 (top), and of a possible disordered arrangement of face-sharing and isolated polyhedral units in Ba3NbMoO8.5 (bottom). O3 is represented at 6c for simplicity. | ||
Bond-valence site energy (BVSE) calculations were employed to analyse the migration pathway and energy landscape of the oxide ion conduction in the average crystal structure of Ba3VWO8.5. Bond-valence based methods have been shown to reproduce well the ionic conduction pathway in several ionic conductors17,45–50 and have successfully been employed for the screening and discovery of new oxide ion conducting materials.51–54 Despite being not as accurate, BVSE barriers generally show a good agreement with results from density functional theory (DFT) calculations.35 Therefore, BVSE analysis can provide useful general indicators on the energetics of ionic migration of inherently complex crystal structures, at a fraction of the computational cost, and the results can be used as a proxy, in relative terms, for more detailed studies.35,47 This approach has recently been employed to further understand the conduction mechanism and relative energetics of migration in V-doped Ba3NbMoO8.5.26 BVSE calculations on Ba3VWO8.5 were performed with the softBV program. Bond-valence site energy landscapes describing connecting local minima, saddle points and relative energy barriers for the interaction of a probe oxide ion were calculated from the refined structural model. Two-dimensional O2–O3 conduction pathways (on the ab plane) corresponding with the lowest bond-valence site energy barriers (<0.5 eV) are evidenced along the palmierite-like layers (Fig. 7a, b), in accordance with previous reports.23,25,26 The oxygen migration along the palmierite-like layers occurs through two saddle points. The saddle point S1 is associated with oxide ion exchange along the connected distribution of partially occupied oxygen positions (Fig. 7a). The saddle point S2 is perpendicular to the undulating distribution of crystallographic oxygen sites (Fig. 7b); its relative energy barrier (E2) can be associated with the energy necessary for the relaxation of the oxygen atoms during migration, and is linked to the average distortion of the M1Ox polyhedral units, σ(R).26 In Ba3M′M′′O8.5, the average metal polyhedra are distorted due to the out-of-centre displacement resulting from the second-order Jahn–Teller distortion induced by the d0 cations.21,23,55 The magnitude of this average distortion depends on the nature and combination of the M′M′′ cations.20,28,30,56 Ba3VWO8.5 presents σ(R) similar to Ba3NbMoO8.5 (∼0.0753 Å and ∼0.0759 Å respectively), and the two systems shows analogous E2 energy barriers (Fig. 7c). The relative energy barrier E1 is influenced by the populations of the average O2/O3 sites, and its height decreases when the fractional occupancy of O3 increases,26 since a larger number of lower coordination geometries provides more favourable and dynamic environments for oxygen transfer.13,14,57Fig. 7c shows that Ba3VWO8.5 and Ba3NbMoO8.5 have comparable relative BVSE energy barriers for the 2-dimensional ionic conduction along the palmierite-like layers, which are considerably smaller than the barrier for Ba3NbWO8.5. However, the bulk conductivity of Ba3VWO8.5 is significantly lower than the bulk conductivity of both Ba3NbMoO8.5 and Ba3NbWO8.5 (see Fig. 3d). Therefore, it would seem that other factors are influencing the ionic transport in these systems.
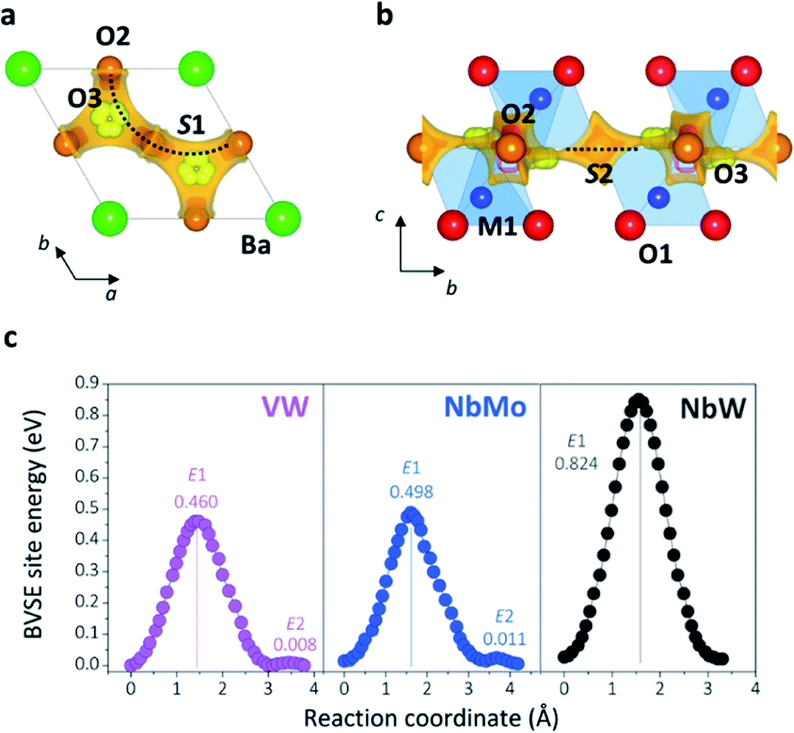 | ||
| Fig. 7 BVSE map of Ba3VWO8.5 showing the 2-dimensional ionic conduction pathway along the palmierite-like layer as seen down the c-axis (a) and a-axis (b). Isosurface levels are drawn at < 0.5 eV (over the global minimum). The dotted lines represent the saddle points as identified by the softBV software. (c) BVSE models of migration barriers along the palmierite-like layers for Ba3VWO8.5 (VW), Ba3NbMoO8.5 (NbMo), and Ba3NbWO8.5 (NbW). Relative bond-valence energy barriers are reported in eV. Note that the minimum site energies have relative values. | ||
The softBV approach reveals all the possible path segments that exist for a given structure,36 therefore further examination and comparison of the BVSE maps and pathways besides the 2-dimensional lowest energy pathway for Ba3VWO8.5, Ba3NbMoO8.5 and Ba3NbWO8.5 can be particularly informative. The BVSE maps calculated for the average structures of Ba3NbMoO8.5 and Ba3NbWO8.5 demonstrate sizeable connectivity between adjacent O1–O2 and O1–O1 positions (Fig. 8, right, and ESI Fig. S5†), implying oxygen motion along the edges of the M1Ox and M2O16 polyhedra, similar to the migration mechanism in cubic ABO3 perovskites.58 The O1–O2 and O1–O1 pathway segments in Ba3NbMoO8.5 have relative BVSE energy barriers of 0.675 eV and 1.257 eV respectively, and of 1.058 eV and 1.491 eV in Ba3NbWO8.5. The BVSE map of Ba3VWO8.5, while showing O1–O2 exchange (with a relative energy barrier of 0.780 eV), does not present any O1–O1 connectivity along the edges of the (vac)O16 pseudo-octahedron (Fig. 8, left). In a previous study, a bond-valence energy landscape (BVEL) map calculated on a refined structure of Ba3NbMoO8.5 from single crystal X-ray diffraction data, modified to contain an ordered M1-vac-M1 stacking configuration, showed connectivity parallel to the c-axis via the O1 sites and the vacant M2 polyhedra.23 It was also proposed that occupation of the M2 site reduces the ease of oxide ion exchange between the O1 layers and the BaO2.5 layers. We performed a similar BVS analysis with softBV on an average Ba3NbMoO8.5 structural model with the M2 site occupancy set to zero. While the calculated BVSE map evidenced sizeable connectivity between O1–O1 sites (Fig. S6 in ESI†), in agreement with the previous single crystal study,23 further examination of the specific pathway segments identified by softBV reveals that this connectivity does not correspond to a viable diffusion pathway. The BVS density within the empty M2 octahedral site is the result of the presence of calculated interstitial energy minimum sites and the associated exchange between these sites and O1. There are no such interstitial oxygen present at the vacant M2 site in Ba3VWO8.5 which would result in unrealistic O–O separations (<2 Å). Hence the calculated BVS density is chemically infeasible.
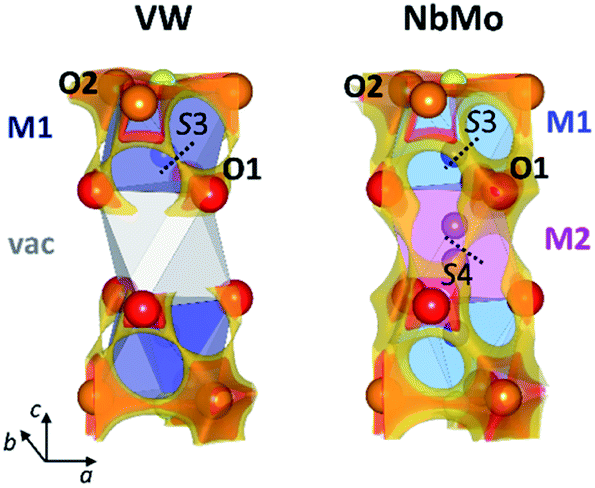 | ||
| Fig. 8 Bond-valence site energy maps calculated for Ba3VWO8.5 (VW) and Ba3NbMoO8.5 (NbMo). The cation vacancy (vac) in VW is represented by the grey octahedron. The dotted lines represent the saddle points at the median of the O1–O2 (S3) and O1–O1 (S4) distances, with bottlenecks constituted by triangles formed by two Ba cations and one M cation, as identified by the softBV software. Darker colours indicate the lower isosurface levels (<0.5 eV over the global minimum), while the lighter colours are for the highest isosurface levels (between 0.7 eV and 1.4 eV over the global minimum). | ||
The presence of connectivity between oxygen sites is certainly a prerequisite for the evaluation of a possible conduction pathway. However, its likelihood as a migration route needs to be rigorously assessed by examination of both the relative energy barrier and the characteristics of the connectivity within the specific pathway segment, to ensure that the pathway is physically viable.36,59
Amongst all the possible pathway segments calculated from BVSE analysis of the average structure, the 2-dimensional O2–O3 pathway along the palmierite-like layer presents the lowest relative BVS energy barrier and is most likely the dominant route contributing to the overall conduction. However, the presence of a continuous connectivity network formed by O1–O2 and O1–O1 pathway segments with accessible relative energy barriers implies long-range diffusion parallel to the c-axis, so that the ionic conductivity in the hybrid structure of Ba3M′M′′O8.5 (M′ = Nb; M′′ = Mo, W) is 3-dimensional.23 The disorder in the cation sub-lattice, together with the inherent ability of the cation defects to rearrange themselves in the hybrid structure,22,24 allow for the formation of a complex and dynamic 3-dimensional ionic percolation network, the topology of which will depend on the average temperature- and time-dependent distribution of cationic and anionic vacancies. In contrast, ordering of the cationic vacancies on the M2 sites in Ba3VWO8.5 disrupts the 3-dimensional conduction/percolation network by hindering any long-range oxygen diffusion (O1–O2 + O1–O1) parallel to the c-axis59,60 so that the conductivity mechanism is two-dimensional. The reduction in dimensionality results in a dramatic reduction in the bulk conductivity (see Arrhenius plot in Fig. 3d). Cation ordering in pyrochlore Gd2(Ti, Zr)2O7 systems has been reported to have a similar negative impact on the conductivity and mobility of the oxygen vacancies, with an analogous reduction in the dimensionality of oxide ion conduction.61,62 We note that the different chemical characteristics of the M′M′′ cations may also influence the ionic conductivity. If we examine the average M1–O2 and M1–O3 bond lengths, we notice that these are considerably shorter in Ba3VWO8.5: respectively 2.1160 (9) Å and 1.455 (2) Å, compared to 2.2065 (8) Å and 1.766 (4) Å in Ba3NbMoO8.5.21 These short distances are typical of V5+Ox polyhedra containing vanadyl bonds63 and may act as a trap for the mobile oxygen atoms along the palmierite-like layers, further reducing the oxide ionic conductivity. Such trapping of the mobile oxide ions due to local structural relaxation/distortion has previously been postulated also for Ba3Nb1−xVxMoO8.5.26
Conclusions
In summary, we have reported the successful synthesis of a new oxide ion conductor Ba3VWO8.5. Similar to Ba3M′M′′O8.5 (M′ = Nb; M′′ = Mo, W), the average crystal structure of Ba3VWO8.5 contains palmierite-like layers with considerable positional oxygen disorder leading to the formation of average mixed coordination M1Ox polyhedral units. However, the cationic vacancies are ordered on the M2 sites in Ba3VWO8.5, thus resulting in an ordered stacking of palmierite-like layers separated by empty octahedral cavities. Electrical characterization reveals that Ba3VWO8.5 is an oxide ion conductor at high pO2. However, the bulk conductivity of this compound is substantially lower than the conductivities of other members of the Ba3M′M′′O8.5 family. BVSE analysis on the average structure of Ba3VWO8.5 reveals that the lowest-energy ionic conduction pathway is along the 2-dimensional palmierite-like layers, in agreement with previous studies on Ba3M′M′′O8.5 (M′ = Nb; M′′ = Mo, W), implying cooperative oxide ion transport via exchange between the partially occupied average oxygen sites within the BaO2.5 layers. The BVSE analysis also shows the absence of any long-range oxygen connectivity parallel to the c-axis, in contrast with what has been reported for Ba3NbMoO8.5 and Ba3NbWO8.5 These results suggest that, although the 2-dimensional conduction pathway along the palmierite-like layers is the predominant diffusion route, long-range ionic migration parallel to the c-axis also provides a relevant contribution to the conductivity of Ba3M′M′′O8.5 systems, thus resulting in a complex 3-dimensional ionic percolation network.23 Ordering of the cation vacancies on the M2 sites most likely disrupts such a network in Ba3VWO8.5, so that the reduction in conductivity can be ascribed to the reduction in the dimensionality of ionic diffusion. These findings highlight how the chemical characteristics of the M′M′′ couple and the presence of disorder in the cationic sub-lattice can have a profound influence on the ionic conducting properties of Ba3M′M′′O8.5 derivatives. Various reports have demonstrated how the presence of disorder and the particular distribution of cations and defects in the cation sub-lattice can affect the conductivity of different oxide ion conductors, and how these generally depend on the composition and chemistry of the cations.61–67 These findings highlight the importance of the oxygen and cation vacancy disorder in defining the ionic conducting properties of Ba3M′M′′O8.5 hexagonal perovskite derivatives. The results also demonstrate that it is possible to obtain oxide ion conducting Ba3M′M′′O8.5 with a range of different cations and synthesis of novel phases with different combinations of M′M′′ are warranted.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the UK Science and Technology Facilities Council (STFC) for provision of Xpress Access neutron beamtime at ISIS. This research was supported by the Leverhulme trust (RPG-2017-351) and the Commonwealth Scholarship Commission for provision of a split-site studentship for AG.References
- A. J. Jacobson, Chem. Mater., 2009, 22, 660 CrossRef.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935 CrossRef CAS PubMed.
- T. A. Adams, J. Nease, D. Tucker and P. I. Barton, Ind. Eng. Chem. Res., 2012, 52, 3089 CrossRef.
- D. J. L. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Chem. Soc. Rev., 2008, 37, 1568 RSC.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602 RSC.
- L. Malavasi, C. A. J. Fisher and M. S. Islam, Chem. Soc. Rev., 2010, 39, 4370 RSC.
- C. R. A. Catlow, Solid State Ionics, 1984, 12, 67 CrossRef CAS.
- X. Kuang, J. L. Payne, M. R. Johnson and I. R. Evans, Angew. Chem., Int. Ed., 2012, 51, 690 CrossRef CAS PubMed.
- C. E. Mohn, S. Stølen, S. T. Norberg and S. Hull, Phys. Rev. Lett., 2009, 102, 155502 CrossRef PubMed.
- E. Kendrick, M. S. Islam and P. R. Slater, J. Mater. Chem., 2007, 17, 3104 RSC.
- P. Lacorre, F. Goutenoire, O. Bohnke, R. Retoux and Y. Laligant, Nature, 2000, 404, 856 CrossRef CAS PubMed.
- F. Abraham, M. Debreuille-Gresse, G. Mairesse and G. Nowogrocki, Solid State Ionics, 1988, 28, 529 CrossRef.
- E. Kendrick, J. Kendrick, K. S. Knight, M. S. Islam and P. R. Slater, Nat. Mater., 2007, 6, 871 CrossRef CAS PubMed.
- X. Kuang, M. A. Green, H. Niu, P. Zajdel, C. Dickinson, J. B. Claridge, L. Jantsky and M. J. Rosseinsky, Nat. Mater., 2008, 7, 498 CrossRef CAS PubMed.
- T. Ishihara, H. Matsuda and Y. Takita, J. Am. Chem. Soc., 1994, 116, 3801 CrossRef CAS.
- M. Li, M. J. Pietrowski, R. A. De Souza, H. Zhang, I. M. Reaney, S. N. Cook, J. A. Kilner and D. C. Sinclair, Nat. Mater., 2014, 13, 31 CrossRef CAS PubMed.
- K. Fujii, Y. Esaki, K. Omoto, M. Yashima, A. Hoshikawa, T. Ishigaki and J. R. Hester, Chem. Mater., 2014, 26, 2488 CrossRef CAS.
- J. B. Goodenough, J. E. Ruiz-Diaz and Y. S. Zhen, Solid State Ionics, 1990, 44, 21 CrossRef CAS.
- J. Darriet and M. A. Subramanian, J. Mater. Chem., 1995, 5, 543 RSC.
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle and A. C. Mclaughlin, Chem. Commun., 2019, 55, 2127 RSC.
- S. Fop, J. M. S. Skakle, A. C. McLaughlin, P. A. Connor, J. T. S. Irvine, R. I. Smith and E. J. Wildman, J. Am. Chem. Soc., 2016, 138, 16764 CrossRef CAS PubMed.
- S. Fop, E. J. Wildman, J. T. S. Irvine, P. A. Connor, J. M. S. Skakle, C. Ritter and A. C. Mclaughlin, Chem. Mater., 2017, 29, 4146 CrossRef CAS.
- J. E. Auckett, K. L. Milton and I. R. Evans, Chem. Mater., 2019, 31, 1715 CrossRef CAS.
- M. S. Chambers, K. S. McCombie, J. E. Auckett, A. C. McLaughlin, J. T. S. Irvine, P. A. Chater, J. S. O. Evans and I. R. Evans, J. Mater. Chem. A, 2019, 7, 25503 RSC.
- M. Yashima, T. Tsujiguchi, K. Fujii, E. Niwa, S. Nishioka, J. R. Hester and K. Maeda, J. Mater. Chem. A, 2019, 7, 13910 RSC.
- S. Fop, K. S. McCombie, R. I. Smith and A. C. Mclaughlin, Chem. Mater., 2020, 32, 4724 CrossRef CAS.
- K. S. McCombie, E. J. Wildman, S. Fop, R. I. Smith, J. M. S. Skakle and A. C. Mclaughlin, J. Mater. Chem. A, 2018, 6, 5290 RSC.
- K. S. McCombie, E. J. Wildman, C. Ritter, R. I. Smith, J. M. S. Skakle and A. C. Mclaughlin, Inorg. Chem., 2018, 57, 11942 CrossRef CAS PubMed.
- A. Bernasconi, C. Tealdi and L. Malavasi, Inorg. Chem., 2018, 57, 6746 CrossRef CAS PubMed.
- S. Fop, E. J. Wildman, J. M. S. Skakle, C. Ritter and A. C. Mclaughlin, Inorg. Chem., 2017, 56, 10505 CrossRef CAS PubMed.
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle, J. T. S. Irvine, P. A. Connor, C. Savaniu, C. Ritter and A. C. Mclaughlin, Nat. Mater., 2020, 19, 752 CrossRef CAS PubMed.
- B. Mössner and S. Kemmler-Sack, J. Less-Common Met., 1985, 114, 333 CrossRef.
- R. I. Smith, S. Hull, M. G. Tucker, H. Y. Playford, D. J. McPhail, S. P. Waller and S. T. Norberg, Rev. Sci. Instrum., 2019, 90, 115101, DOI:10.1063/1.5099568.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- H. Chen, L. L. Wong and S. Adams, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 18 CrossRef CAS.
- H. Chen and S. Adams, IUCrJ, 2017, 4, 614 CrossRef CAS PubMed.
- J. Cumby and J. P. Attfield, Nat. Commun., 2017, 8, 14235 CrossRef CAS PubMed.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132 CrossRef CAS.
- H. Kim, S. Kim, K. Lee, H. Lee and K. Lee, J. Power Sources, 2014, 267, 723 CrossRef CAS.
- G. B. Zhang and D. M. Smyth, Solid State Ionics, 1995, 82, 161 CrossRef CAS.
- Y. Yamazaki, R. Hernandez-Sanchez and S. M. Haile, J. Mater. Chem., 2010, 20, 8158 RSC.
- S. Wang, F. Zhao, L. Zhang and F. Chen, Solid State Ionics, 2012, 213, 29 CrossRef CAS.
- A. C. Mclaughlin, STFC ISIS Neutron and Muon Source, 2019, https://doi.org/10.5286/ISIS.E.RB1890374-1 Search PubMed.
- V. F. Sears, Neutron News, 1992, 3, 29 CrossRef.
- X. Yang, S. Liu, F. Lu, J. Xu and X. Kuang, J. Phys. Chem. C, 2016, 120, 6416 CrossRef CAS.
- W. Uno, K. Fujii, E. Niwa, S. Torii, P. Miao, T. Kamiyama and M. Yashima, J. Ceram. Soc. Jpn., 2018, 126, 341 CrossRef CAS.
- M. L. Tate, C. A. Fuller, M. Avdeev, H. E. A. Brand, G. J. McIntyre and I. Radosavljevic Evans, Dalton Trans., 2017, 46, 12494 RSC.
- M. Duchardt, S. Neuberger, U. Ruschewitz, T. Krauskopf, W. G. Zeier, J. Schmedt auf der Günne, S. Adams, B. Roling and S. Dehnen, Chem. Mater., 2018, 30, 4134 CrossRef CAS.
- L. Zhou, A. Assoud, A. Shyamsunder, A. Huq, Q. Zhang, P. Hartmann, J. Kulisch and L. F. Nazar, Chem. Mater., 2019, 31, 7801 CrossRef CAS.
- K. Park, K. Kaup, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, ACS Energy Lett., 2020, 5, 533 CrossRef CAS.
- R. Inoue, K. Fujii, M. Shiraiwa, E. Niwa and M. Yashima, Dalton Trans., 2018, 47, 7515 RSC.
- Y. Yasui, E. Niwa, M. Matsui, K. Fujii and M. Yashima, Inorg. Chem., 2019, 58, 9460 CrossRef CAS PubMed.
- E. Niwa and M. Yashima, ACS Appl. Energy Mater., 2018, 1, 4009 CrossRef CAS.
- W. Zhang, K. Fujii, E. Niwa, M. Hagihala, T. Kamiyama and M. Yashima, Nat. Commun., 2020, 11, 1224 CrossRef CAS PubMed.
- M. Kunz and I. D. Brown, J. Solid State Chem., 1995, 115, 395 CrossRef CAS.
- K. M. Ok, P. S. Halasyamani, D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. Mater., 2006, 18, 3176 CrossRef CAS.
- X. Yang, A. J. Fernández-Carrión, J. Wang, F. Porcher, F. Fayon, M. Allix and X. Kuang, Nat. Commun., 2018, 9, 4484 CrossRef PubMed.
- M. Cherry, M. S. Islam and C. R. A. Catlow, J. Solid State Chem., 1995, 118, 125 CrossRef CAS.
- S. Adams, Solid State Ionics, 2000, 136–137, 1351 CrossRef CAS.
- S. Adams, Solid State Ionics, 2006, 177, 1625 CrossRef CAS.
- B. P. Uberuaga, R. Perriot and G. Pilania, Phys. Chem. Chem. Phys., 2019, 21, 5956 RSC.
- R. Perriot, B. P. Uberuaga, R. J. Zamora, D. Perez and A. F. Voter, Nat. Commun., 2017, 8, 618 CrossRef PubMed.
- M. Schindler, F. C. Hawthorne and W. H. Baur, Chem. Mater., 2000, 12, 1248 CrossRef CAS.
- B. P. Uberuaga and R. Perriot, Phys. Chem. Chem. Phys., 2015, 17, 24215 RSC.
- B. P. Uberuaga and G. Pilania, Chem. Mater., 2015, 27, 5020 CrossRef CAS.
- P. K. Moon and H. L. Tuller, Solid State Ionics, 1988, 28–30, 470 CrossRef.
- H. L. Tuller, J. Phys. Chem. Solids, 1994, 55, 1393 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta05581f |
| This journal is © The Royal Society of Chemistry 2020 |