
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChelation with a twist: a bifunctional chelator to enable room temperature radiolabeling and targeted PET imaging with scandium-44†
Brett A.
Vaughn
a,
Shin Hye
Ahn
a,
Eduardo
Aluicio-Sarduy
b,
Justin
Devaraj
a,
Aeli P.
Olson
b,
Jonathan
Engle
b and
Eszter
Boros
 *a
*a
aDepartment of Chemistry, Stony Brook University, 100 Nicolls Road, Stony Brook, 11790, New York, USA. E-mail: eszter.boros@stonybrook.edu
bMedical Physics Department, University of Wisconsin-Madison, 1111 Highland Avenue, Madison, 53705, Wisconsin, USA
First published on 20th November 2019
Abstract
Scandium-44 has emerged as an attractive, novel PET radioisotope with ideal emission properties and half-life (t1/2 = 3.97 h, Emean β+ = 632 keV) well matched to the pharmacokinetics of small molecules, peptides and small biologics. Conjugates of the current gold-standard chelator for 44Sc, 1,4,7,10-tetraaza-cyclododecane-1,4,7,10-tetraacetic acid (DOTA), require heating to achieve radiochemical complexation, limiting application of this isotope in conjunction with temperature-sensitive biologics. To establish Sc(III) isotopes as broadly applicable tools for nuclear medicine, development of alternative bifunctional chelators is required. To address this need, we characterized the Sc(III)-chelation properties of the small-cavity triaza-macrocycle-based, picolinate-functionalized chelator H3mpatcn. Spectroscopic and radiochemical studies establish the [Sc(mpatcn)] complex as kinetically inert and appropriate for biological applications. A proof-of-concept bifunctional conjugate targeting the prostate-specific membrane antigen (PSMA), picaga-DUPA, chelates 44Sc to form 44Sc(picaga)-DUPA at room temperature with an apparent molar activity of 60 MBq μmol−1 and formation of inert RRR-Λ and SSS-Δ-twist isomers. Sc(picaga)-DUPA exhibits a Ki of 1.6 nM for PSMA, comparable to the 18F-based imaging probe DCFPyL (Ki = 1.1 nM) currently in phase 3 clinical trials for imaging prostate cancer. Finally, we successfully employed 44Sc(picaga)-DUPA to image PSMA-expressing tumors in a preclinical mouse model, establishing the picaga bifunctional chelator as an optimal choice for the 44Sc PET nuclide.
Introduction
The concept of personalized medicine, where patient treatment is performed according to an individually tailored treatment regime, is an emerging clinical management paradigm. In nuclear medicine, this approach is realized by exploiting diagnostic techniques, such as non-invasive imaging by means of Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT), followed by individualized radiotherapeutic treatment. If the same molecular targeting vector is labeled with a diagnostic and a therapeutic radionuclide and utilized for sequential imaging and treatment, the approach is considered theragnostic.1–3 Ideally, the employed radionuclides represent a matched pair, where both are radioisotopes of the same chemical element;4 however, only few elements have isotopes with suitable emission properties for this purpose, thus clinical applications have focused on perceived chemical homologues 68Ga(III) for PET imaging and 177Lu(III) for subsequent radiotherapy.5 The vast differences in coordination chemistry of these two metal ions create different chemical and biological behaviors between the corresponding radiolabeled agents: Ga(III) and Lu(III) coordinate to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra-acetic acid (DOTA)-complexes, but while Lu(III) is coordinated by all carboxylic acid groups of the octadentate DOTA, Ga(III) forms a six-coordinate complex, leaving two carboxylates in the Ga–DOTA-complex uncoordinated.6,7 This results in well-documented differences between the 68Ga and 177Lu-complexes' lipophilicity, receptor binding affinity and in vivo biodistribution.6,8–10 Consequently, 68Ga-complexes' biodistribution is an imperfect predictor of 177Lu compounds' dosimetry.11,12 Better theragnostic radionuclide pairs are needed (Table 1), and 44Sc/47Sc are ideally suited for this purpose.| Isotope (half-life) | E 〈β+/−〉 | Production route |
|---|---|---|
| 68Ga (67.71 min) | 830 keV (β+) | Generator: 68Ge/68Ga |
| Cyclotron (15–6 MeV) 68Zn(p,n)68Ga | ||
| 44Sc (3.97 h) | 632 keV (β+) | Generator: 44Ti/44Sc |
| Cyclotron (14–6 MeV): 44Ca(p,n)44Sc | ||
| 47Sc (80.38 h) | 162 keV (β−) | Cyclotron (30–10 MeV): 50Ti(p,α)47Sc |
| Reactor: 46Ca(n,γ)47Ca → 47Sc | ||
| 177Lu (6.647 d) | 134 keV (β−) | Reactor: 176Lu(n,γ)177Lu |
| Reactor: 176Yb(n,γ)177Yb → 177Lu |
44Sc (E〈β+〉 = 632 keV, t1/2 = 3.97 h) has ideal properties for PET imaging. The Sc(III) ion is a closer chemical match to Lu(III) than Ga(III); therefore 44Sc represents a better diagnostic partner to therapeutic 177Lu,13 as for instance, both trivalent ions prefer octacoordination of DOTA-type ligands. Additionally, the emission properties of 47Sc (E〈β−〉 = 162 keV, t1/2 = 80.38 h, Table 1)14 are comparable to 177Lu (E〈β−〉 = 134 keV, t1/2 = 159.5 h).15 Like 177Lu, 47Sc emits characteristic γ-rays with an ideal energy (Eγ = 159 keV) for SPECT imaging.4 The increased availability of 44Sc has prompted preclinical in vitro and in vivo studies with DOTA-conjugated biomolecules.13,16 DOTA is currently considered the gold standard scandium(III) chelator, but the slow complexation kinetics require radiolabeling at 70–95 °C for at least 30 minutes,17 which is incompatible with the labeling of antibodies.18 Functionalized DOTA is not ideal for the complexation of scandium isotopes with respect to applications with temperature-sensitive biologics and room temperature-compatible kit formulations.17,19,20 Furthermore, a strong preference for the formation of kinetically labile “out-of-cage” complexes with various DOTA-type derivatives has been observed.20,21 Hexadentate and acyclic chelators coordinate Sc(III) more rapidly than DOTA,22 but with a markedly reduced inertness of corresponding complexes and rapid release of Sc(III).19
The coordination chemistry of the Sc(III) ion resembles that of Y(III) and Lu(III) but is dominated by its smaller ionic radius and preference for chemically hard donor ions (carboxylates, aliphatic amines).19,23 The formation of stable hydroxide coordinating complex species has been documented.19 The significantly smaller ionic radius of Sc(III) indicates a preference for chelators with smaller ion binding cavities, disfavoring the formation of labile out-of-cage complexes. Small-cavity chelators may be better suited to coordinate 44Sc(III) under mild, low temperature conditions without formation of the problematic “out-of-cage” intermediate species.
Recently, the feasibility of this approach has been demonstrated using the heptadentate chelator 1,4-bis(carboxymethyl)-6-[bis(carboxymethyl)]amino-6-methyl-perhydro-1,4-diazepine (AAZTA, Fig. 1), which forms kinetically inert and thermodynamically stable Sc(III)-complexes at room temperature and provides a viable alternative to DOTA.24,25 However, the hydrophilicity of the corresponding complexes can result in accelerated excretion and diminished accumulation in the site of interest. We sought to develop alternative, more lipophilic chelator structures that would enable robust room temperature radiolabeling with Sc(III) isotopes and form kinetically inert complexes, especially in their bifunctional form.
 | ||
| Fig. 1 (Top) Ligand structures DOTA, AAZTA and NOTA, previously explored with Sc(III) isotopes. (Bottom) Chelator library explored in this work. | ||
Inspired by lanthanide chemistry, we designed bifunctional chelators for scandium isotopes using the following criteria: (1) radiolabeling must be rapid at room temperature and pH 4, to enable kit formulations and the labeling of temperature-sensitive biomolecules (2) the chelator must disfavor the formation of an out-of-cage complex to prevent the formation of labile, intermediate complex species (3) the formed complex must be kinetically inert to transchelation in vitro and in vivo, (4) functionalization of the ligand must be feasible without negatively impacting radiolabeling properties or the kinetic inertness of the formed complex24 and (5) integration of a lipophilic moiety to enhance binding affinity to biological targets and slow renal clearance.
Here, we show that a small-cavity, triaza-macrocycle based chelator exhibits excellent properties for the chelation of Sc(III) and corresponding targeted nuclear medicine applications with the PET isotope 44Sc.
Experimental section
Materials and methods
All starting materials were purchased from Acros Organics, Alfa Aesar, Sigma Aldrich, or TCI America and used without further purification. NMR spectra (1H, 13C, HSQC, HMBC) were collected on a 700 MHz Avance III Bruker, 500 MHz or 400 MHz Bruker instrument at 25 °C and processed using TopSpin 3.5pl7. 45Sc NMR was recorded on a 400 MHz Avance III Bruker instrument at 25 °C. Chemical shifts are reported as parts per million (ppm). Mass spectrometry: low-resolution electrospray ionization (ESI) mass spectrometry and high-resolution (ESI) mass spectrometry were carried out at the Stony Brook University Institute for Chemical Biology and Drug Discovery (ICB&DD) Mass Spectrometry Facility with an Agilent LC/MSD and Agilent LC-UV-TOF spectrometers, respectively. UV-Vis spectra were collected with the NanoDrop 1C instrument (AZY1706045). Spectra were recorded from 190 to 850 nm in a quartz cuvette with 1 cm path length. HPLC: preparative HPLC was carried out using a Shimadzu HPLC-20AR equipped with a Binary Gradient, pump, UV-Vis detector, manual injector on a Phenomenex Luna C18 column (250 mm × 21.2 mm, 100 Å, AXIA packed). Method A (preparative purification method): A = 0.1% TFA in water, B = 0.1% TFA in MeCN. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B gradient. Method B (preparative purification method): A = 10−2 M ammonium formate in water, B = 10% 10 mM ammonium formate in water, 90% MeCN. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B gradient. Radio-HPLC analysis was carried out using a Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-Vis detector, autoinjector and Laura radiodetector on a Gemini-NX C18 column (100 mm × 3 mm, 110 Å, AXIA packed). Method C: A = 0.1% TFA in water, B = 0.1% TFA in MeCN with a flow rate of 0.8 mL min−1, UV detection at 260 and 280 nm or using method D, performed on a DIONEX Ultimate 300 HPLC system controlled with the Chromeleon v6.80 software. A reverse-phase 250 × 4.6 mm C18 5 μ 120 A column (DIONEX) was employed with a gradient of A = 0.1% TFA in water, B = 0.1% TFA in MeCN. The following gradient was employed: 0–3.5 min: 5% B; 3.5–13 min: 5–35% B; 13–15 min: 35–90% B; 15–19 min: 90% B; 15–19 min: 90–5% B; 20–24 min: 5% B; and 1 mL flow rate and UV detection at 220 and 254 nm. Benzyl tert-butyl-2-(methylsulfonyloxy)glutarate and tert-butyl-6-(bromomethyl)-2-pyridinecarboxylate were synthesized according to previously published procedures. A detailed account of the chemical synthesis of mpatcn, picaga-DUPA and the DOTA-DUPA derivatives including NMR spectral assignments and corresponding data is provided in the ESI.†Scandium complexation
To obtain a single Sc-species in solution, the following general protocol was employed: ligand (0.02 mmol) previously deprotected under acidic conditions was dissolved in DI H2O (1 mL). Solutions of ScCl3·6H2O (0.02 mmol) were dissolved in H2O (1 mL) and one-half molar equivalent added to each ligand solution. The pH of the resulting acidic solution was subsequently adjusted from pH 3 to 6 by drop-wise addition of 1 M NaOH. The mixture was subsequently heated at 80 °C for 0.5 hours to ensure complete complexation. The resulting aqueous solutions were lyophilized overnight to afford the scandium complexes as an off-white powder.Acid-mediated dissociation kinetics
Solutions of ScCl3 were prepared by dissolving ScCl3·6H2O in 0.2 M HCl at a concentration of 35 mM. The solutions of the [Sc(mpatcn)] complex for kinetic measurements were prepared by mixing the ligand (L) and ScCl3 stock solutions to give a final 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 L:Sc molar ratio and adjustment of the pH to 5.5 with 0.1 M KOH. Complex formation was confirmed using 1H and 45Sc NMR spectroscopy. Dissociation of [Sc(mpatcn)] complex was followed by using UV-Vis spectroscopy. Decomplexation was quantified by plotting the change in absorbance over time at 284 nm. Complex concentration was 0.1 mM and dissociation experiments were performed in aq. HCl (0.1–3.0 M).19
:1 L:Sc molar ratio and adjustment of the pH to 5.5 with 0.1 M KOH. Complex formation was confirmed using 1H and 45Sc NMR spectroscopy. Dissociation of [Sc(mpatcn)] complex was followed by using UV-Vis spectroscopy. Decomplexation was quantified by plotting the change in absorbance over time at 284 nm. Complex concentration was 0.1 mM and dissociation experiments were performed in aq. HCl (0.1–3.0 M).19
Radioactive scandium complexes
:9 EtOH:H2O mixture. The eluate was collected and concentrated in vacuo and reconstituted in PBS for in vivo injection. Rt (method D): 44Sc(picaga)-DUPA: stereoisomers elute at 10.41/10.95 min; 44Sc(DOTA)-DUPA: 9.24 min; free 44Sc: 2.99 min.
Radioactive displacement assay
The inhibition constant (Ki) of Sc-labeled PSMA inhibitors was determined by a radiochemical displacement assay using the known, metal-based PSMA-inhibitor MIP-1427. Sc-complexed compounds were dissolved in H2O and 10 concentrations were prepared ranging from 10−13 to 10−5 M. PC-3 PiP cells (2 × 105 cells per tube in 2 mL Eppendorf tubes) were incubated for 40 min with 17.9 μCi (0.662 MBq) 99mTc-MIP-1427 and 0.01 nM to 100 μM Sc(picaga)-DUPA, Sc(DOTA)-DUPA, or DCFPyL PSMA inhibitors in PBS pH 7.4 with 1% FBS at 37 °C. Nonspecific binding was determined by adding 10 mM DCFPyL and subtracting measured residually bound activity from total binding values obtained for subsequent samples. The cells were then washed with PBS, centrifuged, and the amount of radioactivity remaining in the cell pellet was measured on a gamma counter. Specific binding was calculated as the difference between total binding and nonspecific binding. The Ki was determined by nonlinear regression analysis using GraphPad Prism software.PET imaging and biodistribution
All animal experiments and procedures were performed in accordance with the National Institutes of Health's “Guide for the Care and Use of Laboratory Animals” and approved by Institutional Animal Care and Use Committee (IACUC) at Stony Brook Medicine. Male NCr nude mice (6 weeks, Taconic Biosciences, Rensselaer, NY) were implanted subcutaneously on the right shoulder with 0.7–0.9 × 106 PC-3 PiP cells and on the left shoulder with 0.7–0.9 × 106 PC-3 flu cells suspended in Matrigel (1:1). When the tumors reached 50–100 mm3, the mice were anesthetized with isoflurane, and 0.6–3.0 MBq (15–82 μCi) of the tracer (3–30 μg) was intravenously injected via tail vein catheter. Mice were imaged at 90 min post injection (p.i.) using Siemens Inveon PET/CT Multimodality System, and images were reconstructed using ASIPro software. Region of interest (ROI) analyses on all images were performed using AMIDE. Upon completion of imaging at 120 min p.i., mice were sacrificed, and select organs were harvested. Radioactivity was counted by using a gamma counter, and the radioactivity associated with each organ was expressed as % ID per g. Biodistribution data were assessed by unpaired t-tests using GraphPad Prism to determine if differences between groups were statistically significant (p < 0.05).
Results and discussion
Ligand design and screening with 44Sc
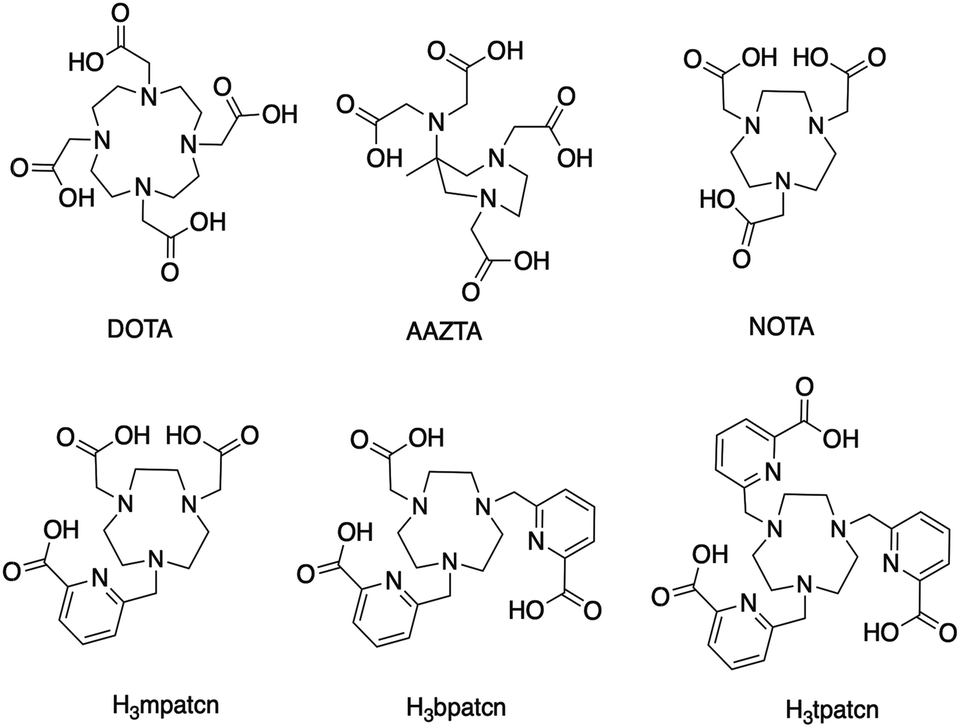
We hypothesized that an octa- or heptadentate, triaza-macrocycle based chelator would exhibit suitable complexation properties with Sc(III) without significant formation of the labile out-of-cage complex and form robust, kinetically inert radiochemical complexes compatible with in vivo applications. The introduction of picolinic acid donor arms increases the number of coordinating donors and imparts additional rigidity to formed complexes; the corresponding picolinate-functionalized triaza-macrocycle chelates show high kinetic inertness and slow interconversion of RRR-Λ and SSS-Δ complex isomers, especially with small lanthanides.28–30 We synthesized a chelator library based on picolinic acid-functionalized triaza-cyclononane (ESI†). H3mpatcn, H3bpatcn and H3tpatcn (Fig. 1) were tested for their radiolabeling properties at room temperature and 80 °C with cyclotron-produced 44ScCl3 (0.2 mL reaction volume, 0.25 M sodium acetate, pH 4.5). We identified one lead structure, the heptadentate chelator, H3mpatcn (Fig. 1, radiolabeling trace Fig. 2). H3mpatcn compared favorably to the DOTA chelator with respect to radiolabeling properties and plasma stability, producing comparable apparent molar activities (0.03 MBq nmol−1) at room temperature and 80 °C (Table S4†). Assessment of inertness in rat plasma at 37 °C revealed the corresponding [Sc(mpatcn)] complex to exhibit minimal detectable transchelation after 2 hours (94.2 ± 1.6% intact complex) comparing well to plasma stability of the DOTA chelator (95.8 ± 2.8% intact complex, Table S5†).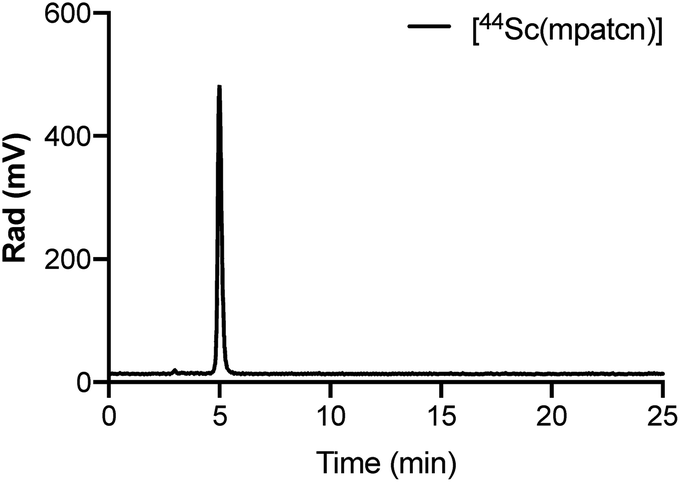 | ||
| Fig. 2 Representative radioHPLC chromatogram of [44Sc(mpatcn)] after 10 min incubation at 80 °C, method D. | ||
[Sc(mpatcn)] complex characterization
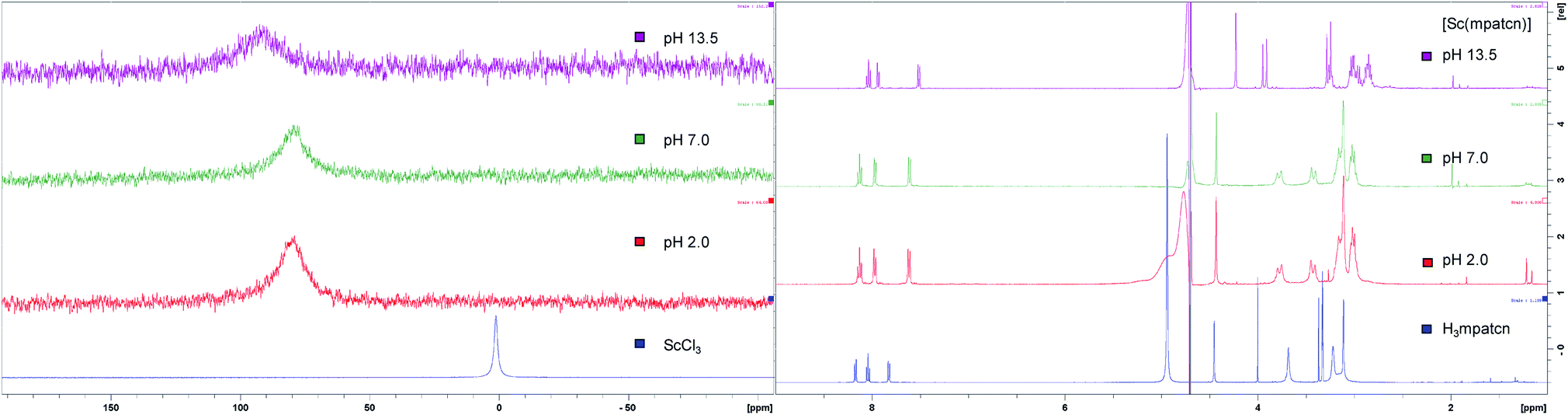
We employed 1H and 45Sc NMR studies to study and compare the complexation of Sc(III) with DOTA, EDTA and mpatcn chelators. 45Sc NMR provides useful complementary information on shielding of the metal ion by the ligand environment.20,24Fig. 3 and S35† show 45Sc NMR spectra of [Sc(DOTA)]−, [Sc(EDTA)(OH2)2]− and [Sc(mpatcn)]. The corresponding peak chemical shifts were found at 91, 51 and 80 ppm respectively at pH 7. The Sc(III)-aqua ion in dilute acid produces a sharp signal at 1.3 ppm, while out-of-cage coordination complexes typically result in 45Sc chemical shifts of 20–30 ppm.20 Low relative chemical shift indicates poor shielding of the 45Sc-nucleus from solvent molecules as evidenced by the [Sc(EDTA)(H2O)2]− complex (Fig. S36†). Our 1H and 45Sc NMR data indicates that the Sc(mpatcn) complex does not exhibit significant metal-centered isomerism on the NMR timescale and appears to behave comparably to the previously reported [Lu(mpatcn)] complex;29 while it is not possible to determine the number of waters bound to the metal ion, NMR experiments indicate that the H3mpatcn ligand can shield Sc(III) from the solvent environment more efficiently than EDTA, but less efficiently than DOTA, hinting at the presence of one inner-sphere water molecule.These results are in accordance with NMR-spectroscopic studies on the 8-coordinate, ternary [Sc(AAZTA)(OH2)]− complex.24 To further elucidate the speciation of the [Sc(mpatcn)] complex under radiochemically and physiologically relevant pH conditions, we carried out pH-titrations followed by NMR (1H and 45Sc, Fig. 3 and 4) and spectrophotometric methods from pH 1.15 to 13.4.
 | ||
| Fig. 3 Representative 45Sc (left) and 1H NMR (right) spectra of H3mpatcn/Sc3+ (blue), [Sc(mpatcn)] (red – pH 2, green – pH 7) and [Sc(mpatcn)(OH)]− (purple). | ||
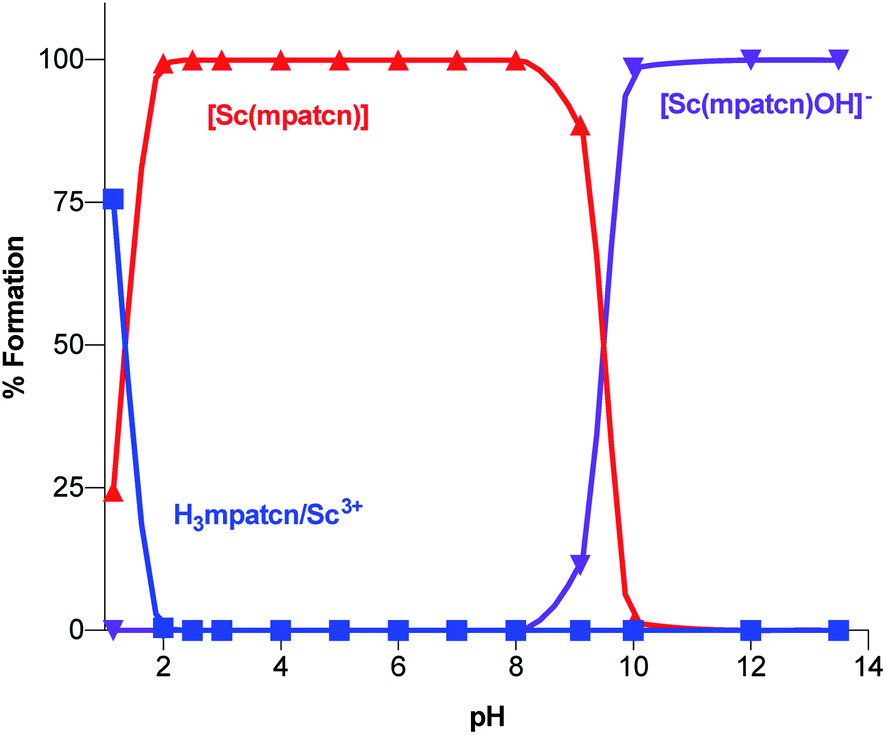 | ||
| Fig. 4 Complex speciation diagram based on 1H NMR data acquired at pH values ranging from 1.15–13.4, connecting curves are drawn between individual points for guidance. | ||
As characteristic absorbance bands of the ligand exhibit only a small bathochromic shift upon complexation (Fig. S38†), we utilized 1H NMR spectral data to quantitate the presence of different ligand and complex species; Table S1† summarizes characteristic chemical shifts and corresponding structural assignments. Both methods identified the presence of two speciation events. At pH 2, the formation of the [Sc(mpatcn)] complex was observed by an increase in absorbance at 280 nm (Fig. S38†), coinciding with an upfield shift of the picolinate protons and appearance of diastereotopic methylene proton signals in the 2–4 ppm range in the 1H NMR spectrum (Fig. 3). The corresponding 45Sc NMR shows formation of the in-cage complex at 80 ppm. We did not observe formation of an intermediate, protonated complex [Sc(Hmpatcn)]+ by 1H NMR or formation of detectable out-of-cage 45Sc-species at pH 2. This behavior contrasts observations made for DOTA, DTPA and AAZTA species under low pH conditions.19,24 We cautiously hypothesize that the lack of the protonated intermediate also favors the rapid in-cage-complex formation to produce the [Sc(mpatcn)] complex, while disfavoring the formation of a stable out-of-cage species. Following the formation of the [Sc(mpatcn)] complex species, no formation of new species was observed below pH 9, where evidence for the [Sc(mpatcn)(OH)]− species arises in the form of an upfield shift of the aromatic proton signals and more pronounced diastereotopic splitting of the methylene and azamacrocycle ring protons (Fig. 3 and Table S1†). The [Sc(mpatcn)(OH)]− species dominates above pH 10 and exhibits a 45Sc chemical shift of 89 ppm, clearly distinct and more downfield from the corresponding aqua-species. The formation of a hydroxo-species is typical for a hydrolytic, strongly Lewis acidic ion such as Sc(III) but represents a stark contrast to the speciation of analogous lanthanide complexes, where the formation of hydroxo-complex species is less prevalent. Indeed, the previously characterized [Gd(mpatcn)(OH2)2] complex does not form below pH 4, nor does it exhibit formation of the hydroxo-bound species even at pH > 11.28,31 Our experiments indicate that under radiochemically and physiologically relevant conditions, the desired [Sc(mpatcn)] complex species dominates.
Acid-catalyzed complex dissociation
Under physiological pH conditions, the [Sc(mpatcn)] complex remains inert and no complex dissociation can be detected over extended periods of time. In order to compare the inertness of [Sc(mpatcn)] with [Sc(DOTA)]− under more forcing conditions, we carried out UV-Vis spectroscopy-assisted measurement of dissociation constants and half-lives at proton concentrations ranging from 3–0.1 M (Table S3†).19 Rates of dissociation decrease rapidly as proton concentration is diminished, with a kobs of 6.5 × 10−5 s−1 and a t1/2 of 179.6 min at 0.1 M, indicating that even at pH 1, the acid-mediated decomplexation rate is slow. At proton concentrations below 0.1 M, the acid-mediated dissociation was too slow to be measured by UV-Vis spectroscopic methods.These experiments allow us to conclude that acid mediated decomplexation poses no significant concerns for in vivo applications of [Sc(mpatcn)] at physiological conditions.
Functionalization of mpatcn:picaga
Based on the promising results obtained with the non-functionalized chelator, we synthesized a proof-of-concept bioconjugate, to incorporate a biological targeting vector, 2-[3-(1,3-dicarboxypropyl)ureido]pentanedioic acid (DUPA).32,33 DUPA targets the prostate specific membrane antigen (PSMA) overexpressed in a large fraction of metastasizing prostate cancers;34 additionally, the growing number of studies with PSMA-targeting nuclear probes evaluated in preclinical and human data provides a suitable reference to probe the efficacy of a novel conjugate.8,9,35–39 We elected to substitute the mpatcn ligand via a functionalized glutarate in close analogy to the 6-coordinate NODAGA and 8-coordinate DOTAGA chelators to preserve the original ligand donor set as closely as possible.6,40,41 This contrasts the mono-amidation conjugation strategy commonly employed for PSMA-targeted DOTA-conjugates, which converts one of the coordinating carboxylates into an amide.10 Amide coordination to the Sc(III) ion would impart a significantly altered coordination environment with a greater, not directly predictable impact on the radiolabeling properties and corresponding kinetic inertness. The functionalized mpatcn derivative, picaga-DUPA (Fig. 5), was synthesized by alkylation of triazacyclononane with benzyl(methyl-sulfonyloxy)-glutarate, followed by step-wise introduction of tert-butyl-6-(bromomethyl)-2-pyridinecarboxylate, followed by alkylation with tert-butyl-bromoacetate (Scheme 1). Each alkylated intermediate was isolated and purified using reverse-phase preparative HPLC (method A). Following the complete assembly of the orthogonally protected ligand, the benzyl-ester was deprotected and amidated with the t-butyl ester protected form of aminopentyl-functionalized DUPA.32,42,43 A direct DO3A-monoamide DUPA conjugate analogue was also synthesized (ESI,† compound 17). The fully assembled conjugates picaga-DUPA (15) and DOTA-DUPA (17) were deprotected and characterized using 1H NMR, 13C NMR and mass spectrometry. Following complete characterization, the non-radioactive scandium complexes were formed and characterized using HPLC-analysis and mass spectrometry. The glutarate arm is introduced as the R-enantiomer, with the alpha carbon subsequently introducing a stereocenter to the ligand.44,45 Upon coordination to Sc(III) this results in the formation of kinetically inert diastereomers with distinct HPLC-retention times (Fig. 6A). The formation of coordinative diastereomers has been observed and characterized for similar molecules of this bifunctional compound class, specifically for DOTAGA-type derivatives which display not only RRR-Λ and SSS-Δ, but also SAP and TSAP-isomerism on the NMR timescale.46 The Sc(III) RRR-Λ,R- and SSS-Δ,R-diastereomers were chromatographically separated and subsequently analyzed for re-isomerization in solution; no interconversion was observed after 18 hours (Fig. S39†), exemplifying the high kinetic inertness of the corresponding diastereomers with respect to isomerization and reaffirming the conformational rigidity of the bifunctional, picolinate-functionalized triaza-macrocycle.47 | ||
| Fig. 5 PSMA-conjugates synthesized and utilized to determine comparative receptor binding affinity and biodistribution. | ||
 | ||
| Scheme 1 Step-wise chemical synthesis of picaga-DUPA (15) including isolated yields. | ||
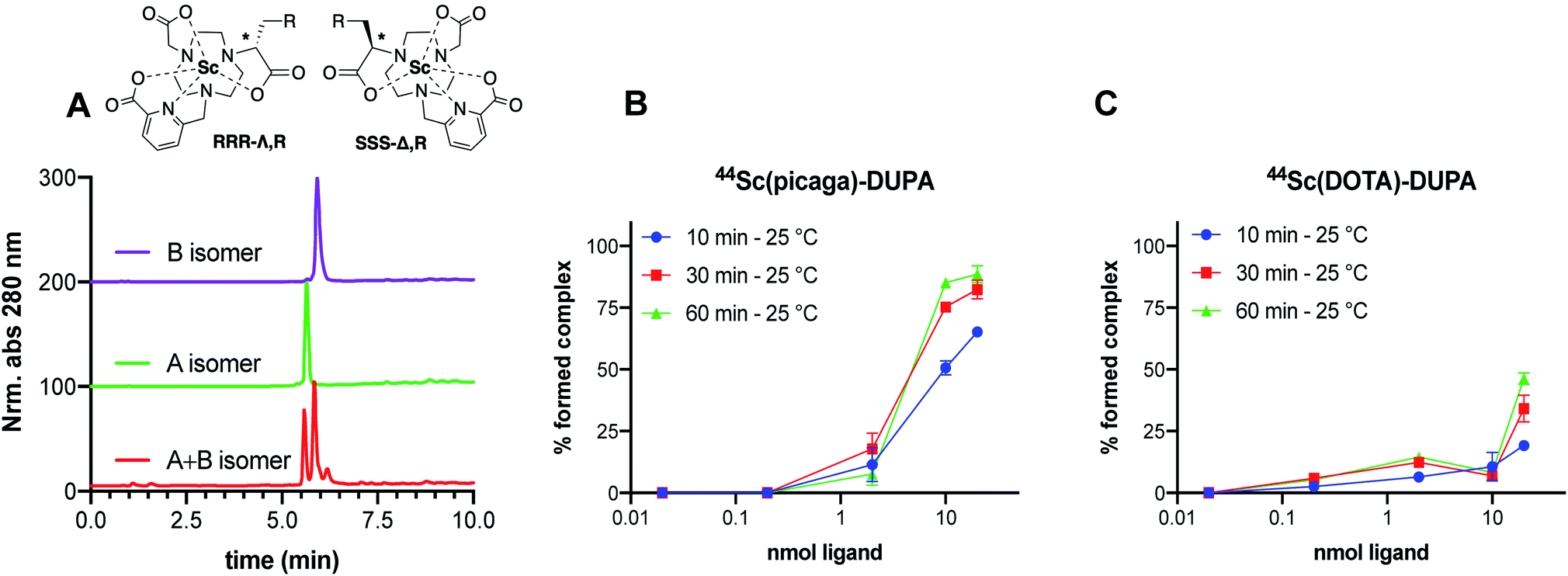 | ||
| Fig. 6 (A) Characterization of non-interconverting RRR-Λ and SSS-Δ-isomers of Sc(picaga)-DUPA using chromatography method C. (B and C) Radiolabeling yields obtained in dependence of increasing amounts of DUPA-conjugate demonstrate superior radiolabeling properties at room temperature for the bifunctional picaga chelator when compared with the functionalized DO3A-monoamide structure. | ||
Complexation with 44Sc
DOTA-DUPA and picaga-DUPA conjugates were subjected to radiochemical complexation experiments, testing both time and temperature dependence of complexation reactions. 44Sc(picaga)-DUPA produces high radiochemical yields with 10 nmol of conjugate: 83% complexation was achieved at room temperature and 96% at 80 °C (Fig. 6B and S41†). This result is in contrast with radiolabeling results obtained for the non-functionalized mpatcn ligand. We hypothesize that the efficient structural preorganization of the functionalized derivative accelerates room temperature complexation. Results obtained with picaga-DUPA at room temperature compare favorably to radiolabeling of 44Sc with DOTA-conjugates at 95 °C (Fig. S41,† 87% yield with 10 nmol ligand). The radiochemical complexes also form the characteristic diastereomers, with a minor and major stereoisomer separated by 0.5 minute retention time using chromatographic separation. For radiochemical and subsequent in vivo imaging studies, no attempts were made to separate the two diastereomers as the two complexes co-elute when separated from free 44Sc by reverse-phase solid phase extraction.Binding affinity to PSMA
In order to produce a bioconjugate with potential to image a biological target, a high binding affinity of the imaging probe is paramount. Picaga-DUPA does not incorporate additional secondary affinity tags by modification of the linker structure as is the case for PSMA-617 complexes.48 We reasoned that Sc(picaga)-DUPA exhibited sufficiently lipophilic character and the coordination complex could suffice to produce high binding affinity to the PSMA target without additional linker modifications in analogy with the 68Ga-based PET probe PSMA-11.49 The difference in lipophilicity is evidenced by the increase in HPLC retention times from [Sc(DOTA)]− (3.7 min) to Sc(mpatcn) (5.0 min), with the same trend observed for the corresponding DUPA conjugate complexes (Table S4†). In order to determine the binding affinity of both conjugates, we carried out the chemical synthesis of MIP-1427, a 99mTc-radiolabeled, well-established PSMA-targeting imaging probe which has already been evaluated in patients.32,50 Following successful synthesis, we carried out a radioactive displacement assay by challenging constant amounts of MIP-1427 with a varying concentration of non-radioactive Sc(III) complex. Non-radioactive DCFPyL was employed as an assay standard and to calculate relative Ki's based on the obtained IC50 values. Our experiments revealed a Ki value of 1.6 ± 0.4 nM for Sc(picaga)-DUPA. This value compares well to the compounds with high PSMA-affinity reported in literature, specifically DCFPyL and MIP-1427 (Table 2).Sc(DOTA)-DUPA exhibits a two orders of magnitude lower binding affinity, which is in accordance with previously investigated DOTA conjugates49 that do not bear a modified lipophilic linker to enhance affinity, in contrast to PSMA-617. As we are able to separate the non-interconverting diastereomers, we also determined the Ki of the A isomer and the B isomer separately. We found that the B isomer exhibited a 10-fold greater binding affinity (0.2 ± 0.2 nM) than the A isomer (3.6 ± 2.0 nM). This discrepancy in Ki values indicates that the structure Sc coordination complex plays a significant role in bonding of the conjugate with the target.
In vivo imaging and biodistribution experiments
To assess the in vivo behavior of 44Sc(picaga)-DUPA and applicability to targeted in vivo imaging, we administered the radiolabeled compound to mice bearing PSMA+ (PC-3 PiP) and PSMA− (PC-3 Flu) tumor xenografts on the right and the left flank respectively.37,42 As a reference, the corresponding 44Sc-DOTA-DUPA and 99mTc-labeled MIP-1427 complexes were also synthesized and evaluated in vivo. Mice were imaged at 90 minutes post injection using PET-CT for 44Sc-labeled compounds (Fig. 7A and B). All cohorts were sacrificed at 120 minutes post injection for biodistribution analysis. | ||
| Fig. 7 Coronal PET-CT slices 90 min post injection show the superior tumor uptake achieved with 44Sc(picaga)-DUPA (panel B) in comparison to 44Sc-DOTA-DUPA (panel A), confirmed by the results obtained from the biodistribution analysis 120 min post injection (panel C). | ||
The animal studies reveal that 44Sc(picaga)-DUPA exhibits excellent properties for in the vivo imaging of PSMA+ cancer xenografts; the uptake achieved with 44Sc(picaga)-DUPA was 13.8 ± 0.6% ID per g, which supersedes uptake observed for the corresponding DOTA conjugate (2.8 ± 1.3% ID per g) evaluated in this study and compares well to the tumor uptake observed with MIP-1427 (21.2 ± 6.2% ID per g) (Fig. 6C). The difference in uptake between the DOTA and picaga conjugates likely arises from the enhanced binding affinity to the target for 44Sc(picaga)-DUPA, and possibly also from slower blood clearance of the latter. The increased lipophilicity of 44Sc(picaga)-DUPA conjugate also affects the path of excretion as evidenced by enhanced hepatic clearance, contrasting the almost exclusively renally clearing DOTA conjugate; other picolinate-derivatized complex conjugates have been reported to exhibit similar in vivo behavior.52,53 Notably, in comparison with MIP-1427, kidney and splenic uptake are low; this bodes especially well for prospective therapeutic applications with 47Sc where elevated kidney uptake can become a dosimetric concern.
Conclusions
We have employed lanthanide-chemistry based design principles and a medicinal inorganic chemistry screening approach to identify the picolinate-functionalized, triaza-macrocycle-based chelator H3mpatcn as a suitable ligand to coordinate trivalent scandium rapidly and form kinetically inert coordination complexes under macroscopic and tracer conditions. Investigations of the complex formation using optical and NMR spectroscopy, as well as pH dependent speciation experiments revealed that H3mpatcn complexes Sc at pH 2 to form the desired in-cage complex species. The corresponding, neutral, heptacoordinate [Sc(mpatcn)] complex species dominates in the pH range from 2 to 9 with putative coordination of a single water molecule as the eighth donor within the inner sphere, in close analogy to [Sc(AAZTA)]−. A bifunctional chelate was furnished and appended to the PSMA-targeting dipeptide DUPA, a targeting vector which has been clinically employed for the imaging of PSMA-expressing prostate cancers. The corresponding conjugate picaga-DUPA forms inert, diastereotopic RRR-Λ,R and SSS-Δ,R-twist–isomer complexes with Sc(III). The preorganized structure of picaga enables radiolabeling at room temperature with 60 MBq μmol−1 apparent molar activity while retaining complex inertness in rat plasma. A competitive binding assay was carried out to evaluate the affinity of the Sc(picaga)-DUPA conjugate to the PSMA-target; an exceptionally high binding affinity of the complex (Ki = 1.6 ± 0.4 nM) was affirmed. Finally, an in vivo imaging and biodistribution study with 44Sc(picaga)-DUPA demonstrates efficacy of the conjugate to image PSMA+ tumors. The high target tumor uptake and favorable biodistribution profile comparable to clinically validated MIP-1427 qualifies 44Sc(picaga)-DUPA as a tracer with excellent characteristics for future clinical translation and suitable for tandem radiotherapeutic applications with 47Sc.In summary, we demonstrate that H3mpatcn and its bifunctional analogue picaga represent excellent additions to the chelator toolbox for the emerging 44/47Sc theragnostic isotope pair. Future studies are focused on expanding the scope of diseases imaged with picaga-conjugates, treatment studies with 47Sc(picaga)-DUPA and evaluation of H3mpatcn for the complexation of other lanthanides and pseudolanthanides of interest for biomedical imaging applications.
Ethical statement
All animal experiments and procedures were performed in accordance with the National Institutes of Health's “Guide for the Care and Use of Laboratory Animals” and approved by Institutional Animal Care and Use Committee (IACUC) at Stony Brook Medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Institutes of Health (NIH) via a Pathway to Independence Award (NHLBI R00HL125728-04), a LIBH REACH grant (U01HL127522-18150031) and Stony Brook University for startup funds. Peter Smith-Jones is acknowledged for providing DCFPyL.References
- J. Notni and H. J. Wester, J. Labelled Compd. Radiopharm., 2018, 61, 141–153 CrossRef CAS PubMed.
- F. Rösch and R. P. Baum, Dalton Trans., 2011, 40, 6104–6111 RSC.
- S. M. Qaim, B. Scholten and B. Neumaier, J. Radioanal. Nucl. Chem., 2018, 318, 1493–1509 CrossRef CAS.
- C. Müller, M. Bunka, S. Haller, U. Köster, V. Groehn, P. Bernhardt, N. van der Meulen, A. Türler and R. Schibli, J. Nucl. Med., 2014, 55, 1658–1664 CrossRef PubMed.
- D. J. Kwekkeboom, W. W. de Herder, B. L. Kam, C. H. van Eijck, M. van Essen, P. P. Kooij, R. A. Feelders, M. O. van Aken and E. P. Krenning, J. Clin. Oncol., 2008, 26, 2124–2130 CrossRef CAS PubMed.
- A. Heppeler, S. Froidevaux, H. Mäcke, E. Jermann, M. Béhé, P. Powell and M. Hennig, Chem.–Eur. J., 1999, 5, 1974–1981 CrossRef CAS.
- M. V. Deshmukh, G. Voll, A. Kühlewein, H. Mäcke, J. Schmitt, H. Kessler and G. Gemmecker, J. Med. Chem., 2005, 48, 1506–1514 CrossRef CAS.
- M. Weineisen, M. Schottelius, J. Simecek, R. P. Baum, A. Yildiz, S. Beykan, H. R. Kulkarni, M. Lassmann, I. Klette and M. Eiber, J. Nucl. Med., 2015, 56, 1169–1176 CrossRef CAS PubMed.
- M. Schottelius, M. Wirtz, M. Eiber, T. Maurer and H.-J. Wester, EJNMMI Res., 2015, 5, 68 CrossRef PubMed.
- C. A. Umbricht, M. Benešová, R. M. Schmid, A. Türler, R. Schibli, N. P. van der Meulen and C. Müller, EJNMMI Res., 2017, 7, 9 CrossRef PubMed.
- G. Flux, M. Bardies, M. Monsieurs, S. Savolainen, S.-E. Strand and M. Lassmann, Z. Med. Phys., 2006, 16, 47–59 CrossRef PubMed.
- D. McGowan and M. Guy, Br. J. Radiol., 2015, 88, 20140720 CrossRef CAS.
- C. A. Umbricht, M. Benešová, R. M. Schmid, A. Türler, R. Schibli, N. P. van der Meulen and C. Müller, EJNMMI Res., 2017, 7, 9 CrossRef PubMed.
- I. F. Chaple and S. E. Lapi, J. Nucl. Med., 2018, 59, 1655–1659 CrossRef CAS PubMed.
- K. A. Domnanich, C. Müller, M. Benešová, R. Dressler, S. Haller, U. Köster, B. Ponsard, R. Schibli, A. Türler and N. P. van der Meulen, EJNMMI Radiopharmacy and Chemistry, 2017, 2, 5 CrossRef PubMed.
- K. A. Domnanich, C. Müller, R. Farkas, R. M. Schmid, B. Ponsard, R. Schibli, A. Türler and N. P. van der Meulen, EJNMMI Radiopharmacy and Chemistry, 2017, 1, 8 CrossRef PubMed.
- M. Pruszyński, A. Majkowska-Pilip, N. S. Loktionova, E. Eppard and F. Roesch, Appl. Radiat. Isot., 2012, 70, 974–979 CrossRef.
- R. Hernandez, H. F. Valdovinos, Y. Yang, R. Chakravarty, H. Hong, T. E. Barnhart and W. Cai, Mol. Pharmaceutics, 2014, 11, 2954 CrossRef CAS PubMed.
- M. Pniok, V. Kubíček, J. Havlíčková, J. Kotek, A. Sabatie-Gogová, J. Plutnar, S. Huclier-Markai and P. Hermann, Chem.–Eur. J., 2014, 20, 7944–7955 CrossRef CAS PubMed.
- R. Kerdjoudj, M. Pniok, C. Alliot, V. Kubíček, J. Havlíčková, F. Rösch, P. Hermann and S. Huclier-Markai, Dalton Trans., 2016, 45, 1398–1409 RSC.
- M. Pérez-Malo, G. Szabó, E. Eppard, A. Vagner, E. Brücher, I. Tóth, A. Maiocchi, E. H. Suh, Z. n. Kovács and Z. Baranyai, Inorg. Chem., 2018, 57, 6107–6117 CrossRef PubMed.
- A. Majkowska-Pilip and A. Bilewicz, J. Inorg. Biochem., 2011, 105, 313–320 CrossRef CAS PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- G. Nagy, D. Szikra, G. Trencsényi, A. Fekete, I. Garai, A. M. Giani, R. Negri, N. Masciocchi, A. Maiocchi, F. Uggeri, I. Tóth, S. Aime, G. Giovenzana and Z. Baranyai, Angew. Chem., Int. Ed., 2017, 56, 2118–2122 CrossRef CAS PubMed.
- J.-P. Sinnes, J. Nagel and F. Rösch, EJNMMI Radiopharmacy and Chemistry, 2019, 4, 18 CrossRef PubMed.
- C. Alliot, R. Kerdjoudj, N. Michel, F. Haddad and S. Huclier-Markai, Nucl. Med. Biol., 2015, 42, 524–529 CrossRef CAS PubMed.
- E. Aluicio-Sarduy, R. Hernandez, A. P. Olson, T. E. Barnhart, W. Cai, P. A. Ellison and J. W. Engle, Sci. Rep., 2019, 9, 10658 CrossRef PubMed.
- A. Nonat, M. Giraud, C. Gateau, P. H. Fries, L. Helm and M. Mazzanti, Dalton Trans., 2009, 8033–8046 RSC.
- A. M. Nonat, C. Gateau, P. H. Fries, L. Helm and M. Mazzanti, Eur. J. Inorg. Chem., 2012, 2049–2061 CrossRef CAS.
- J. W. Walton, R. Carr, N. H. Evans, A. M. Funk, A. M. Kenwright, D. Parker, D. S. Yufit, M. Botta, S. De Pinto and K.-L. Wong, Inorg. Chem., 2012, 51, 8042–8056 CrossRef CAS PubMed.
- Y. Bretonnière, M. Mazzanti, J. Pécaut, F. A. Dunand and A. E. Merbach, Chem. Commun., 2001, 621–622 RSC.
- S. M. Hillier, K. P. Maresca, G. Lu, R. D. Merkin, J. C. Marquis, C. N. Zimmerman, W. C. Eckelman, J. L. Joyal and J. W. Babich, J. Nucl. Med., 2013, 54, 1369–1376 CrossRef CAS PubMed.
- C. M. Zechmann, A. Afshar-Oromieh, T. Armor, J. B. Stubbs, W. Mier, B. Hadaschik, J. Joyal, K. Kopka, J. Debus and J. W. Babich, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 1280–1292 CrossRef CAS PubMed.
- A. Ghosh and W. D. Heston, J. Cell. Biochem., 2004, 91, 528–539 CrossRef CAS PubMed.
- A.-C. Baranski, M. Schäfer, U. Bauder-Wüst, M. Roscher, J. Schmidt, E. Stenau, T. Simpfendörfer, D. Teber, L. Maier-Hein and B. Hadaschik, J. Nucl. Med., 2018, 59, 639–645 CrossRef.
- R. P. Baum, H. R. Kulkarni, C. Schuchardt, A. Singh, M. Wirtz, S. Wiessalla, M. Schottelius, D. Mueller, I. Klette and H.-J. Wester, J. Nucl. Med., 2016, 115, 168443 Search PubMed.
- Z. Szabo, E. Mena, S. P. Rowe, D. Plyku, R. Nidal, M. A. Eisenberger, E. S. Antonarakis, H. Fan, R. F. Dannals, Y. Chen, R. C. Mease, M. Vranesic, A. Bhatnagar, G. Sgouros, S. Y. Cho and M. G. Pomper, Mol. Imaging Biol., 2015, 17, 565–574 CrossRef CAS PubMed.
- M. Dietlein, C. Kobe, G. Kuhnert, S. Stockter, T. Fischer, K. Schomäcker, M. Schmidt, F. Dietlein, B. D. Zlatopolskiy and P. Krapf, Mol. Imaging Biol., 2015, 17, 575–584 CrossRef CAS PubMed.
- M. Benešová, M. Schäfer, U. Bauder-Wüst, A. Afshar-Oromieh, C. Kratochwil, W. Mier, U. Haberkorn, K. Kopka and M. Eder, J. Nucl. Med., 2015, 56, 914–920 CrossRef PubMed.
- K.-P. Eisenwiener, P. Powell and H. R. Mäcke, Bioorg. Med. Chem. Lett., 2000, 10, 2133–2135 CrossRef CAS PubMed.
- K.-P. Eisenwiener, I. M. Prata, I. Buschmann, H. W. Zhang, A. C. Santos, S. Wenger, J. C. Reubi and H. R. Mäcke, Bioconjugate Chem., 2002, 13, 530–541 CrossRef CAS PubMed.
- S. R. Banerjee, M. Pullambhatla, C. A. Foss, S. Nimmagadda, R. Ferdani, C. J. Anderson, R. C. Mease and M. G. Pomper, J. Med. Chem., 2014, 57, 2657–2669 CrossRef CAS PubMed.
- Y. Chen, M. Pullambhatla, S. R. Banerjee, Y. Byun, M. Stathis, C. Rojas, B. S. Slusher, R. C. Mease and M. G. Pomper, Bioconjugate Chem., 2012, 23, 2377–2385 CrossRef CAS PubMed.
- S. G. Levy, V. Jacques, K. L. Zhou, S. Kalogeropoulos, K. Schumacher, J. C. Amedio, J. E. Scherer, S. R. Witowski, R. Lombardy and K. Koppetsch, Org. Process Res. Dev., 2009, 13, 535–542 CrossRef CAS.
- K. Overoye-Chan, S. Koerner, R. Looby, A. Kolodziej, S. Zech, Q. Deng, J. Chasse, T. McMurry and P. Caravan, J. Am. Chem. Soc., 2008, 130, 6025–6039 CrossRef CAS PubMed.
- S. Aime, M. Botta, Z. Garda, B. E. Kucera, G. Tircso, V. G. Young and M. Woods, Inorg. Chem., 2011, 50, 7955–7965 CrossRef CAS PubMed.
- J. W. Walton, L. Di Bari, D. Parker, G. Pescitelli, H. Puschmann and D. S. Yufit, Chem. Commun., 2011, 47, 12289–12291 RSC.
- M. Benešová, U. Bauder-Wüst, M. Schäfer, K. D. Klika, W. Mier, U. Haberkorn, K. Kopka and M. Eder, J. Med. Chem., 2016, 59, 1761–1775 CrossRef PubMed.
- M. Eder, M. Schäfer, U. Bauder-Wüst, W.-E. Hull, C. Wängler, W. Mier, U. Haberkorn and M. Eisenhut, Bioconjugate Chem., 2012, 23, 688–697 CrossRef CAS PubMed.
- H. Rathke, A. Afshar-Oromieh, F. L. Giesel, C. Kremer, P. Flechsig, S. Haufe, W. Mier, T. Holland-Letz, M. De Bucourt and T. Armor, J. Nucl. Med., 2018, 117, 200220 Search PubMed.
- E. Eppard, A. de la Fuente, M. Benešová, A. Khawar, R. A. Bundschuh, F. C. Gärtner, B. Kreppel, K. Kopka, M. Essler and F. Rösch, Theranostics, 2017, 7, 4359 CrossRef CAS PubMed.
- E. M. Gale, I. P. Atanasova, F. Blasi, I. Ay and P. Caravan, J. Am. Chem. Soc., 2015, 137, 15548–15557 CrossRef CAS PubMed.
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. Robertson, C. Rodríguez-Rodríguez, P. Schaffer, C. J. Williams, J. Babich, V. Radchenko and J. Wilson, Angew. Chem., Int. Ed., 2017, 56, 14712–14717 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04655k |
| This journal is © The Royal Society of Chemistry 2020 |