
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical generation of singlet oxygen†
Abdurrahman Turksoy‡
a,
Deniz Yildiz‡ a,
Simay Aydonat‡a,
Tutku Beduka,
Merve Canyurta,
Bilge Baytekin*a and
Engin U. Akkaya*b
a,
Simay Aydonat‡a,
Tutku Beduka,
Merve Canyurta,
Bilge Baytekin*a and
Engin U. Akkaya*b
aDepartment of Chemistry, Bilkent University, 06800 Ankara, Turkey
bState Key Laboratory of Fine Chemicals, Department of Pharmacy, Dalian University of Technology, 2 Linggong Road, 116024, Dalian, China
First published on 2nd March 2020
Abstract
Controlled generation of singlet oxygen is very important due to its involvement in scheduled cellular maintenance processes and therapeutic potential. As a consequence, precise manipulation of singlet oxygen release rates under mild conditions, is crucial. In this work, a cross-linked polyacrylate, and a polydimethylsiloxane elastomer incorporating anthracene-endoperoxide modules with chain extensions at the 9,10-positions were synthesized. We now report that on mechanical agitation in cryogenic ball mill, fluorescence emission due to anthracene units in the PMA (polymethacrylate) polymer is enhanced, with a concomitant generation of singlet oxygen as proved by detection with a selective probe. The PDMS (polydimethylsiloxane) elastomer with the anthracene endoperoxide mechanophore, is also similarly sensitive to mechanical force.
Introduction
Singlet oxygen refers to the lowest energy excited state of the molecular oxygen. Since the direct excitation of the ground state oxygen is forbidden by spin, symmetry and parity rules, it is most commonly generated by photosensitization, making use of the intermediacy of photosensitizer organic compounds with high triplet efficiencies.1 Such photosensitized generation of singlet oxygen is of particular importance due to its involvement in photodynamic therapy (PDT) of cancer.2 Thus, a singlet oxygen releasing mechanophoric group would be a valuable addition to the growing list of mechanically activatable functionalities.Reversible storage and delivery of singlet oxygen may provide a viable alternative to photosensitized generation of singlet oxygen.3 Photodynamic therapy has a number of advantages and superiorities compared to the classical therapeutic regimens, yet it is not considered to be a first line therapy. Two easily identifiable reasons for this reluctance are; first, the fact that light is required for the sensitization, but tissue penetration is limited,4 and second, tumor oxygen concentrations are typically low,5 and especially in the most aggressive tumors where a hypoxic region with a very low oxygen concentration develops.6 These lingering problems, unfortunately limit the practice of PDT to mostly superficial tumors. In our previous work, we presented a different strategy to circumvent these issues.7
The stability of the endoperoxides differ widely, and in a large number of 2-pyridone, naphthalene and anthracene endoperoxides, high yield release of singlet oxygen without side reactions was documented.8 As a matter of fact, the diversity in the rates of cycloreversion reactions may eventually prove to be very useful for fine tuning a biologically relevant singlet oxygen release. Release of singlet oxygen in cancer cell cultures were shown to induce apoptotic response.9
Mechanochemistry provides various avenues for transduction of mechanical stress towards selective chemical transformations.10,11 Bonds or molecular modules sensitive to mechanical stress are known as “mechanophores”. In polymeric systems, the shear forces to induce, or facilitate selective bond cleavage at a weak bond. An anthracene–dienophile adduct cycloreversion reported by Craig and coworkers12 and generation of chemiluminescence as a result of dioxetane cleavage.13 are two recent relevant examples.
In this work, we wanted to demonstrate that a reactive oxygen species with a microsecond lifetime could be generated on a polymer support when mechanical shear forces are applied.
Experimental
Materials and instrumentation
All chemicals and reaction solvents purchased from Sigma Aldrich, Acros Organics and ABCR were used without purification. Column chromatography purifications were performed with glass columns using Merck Silica gel 60 (particle size: 0.040–0.063 mm, 230–400 mesh ASTM) and reactions were monitored by thin layer chromatography (TLC) using precoated silica gel plates (Merck Silica Gel PF-254). Chromatography solvents (DCM, n-hexane, EtOAc) were purchased as technical grade and were purified employing fractional distillation before use. Anhydrous THF was used freshly after refluxing over Na in the presence of benzophenone under Ar.NMR spectra were recorded on Bruker Spectrospin Avance DPX 400 spectrometer (operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR) using deuterated solvents (CDCl3, DMSO-d6) with tetramethylsilane (TMS) as internal standard purchased from Merck. Spin multiplicities are reported as following: s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext (sextet), dd (doublet of doublets), dt (doublet of triplets), td (triplet of doublets), m (multiplet), bs (broad signal). High Resolution Mass Spectroscopy (HRMS) experiments were done on an Agilent Technologies-6530 Accurate-Mass Q-TOF-LC/MS. The thermogravimetric analysis (TGA) was performed with TA – Q500 TGA. For rheological analysis, hexagonal-squared shaped PDMS-EPO pieces were cut to help the measurements by rheometer. The mechanical characterization was firstly performed by using Anton Paar MCR-301 Rheometer. PP08-SN18311 measuring device with 8 mm diameter was used. Further data were acquired with dynamic mechanical analyzer (DMA, Q800 TA Instruments) equipped with oscillation mode.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mL) and cooled in ice bath. A pinch of methylene blue was added to the solution, and the mixture was irradiated with halogen lamp (500 W) while O2 (g) was passing through the system for 2 h. The progress of reaction was monitored with TLC (eluent:EtOAc). After removal of the solvent under reduced pressure, the residue was purified with column chromatography over silica gel with EtOAc as eluent. Compound 4 was obtained as white solid (0.13 g, 60%). 1H NMR (400 MHz, DMSO-d6): δ 7.65 (d, J = 8.1 Hz, 4H), 7.59 (d, J = 8.1 Hz, 4H), 7.33–7.27 (m, 4H), 7.12–7.05 (m, 4H), 5.38 (t, J = 5.7 Hz, 2H), 4.68 (d, J = 5.7 Hz, 4H). 13C NMR (100 MHz, DMSO-d6): δ 143.3, 140.4, 131.0, 128.3, 127.3, 127.0, 123.5, 83.8, 63.1.:60 mL) and the slurry mixture was stirred at room temperature for 24 h under Ar. The DCU was removed by vacuum filtration and the filtrate was collected. The solvent was removed under reduced pressure and the residue was purified with column chromatography over silica gel with DCM as eluent. Compound 5 was obtained as yellow solid (0.14 g, 38%). 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 8.4 Hz, 4H), 7.67 (d, J = 8.4 Hz, 4H), 7.26–7.22 (m, 4H), 7.22–7.17 (m, 4H), 6.57 (dd, J12 = 17.3 Hz J23 = 1.4 Hz, 2H), 6.29 (dd, J12 = 17.3 Hz J34 = 10.4 Hz, 2H), 5.95 (dd, J23 = 1.4 Hz J34 = 10.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 166.1, 140.1, 136.1, 133.0, 131.4, 128.3, 128.0, 127.8, 127.7, 123.5, 84.1, 65.9.:1 ratio in a plastic vessel with a wood stirrer until whitish colored foam is seen (at least 2–3 min is a necessity). PDMS-EPO (75 mg, 0.14 mmol) which was previously dissolved in DCM (0.5 mL) was mixed with whitish foam Sylgard® 184 until complete dissolution is established. Part of the resulting mixture (5.0 g) was placed and spread evenly in a Petri dish. Then, it put in vacuum desiccator and vacuum was applied for 1 h. Then, Petri dishes left to stand for 2 days at RT. Once cured, the films were peeled away and cut into strips for testing.
:5 mL) and cooled in ice bath. A pinch of methylene blue was added to the solution, and the mixture was irradiated with halogen lamp (500 W) while O2 (g) was passing through the system for 2 h. The progress of reaction was monitored with TLC (eluent:EtOAc). After removal of the solvent under reduced pressure, the residue was purified with column chromatography over silica gel with EtOAc as eluent. Compound 4 was obtained as white solid (0.13 g, 60%). 1H NMR (400 MHz, DMSO-d6): δ 7.65 (d, J = 8.1 Hz, 4H), 7.59 (d, J = 8.1 Hz, 4H), 7.33–7.27 (m, 4H), 7.12–7.05 (m, 4H), 5.38 (t, J = 5.7 Hz, 2H), 4.68 (d, J = 5.7 Hz, 4H). 13C NMR (100 MHz, DMSO-d6): δ 143.3, 140.4, 131.0, 128.3, 127.3, 127.0, 123.5, 83.8, 63.1.:60 mL) and the slurry mixture was stirred at room temperature for 24 h under Ar. The DCU was removed by vacuum filtration and the filtrate was collected. The solvent was removed under reduced pressure and the residue was purified with column chromatography over silica gel with DCM as eluent. Compound 5 was obtained as yellow solid (0.14 g, 38%). 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 8.4 Hz, 4H), 7.67 (d, J = 8.4 Hz, 4H), 7.26–7.22 (m, 4H), 7.22–7.17 (m, 4H), 6.57 (dd, J12 = 17.3 Hz J23 = 1.4 Hz, 2H), 6.29 (dd, J12 = 17.3 Hz J34 = 10.4 Hz, 2H), 5.95 (dd, J23 = 1.4 Hz J34 = 10.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 166.1, 140.1, 136.1, 133.0, 131.4, 128.3, 128.0, 127.8, 127.7, 123.5, 84.1, 65.9.:1 ratio in a plastic vessel with a wood stirrer until whitish colored foam is seen (at least 2–3 min is a necessity). PDMS-EPO (75 mg, 0.14 mmol) which was previously dissolved in DCM (0.5 mL) was mixed with whitish foam Sylgard® 184 until complete dissolution is established. Part of the resulting mixture (5.0 g) was placed and spread evenly in a Petri dish. Then, it put in vacuum desiccator and vacuum was applied for 1 h. Then, Petri dishes left to stand for 2 days at RT. Once cured, the films were peeled away and cut into strips for testing.Results and discussion
The mechanophoric monomer that we targeted for synthesis (Fig. 1) was an acrylate ester of 9,10-diphenylanthracene endoperoxide (EPO). We aimed for two different polymers with different mechanical properties; a cross-linked polyacrylate gel (PMA-EPO) and an elastomeric poly(dimethylsiloxane) (PDMS-EPO). Of course, to minimize the thermal decomposition of endoperoxides, we avoided any polymerization procedures which necessitate prolonged heating above 60 °C. In both polymers, the chain growth/cross-linking originates from substituents placed at 9,10 positions of the anthracene core, which is also the site of labile endoperoxide linkage. The parent anthracene endoperoxide is stable at room temperature, any discernible cycloreversion requires heating at temperatures above 100 °C, which makes this module, an optimal storage compound for singlet oxygen at ambient temperatures. The monomer was synthesized in a few steps from commercially available compounds in accord with previous literature.7,13 For the preparation of PMA-EPO, cross-linking copolymerization reaction (Fig. 2) was carried out at 0 °C, with methyl acrylate, in the presence of radical initiator AIBN and under UV irradiation (360 nm). The polymer was then triturated in THF and dried in vacuo to yield a translucent gel-like solid.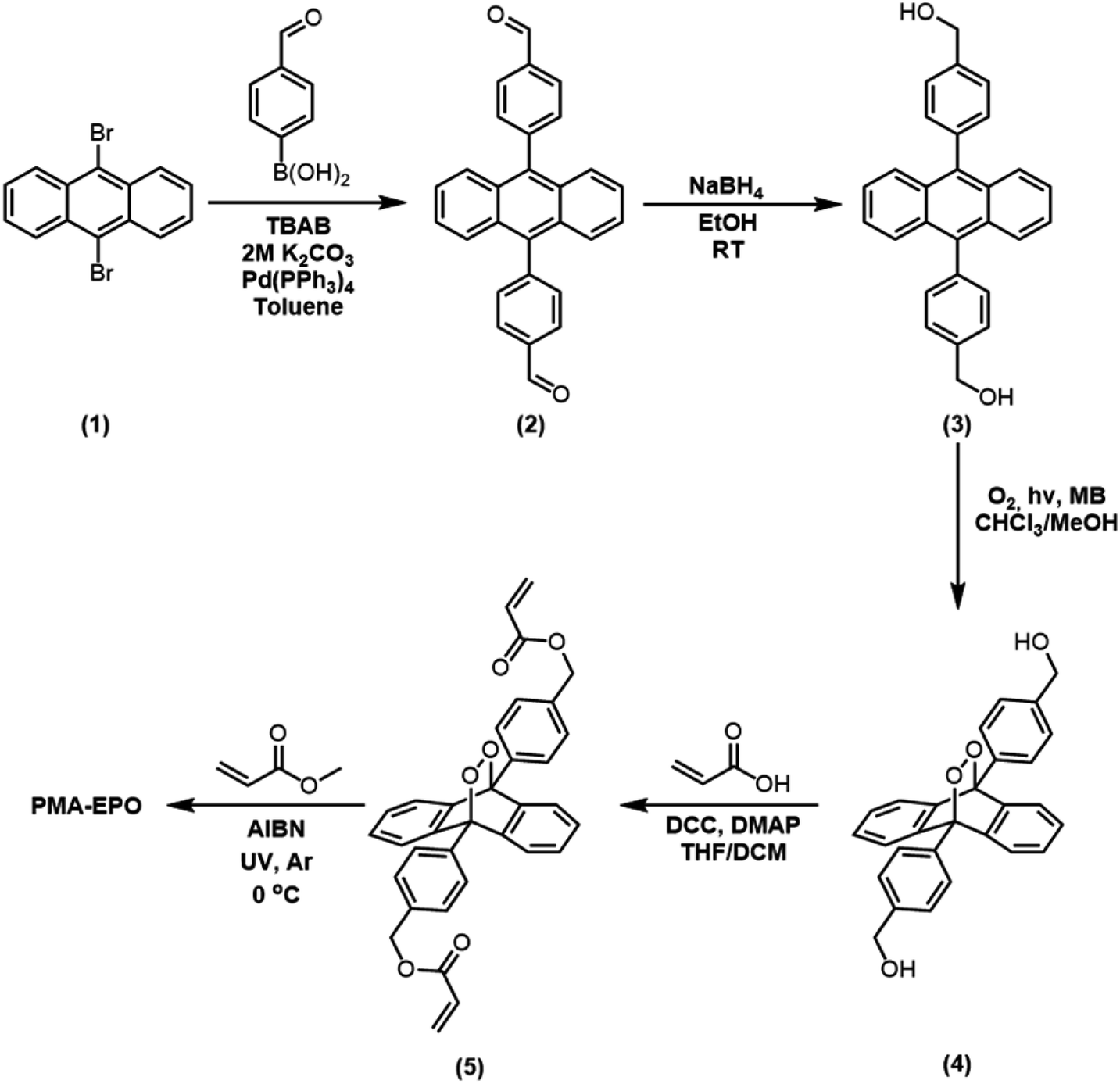 | ||
| Fig. 1 Synthesis scheme for the monomer 5. | ||
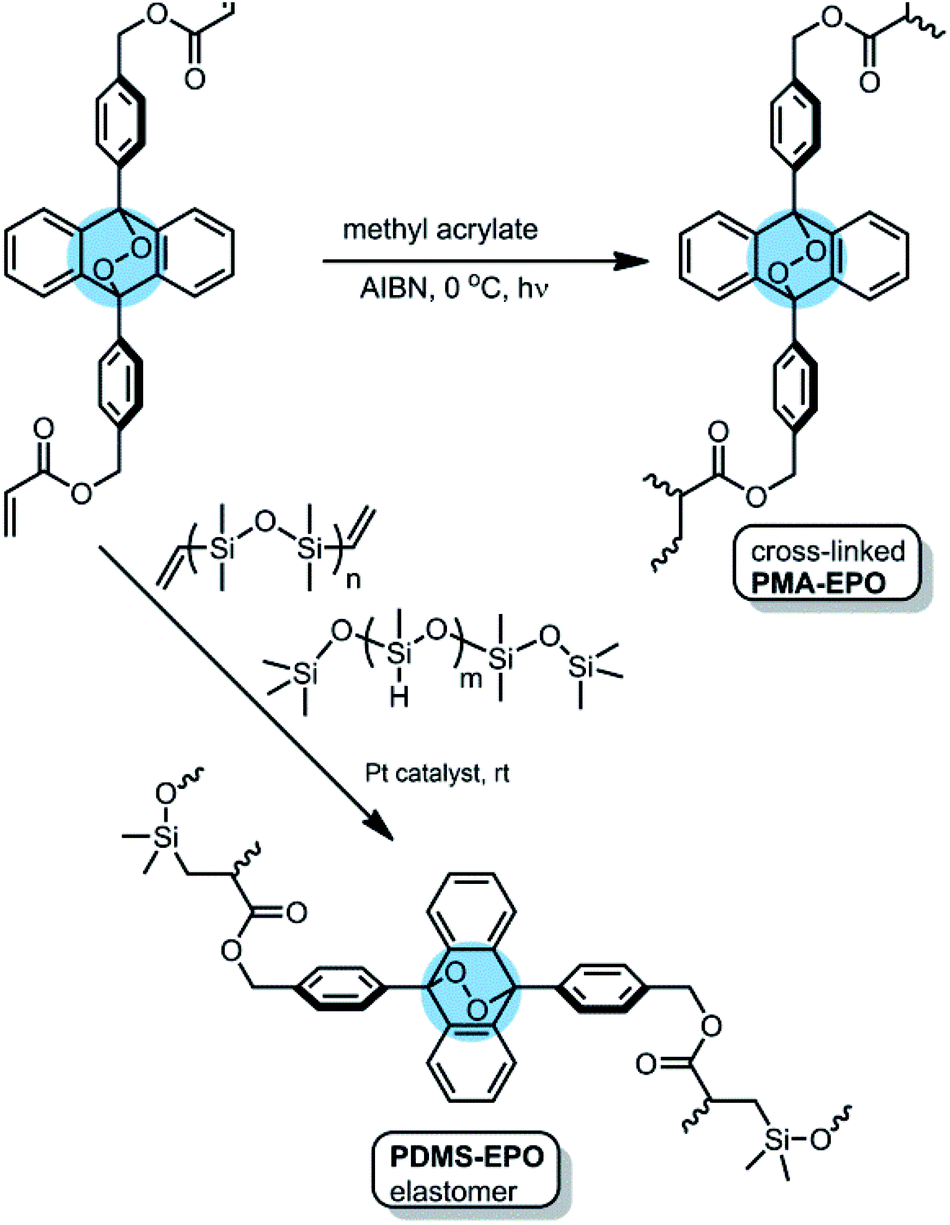 | ||
| Fig. 2 The structure and the preparation of PMA-EPO (anthracene-endoperoxide polymethacrylate copolymer) and PDMS-EPO elastomer. | ||
To eliminate the possibility of thermal decomposition during mechanical treatment, a cryogenic ball mill was used. Cryogenic temperatures minimize thermal processes, and at the same time, with impact and shear forces typically produced in internally agitated ball mills, a mechanochemical reaction can be triggered.
Both heating the PMA-EPO gel at 150 °C for 15 seconds, and cryomilling resulted in a very bright emission under irradiation at 360 nm. This is due to the cycloreversion process which leads to the formation of the fluorescent anthracene core (Fig. 3 and inset pictures). However, it is equally important to show that singlet oxygen is produced concomitantly. To that end, we repeated cryomilling in the presence of selective singlet oxygen probe “Singlet Oxygen Sensor Green” (SOSG). Even a short 10 or 20 minute cryomilling generates significant (100% and 150%, respectively) singlet oxygen compared to the background (Fig. 3).
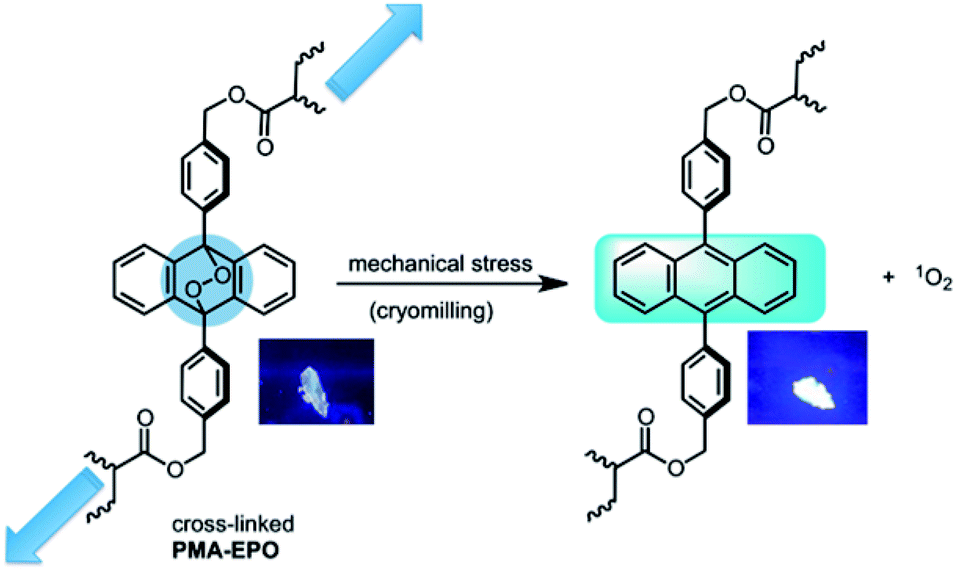 | ||
| Fig. 3 Application of mechanical stress leads to the emergence of fluorescent anthracene cores presumably via cycloreversion with a concomitant singlet oxygen release. Distortion of the endoperoxide bridgehead positions is expected initiate cycloreversion. Inset pictures: appearance of the polymer under irradiation with a hand held-UV lamp at 360 nm, left before (left), and after (right) cryomilling. | ||
We also wanted to demonstrate the applicability of the new mechanophoric unit in an elastomer, and to that end we targeted a poly(dimethylsiloxane) (PDMS-EPO) with anthracene endoperoxide crosslinkers for synthesis (Fig. 1). The preparation was carried out in analogy to previous literature12 by mixing vinyl and TMS-terminated siloxanes under platinum catalysis followed by curing under vacuum at 40 °C for 4 hours (ESI).† The elastomer obtained this way has a very weak fluorescence, but on heating fluorescence emission intensity increases due to the cycloreversion reaction which produces 9,10-diphenylanthracene cores (Fig. 4).
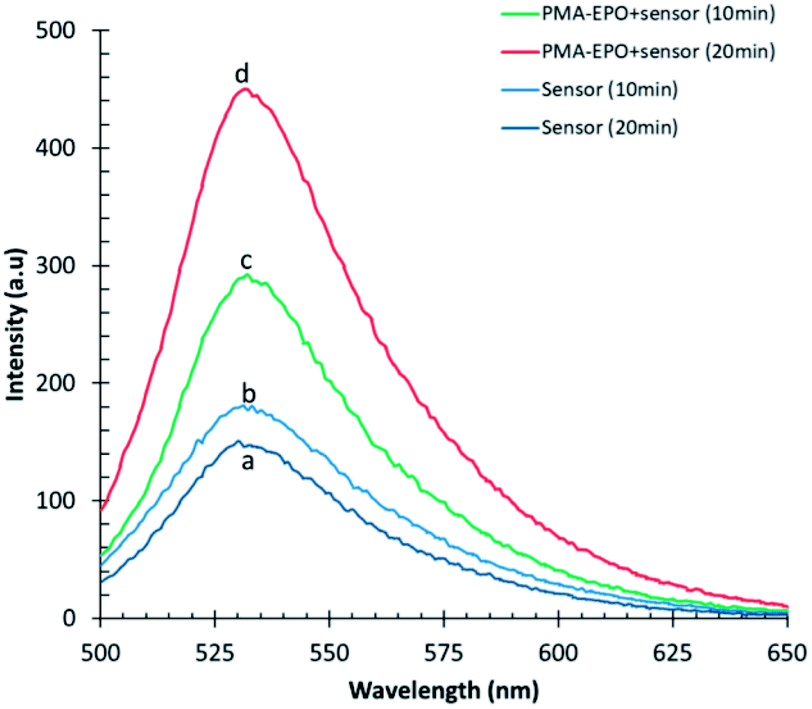 | ||
| Fig. 4 Evolution of fluorescence emission spectra of the singlet oxygen probe SOSG in a cryomill: (a) singlet oxygen sensor milled alone for 10 min, and (b) 20 min; PMA-EPO (240 mg) together with singlet oxygen sensor (c) after 10 min, (d) and 20 min of milling. Singlet oxygen sensor concentration was 5 μM, in 3 mL DMSO. Excitation wavelength is 480 nm. | ||
The mechanical response of the PDMS-EPO polymer was studied by both dynamic mechanical analysis (DMA) and also a rotational rheometer. In DMA experiment, rectangular shaped strip samples were prepared with dimensions of around 6.1–7.4 mm (length) × 3.5–4.0 mm (width) × 1.5–1.8 mm (thickness). Frequency sweep between 1 Hz and 100 Hz of the samples were performed at 25 °C and 45 °C, while the amplitude was adjusted to 100 μm. The sample polymer strip to which strain was applied became clearly more fluorescent under irradiation at 360 nm (Fig. 5a at 25 °C and Fig. 5b at 45 °C). Mechanical force applied on the PDMS-EPO strips in a rheometer provided similar results. Amplitude sweep of the samples was performed at 25 °C by adjusting the angular frequency and strain magnitudes as ω = 10 rad s−1 and 0.01 to 100%, respectively. Pictures of PDMS-EPO spieces before and after the application of mechanical force show a significant enhancement of fluorescence emission of the sample after the mechanical induction (Fig. 5c).
 | ||
| Fig. 5 Application of mechanical stress on PDMS-EPO strips leads to the enhancement of fluorescence as mechanically triggered cycloreversion generates singlet oxygen and diphenylanthracene cores. (a) Strips before (left) and after (right) being subjected to oscillating force in DMA at 25 °C, (b) strips before (left) and after (right) being subjected to oscillating force in DMA at 45 °C, (c) mechanical force introduced in rheometer also triggers an enhancement of the fluorescence emission at 25 °C (before, left; after, right). Pictures were taken while the strips were illuminated at 360 nm with a hand-held UV lamp. | ||
Conclusion
We successfully obtained crosslinked polymers with novel mechanophores which can release singlet oxygen under mechanical stress. Considering the utility of PDT approach in various skin conditions including melanomas, psoriasis or acne,14 we are confident that polymeric singlet oxygen generators at ambient temperature on mild application of pressure will prove to be highly valuable. Our work towards that goal is in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from Dalian University of Technology, Grant No. DUT18RC(3)062 (E. U. A.) A. T. acknowledges graduate scholarship support from TUBITAK (2210-E).References
- P. Ogilby, Photochem. Photobiol. Sci., 2010, 9, 1543–1560 RSC; G. Herzberg, Molecular Spectra and Molecular Structure. I. Spectra of Diatomic Molecules, Van Nostrand Reinhold, New York, 2nd edn, 1950 Search PubMed.
- T. J. Dougherty, J. E. Kaufman, A. Goldfarb, K. R. Weishaupt, D. Boyle and A. Mittleman, Cancer Res., 1978, 38, 2628–2635 Search PubMed; J. Moan and Q. Peng, Anticancer Res., 2003, 23, 591–600 Search PubMed; D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- E. Ucar, D. Xi, O. Seven, C. Kaya, X. J. Peng, W. Sun and E. U. Akkaya, Chem. Commun., 2019, 55, 13808–13811 RSC; I. S. Turan, D. Yildiz, A. Turksoy, G. Gunaydin and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 2875–2878 CrossRef CAS PubMed.
- Z. Huang, Technol. Cancer Res. Treat., 2005, 4, 283–293 CrossRef CAS PubMed; M. G. Bredell, E. Besic, C. Maake and H. Walt, J. Photochem. Photobiol., B, 2010, 101, 185–190 CrossRef PubMed; N. C. Zeitouni, A. R. Oseroff and S. Shieh, Mol. Immunol., 2003, 39, 1133–1136 CrossRef PubMed; S. Stolik, J. A. Delgado, A. Perez and L. Anasagasti, J. Photochem. Photobiol., B, 2000, 57, 90–93 CrossRef; C. A. Morton, K. E. McKenna and L. E. Rhodes, Br. J. Dermatol., 2008, 159, 1245–1266 CrossRef PubMed.
- Y. Liu, Y. Liu, W. Bu, C. Cheng, C. Zuo, Q. Xiao, Y. Sun, D. Ni, C. Zhang and J. Liu, Angew. Chem., Int. Ed., 2015, 54, 8105–8109 CrossRef CAS PubMed; J. Xu, S. Sun, Q. Li, Y. Yue, Y. Li and S. Shao, Analyst, 2015, 140, 574–581 RSC; S. Wang, H. Liu, J. Mack, J. Tian, B. Zou, H. Lu, Z. Li, J. Jiang and Z. Shen, Chem. Commun., 2015, 13389–13392 RSC; K. Kiyose, K. Hanaoka, D. Oushiki, T. Nakamura, M. Kajimura, M. Suematsu, H. Nishimatsu, T. Yamane, T. Terai and Y. Hirata, J. Am. Chem. Soc., 2010, 132, 15846–15848 CrossRef PubMed; G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef PubMed; J. Pouyssegur, F. Dayan and N. M. Mazure, Nature, 2006, 441, 437–443 CrossRef PubMed; X. Zheng, X. Wang, H. Mao, W. Wu, B. Liu and X. Jiang, Nat. Commun., 2015, 6, 5834 CrossRef PubMed; W. Gallagher, L. Allen, C. O'Shea, T. Kenna, M. Hall, A. Gorman, J. Killoran and D. O'Shea, Br. J. Cancer, 2005, 92, 1702–1710 CrossRef PubMed; M. I. Koukourakis, A. Giatromanolaki, J. Skarlatos, L. Corti, S. Blandamura, M. Piazza, K. C. Gatter and A. L. Harris, Cancer Res., 2001, 61, 1830–1832 Search PubMed.
- R. Sullivan and C. H. Graham, Cancer Metastasis Rev., 2007, 26, 319–331 CrossRef CAS PubMed.
- S. Kolemen, T. Ozdemir, D. Lee, G. M. Kim, T. Karatas, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 3606–3610 CrossRef CAS PubMed.
- J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed; H. H. Wasserman, K. B. Wiberg, D. L. Larsen and J. Parr, J. Org. Chem., 2005, 70, 105–109 CrossRef PubMed; S. Benz, S. Notzli, J. S. Siegel, D. Eberli and H. J. Jessen, J. Med. Chem., 2013, 56, 10171–10182 CrossRef PubMed; J. Poully, J. Schermann, N. Nieuwjaer, F. Lecomte, G. Gregoire, C. Desfrançois, G. Garcia, L. Nahon, D. Nandi and L. Poisson, Phys. Chem. Chem. Phys., 2010, 12, 3566–3572 RSC; M. Matsumoto, M. Yamada and N. Watanabe, Chem. Commun., 2005, 483–485 RSC; I. S. Turan, F. P. Cakmak, D. C. Yildirim, R. Cetin-Atalay and E. U. Akkaya, Chem.–Eur. J., 2014, 49, 16088–16092 CrossRef PubMed.
- T. Torring, S. Helmig, P. R. Ogilby and K. V. Gothelf, Acc. Chem. Res., 2014, 47, 1799–1806 CrossRef CAS PubMed; E. J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef PubMed; K. Otsu, K. Sato, Y. Ikeda, H. Imai, Y. Nakagawa, Y. Ohba and J. Fujii, Biochem. J., 2005, 389, 197–206 CrossRef PubMed; K. Otsu, K. Sato, M. Sato, H. Ono, Y. Ohba and Y. Katagata, Cell Biol. Int., 2008, 32, 1380–1387 CrossRef PubMed; D. Posavec, M. Zabel, U. Bogner, G. Bernhardt and G. Knor, Org. Biomol. Chem., 2012, 10, 7062–7069 RSC.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed; J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef.
- Y. Teki, S. Miyamoto, M. Nakatsuji and Y. Miura, J. Am. Chem. Soc., 2001, 123, 294–305 CrossRef CAS; H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556–559 CrossRef PubMed; N. Mase, T. Ando, F. Shibagaki, A. Sugita, T. Narumi, M. Toda, N. Watanabe and F. Tanaka, Tetrahedron Lett., 2014, 55, 1946–1948 CrossRef; K. J. Rodriguez, A. M. Hanlon, C. K. Lyon, J. P. Cole, B. T. Tuten, C. A. Tooley, E. Berda and S. Pazicni, Inorg. Chem., 2016, 55, 9493–9496 CrossRef PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559–562 CrossRef CAS PubMed.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497–508 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00831a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |