
Copper-catalyzed asymmetric silyl addition to alkenyl-substituted N-heteroarenes†
Ya-Li
Zeng
a,
Bo
Chen
a,
Ya-Ting
Wang
a,
Cheng-Yu
He
a,
Zi-Yuan
Mu
a,
Ji-Yuan
Du
 b,
Long
He
c,
Wen-Dao
Chu
*a and
Quan-Zhong
Liu
*a
b,
Long
He
c,
Wen-Dao
Chu
*a and
Quan-Zhong
Liu
*a
aChemical Synthesis and Pollution Control, Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, No. 1, Shida Road, Nanchong 637002, China. E-mail: chuwendaonpo@126.com; quanzhongliu@cwnu.edu.cn
bCollege of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng, Shandong 252059, China
cCollege of Chemistry and Materials Engineering, Guiyang University, Guiyang 550005, China
First published on 6th January 2020
Abstract
Asymmetric conjugate addition of PhMe2SiBPin to a wide range of N-heteroaryl alkenes proceeded in the presence of a copper catalyst coordinated with an easily accessible chiral phosphoramidite ligand to afford useful β-silyl N-heteroarenes in high yields (up to 96%) and excellent enantioselectivities (up to 97% ee).
N-Containing heteroarenes are very important structural units, which are commonly found in a wide variety of natural products and pharmaceutically active compounds. Although elaboration of the N-containing heteroarenes continues to be the main approach to access functionalized heterocyclic aromatic compounds, exploration of asymmetric conjugate addition of nucleophiles to alkenes activated by adjacent N-containing heteroarenes has gained prominence in the last decade (Scheme 1a).1 Key contributions in the area include transition metal-catalyzed asymmetric arylation and hydrogenation of alkenyl azaarenes, reported by Lam and co-workers.2 In addition, other groups presented elegant, highly enantioselective conjugate additions to N-heteroaryl alkenes using pyrazoles,3c Grignard reagents,3d,e B2(pin)23f and thiols3h as nucleophiles, respectively. However, contrary to common Michael acceptors, such as α,β-unsaturated carbonyl, nitro, nitrile and sulfonyl compounds, which have been widely studied in asymmetric conjugate addition,4 alkenyl azaarenes have received little attention.3 The scarcity of available asymmetric approaches is attributed to the low activation from the N-heteroaryl group and the generation of unfavorable intermediates through a transient dearomatization step.
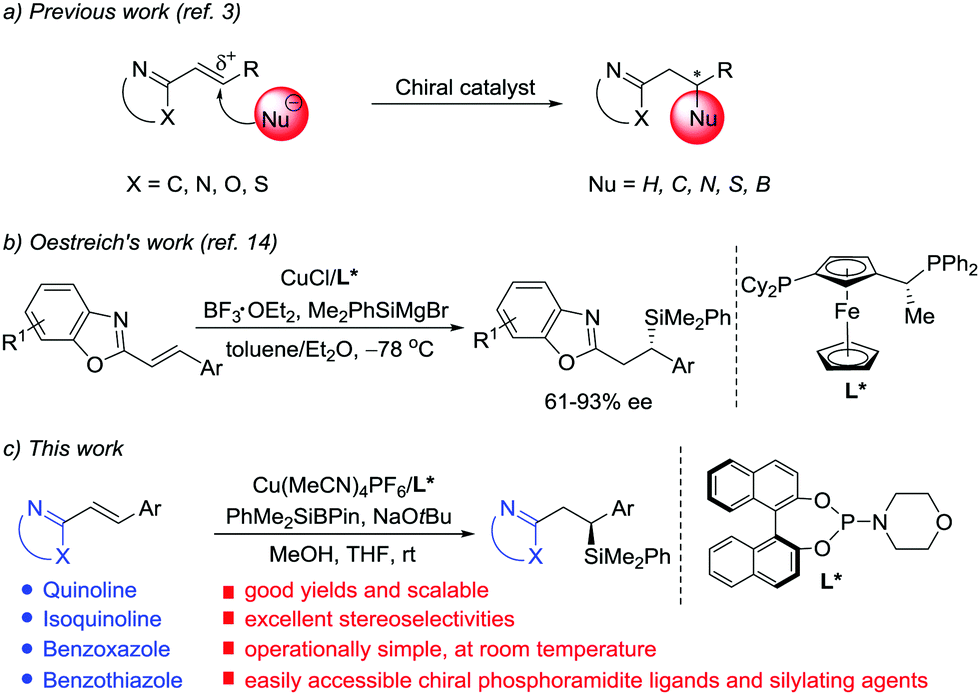 | ||
| Scheme 1 Catalytic asymmetric nucleophilic addition to alkenyl azaarenes. | ||
Chiral silanes are versatile intermediates in organic synthesis.5 The silicon motif is widely used as a masked hydroxyl group,6 and the C–Si bond can also be readily transformed into C–C bonds.7 Accordingly, the vital objective in this area of research is to develop efficient and practical routes to chiral silanes. Among the available approaches to provide chiral organosilicon compounds, asymmetric silyl conjugate addition to electron-deficient alkenes is one of the most reliable ways.8–13 In 1988, Hayashi and Ito's group pioneered the development of enantioselective disilylation of enones using palladium catalysis.8 Subsequently, the reaction scope and catalytic systems were expanded by Oestreich,9 Hoveyda,10 Córdova,11 Procter12 and Kobayashi.13 Although significant progress has been achieved with regard to asymmetric silylation of a wide range of cyclic and acyclic α,β-unsaturated carbonyl systems, a few studies on silyl transfer to alkenes conjugated to weaker electron-withdrawing groups, and in particular alkenyl-substituted N-heterocycles, have been described. The topic still represents a challenge. Very recently, Oestreich reported the first Cu(I)-catalyzed asymmetric conjugate addition of silicon Grignard reagents to poorly reactive alkenyl heteroarenes with Lewis acid activation (Scheme 1b).14 Because of the high activity of the silicon Grignard reagents, the reactions must be carried out at very low temperature.14,15 Although this work marks a significant breakthrough, the enantioselective reaction is limited to benzoxazole-derived substrates and the ee values are moderate in most cases. Thus, further development of catalytic highly enantioselective silyl addition to alkenes activated by other azaaryl groups with an easily obtained catalyst is urgently demanded. Herein, we describe an operationally simple, highly chemo- and enantioselective silylation protocol of a wide range of N-heteroaryl-substituted alkenes using PhMe2SiBpin16,17 as the nucleophile and chiral phosphoramidite/Cu(I) as the catalyst (Scheme 1c). PhMe2SiBpin is readily available, bench-stable, and easy-to-handle. Since Suginome and Ito first introduced it in the Pd-catalyzed silaboration of alkynes,16 the development of PhMe2SiBpin chemistry has been the subject of intense research in recent years.17
Initially, we selected 2-alkenylquinoline 1a and PhMe2SiBpin as model substrates to study the silylation reaction (Table 1).18 Racemic product 2a was produced in low yields in the presence of Cu(MeCN)4PF6, the bidentate chiral phosphine ligand (L1 or L2) and NaOtBu in THF at room temperature (entries 1 and 2). To our delight, the chiral phosphoramidite ligand L3 was found to be effective in asymmetric induction, presenting 57% enantioselectivity and 19% yield (entry 3). Encouraged by this result, the substituents on the nitrogen atom were examined. The morpholine moiety (L9) was superior to others, such as other acyclic amines (L4–L6), pyrrolidine (L7) and indoline (L8), affording 2a in 86% yield with 93% ee (entries 4–9). In addition, we evaluated the effect of the solvent on the reaction. It was found that the reactions in Et2O, hexane, toluene and DMF all provided the product 2a with similarly high ee, but the yields varied greatly and the reaction in THF gave the highest yield (entries 9–13).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | eec (%) |
a Reaction conditions: 1a (0.1 mmol), PhMe2SiBpin (0.2 mmol), NaOtBu (0.2 equiv.), Cu(MeCN)4PF6 (10 mol%), ligand (22 mol%), MeOH (4.0 equiv.), and the solvent (0.05 M).
b Isolated yields.
c Determined by chiral HPLC.
d 0.11 equiv. of ligand was employed.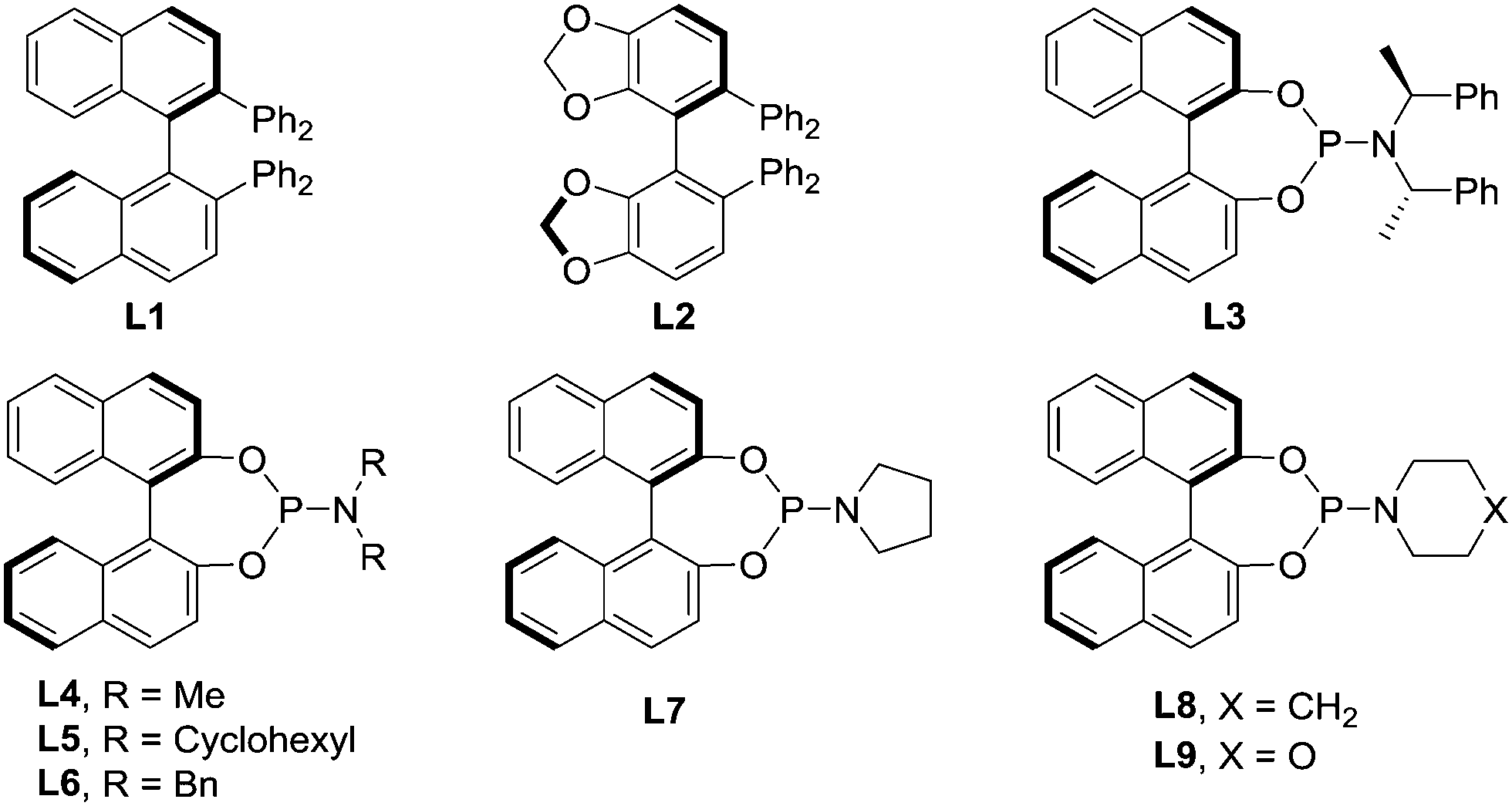
|
||||
| 1d | L1 | THF | 44 | 0 |
| 2d | L2 | THF | 39 | 5 |
| 3 | L3 | THF | 19 | 57 |
| 4 | L4 | THF | 48 | 83 |
| 5 | L5 | THF | 32 | 85 |
| 6 | L6 | THF | 22 | 85 |
| 7 | L7 | THF | 64 | 89 |
| 8 | L8 | THF | 49 | 91 |
| 9 | L9 | THF | 86 | 93 |
| 10 | L9 | Et2O | 69 | 93 |
| 11 | L9 | Hexane | 66 | 90 |
| 12 | L9 | Toluene | 58 | 93 |
| 13 | L9 | DMF | 48 | 89 |
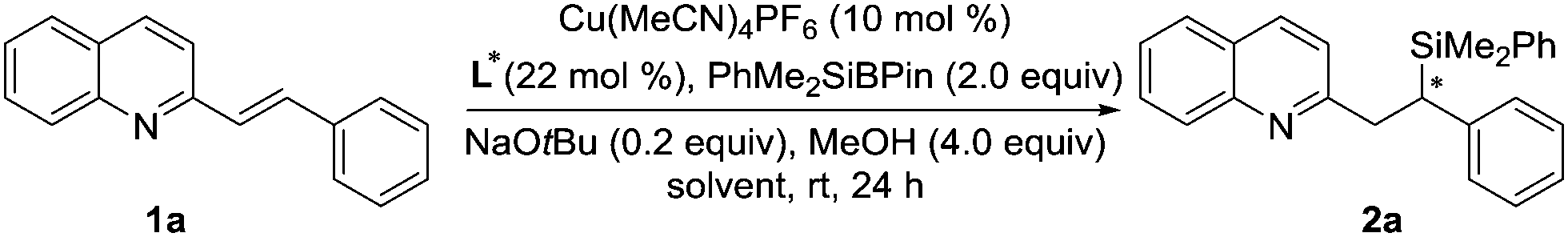
With the optimal conditions in hand, the substrate scope was then examined (Table 2). A wide range of alkenyl quinolines bearing electron-neutral, -deficient or -rich β-aromatic substituents reacted efficiently with PhMe2SiBpin to afford the corresponding products 2a–q in 80–95% yields and 88–97% ee. The absolute configuration of 2b was unambiguously established by X-ray crystallographic analysis.19 Substrates with 2- and 3-thienyl groups were also amenable to this protocol to produce the corresponding products 2r–s with high yields and enantioselectivities. Interestingly, when the silylation of the conjugate divinyl quinoline 1t was investigated, there was only 1,4-addition and the corresponding adduct 2t was isolated in 76% yield with 85% ee. The substituents (such as Br, Me, OMe or Cl) on the quinoline ring of 1 did not compromise the enantioselectivities of 2u–x. Other heteroaromatic substrates, such as isoquinoline (1y), benzoxazole (1z) and benzothiazole (1za), were also suitable for the reaction and the corresponding silylated products 2y–2za were obtained with excellent results (81–89% yields, 85–95% ee). Finally, the reactions between PhMe2SiBpin and alkyl-substituted 1zb, α,β-disubstituted 1zc or β,β-disubstituted 1zd were also attempted, but they were unsuccessful.
| a Reaction conditions: 1 (0.1 mmol), PhMe2SiBpin (0.2 mmol), NaOtBu (0.2 equiv.), Cu(MeCN)4PF6 (10 mol%), L9 (22 mol%), MeOH (4.0 equiv.), and THF (0.05 M). Yields of the isolated product 2. ee determined by HPLC analysis. N.D. = not detected. |
|---|

|
The reaction on a gram scale (2.5 mmol of 1i) was also performed to obtain the 1,4-adduct 2i with 94% ee and 94% yield (Scheme 2a). A key synthetic transformation of the silicon motif is that it can be used as a masked hydroxyl group.6 Compound 2i could be transformed into alcohol 3 through Tamao–Fleming oxidation20 without the erosion of stereoselectivity (Scheme 2b). Furthermore, the treatment of 2i in the presence of TBAT and CO2 could provide the desired ester 4 in 50% yield.17k Unfortunately, no transformation in chirality was observed in this reaction (Scheme 2c).
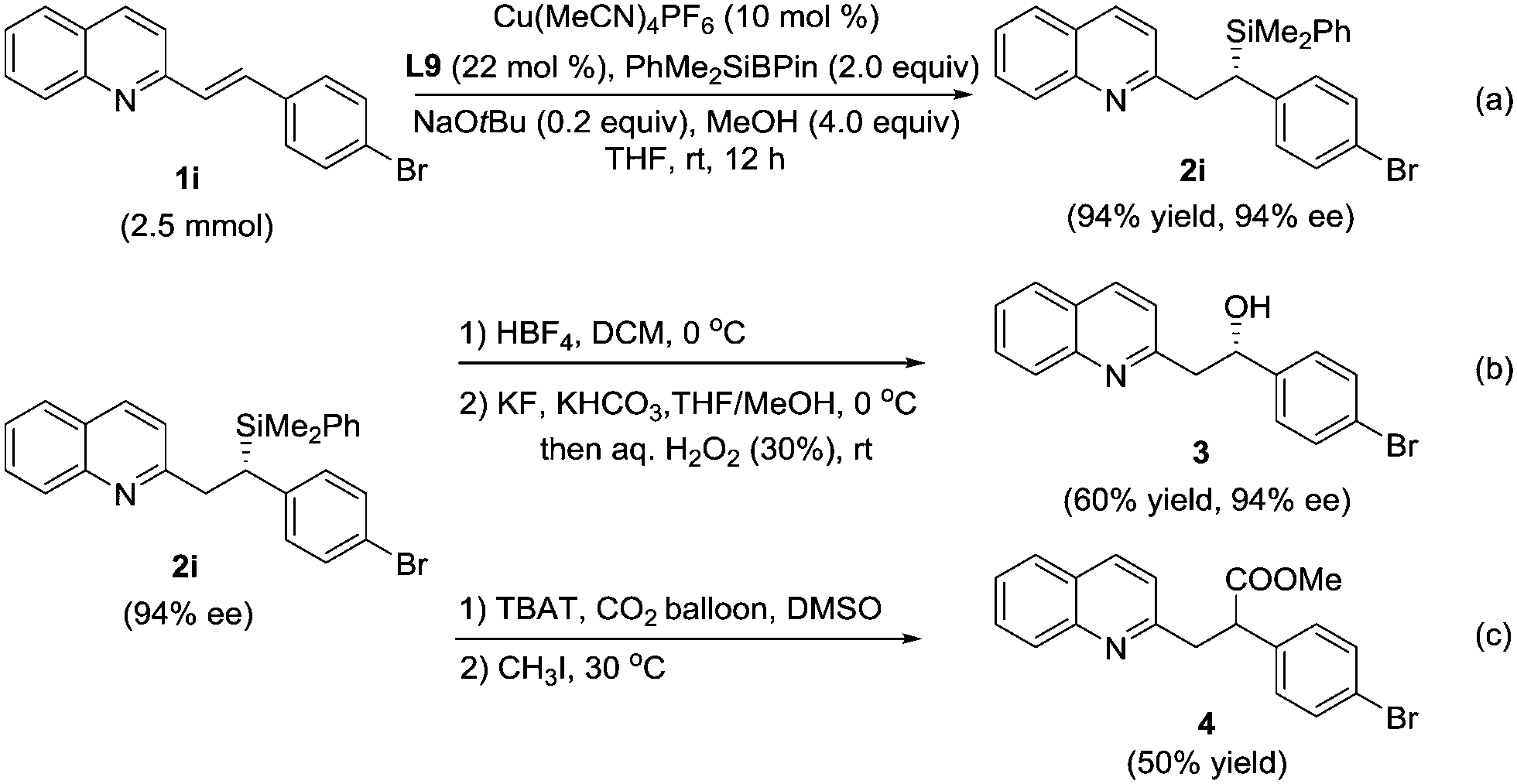 | ||
| Scheme 2 Scale-up experiment and synthetic applications of compound 2i. | ||
A possible mechanism for the reactions is illustrated in Scheme 3. LnCu-alkoxides react with PhMe2SiBpin to produce the derived LnCu–SiMe2Ph I, which can undergo an asymmetric conjugate addition reaction with alkenyl-substituted quinoline 1a to obtain intermediate III through transition state II. The resulting aza-π-allylcopper intermediate III reacts with methanol to afford the desired product 2a and regenerate the active copper catalysts for the next catalytic cycle.
 | ||
| Scheme 3 Proposed catalytic cycle. | ||
In conclusion, we have developed an operationally simple and highly enantioselective Cu(I)-catalyzed conjugate addition of PhMe2SiBPin to a wide range of alkenyl N-heteroarenes with easily accessible chiral phosphoramidite as the ligand. A series of useful β-silyl N-heteroarenes were achieved in high yields and ees. Moreover, the reaction can be effectively performed at room temperature on a gram scale.
We gratefully acknowledge the Department of Science and Technology of Sichuan Province (2019YJ0339) and the NSFC (21572183, 21772158, and 21801208) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Reviews: (a) D. Best and H. W. Lam, J. Org. Chem., 2014, 79, 831 CrossRef CAS PubMed; (b) D. A. Klumpp, Synlett, 2012, 1590 CrossRef CAS.
- (a) L. Rupnicki, A. Saxena and H. W. Lam, J. Am. Chem. Soc., 2009, 131, 10386 CrossRef CAS; (b) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373 CrossRef CAS; (c) A. Saxena and H. W. Lam, Chem. Sci., 2011, 2, 2326 RSC; (d) A. Saxena, B. Choi and H. W. Lam, J. Am. Chem. Soc., 2012, 134, 8428 CrossRef CAS PubMed; (e) I. D. Roy, A. R. Burns, G. Pattison, B. Michel, A. J. Parker and H. W. Lam, Chem. Commun., 2014, 50, 2865 RSC; (f) B. Choi, A. Saxena, J. J. Smith, G. H. Churchill and H. W. Lam, Synlett, 2015, 350 CAS; (g) J. J. Smith, D. Best and H. W. Lam, Chem. Commun., 2016, 52, 3770 RSC.
- (a) A. A. Friedman, J. Panteleev, J. Tsoung, V. Huynh and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 9755 CrossRef CAS PubMed; (b) S. Wang, X. Li, H. Liu, L. Xu, J. Zhuang, J. Li, H. Li and W. Wang, J. Am. Chem. Soc., 2015, 137, 2303 CrossRef CAS; (c) Y.-Y. Wang, K. Kanomata, T. Korenaga and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 927 CrossRef CAS; (d) R. P. Jumde, F. Lanza, M. J. Veenstra and S. R. Harutyunyan, Science, 2016, 352, 433 CrossRef CAS PubMed; (e) R. P. Jumde, F. Lanza, T. Pellegrini and S. R. Harutyunyan, Nat. Commun., 2017, 8, 2058 CrossRef; (f) L. Wen, Z. Yue, H. Zhang, Q. Chong and F. Meng, Org. Lett., 2017, 19, 6610 CrossRef CAS PubMed; (g) Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083 CrossRef CAS PubMed; (h) M. Formica, G. Sorin, A. J. M. Farley, J. Díaz, R. S. Paton and D. J. Dixon, Chem. Sci., 2018, 9, 6969 RSC.
- For selected recent reviews, see: (a) G. J. Reyes-Rodríguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221 CrossRef; (b) L. Wu, J. Shen, G. Yang and W. Zhang, Tetrahedron Lett., 2018, 59, 4055 CrossRef CAS; (c) K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586 CrossRef CAS PubMed; (d) T. Jia, P. Cao and J. Liao, Chem. Sci., 2018, 9, 546 RSC; (e) B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef CAS PubMed; (f) C. Hui, F. Pu and J. Xu, Chem. – Eur. J., 2017, 23, 4023 CrossRef CAS; (g) M. Hayashi and R. Matsubara, Tetrahedron Lett., 2017, 58, 1793 CrossRef CAS; (h) T. N. Nguyen and J. A. May, Tetrahedron Lett., 2017, 58, 1535 CrossRef CAS; (i) A. Mondal, S. Bhowmick, A. Ghosh, T. Chanda and K. C. Bhowmick, Tetrahedron: Asymmetry, 2017, 28, 849 CrossRef CAS; (j) D. A. Alonso, A. Baeza, R. Chinchilla, C. Gómez, G. Guillena, I. M. Pastor and D. J. Ramón, Molecules, 2017, 22, 895 CrossRef PubMed; (k) M. M. Heravi, M. Dehghani and V. Zadsirjan, Tetrahedron: Asymmetry, 2016, 27, 513 CrossRef CAS.
- Reviews: (a) J. R. Wilkinson, C. E. Nuyen, T. S. Carpenter, S. R. Harruff and R. Van Hoveln, ACS Catal., 2019, 9, 8961 CrossRef CAS; (b) S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16 CrossRef; (c) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221 CrossRef CAS PubMed.
- (a) D. J. Ager and I. Fleming, J. Chem. Soc., Chem. Commun., 1978, 177 RSC; (b) K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694 CrossRef CAS; (c) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29 RSC; (d) K. Tamao, T. Tanaka, T. Nakajima, R. Sumiya, H. Arai and Y. Ito, Tetrahedron Lett., 1986, 27, 3377 CrossRef CAS; (e) I. Fleming and P. E. J. Sanderson, Tetrahedron Lett., 1987, 28, 4229 CrossRef CAS ; For selected reviews, see: ; (f) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed; (g) I. Fleming, in Science of Synthesis, ed. I. Fleming, Thieme, Stuttgart, 2002, vol. 4, pp. 927–946 Search PubMed.
- (a) L.-W. Xu, L. Li, G.-Q. Lai and J.-X. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC; (b) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS.
- T. Hayashi, Y. Matsumoto and Y. Ito, J. Am. Chem. Soc., 1988, 110, 5579 CrossRef CAS.
- (a) C. Walter, G. Auer and M. Oestreich, Angew. Chem., Int. Ed., 2006, 45, 5675 CrossRef CAS PubMed; (b) C. Walter and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 3818 CrossRef CAS PubMed; (c) E. Hartmann and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 6195 CrossRef CAS PubMed; (d) E. Hartmann and M. Oestreich, Org. Lett., 2012, 14, 2406 CrossRef CAS PubMed.
- (a) K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898 CrossRef CAS PubMed; (b) J. M. O’Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712 CrossRef PubMed; (c) K.-s. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823 CrossRef CAS.
- I. Ibrahem, S. Santoro, F. Himo and A. Córdova, Adv. Synth. Catal., 2011, 353, 245 CrossRef CAS.
- (a) V. Pace, J. P. Rae, H. Y. Harb and D. J. Procter, Chem. Commun., 2013, 49, 5150 RSC; (b) V. Pace, J. P. Rae and D. J. Procter, Org. Lett., 2014, 16, 476 CrossRef CAS.
- T. Kitanosono, L. Zhu, C. Liu, P. Xu and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 15422 CrossRef CAS.
- W. Mao, W. Xue, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 10723 CrossRef CAS PubMed.
- (a) W. Xue, R. Shishido and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 12141 CrossRef CAS PubMed; (b) W. Xue and M. Oestreich, Synthesis, 2019, 233 CAS; (c) H. Yi and M. Oestreich, Chem. – Eur. J., 2019, 25, 6505 CrossRef CAS PubMed.
- M. Suginome, H. Nakamura and Y. Ito, Chem. Commun., 1996, 2777 RSC.
- For reviews on the utility of silylborane reagents in chemical synthesis, see: (a) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29 CrossRef CAS; (b) E. Hartmann and M. Oestreich, Chim. Oggi, 2011, 29, 34 CAS; (c) E. Hartmann, D. J. Vyas and M. Oestreich, Chem. Commun., 2011, 47, 7917 RSC; (d) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402 CrossRef CAS PubMed , and recent asymmetric examples see: ; (e) Z.-L. Liu, C. Yang, Q.-Y. Xue, M. Zhao, C.-C. Shan, Y.-H. Xu and T. Loh, Angew. Chem., Int. Ed., 2019, 58, 16538 CrossRef CAS; (f) C.-Y. He, L.-B. Xie, R. Ding, P. Tian and G.-Q. Lin, Tetrahedron, 2019, 75, 1682 CrossRef CAS; (g) L. Zhang and M. Oestreich, Chem. – Eur. J., 2019, 25, 14304 CrossRef CAS PubMed; (h) K. Yabushita, A. Yuasa, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 113 CrossRef CAS PubMed; (i) Y. Shi, Q. Gao and S. Xu, J. Org. Chem., 2018, 83, 14758 CrossRef CAS PubMed; (j) B.-C. Da, Q.-J. Liang, Y.-C. Luo, T. Ahmad, Y.-H. Xu and T.-P. Loh, ACS Catal., 2018, 8, 6239 CrossRef CAS; (k) F.-F. Meng, J.-H. Xie, Y.-H. Xu and T.-P. Loh, ACS Catal., 2018, 8, 5306 CrossRef CAS.
- For screening of bases, additives and copper salts, see the ESI†.
- CCDC 1955733 (2b)† contains the supplementary crystallographic data for this paper.
- R. Shintani, K. Okamoto and T. Hayashi, Org. Lett., 2005, 7, 4757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, and copies of NMR and HPLC spectra, including X-ray crystal structures of 2b. CCDC 1955733. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08910a |
| This journal is © The Royal Society of Chemistry 2020 |