DOI:
10.1039/D0RA07579E
(Review Article)
RSC Adv., 2020,
10, 39478-39484
Nitroacetonitrile as a versatile precursor in energetic materials synthesis
Received
4th September 2020
, Accepted 19th October 2020
First published on 28th October 2020
Abstract
Nitroacetonitrile is the simplest α-nitronitrile; it possesses a single central carbon attached to two strong electronegative, electron-withdrawing groups allowing extensive chemistry through the active methylene center. Free nitroacetonitrile has purification and stability issues, however stable salts of nitroacetonitrile possess the same reactivity as the free acid and are much more stable. Nitroacetonitrile serves as a versatile synthetic precursor in the formation of heterocyclic and polyfunctional aliphatic products and can allow for straightforward conversion to amino, acyl, and other functional groups. A main advantage of using nitroacetonitrile in the formation of heterocyclic-based energetics is its ability to add vicinal amino and nitro moieties onto fused ring structures, a common structural motif in insensitive energetic materials. In this minireview we discuss the preparation of nitroacetonitrile and its stable salts, as well as discuss the range of energetic materials this versatile precursor has found use in.
 Shannon Creegan | Shannon Creegan received her B. S. (Honors) in Chemical Engineering from New Mexico State University. She is currently pursuing a PhD in Materials Engineering at Purdue University. |
 Davin Piercey | Prof. Dr Davin Piercey received his PhD from the Ludwig Maximilian University of Munich in Germany in 2013 under the direction of Prof. Dr Thomas M. Klapötke and completed his postdoctoral research at Los Alamos National Laboratory in New Mexico under Dr David E Chavez. Since 2018 he has been faculty at Purdue University directing his research group on the synthesis of new energetic materials. |
1 Introduction
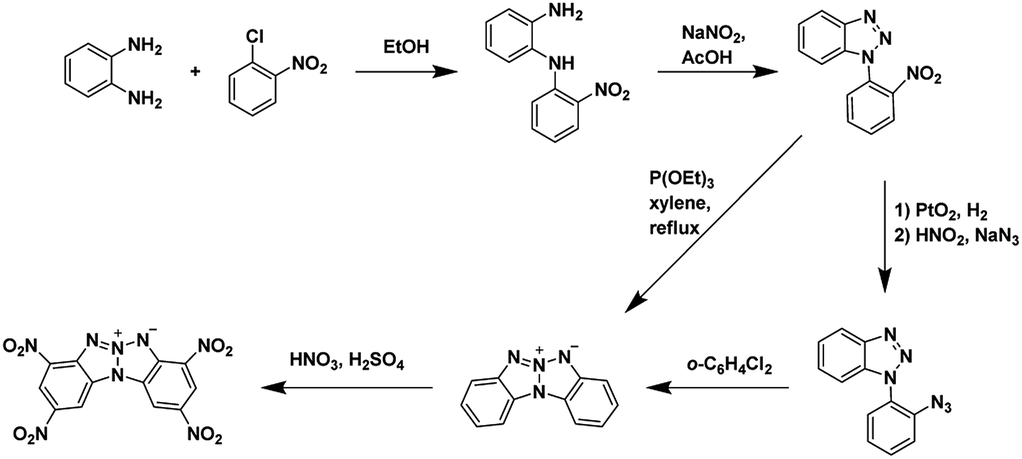
Modern energetic materials need to meet performance, environmental, and economic requirements in order to be considered for practical use. Synthetic transformations which produce multiple energetic functionalities (aka ‘explosophore’) in a single step are of special interest in the field. The nitrogen-rich heterocycles including triazoles,1–7 tetrazole,1,8–13 triazines,6,14–17 and tetrazines16,18–26 have found extensive use as the backbone in new energetic materials. Annulated heterocyclic systems are arguably some of the most important backbones; when designing new energetic materials as they often have some of the most desirable properties including higher thermal stabilities, low sensitivities, and higher densities when compared to the related non-annulated materials;27,28 however, the syntheses of many annulated heterocyclic systems are complex and consist of many steps (examples shown Schemes 1 and 2).
 |
| | Scheme 1 Synthesis of 2,9-dinitro-bis[1,2,4]triazolo[1,5-d:50,10-f][1,2,3,4]tetrazine, an annulated energetic system.26,28,32 | |
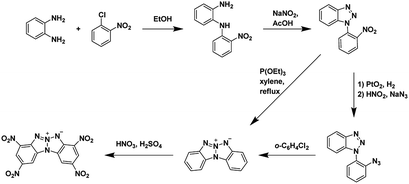 |
| | Scheme 2 Synthesis of 2,4,8,10-tetranitrobenzo [4′,5′][1,2,3]triazolo[2′,1′:2,3][1,2,3]triazolo[4,5-b]pyridin-6-ium-5-ide (TACOT), an annulated energetic system.33 | |
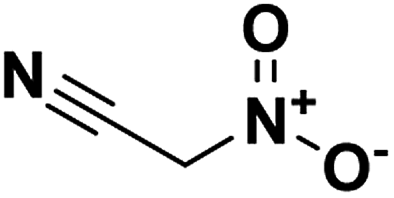
The main utility of nitroacetonitrile in the synthesis of annulated heterocyclic compounds is their ability to produce annulated 1,2,4-triazines bearing a vicinal amino and nitro group in a single step. In this, a diazonium-containing compound is reacted with either nitroacetonitrile or a salt thereof, and the produced nitrocyanohydrazone then cyclizes through the nitrile giving a heterocycloaminonitro-1,2,4-triazine. Pure acid nitroacetonitrile as a precursor suffers from numerous drawbacks including lack of high-yield high-purity syntheses, and chemical instability including the chance of spontaneous explosion.29 Stable salts of nitroacetonitrile, such as the potassium salt of nitroacetonitrile which is easily prepared via an indirect route and does not pose a lab hazard,30,31 have made the use of nitroacetonitrile practical for energetics chemistry on both the lab and large scale (Fig. 1).
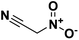 |
| | Fig. 1 Neutral nitroacetonitrile. | |
2 Nitroacetonitrile
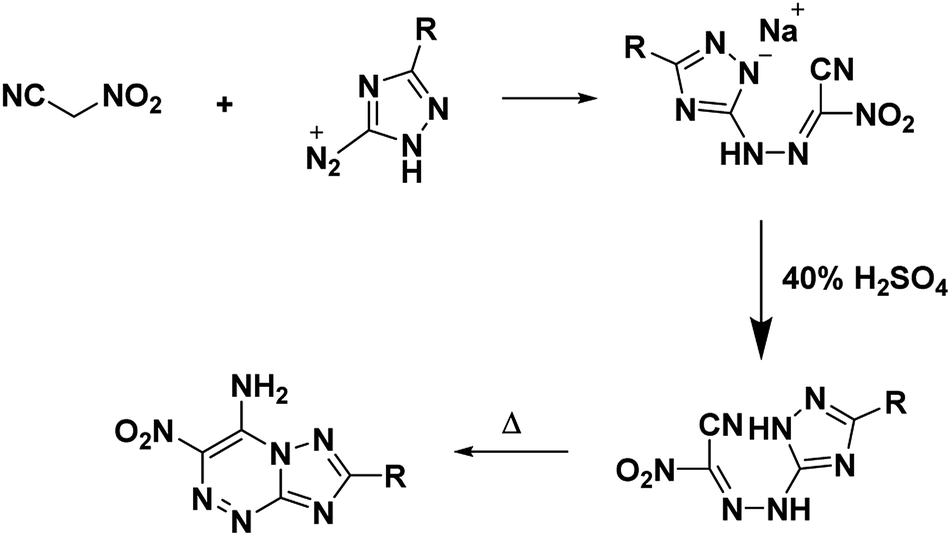
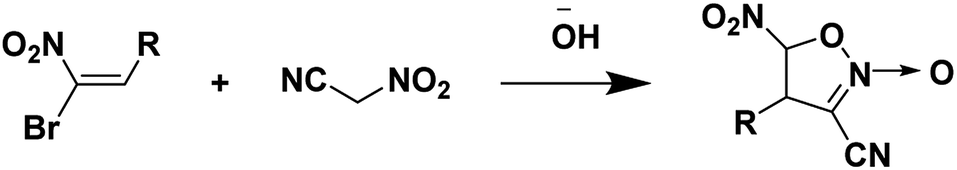
Nitroacetonitrile serves as a versatile synthetic precursor in organic synthesis as an intermediate in the formation of heterocyclic and polyfunctional aliphatic products.34 (Schemes 3 and 4).
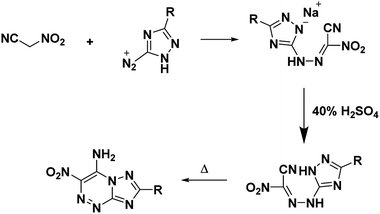 |
| | Scheme 3 Synthesis of 7-amino-6-nitro-1,2,4-triazolo[5,1-c][1,2,4]triazines. | |
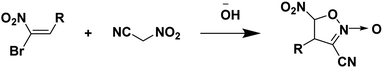 |
| | Scheme 4 Synthesis of 4-alkyl(aryl)-3-cyano-5-nitroisoxazolines.34 | |
Nitroacetonitrile is the simplest α-nitronitrile compound and possesses two strongly electronegative, electron-withdrawing groups directly attached to the same α-carbon, making the protons of the central carbon (CH2 group) acidic.34 Nitroacetonitrile's role as a cyano(nitro)methylation agent is invaluable as it alters compounds' structures to allow for the straightforward conversion into target molecules with numerous functional groups.35,36 This is possible due to the highly reactive nature of the cyano group, which enables conversion to amino, acyl, and other functional groups37 and the nitro group's capability of converting into an amino or carbonyl group35 and the active methylene center's ability to form C–C or N–C bonds.38,39
Nitroacetonitrile is thermodynamically unstable, and a common practice is to maintain reactions in which it serves as a reagent under 50 °C (max).40 Dr Craig Thomas of the National Institutes of Health Chemical Genomics Center, in 2009, reported the explosion of a flask containing nitroacetonitrile allegedly due to decomposition.29 Now, a widely stated hazard, one of the earliest references of nitroacetonitrile possessing energetic properties, was made by Grivas and Taurins.40 The decomposition of nitroacetonitrile at 109 °C, measured by DSC, released 874 J g−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 of energy.
41 of energy.
3 Synthesis
3.1. Neutral nitroacetonitrile
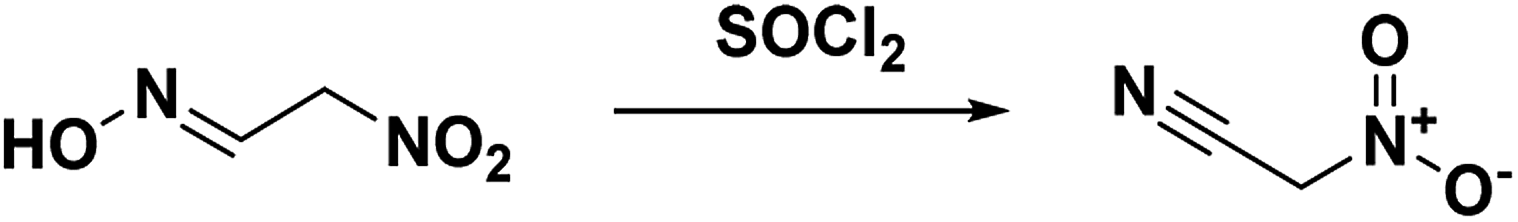
The first synthesis of nitroacetonitrile was reported by Steinkopf and Bohrmann in 190842 where methazonic acid (nitroacetaldehyde oxime) was dehydrated by thionyl chloride (Scheme 5). In this synthesis thionyl chloride was added to a gently refluxing solution of methazonic acid in diethyl ether and heated until there was no further production of hydrogen chloride and sulfur dioxide. The mixture was subsequently filtered and concentrated under pressure to yield a yellow or brown oil.43–45 Many authors used this preparation method or variations of over the following years. This method results in low yields, 13–47%, with varying degrees of purity. The oil can also be purified via a silica gel column with benzene as the eluent.43 Another purification option reported is to decolorize the oil with activated charcoal, then turn the compound into a salt with gaseous ammonia before acidifying and extracting into ether.45
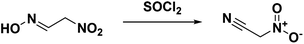 |
| | Scheme 5 Synthesis of neutral nitroacetonitrile from methazonic acid (nitroacetaldehyde oxime). | |
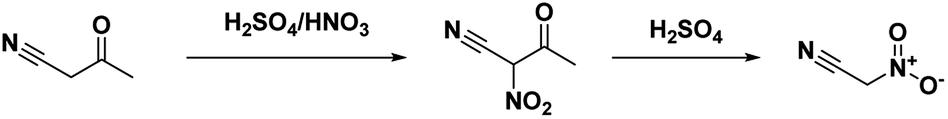
In 1994, Kislyi et al.46 reported an alternate method for the synthesis of nitroacetonitrile from nitrocyanoacetone with a yield of 70–85% via deacylation. Nitrocyanoacetone was prepared by the nitration of cyanoacetone with a sulfuric acid/nitric acid mixture in chloroform (Scheme 6). The chloroform layer was then removed and evaporated, leaving a residue of nitrocyanoacetone. The deacylation was performed by dissolving nitrocyanoacetone in ethanol (or methanol) and acidifying with 10% sulfuric acid before neutralizing with sodium bicarbonate and pouring into water. Nitroacetonitrile was then isolated as a residue via extraction and evaporation of the organic solvent.46
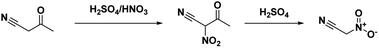 |
| | Scheme 6 Synthesis of neutral nitroacetonitrile from cyanoacetone. | |
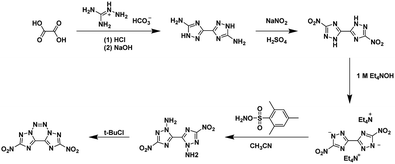
3.2. Potassium salt of nitroacetonitrile
Due to the low thermal stability, low yield and purity, and hazardous nature of nitroacetonitrile decomposition process, the work done with nitroacetonitrile was hampered by this inconvenient precursor until the development of stable salts as synthetic equivalents by alternative synthesis routes was developed. The advantage of using salts and avoiding pure neutral nitroacetonitrile is the decreased risk due to by-passing the generation of free neutral nitroacetonitrile during a reaction. These alternatives allow work without the hazard of explosion and allow for purer precursors to be used in synthesis using nitroacetonitrile as a reagent.
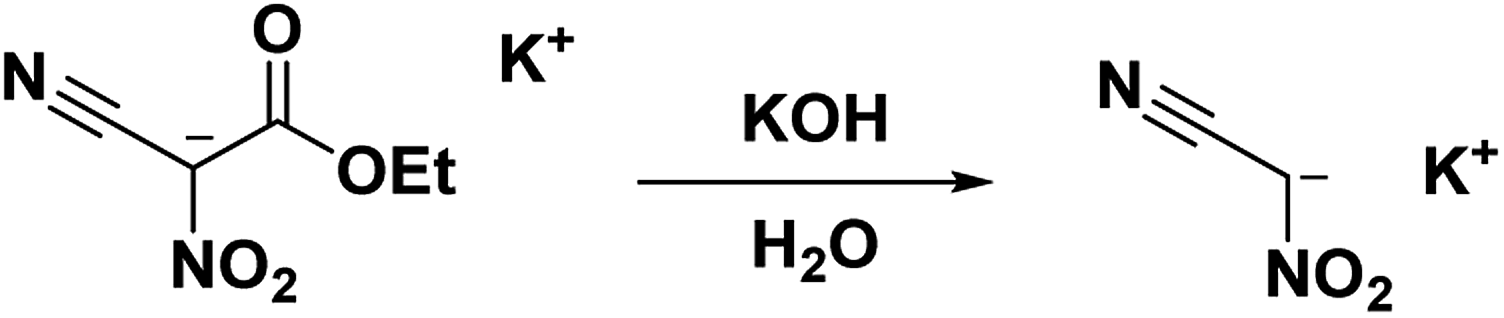
The potassium salt of nitroacetonitrile is an excellent alternative to neutral nitroacetonitrile being thermodynamically stable, chemically equivalent, and water-soluble; in fact, many syntheses using nitroacetonitrile begin with the formation of a nitroacetonitrile salt in situ6,14,15,27,47–50 The potassium salt of nitroacetonitrile can be obtained from the hydrolysis of the potassium salt of ethyl nitrocyanoacetate into the unstable dipotassium salt of nitrocyanoacetic acid (cyano-aci-nitroacetate) which readily converts, via decarboxylation, into the potassium salt of nitroacetonitrile (Scheme 7) with a 45% yield.31 The nitroacetonitrile potassium salt has also been obtained with a 79% yield by the decarboxylation of an acidic aqueous solution of dipotassium nitrocyanoacetic acid through an ion-exchange resin (IRC-50).51 This route entirely avoids unstable intermediates such as methazonic acid or nitroacetonitrile.
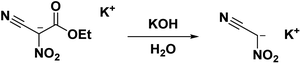 |
| | Scheme 7 Synthesis route for the stable potassium salt of nitroacetonitrile. | |
Voinkov et al.31 showed the ability of the potassium salt to replace nitroacetonitrile by reacting nitroacetonitrile potassium salt with aromatic and heterocyclic aldehydes to form 3-(het)aryl-2-nitroacrylonitriles and demonstrating the ability of nitroacetonitrile potassium salt to serve as an azocoupling reagent with the production of 2-nitro-2-phenylhydrazonoacetonitrile (Scheme 8).31 Ulomskiy et al.30 also provided verification by replicating a 1985 study done by Rusinov et al.47 (refer Scheme 3) to synthesize azolo[5,1-c][1,2,4]triazines using neutral nitroacetonitrile (Scheme 9).
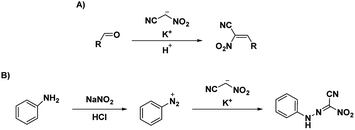 |
| | Scheme 8 (A) Formation of 3-(het)aryl-2-nitroacrylonitriles and (B) synthesis of 2-nitro-2-phenylhydrazonoacetonitrile.31 | |
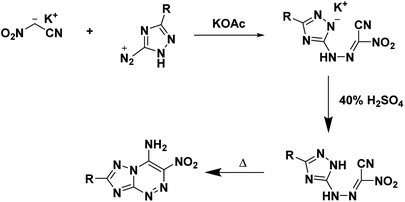 |
| | Scheme 9 Synthesis of azolo[5,1-c][1,2,4]triazines using the potassium salt of nitroacetonitrile.30 | |
4 Nitroacetonitrile in energetic synthesis
The advantage of using nitroacetonitrile in the synthesis of heterocyclic energetics is its ability to create annulated 1,2,4-triazines containing vicinal amino and nitro groups. The addition of the vicinal amino and nitro group structural motif stabilizes via intra- and intermolecular hydrogen bonding.6,50 The presence of amine and nitro groups can lead to conjugation resulting in a planar structure with π–π stacking interactions. These interactions are also seen in fused-rings with flat molecular geometry.15 Often, energetics with the lowest thermal and mechanical sensitivities are flat molecules with π–π interactions and hydrogen bonding. The flat geometry can result in a crystal lattice with parallel planar52 packing that allows slip between the layers. The ability for slip decreases the resistance experienced by the compound during compression initiated by shock or impact52 this, in turn, reduces the probability of hotspot generation and inadvertent initiation.15
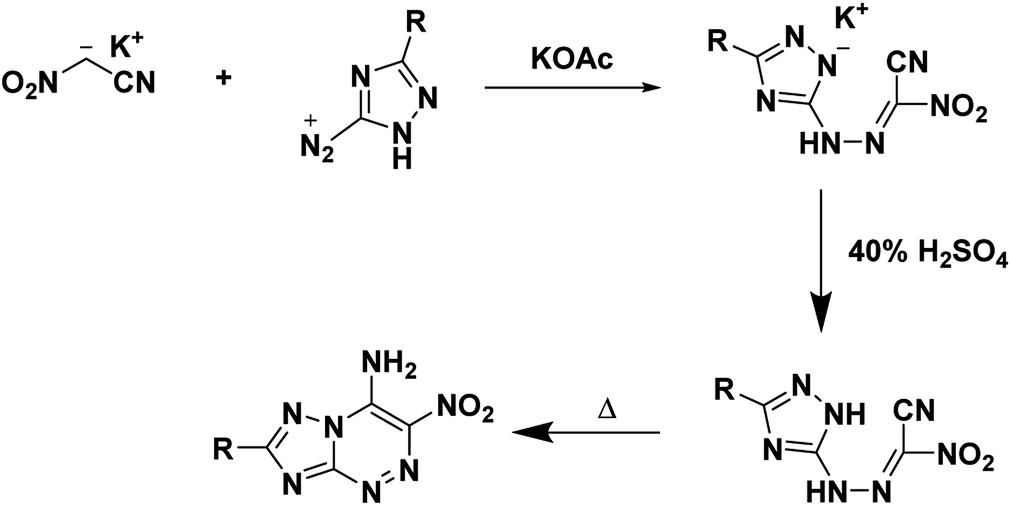
The typical synthesis of heterocyclic energetics using free nitroacetonitrile begins with the conversion of a parent compound into its corresponding diazonium salt. The diazonium product is then treated with a solution containing a salt of nitroacetonitrile in acid yielding a hydrazone. The resulting molecule is then cyclized by heating in an organic solvent or mixture if it does not cyclize during synthesis spontaneously.
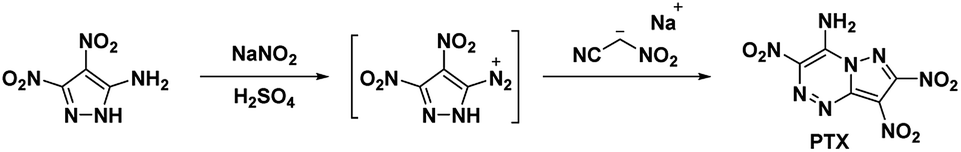
4.1. 7-Amino-2,3,6-trinitropyrazolo[5,1-c][1,2,4]triazine (PTX)
In 2010, Dalinger et al.53 published a seven-step procedure for the formation of 7-amino-2,3,6-trinitropyrazolo[5,1-c][1,2,4]triazine a fused pyrazolo-triazine explosive later synthesized in two-steps by Chavez et al.,15 in 2015. The synthesis of PTX (Scheme 10) starts from 5-amino-3,4-dinitropyrazole and begins by using sodium nitrite and acid to form the diazonium. The diazodinitropyrazole was then coupled, in situ, with nitroacetonitrile to yield PTX. Chavez et al. investigated PTX further based on its potential to match TATB's insensitivity while providing increased performance as it is a fused ring system with a flat molecular geometry, π–π interaction, and intra/inter-molecular hydrogen bonding and was anticipated to possess a planar crystal structure. Analysis of the crystal structure supported the predicted structural characteristics but showed four unique plane stacking orientations which prevent interlayer sliding within the crystal lattice. According to Chavez et al. this structure is classified as herringbone crystal packing. It was concluded, the inability to slip is what prevented PTX from matching TATB's insensitivity while still having a lower sensitivity than HMX. Despite this, PTX compares favourably with RDX as it has a calculated detonation velocity of 8998 m s−1 and a detonation pressure of 36.04 GPa (Table 1).
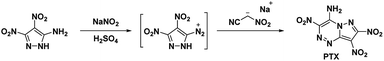 |
| | Scheme 10 Synthesis of PTX. | |
Table 1 Physical and energetic properties of baseline energetics and those developed with nitroacetonitrilea
| Compound |
Tdb [°C] |
ρc [g cm−1] |
ΔHf d [kJ mol−1] |
ISe [J] |
FSf [N] |
Sparkg [J] |
PCJh [GPa] |
VDi [m s−1] |
| Conditions, instrumentation, and equations vary depending on the individuals reporting; please refer to sources. DSC thermal decomposition temperature (onset) under nitrogen gas. Density was calculated or measured via crystal analysis or gas pycnometery at literature temperature. Calculated heat of formation. Impact sensitivity, LANL type 12, 50% drop height, 2.5 kg. Friction sensitivity, 50% load Bruceton up/down method. ABL spare 3.4% threshold initiation level. Calculated detonation pressure. Calculated detonation velocity. |
| TATB14,27,50 |
350 |
1.93 |
−139.7 |
50 |
>360 |
— |
30.5 |
8179 |
| RDX6,14 |
204 |
1.80 |
92.6 |
4.6 |
157 |
0.062 |
34.9 |
8795 |
| HMX15 |
265 |
1.89 |
75 |
6.1 |
150 ± 40 |
0.025–0.125 |
39.0 |
9110 |
| LLM-105 (ref. 50) |
342 |
1.92 |
11 |
20 |
360 |
— |
31.7 |
8639 |
| PETN6,55,56 |
164 |
1.77 |
−501.24 |
2.5 |
92 |
0.062 |
33.2 |
8260 |
| PTX15 |
246 |
1.95 |
370 |
14.3 |
324–360 |
0.0625 |
36.04 |
8998 |
| DNPTDA50 |
355 |
1.90 |
344 |
>60 |
>360 |
— |
32.6 |
8727 |
| DPX-266 |
232 |
1.86 |
387 |
29 |
>360 |
0.125 |
32 |
8700 |
| DPX-276 |
138 |
1.90 |
378 |
10.3 |
258 |
0.062 |
35.4 |
8970 |
| ANCTT49 |
229 |
1.80 |
533.34 |
— |
— |
— |
23.05 |
7760 |
| ANTTT49 |
305 |
1.82 |
723.43 |
>78.5 |
>360 |
0.125 |
27.04 |
8310 |
| AANT54 |
162 |
1.781 |
494.3 |
7 |
120 |
— |
28.88 |
8310 |
| TNTHBPTDA27 |
315 |
1.85 |
899 |
>60 |
>360 |
— |
31.4 |
8572 |
4.2. 3,8-Dinitropyrazolo[5,1-c][1,2,4]triazine-4,7-diamine (DNPTDA)
The goal of Shreeve et al.50 was to create azolotriazine systems with vicinal amine and nitro moieties based on the trend of annulated systems having a higher thermal stability than the single ring parent structure. The expectation was to be able to use the relationship between crystal structure and properties of a compound to increase thermal stability. The starting compound 3,5-diamino-4-nitropyrazole was converted into its diazonium salt at one amine group using tert-butyl nitrite before treatment with the sodium salt of nitroacetonitrile in 20% sulfuric acid. The formed hydrazone intermediate was then cyclized by refluxing in a mixture of methanol and water (refer Scheme 11). The resulting compound DNPTDA proved impervious to further oxidation with either HOF or TFAA and 50% hydrogen peroxide. The overall results were as hoped with the fused ring, amino, and nitro groups essentially occupying the same plane with extensive hydrogen bonding. DNPTDA possessed higher thermal stability than TATB with an onset decomposition temperature of 355 °C and is less sensitive than TATB. Additionally, DNPTDA is expected to have a comparative performance with RDX (Table 1).
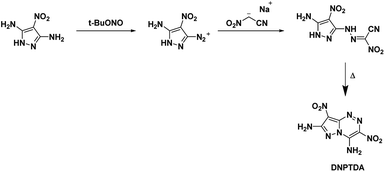 |
| | Scheme 11 Synthesis of DNPTDA. | |
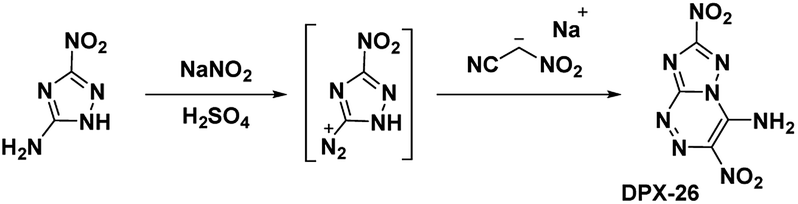
4.3. 4-Amino-3,7-dinitrotriazolo-[5,1-c][1,2,4] triazine (DPX-26)
Building upon the work with PTX, which was stabilized by vicinal amino and nitro groups, Chavez et al.6 synthesized the compound 4-amino-3,7-dinitrotriazolo-[5,1-c][1,2,4] triazine (Scheme 12). The precursor molecule 3-amino-5-nitro-triazole (ANTA) was diazotized and reacted with nitroacetonitrile to produce an acyclic hydrazone compound which spontaneously cyclized to produce DPX-26. Analysis of the crystal structure shows DPX-26 possesses a nearly planar structure, with intra- and intermolecular hydrogen bonding and strong nitro–π interactions between nitro groups and both rings. The compound was similarly synthesized by Shreeve et al.,14 however the compound did not cyclize in situ and the acyclic hydrazone needed to be refluxed in a methanol/water mixture to complete the cyclization.
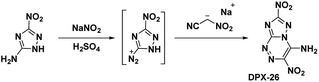 |
| | Scheme 12 Synthesis of DPX-26. | |
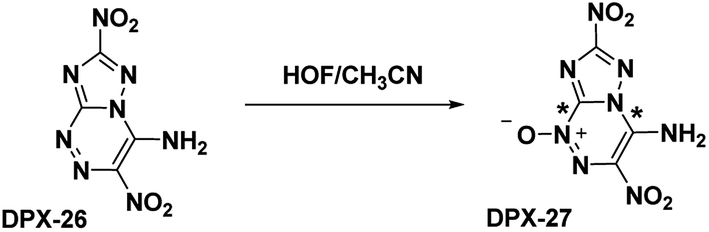
The oxidation of DPX-26 with hypofluorous acid in acetonitrile yielded 4-amino-3,7-dinitrotriazolo-[5,1-c][1,2,4] triazine 4-oxide (DPX-27) with the N-oxide installed on the N1 position of the triazine (refer Scheme 13). Similar to PTX, the DPX-27 water complex possesses a herringbone crystal lattice structure with eight molecules per unit cell. Crystal analysis of DPX-27 solvates from water and nitromethane showed an increase in bond-length between C1–N5 and C4–N4 (indicated * in Scheme 13), compared to DPX-26, leading to an increase in separation between the 1,2,4-triazine ring and the triazole ring. This separation is not seen in DPX-26 and was concluded to be the reason behind the decrease in decomposition temperature from 232 °C (DPX-26) to 138 °C (DPX-27). DPX-26 has improved thermal stability, heat of formation, and insensitivity over RDX (Table 1) while DPX-27 has increased resistance to impact and friction and higher calculated performance with a detonation velocity of 8970 m s−1 and a detonation pressure of 35.4 GPa.
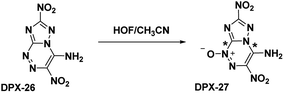 |
| | Scheme 13 Synthesis of DPX-27. | |
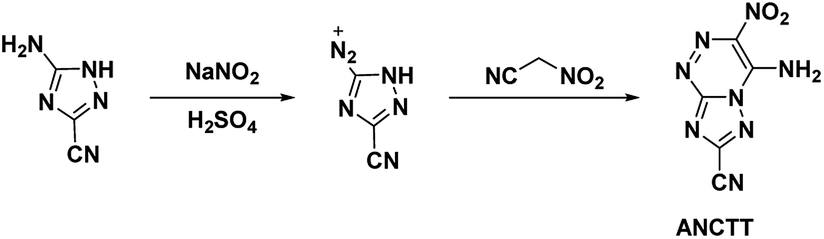
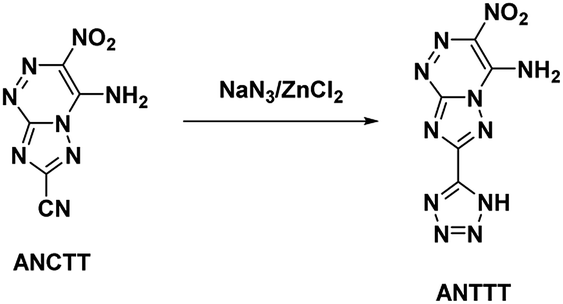
4.4. 4-Amino-3-nitro-7-cyanotriazolo-[5,1-c][1,2,4] triazine (ANCTT) and 4-amino-3-nitro-7-(5-1H-tetrazolyl)triazolo-[5,1-c][1,2,4] triazine (ANTTT)
Reviewing the research done on the stabilizing effects of vicinal amine and nitro moieties, Snyder et al.49 sought to improve sensitivities and thermal stability by replacing azolo-bound nitro groups with functional groups which express the traits themselves. Tetrazolyl moieties satisfy this requirement by possessing high thermal stability and endothermicity with low sensitivity. Snyder et al. developed an energetic precursor to which a tetrazolyl moiety could be added by azocoupling diazotized 3-amino-5-cyano-1,2,4-triazole (ACT) with nitroacetonitrile (Scheme 14). Crystal analysis of ANCTT showed herringbone packing and π-interactions. Bond lengths indicated delocalization on the bicyclic system and high delocalization of the amine lone-pair in the π-ring system. While ANCTT has greater thermal stability than RDX, the calculated performance of ANCTT was seen to be significantly less, with a predicted detonation velocity of 7760 m s−1 and detonation pressure of 23.05 GPa. The low performance was concluded to be the result of poor oxygen balance. ANTTT was made by reacting ANCTT with sodium azide in the presence of zinc chloride (Scheme 15). The addition of the tetrazolyl moiety improved performance (VD = 8310 m s−1; PCJ = 27.04 GPa), though still less than that of RDX, and increased the thermal stability. The reported impact and friction sensitivity of ANTTT are higher than that of TATB (Table 1).
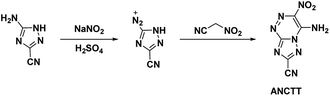 |
| | Scheme 14 Synthesis of ANCTT. | |
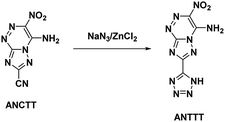 |
| | Scheme 15 Synthesis of ANTTT. | |
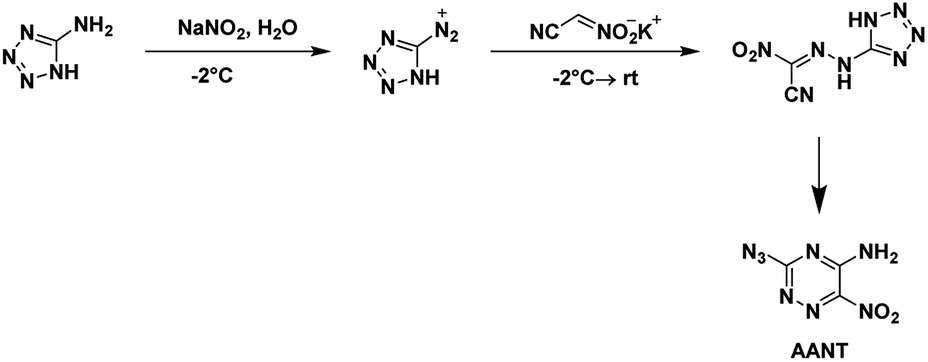
4.5. 3-Azido-5-amino-6-nitro-1,2,4-triazine (AANT)
AANT was first synthesized by Rusinov et al.48 in 1984 using free nitroacetonitrile. It was later synthesized in 2020 by Piercey et al.54 using the potassium salt of nitroacetonitrile and characterized, for the first time, as an energetic. The commercial precursor 5-aminotetrazole was diazotized and reacted with the potassium salt of nitroacetonitrile (Scheme 16). Two separate precipitation events occurred with the first yielding the nitroacetamide and the second the azido-triazine. The nitroacetamide crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the inclusion of a single hydration water per molecule with two formula units within the unit cell and a density of 1.781 g cm−3. AANT crystallizes in the orthorhombic space group P212121 with four formula units in the unit cell and a density of 1.781 g cm−3. The compound is less sensitive to impact and friction than RDX but is less thermally stable. The calculated performance is also less with a detonation pressure of 28.9 GPa and detonation velocity of 8310 m s−1.
with the inclusion of a single hydration water per molecule with two formula units within the unit cell and a density of 1.781 g cm−3. AANT crystallizes in the orthorhombic space group P212121 with four formula units in the unit cell and a density of 1.781 g cm−3. The compound is less sensitive to impact and friction than RDX but is less thermally stable. The calculated performance is also less with a detonation pressure of 28.9 GPa and detonation velocity of 8310 m s−1.
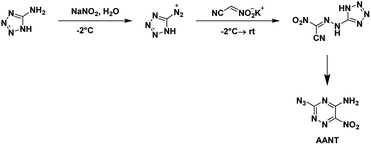 |
| | Scheme 16 Synthesis of AANT. | |
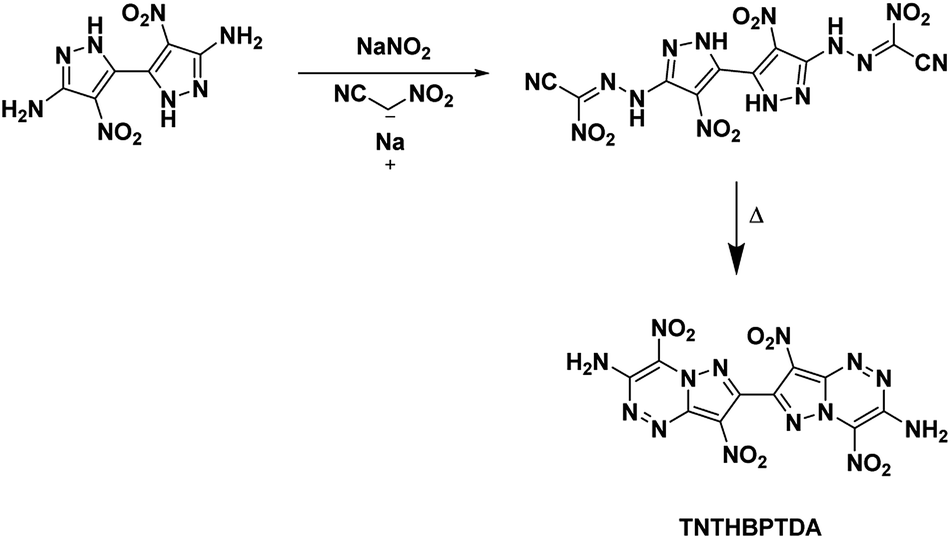
4.6. 4,4′,8,8′-Tetranitro-6,6′,8a,8′a-tetrahydro-[7,7′-bipyrazolo[5,1-c][1,2,4]triazine]-3,3′-diamine (TNTHBPTDA)
In this study, Shreeve et al.27 characterized a newly synthesized energetic bicyclic C–C bonded amino-, nitro-pyrazole system, 4,4′-dinitro-2H,2′H-[3,3′-bipyrazole]-5,5′-diamine. Reacting the bipyrazole with nitroacetonitrile yielded a hydrazone intermediate which cyclized into the annulated structure TNTHBPTDA upon refluxing in a solution of water and methanol (Scheme 17). The crystal structure of TNTHBPTDA showed an increase in bond lengths between nitro groups and their rings when compared to the precursor compound (1.42 Å from 1.39 Å, respectively). Additionally, the structure obtained shows the molecule is not planar with a rotation along the central C–C bond though the amino and nitro groups are coplanar to their associated rings. The 5,6-fused diamine has high thermal stability and is more insensitive than TATB (Table 1). Its performance is less than but still comparable to that of RDX with a detonation velocity of 8572 m s−1 and a detonation pressure of 31.4 GPa.
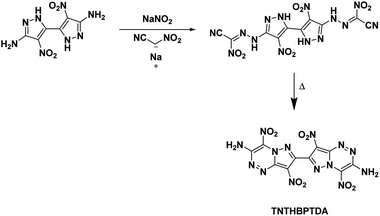 |
| | Scheme 17 Synthesis of TNTHBPTDA. | |
5 Conclusion
Nitroacetonitrile is useful synthetic precursor capable of creating a wide variety of energetic compounds. The development of a stable form has removed the limitations placed on use of the compound due to its energetic nature and the purity and stability challenges. The use of nitroacetonitrile allows for the synthesis of annulated heterocyclic energetic systems with simple routes of preparation. The energetic materials prepared from this versatile precursor are in general, highly performing and highly thermally stable. Of the eight materials presented two are expected to outperform RDX and another two to perform similarly to RDX. In terms of insensitivity, seven are less mechanically and thermally sensitive than RDX while one surpasses TATB.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support of this work was provided by the Office of Naval Research under grant N00014-19-1-2089. Our lab is also supported by Purdue University and The Army Research Office (ARO). We are indebted to and thank Ms. Dominique Wozniak and Mr Matthew Gettings for their support of our work.
Notes and references
- A. A. Dippold, D. Izsák, T. M. Klapötke and C. Pflüger, Chem.–Eur. J., 2016, 22, 1768–1778 CrossRef CAS
 .
. - Y. Huang, H. Gao, B. Twamley and J. M. Shreeve, Eur. J. Inorg. Chem., 2008, 2008, 2560–2568 CrossRef .
- T. M. Klapötke, P. C. Schmid, S. Schnell and J. Stierstorfer, Chem.–Eur. J., 2015, 21, 9219–9228 CrossRef .
- A. S. Kumar, V. D. Ghule, S. Subrahmanyam and A. K. Sahoo, Chem.–Eur. J., 2013, 19, 509–518 CrossRef CAS .
- Q. Ma, G. Fan, L. Liao, H. Lu, Y. Chen and J. Huang, ChemPlusChem, 2017, 82, 474–482 CrossRef CAS .
- D. G. Piercey, D. E. Chavez, B. L. Scott, G. H. Imler and D. A. Parrish, Angew. Chem., Int. Ed., 2016, 55, 15315–15318 CrossRef CAS .
- Y. Tang, S. Dharavath, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem.–Eur. J., 2017, 23, 9185–9191 CrossRef CAS .
- M. Dachs, A. A. Dippold, J. Gaar, M. Holler and T. M. Klapötke, Z. Anorg. Allg. Chem., 2013, 639, 2171–2180 CrossRef CAS .
- D. Fischer, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Eng. Mater. Technol., 2012, 30, 40–54 CAS .
- N. Fischer, D. Izsák, T. M. Klapötke and J. Stierstorfer, Chem.–Eur. J., 2013, 19, 8948–8957 CrossRef CAS .
- T. M. Klapötke, M. Q. Kurz, R. Scharf, P. C. Schmid, J. Stierstorfer and M. Sućeska, ChemPlusChem, 2015, 80, 97–106 CrossRef .
- T. M. Klapötke and J. Stierstorfer, Helv. Chim. Acta, 2007, 90, 2132–2150 CrossRef .
- T. M. Klapötke and J. C. p. Stierstorfer, in Green Energetic Materials, 2014, ch. 6, pp. 133–178, DOI:10.1002/9781118676448.ch06 .
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem.–Eur. J., 2017, 23, 1743–1747 CrossRef CAS .
- M. C. Schulze, B. L. Scott and D. E. Chavez, J. Mater. Chem. A, 2015, 3, 17963–17965 RSC .
- H. Wei, J. Zhang and J. M. Shreeve, Chem.–Asian J., 2015, 10, 1130–1132 CrossRef CAS .
- Q. Wang, Y. Shao and M. Lu, Chem. Commun., 2019, 55, 6062–6065 RSC .
- K. O. Christe, D. A. Dixon, M. Vasiliu, R. I. Wagner, R. Haiges, J. A. Boatz and t. l. H. L. Ammon, Propellants, Explos., Pyrotech., 2015, 40, 463–468 CrossRef CAS .
- T. M. Klapötke, D. G. Piercey, J. Stierstorfer and M. Weyrauther, Propellants, Explos., Pyrotech., 2012, 37, 527–535 CrossRef .
- D. G. Piercey, D. E. Chavez, S. Heimsch, C. Kirst, T. M. Klapötke and J. Stierstorfer, Propellants, Explos., Pyrotech., 2015, 40, 491–497 CrossRef CAS .
- X. Song, J. Li, H. Hou and B. Wang, J. Comput. Chem., 2009, 30, 1816–1820 CrossRef CAS .
- G. Wang, T. Lu, G. Fan, C. Li, H. Yin and F.-X. Chen, Chem.–Asian J., 2018, 13, 3718–3722 CrossRef CAS .
- H. Wei, H. Gao and J. M. Shreeve, Chem.–Eur. J., 2014, 20, 16943–16952 CrossRef CAS .
- C.-C. Ye, Q. An, W. A. Goddard Iii, T. Cheng, W.-G. Liu, S. V. Zybin and X.-H. Ju, J. Mater. Chem. A, 2015, 3, 1972–1978 RSC .
- G.-D. Zhang, Z. Wang and J.-G. Zhang, Z. Anorg. Allg. Chem., 2018, 644, 512–517 CrossRef CAS .
- D. E. Chavez, J. C. Bottaro, M. Petrie and D. A. Parrish, Angew. Chem., Int. Ed., 2015, 54, 12973–12975 CrossRef CAS .
- Y. Tang, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem. Commun., 2018, 54, 10566–10569 RSC .
- H. Gao, Q. Zhang and J. M. Shreeve, J. Mater. Chem. A, 2020, 8, 4193–4216 RSC .
- C. J. Thomas, Chem. Eng. News, 2009, 87, 4 CAS .
- E. K. Voinkov, E. N. Ulomskiy, V. L. Rusinov, O. N. Chupakhin, E. B. Gorbunov, R. A. Drokin and V. V. Fedotov, Chem. Heterocycl. Compd., 2015, 51, 1057–1060 CrossRef CAS .
- E. K. Voinkov, E. N. Ulomskiy, V. L. Rusinov, K. V. Savateev, V. V. Fedotov, E. B. Gorbunov, M. L. Isenov and O. S. Eltsov, Mendeleev Commun., 2016, 26, 172–173 CrossRef CAS .
- A. A. Dippold, T. M. Klapötke and N. Winter, Eur. J. Inorg. Chem., 2012, 2012, 3474–3484 CrossRef CAS .
- L. Türker and S. Variş, Polycyclic Aromat. Compd., 2009, 29, 228–266 CrossRef .
- S. G. Zlotin, G. N. Varnaeva and O. A. Luk'yanov, Russ. Chem. Rev., 1989, 58, 470–478 CrossRef .
- D. Amantini, F. Fringuelli, O. Piermatti, F. Pizzo and L. Vaccaro, Green Chem., 2001, 3, 229–232 RSC .
- H. Asahara, K. Muto and N. Nishiwaki, Tetrahedron, 2014, 70, 6522–6528 CrossRef CAS .
- K. Iwai, H. Asahara and N. Nishiwaki, J. Org. Chem., 2017, 82, 5409–5415 CrossRef CAS .
- Y. Liu and J.-P. Wan, Chem.–Asian J., 2012, 7, 1488–1501 CrossRef CAS .
- X.-P. Hui, C. Yin, J. Ma and P.-F. Xu, Synth. Commun., 2009, 39, 676–690 CrossRef CAS .
- J. C. Grivas and A. Taurins, Can. J. Chem., 1959, 37, 1266–1267 CrossRef CAS .
- N. Nishiwaki, Y. Kumegawa, K. Iwai and S. Yokoyama, Chem. Commun., 2019, 55, 7903–7905 RSC .
- W. Steinkopf and L. Bohrmann, Ber. Dtsch. Chem. Ges., 1908, 41, 1044 CrossRef CAS .
- A. Boguszewska-Czubara, A. Lapczuk-Krygier, K. Rykala, A. Biernasiuk, A. Wnorowski, L. Popiolek, A. Maziarka, A. Hordyjewska and R. Jasiński, J. Enzyme Inhib. Med. Chem., 2016, 31, 900–907 CrossRef CAS .
- E. N. Gate, M. D. Threadgill, M. F. G. Stevens, D. Chubb, L. M. Vickers, S. P. Langdon, J. A. Hickman and A. Gescher, J. Med. Chem., 1986, 29, 1046–1052 CrossRef CAS .
- M. I. Kanishchev, N. V. Korneeva and S. A. Shevelev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 2145–2147 CrossRef .
- V. P. Kislyi, A. L. Laikhter, B. I. Ugrak and V. V. Semenov, Russ. Chem. Bull., 1994, 43, 70–74 CrossRef .
- V. L. Rusinov, A. Y. Petrov, O. N. Chupakhin, N. A. Klyuev and G. G. Aleksandrov, Chem. Heterocycl. Compd., 1985, 21, 576–582 CrossRef .
- V. L. Rusinov, T. V. Dragunova, V. A. Zyryanov, G. G. Aleksandrov, N. A. Klyuev and O. N. Chupakhin, Chem. Heterocycl. Compd., 1984, 20, 455–459 CrossRef .
- C. J. Snyder, T. W. Myers, G. H. Imler, D. E. Chavez, D. A. Parrish, J. M. Veauthier and R. J. Scharff, Propellants, Explos., Pyrotech., 2017, 42, 238–242 CrossRef CAS .
- Y. Tang, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, ACS Appl. Energy Mater., 2019, 2, 2263–2267 CrossRef CAS .
- N. Nishiwaki, Y. Takada, Y. Inoue, Y. Tohda and M. Ariga, J. Heterocycl. Chem., 1995, 32, 473–475 CrossRef CAS .
- P. Politzer and J. S. Murray, in Energetic Materials: From Cradle to Grave, ed. M. K. Shukla, V. M. Boddu, J. A. Steevens, R. Damavarapu and J. Leszczynski, Springer International Publishing, Cham, 2017, pp. 1–22, DOI:10.1007/978-3-319-59208-4_1 .
- I. L. Dalinger, I. A. Vatsadse, T. K. Shkineva, G. P. Popova, B. I. Ugrak and S. A. Shevelev, Russ. Chem. Bull., 2010, 59, 1631–1638 CAS .
- S. E. Creegan, M. Zeller, E. F. C. Byrd and D. G. Piercey, Propellants, Explos., Pyrotech., 2020 DOI:10.1002/prep.202000136 .
- T. W. Myers, C. J. Snyder, D. E. Chavez, R. J. Scharff and J. M. Veauthier, Chem.–Eur. J., 2016, 22, 10590–10596 CrossRef CAS .
- V. W. Manner, M. J. Cawkwell, E. M. Kober, T. W. Myers, G. W. Brown, H. Tian, C. J. Snyder, R. Perriot and D. N. Preston, Chem. Sci., 2018, 9, 3649–3663 RSC .
|
| This journal is © The Royal Society of Chemistry 2020 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Davin G. Piercey
b and
Davin G. Piercey