
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilyloxymethanesulfinate as a sulfoxylate equivalent for the modular synthesis of sulfones and sulfonyl derivatives†
Dae-Kwon
Kim‡
,
Hyun-Suk
Um‡
,
Hoyoon
Park
,
Seonwoo
Kim
,
Jin
Choi
and
Chulbom
Lee
 *
*
Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: chulbom@snu.ac.kr
First published on 22nd October 2020
Abstract
An efficient protocol for the modular synthesis of sulfones and sulfonyl derivatives has been developed utilizing sodium tert-butyldimethylsilyloxymethanesulfinate (TBSOMS-Na) as a sulfoxylate (SO22−) equivalent. TBSOMS-Na, easily prepared from the commercial reagents Rongalite™ and TBSCl, serves as a potent nucleophile in S-alkylation and Cu-catalyzed S-arylation reactions with alkyl and aryl electrophiles. The sulfone products thus obtained can undergo the second bond formation at the sulfur center with various electrophiles without a separate unmasking step to afford sulfones and sulfonyl derivatives such as sulfonamides and sulfonyl fluorides.
Introduction
Synthesis of sulfonyl compounds by means of C–S bond formation is of high importance as sulfonyl linkages constitute mainstay structural motifs in a wide variety of pharmaceuticals, agrochemicals and organic materials.1 The direct installation of the SO2 unit, in particular, has long been practiced employing sulfur dioxide2 and recently underwent notable advancement owing to the development of sulfur dioxide surrogates, such as DABSO3 and metal sulfite salts,4 that enabled facile SO2 insertion in various processes. For the generation of the sulfonyl motif, the amphoteric reactivity of the sulfur atom has been mostly exploited, conjoining a nucleophile and an electrophile to give rise to sulfonyl compounds (Scheme 1A). Broader access to sulfonyl products may be feasible by engaging two electrophiles such as organohalides, which are more readily available than the corresponding nucleophiles. While this approach has been implemented in reductive settings, the scope is limited largely to substrate systems paired up by each of aryl and alkyl halides due to the requirement for distinctive reactivity toward transition metal activation or radical generation.5 The protocol providing more general access to a wider range of sulfonyl products including aliphatic as well as aromatic derivatives from large pools of electrophiles would be of high synthetic value, but remains unexplored. | ||
| Scheme 1 Synthetic strategies for installing sulfonyl units. | ||
From the disconnection vantage point, central to various syntheses of sulfonyl compounds is the intermediacy of an organosulfinate capable of reacting with electrophiles. A variety of sulfonyl derivatives have indeed been shown to function as precursors that form the sulfinate intermediate upon removal of one sulfonyl substituent from the sulfur center.6 For the de novo synthesis enlisting two electrophiles, a sulfinate having a removable masking group already in place can serve as the starting point (Scheme 1B). This strategy based on a dianion equivalent of sulfur dioxide, sulfoxylate (SO22−), has been put into practice by making use of sodium salts of 3-methoxy-3-oxopropane-1-sulfinate (SMOPS),7 benzothiazole-2-sulfinate (BTS),8 hydroxymethanesulfinate (Rongalite™),9 and its acyl derivative (Rongacyl).10 Despite their utility in certain settings, however, a range of shortcomings are associated with the methods using these reagents. For example, SMOPS and BTS are prepared from mephitic thiol and sulfide compounds through rather laborious processes, and release of the sulfinates requires unmasking under strongly basic and nucleophilic conditions, which are unsuitable for sensitive molecules. Direct use of the commercial reagent Rongalite™ is advantageous in terms of accessibility and cost, but has been limited mostly to the formation of sulfonamides in the presence of a large excess of the reagent to avoid a side reaction producing undesired symmetrical sulfones due to the labile hydroxymethyl group. The Rongacyl reagent free from this problem has proven to be quite effective in the preparation of various sulfonyl derivatives, but its utility has been limited to aliphatic substrates.
With the goal of developing an efficient method enabling modular access to a diverse range of sulfonyl products including alkyl, alkenyl, alkynyl, and aryl derivatives, we sought to probe sodium tert-butyldimethylsilyloxymethanesulfinate (TBSOMS-Na, 1) for its potential to work as an effective sulfonylating reagent.11 We envisaged that the potent reactivity of 1 toward π-allylpalladium species could be translated into C–S bond formation with other types of electrophiles. Of particular interest was the prospect of subjecting the resulting TBSOCH2 sulfone directly to the second reaction without a separate unmasking step. It was anticipated that the mildness and mechanistic orthogonality of the fluoride-induced desilylation event would allow for a wide swath of reactions to be viable with a broad range of functional groups being tolerated. Thus, the synthetic sequence from TBSOMS-Na to sulfonyl products may be performed through operationally simple, all-in-one-pot procedures. We report here our studies on the novel sulfinate TBSOMS-Na for use as a versatile sulfoxylate equivalent in the modular and efficient synthesis of sulfones, sulfonamides and sulfonyl fluorides.
Results and discussion
Our studies started with examining the reactivity of TBSOMS-Na (1), readily prepared as a shelf-stable solid from Rongalite™ and TBSCl in 97% yield, in S-alkylation with alkyl electrophiles (Table 1). Gratifyingly, the reaction of 1 (1.5 equiv.) with an assortment of alkyl halides proceeded smoothly to afford the corresponding S-alkylated products in moderate to good yield (in DMSO at ambient temperature, unoptimized). The primary bromide 2a participated well in the reaction to afford the TBSOCH2 sulfone while the β-branched primary bromide 2b produced a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of sulfone and sulfinate ester products. As expected, secondary halides displayed diminished reactivity (2c and 2d), and excellent yields of sulfone products were obtained from the reactions of activated systems such as allylic (2e), benzylic (2f and 2g) and α-carbonyl halides (2h–2j). It should be noted that sulfinate esters arising from O-alkylation were formed as minor products in most cases (S:O = 4:1–6:1), whereas S-alkylation took place predominantly with activated substrates (>10:1).
:1 mixture of sulfone and sulfinate ester products. As expected, secondary halides displayed diminished reactivity (2c and 2d), and excellent yields of sulfone products were obtained from the reactions of activated systems such as allylic (2e), benzylic (2f and 2g) and α-carbonyl halides (2h–2j). It should be noted that sulfinate esters arising from O-alkylation were formed as minor products in most cases (S:O = 4:1–6:1), whereas S-alkylation took place predominantly with activated substrates (>10:1).

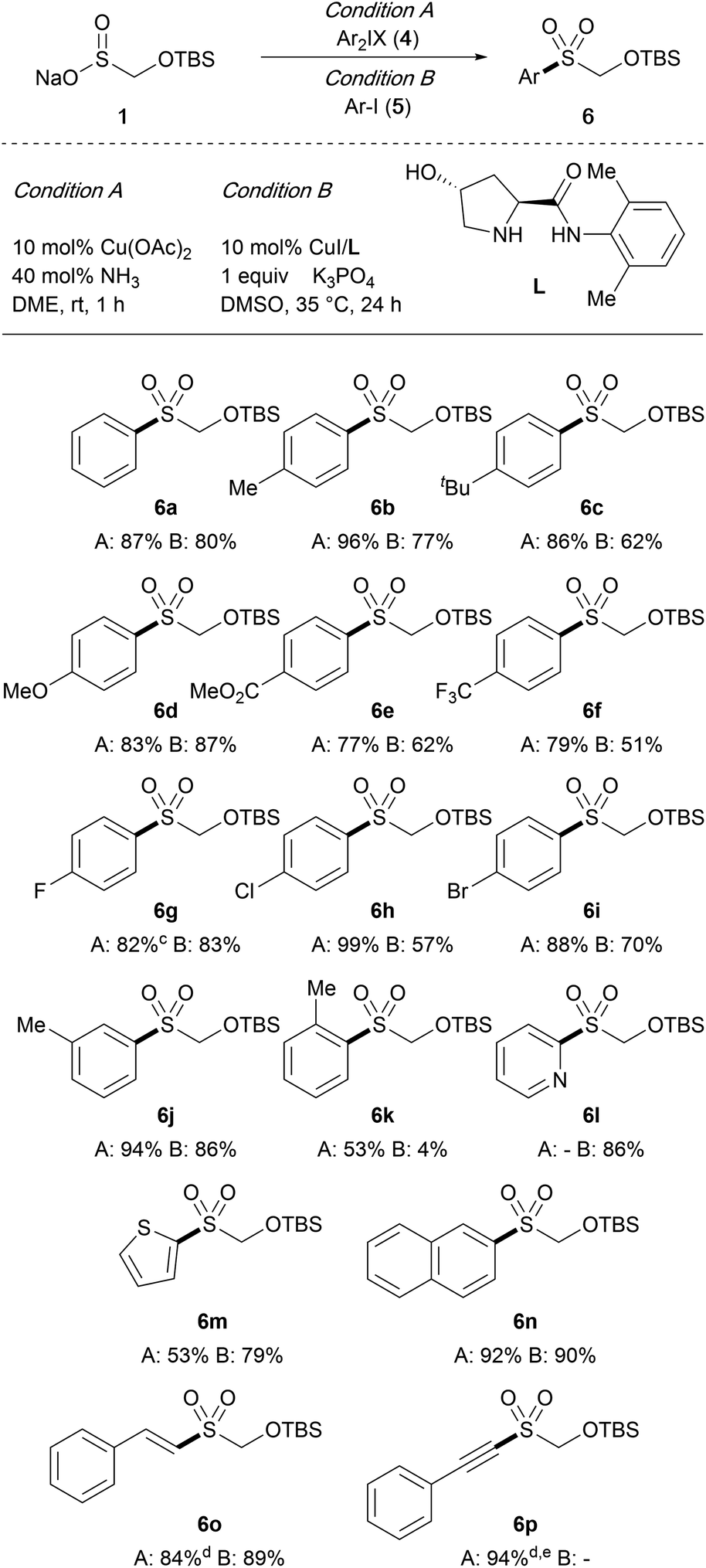
We next probed the feasibility of using TBSOMS-Na as a nucleophile in the S-arylation reactions. For our initial survey, we chose diaryliodonium salts as the arylating agent because of their ability to undergo arylation as well as their accessibility, nontoxic nature, and air and moisture stable properties. The reaction with diphenyliodonium triflate under the reported catalyst-free conditions (DMF, 90 °C, 24 h),12 however, led to decomposition of 1, forming only a trace amount of the S-phenylation product. In light of the infeasibility of the thermal conditions, we elected to explore the possibility of catalysis. To this end, a series of copper catalysts known to be capable of effecting arylation with diaryliodonium salts were screened. Surprisingly, it was found that the S-arylation could be carried out most efficiently with the Cu(II) catalyst system developed for the oxidative cross-coupling of arylboronic acids.13 In the event, in the presence of 10 mol% Cu(OAc)2 and 40 mol% NH3 (7 N in MeOH), the reaction of TBSOMS-Na (1) with diphenyliodonium triflate took place at ambient temperature to furnish the S-phenylation product 6a in 87% yield (Condition A). As illustrated in Table 2, the air and moisture tolerant reaction conditions proved to be efficient with substrates that incorporated a wide range of functional groups at the aryl ring, such as alkyl, ether, ester, trifluoromethyl, and halide groups. In most cases, the reaction was completed within 1 h to generate the TBSOCH2 sulfone products while tolerating significant electronic variation in the aryl ring. On the other hand, ortho-substitution was inimical to this Cu-catalyzed reaction as shown by the relatively lower yield of 6k, forming a contrast to the thermal process,12 in which the sulfone product arose typically from transfer of the sterically more demanding aryl group of a mixed diaryliodonium reagent. In addition to the aryl substrates, heteroaryl iodonium salts were also found to be viable participants of the reaction giving rise to the 2-pyridyl (6l) and thiophenyl (6m) sulfones. Finally, the protocol could be extended to promote S-alkenylation (6o) and S-alkynylation (6p) by using alkenylaryl and alkynylaryl iodonium salts, respectively, the latter of which reacted in the absence of a copper catalyst.14
| a Condition A: TBSOMS-Na (0.22 mmol), iodonium salt (0.2 mmol), Cu(OAc)2 (0.02 mmol) and NH3 (0.08 mmol) in DME (1.0 mL). Condition B: TBSOMS-Na (0.5 mmol), aryl iodide (1.0 mmol), CuI (0.05 mmol), L (0.05 mmol) and K3PO4 (0.5 mmol) in DMSO (3.2 mL). b Isolated yields. c TBSOMS-Na (0.2 mmol) and iodonium salt (0.4 mmol). d Unsymmetrical iodonium salts were incorporated. e Cu(OAc)2 and NH3 were absent in the reaction conditions. |
|---|
|
Having established a mild catalytic protocol for S-arylation using iodonium reagents, we next explored the possibility of obtaining the same products from aryl halides. Among various C(sp2)–S coupling methods for aryl sulfone synthesis,15–20 the copper catalyst supported by the proline-derived ligand L was deemed suitable due to its known ability to promote S-arylation of sulfinates with aryl iodides under mild conditions.15f Indeed, using 10 mol% CuI and ligand L in the presence of K3PO4, the reaction of TBSOMS-Na (1) with aryl iodides 5 in DMSO at 35 °C was completed in 24 h to furnish the corresponding aryl and heteroaryl sulfones in moderate to good yield (Condition B). In general, the same level of the reaction scope was maintained, but the sulfone products were formed in relatively lower yields in comparison to the reaction with iodonium reagents. However electron-rich substrates gave higher yields, mirroring the trends found in this catalyst system, and a more pronounced steric effect was noted in the reaction of the ortho-substituted substrate (6k). The copper-catalyzed reaction was also viable for the S-alkenylation (6o). These results, taken together with those of the reaction with iodonium salts, establish the feasibility of converting TBSOMS-Na (1) to aryl, alkenyl and alkynyl sulfones under the mild conditions we were targeting at the outset.
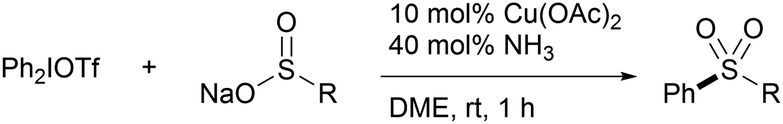
With the observation of the efficient S-arylation of 1 with iodonium salts under remarkably mild reaction conditions, we examined the applicability of the protocol to aryl sulfone synthesis with other sulfinates (Table 3). In stark contrast to 1, sulfinates bearing other removable masking groups did not fare well in the Cu-catalyzed S-arylation, and only BTS provided the phenylated product in low yield (entries 1 vs. 2–4). In addition, both methanesulfinate and p-toluenesulfinate failed to couple with diphenyliodonium triflate under the standard conditions as well (entries 5 and 6). Intriguingly, upon addition of 10 mol% 1, a rapid reaction took place to furnish diarylsulfone 12d (46%) along with sulfone 6a (9%) (entry 7). Furthermore, sulfinate ester 3a′ and sulfone 6a additives (20 mol%) also induced phenylation, albeit with low conversions in these cases (entries 8 and 9). Although the mechanism of the reaction remains unclear, these results indicate involvement of the TBSOCH2 moiety derived from the Rongalite™ architecture in the coordination of copper, playing a critical role for successful S-arylation.
|
|
|||
|---|---|---|---|
| Entry | R | Additive | Yield (%) |
| a Reaction conditions: sodium p-toluenesulfinate (0.22 mmol), diphenyliodonium triflate (0.2 mmol), Cu(OAc)2 (0.02 mmol) and NH3 (0.08 mmol, 7 N in MeOH) in DME (1.0 mL). b Isolated yields. | |||
| 1 | CH2OTBS (1) | — | 87 |
| 2 | 2-Benzothiazole (BTS) | — | 35 |
| 3 | CH2CH2CO2Me (SMOPS) | — | 0 |
| 4 | 2-Pyridyl | — | 0 |
| 5 | Me | — | 0 |
| 6 | p-Tol | — | 0 |
| 7 | p-Tol | 10 mol% 1 | 46 |
| 8 | p-Tol | 20 mol% 3a′ | 21 |
| 9 | p-Tol | 20 mol% 6a | 10 |

|
|||
In order to demonstrate the utility of TBSOMS-Na as a novel sulfoxylate equivalent in the modular synthesis of sulfones, the TBSOCH2 sulfone was probed for its ability to react with second electrophiles. After a set of screening experiments, it was found that the TBSOCH2 group could be replaced directly with various alkyl and aryl groups through the reactions performed in the presence of TBAF or CsF, which likely revealed in situ the requisite sulfinate for C–S bond formation at the sulfur center. We first examined the S-alkylation of alkyl (3a, R = CH2CH2CH2Ph) and aryl (6a, R = Ph) sulfones in their reactions with alkyl electrophiles (Table 4). Treatment of 3a and 6a with alkyl halides at 80 °C in the presence of TBAF gave the dialkyl (8) and alkyl aryl (9) sulfones in good to excellent yield. An array of alkyl halides containing acetal (7b), alkene (7c), aryl (7d), alkyne (7g), and hydroxy (7h) groups all participated well in the reaction. Similar to the alkylation of 1 (cf.Table 1), the reaction with secondary halides was less efficient, and high yields were uniformly obtained from reactive substrates with the exception of the reaction of 6a with benzyl bromide which gave a lower yield of 9d due to the O-alkylation forming the sulfinate ester (25%).

Encouraged by the results of alkylation, we then explored the direct arylation of the TBSOCH2 sulfone 3a (Table 5). We were pleased to find that the desired alkyl aryl sulfones 11 were generated from the reaction of 3a with aryl halides under the conditions employing catalytic CuI and L-proline together with CsF (1.5 equiv.).15b A wide variety of aryl iodides (10a–j and 10p) as well as bromides (10k–o and 10q–r) proved to be competent participants in the coupling reaction, tolerating a range of functional groups in various positions of the aryl ring. The ortho-substituted iodide (10f) that exhibited poor efficiency in the reaction with 1 (cf.6k) gave a reasonable yield of the aryl sulfone product. Interestingly, a precipitous decrease in yield was observed in the reactions with some heteroaryl substrates (10p, 10q and 10r). Noting the poor conversion and sluggishness of these reactions, we speculated that the copper catalyst might be rendered inactive by formaldehyde arising from the fluoride-induced desilylation.21 A control experiment carried out by running an otherwise efficient reaction in the presence of paraformaldehyde led to a significant decrease in the yield of the product (see the ESI†). In light of the effect of formaldehyde on the copper catalytic system, the reactions with heteroaryl halides were performed using an additional equivalent of L-proline, which was expected to trap formaldehyde while serving as the ligand. Gratifyingly, the reactions under these modified conditions gave the heteroaryl sulfone products in substantially increased yield.
| a Reaction conditions for 11: TBSOCH2 sulfone 3a (0.4 mmol), aryl halide (0.48 mmol), CuI (0.04 mmol), L-proline (0.08 mmol), NaOH (0.08 mmol) and CsF (0.6 mmol) in DMSO (0.4 mL), 24 h. b Reaction conditions for 12: TBSOCH2 sulfone 6a (0.4 mmol), aryl halide (0.48 mmol), CuI (0.04 mmol), L-proline (0.48 mmol), NaOH (0.08 mmol) and CsF (0.6 mmol) in DMSO (0.4 mL), 36 h. c Isolated yields. d 36 h. |
|---|

|
Having established suitable conditions for arylation, we then examined the protocol for the synthesis of diaryl sulfones. As the reaction of phenyl sulfone 6a proceeded more slowly than that of alkyl sulfone 3a, susceptible to catalyst deactivation, the arylation was performed employing additional L-proline (Table 5). The copper-catalyzed direct arylation of 6a under the modified conditions displayed broad substrate capacity, accommodating a range of aryl and heteroaryl halides. It is worthy of note that this consecutive S-arylation sequence with TBSOMS-Na constitutes an expeditious entry to unsymmetrical diaryl sulfones from two aryl electrophiles, a transformation that has never been demonstrated with a sulfoxylate synthon. We then further investigated the feasibility of the synthesis of unsymmetrical sulfones through single pot procedures without isolating the TBSOCH2 sulfone intermediates (Scheme 2). When TBSOMS-Na was subjected to the copper-catalyzed arylation with 10a (35 °C, 24 h) and then with 10c (95 °C, 36 h, 1 equiv L-proline), diaryl sulfone 13 was obtained in 53% yield. Moreover, the synthesis of an alkenyl aryl sulfone was also achieved in an atom-economical fashion by making use of both the alkenyl and aryl groups of the mixed iodonium reagent 4o.22 Subsequent to the S-alkenylation of 1 with 4o, the resulting TBSOCH2 sulfone and the iodobenzene byproduct were treated with catalytic CuOAc (10 mol%) along with TBAF and L-proline in DMSO. This two-stage, one-pot procedure afforded the desired alkenyl aryl sulfone 14 in a yield of 77%. This one-pot strategy was also applicable to the synthesis of dialkyl sulfones as exemplified in the gram scale preparation of 8c.
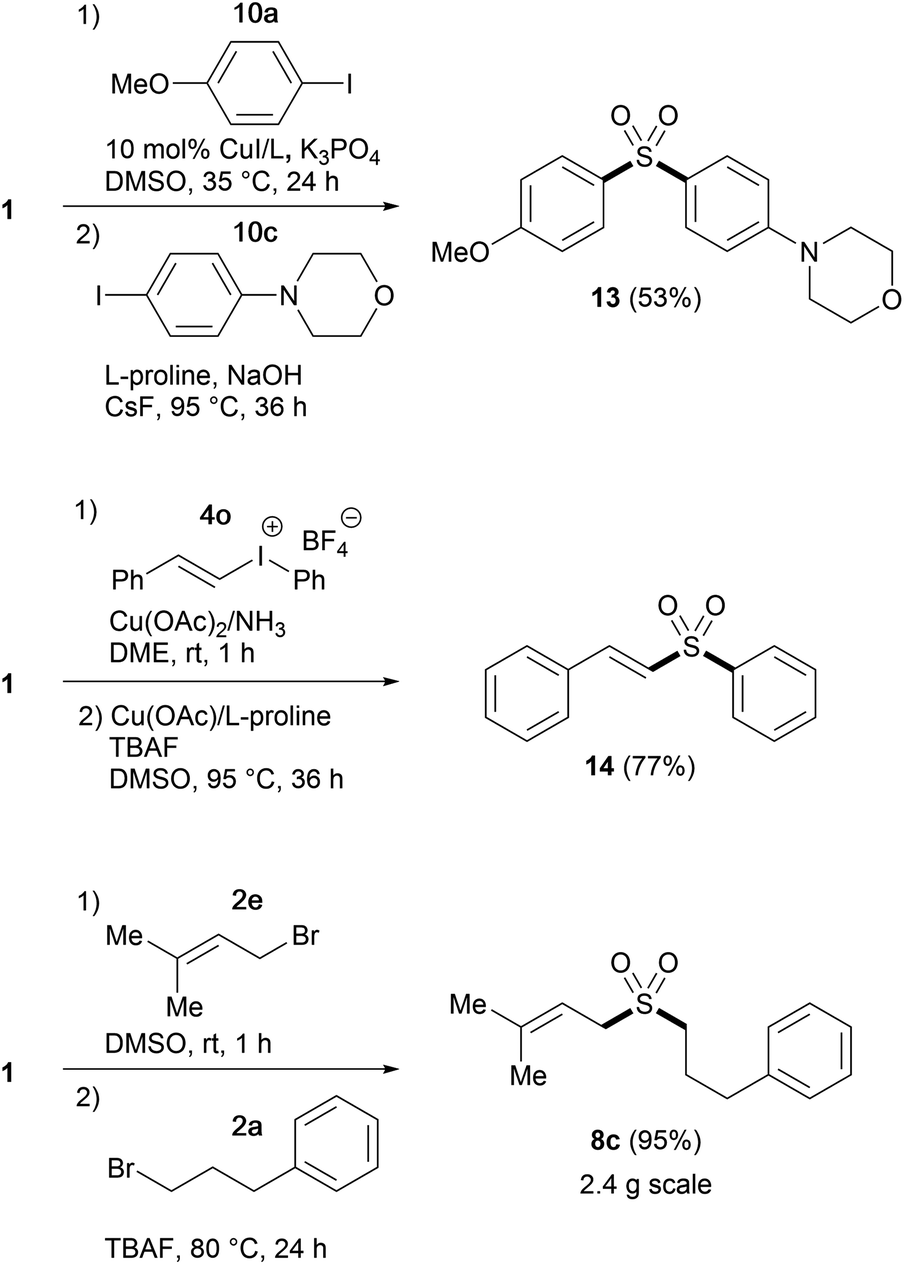 | ||
| Scheme 2 One-pot synthesis of unsymmetrical sulfones. | ||
The versatility of the TBSOCH2 sulfones as masked sulfinates was further demonstrated through the synthesis of a range of sulfonyl derivatives. As outlined in Table 6, sulfones 3a and 6a readily engaged in the reactions with various electrophiles in the presence of CsF or TBAF. The epoxide in cyclohexene was opened with exclusive anti-stereoselectivity upon treatment with sulfones 3a and 6a in water to furnish the trans-sulfonyl alcohols 15 and 16. In addition to epoxides, the strategy of introducing substituents in place of the TBSOCH2 group was amenable for the synthesis of sulfonyl fluorides as exemplified by the direct S-fluorination with NFSI or Selectfluor, both of which gave high yields. While the reaction with HOSA (hydroxylamine O-sulfonic acid) gave the primary sulfonamides (19a and 20a), the secondary (19b and 20b) and the tertiary (19c and 20c) sulfonamides as well as the N-arylsulfonamides (19d and 20d) were all prepared in good yields from the reactions carried out with the aid of NCS.
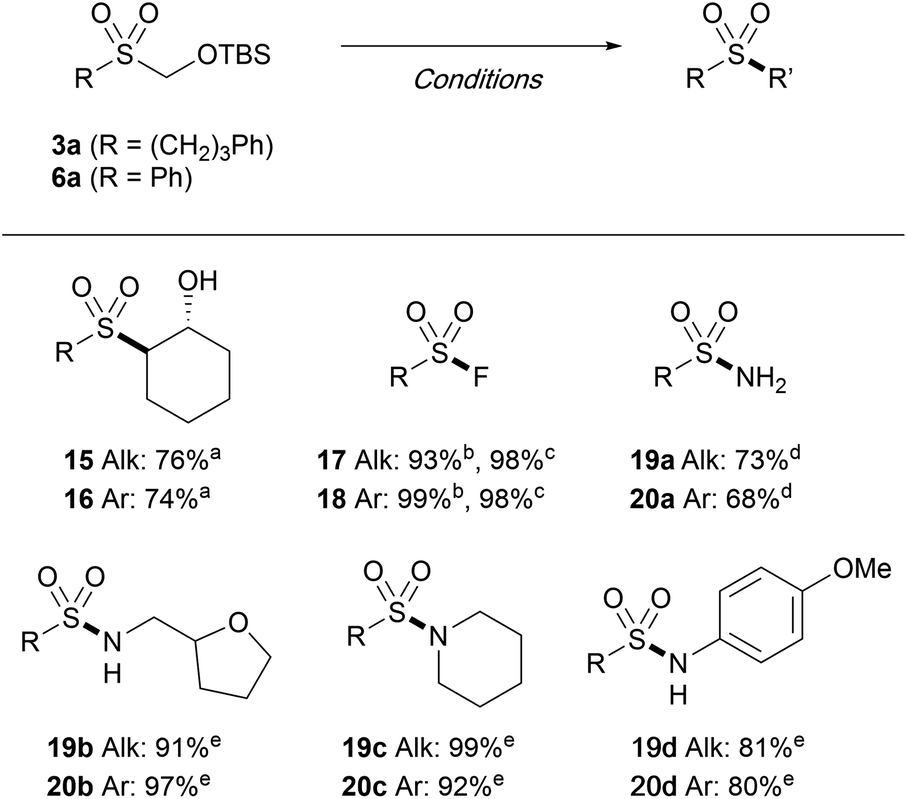
Although a wide variety of sulfones and sulfonyl derivatives are accessed directly from the intermediate sulfone without a discrete unmasking step, isolation of the TBSOM sulfone may be beneficial in case structural elaborations are desired. We thus probed the robustness of the TBSOCH2 moiety in the context of various functionalizations of β-ketosulfone 3j (Scheme 3A).23 When subjected to the alkylation with 1,2-dibromoethane, 3j gave cyclopropane 21a in high yield. Sulfone 3j also sustained a palladium-catalyzed coupling with phenylboronic acid to give rise to biphenyl 21b in nearly quantitative yield. Furthermore, we observed clean reduction of the ketone to β-hydroxysulfone 21c using DIBAL-H, a reagent that might unmask the sulfones derived from SMOPS, BTS, and Rongacyl salts. Subsequently, the functionalized TBSOCH2 sulfones 21a and 21c could be advanced to alkyl and aryl sulfones 22 and 23via direct S-alkylation and -arylation, respectively, thus establishing the divergent synthetic strategy for unsymmetrical sulfones.
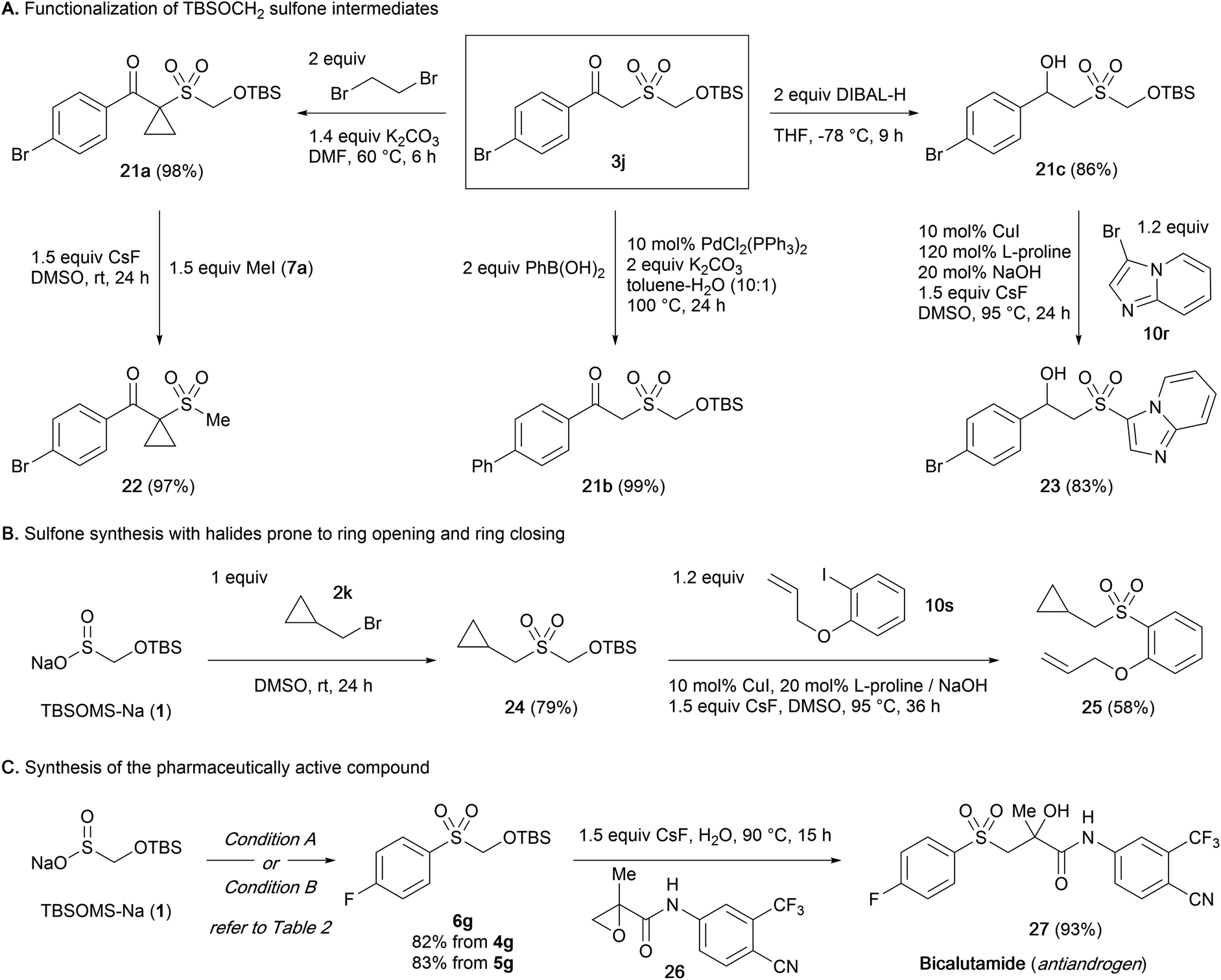 | ||
| Scheme 3 Application of the sulfoxylate strategy for the modular synthesis of sulfonyl derivatives. | ||
Next, we examined the viability of the sulfoxylate strategy with electrophiles whose incorporation in the sulfone synthesis might be complicated due to their sensitive structures (Scheme 3B). Starting from 1, the sequence of S-alkylation with bromide 2k followed by S-arylation with iodide 10s under the standard conditions could be carried out uneventfully to form the alkyl aryl sulfone 25 with the cyclopropane and allyl moieties intact.24
Lastly, the synthetic usefulness of the present sulfoxylate approach was demonstrated through an application in the synthesis of bicalutamide (27), an antiandrogen medication (Scheme 3C).25 The TBSOCH2 sulfone 6g bearing a 4-fluorophenyl group was prepared efficiently from the reaction of 1 with diaryliodonium salt 4g or aryl iodide 5g. Subsequently, treatment of 6g with an aqueous mixture of the known epoxide 26 and CsF afforded bicalutamide in a yield of 93%. The concise synthesis, avoiding the use of an expensive 4-fluorobenzenesulfinate salt or mephitic 4-fluorothiophenol, highlights the practical aspect of our sulfoxylate strategy.
Conclusions
In summary, we have developed an efficient strategy for the modular synthesis of various sulfones and sulfonyl derivatives by using TBSOMS-Na (1) as a novel sulfoxylate equivalent. The TBSOMS-Na salt is shelf-stable and easily prepared in deca-gram scales from commercial reagents Rongalite™ and TBSCl, and has been shown to be a potent S-nucleophile to engage in various C–S bond formations effecting alkylation, alkenylation, alkynylation, and arylation at the sulfur center via the reaction with organohalides and iodonium salts. The resulting TBSOCH2 sulfones, which are robust to sustain a range of elaborations, can undergo the reaction with a second electrophile in the presence of a fluoride anion that directly replaces the TBSOCH2 moiety with alkyl, aryl, fluoro, and amino groups to produce sulfones, sulfonyl fluorides and sulfonamides. This sequence of introducing two discrete electrophiles, which can be carried out in one-pot, will streamline synthetic strategies for the assembly of a wide variety of sulfonyl motifs. We anticipate that this sulfoxylate strategy, complementary to the approaches based on the use of sulfur dioxide, will provide a useful means for the construction of sulfonyl compounds.Conflicts of interest
D.-K. Kim, H.-S. Um, H. Park, and C. Lee are inventors on patent application 10-2019-0126427 (Republic of Korea) submitted by Seoul National University that covers the modular synthesis of sulfones and sulfonyl derivatives using TBSOMS-Na.Acknowledgements
Support for this research was provided by the National Research Foundation (NRF) funded by the Ministry of Science and ICT of Korea (2017R1A2B3002869 and 2020R1A2B5B03002271). H.-S. Um gratefully acknowledges Seoul National University for generous funding through the SNU Fellowship for Fundamental Academic Fields. This paper is dedicated to P. H. Dixneuf for his outstanding contribution to organometallic chemistry and catalysis.Notes and references
- For reviews on the construction of C–S bonds, see:
(a) T. Kondo and T.-A. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS
; (b) N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939 CrossRef CAS
-
(a)
S. D. Burke, in Encyclopedia of Reagents for Organic Synthesis, ed. L. A. Paquette, John Wiley & Sons, Chichester, 1995, vol. 7, p. 4688 Search PubMed
- For a seminal report, see:
(a) H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thomson and M. C. Willis, Org. Lett., 2011, 13, 4876 CrossRef CAS
- S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013 RSC
-
(a) J. Zhang, K. Zhou, G. Qiu and J. Wu, Org. Chem. Front., 2019, 6, 36 RSC
- For reviews on sulfinates, see:
(a) J. Aziz, S. Messaoudi, M. Alami and A. Hamze, Org. Biomol. Chem., 2014, 12, 9743 RSC
- J. M. Baskin and Z. Wang, Tetrahedron Lett., 2002, 43, 8479 CrossRef CAS
- J. J. Day, D. L. Neill, S. Xu and M. Xian, Org. Lett., 2017, 19, 3819 CrossRef CAS
- For reviews on Rongalite, see:
(a) S. Kotha and P. Khedkar, Chem. Rev., 2012, 112, 1650 CrossRef CAS
- A. Shavnya, K. D. Hesp and A. S. Tsai, Adv. Synth. Catal., 2018, 360, 1768 CrossRef CAS
- H.-S. Um, J. Min, T. An, J. Choi and C. Lee, Org. Chem. Front., 2018, 5, 2158 RSC
-
(a) N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 188 CrossRef CAS
- H. Yang, Y. Li, M. Jiang, J. Wang and H. Fu, Chem.–Eur. J., 2011, 17, 5662 Search PubMed
- A. Rodríguez and W. J. Moran, J. Org. Chem., 2016, 81, 2543 CrossRef
- For examples of coupling between sulfinates and aryl halides under copper catalysis, see:
(a) J. M. Baskin and Z. Wang, Org. Lett., 2002, 4, 4423 CrossRef CAS
- For examples of coupling between sulfinates and aryl halides under palladium catalysis, see:
(a) S. Cacchi, G. Fabrizi, A. Goggiamani and L. M. Parisi, Org. Lett., 2002, 4, 4719 CrossRef CAS
- For examples of coupling between sulfinates and aryl halides under nickel catalysis, see: N.-W. Liu, S. Liang, N. Margraf, S. Shaaban, V. Luciano, M. Drost and G. Manolikakes, Eur. J. Org. Chem., 2018, 1208 CrossRef CAS
- For examples of coupling between sulfinates and aryl halides under metal-free conditions, see:
(a) K. M. Maloney, J. T. Kuethe and K. Linn, Org. Lett., 2011, 13, 102 CrossRef CAS
- For examples of coupling between sulfinates and aryl halides under photoredox catalysis, see:
(a) H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371 CrossRef CAS
- For examples of coupling between sulfinates and aryl halides under irradiation, see: L. Chen, J. Liang, Z.-Y. Chen, J. Chen, M. Yan and X.-J. Zhang, Adv. Synth. Catal., 2019, 361, 956 CAS
-
(a) J. J. Byerley and W. K. Teo, Can. J. Chem., 1969, 47, 3355 CrossRef CAS
-
(a) S. G. Modha and M. F. Greaney, J. Am. Chem. Soc., 2015, 137, 1416 CrossRef CAS
- Y. M. Markitanov, V. M. Timoshenko and Y. G. Shermolovich, J. Sulfur Chem., 2014, 35, 188 CrossRef CAS
- The reaction of 2k, 10s and Na2S2O5 under palladium catalysis (ref. 5c) did not produce the unsymmetrical sulfone 25, but led mainly to the formation of 2-iodophenol via deallylation of 10s. See the ESI† for details.
-
(a) B. J. A. Furr, B. Valcaccia, B. Curry, J. R. Woodburn, G. Chesterson and H. Tucker, J. Endocrinol., 1987, 113, R7 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new compounds and other experimental details. See DOI: 10.1039/d0sc02947e |
| ‡ These authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2020 |