
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of charged groups on the cross-linking efficiency and release of guest molecules from thiol–ene cross-linked poly(2-oxazoline) hydrogels†
Julia
Blöhbaum
a,
Ilona
Paulus
a,
Ann-Christin
Pöppler
 b,
Jörg
Tessmar
a and
Jürgen
Groll
*a
b,
Jörg
Tessmar
a and
Jürgen
Groll
*a
aDepartment of Functional Materials in Medicine and Dentistry and Bavarian Polymer Institute (BPI), University of Würzburg, Pleicherwall 2, 97070 Würzburg, Germany. E-mail: juergen.groll@fmz.uni-wuerzburg.de
bInstitute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 13th February 2019
Abstract
We describe the preparation of hydrogels using highly functionalized poly(oxazoline) based polymeric precursors and cross-linking via UV mediated radical thiol–ene chemistry. Random copolymers were synthesized based on the combination of the more hydrophilic 2-methyl-2-oxazoline or the less hydrophilic monomer 2-ethyl-2-oxazoline with 2-(3-butenyl)-2-oxazoline. These copolymers were functionalized via a post-polymerization technique with thiol or cysteine functionality at the side chain. Hence, hydrogels were obtained, for which the thermo-responsive behavior, network density and correlated properties such as swelling and mechanics, as well as the possibility of electrostatic interaction, can be tuned. Cell culture tests demonstrated good cytocompatibility of the synthesized copolymers and hydrogels. A study with two low molecular weight substances, methylene blue and fluorescein sodium, was performed to investigate how the thermo-responsive behavior or the positive charge incorporated by cysteine could influence the interaction with the compounds. It was found that the interaction with the hydrogel network was strongly influenced by the chemical properties of the dye. A hydrophilic and positively charged hydrogel network was shown to be a promising candidate for the uptake and prolonged release of negatively charged low molecular weight substances.
1. Introduction
Hydrogels have become an important tool in biomedical applications as they have been used for the controlled delivery of bioactive molecules, for the encapsulation of living cells or merely as implantable materials for regenerative medicine.1 In regard to these applications, their high water content facilitates the diffusion of drugs, nutrients or cellular waste products.Furthermore, hydrogel networks that are formed by stimuli-responsive polymers are particularly attractive as they allow tuning of swelling kinetics based on external stimuli and the change in release kinetics or dimensions accordingly. One interesting class of polymers that has been investigated as a basis for such hydrogels is poly(2-alkyl-2-oxazoline)s, which belong to the class of thermo-responsive polymers exhibiting a lower critical solution temperature (LCST). Poly(oxazoline)s (POx) are synthesized via “living” cationic ring opening polymerization (CROP) and their low dispersity helps in gaining better control over hydrogel mesh size compared to many synthetic and biological polymers, which are usually highly disperse. A variety of different monomers can be copolymerized, resulting in highly functional POx. In addition, specific functionalities can also be added to the start and end-termini of the polymer via initiators or terminating reagents.2 Since the early 2000s, researchers have worked towards improving the synthesis of POx via microwave heating, decreasing the reaction time from days to several minutes3 and several studies have been performed regarding their comparability to PEG with respect to cytotoxicity, hemocompatibility and immunogenicity.4–7 Overall, POx were found to be quite similar; however, this strongly depends on the chosen chemical composition and on the molecular weight.
The side chain chemistry of POx strongly affects their solubility. Polymers based on the most hydrophilic monomer 2-methyl-2-oxazoline (MeOx) are soluble in water at all temperatures, while only polymers consisting of 2-ethyl-2-oxazoline (EtOx) with a molecular weight below 10 kDa are water soluble regardless of temperature.8 P(EtOx) with much higher molecular weights exhibit cloud points between 61 and 64 °C in aqueous solution,9 and several gradient, block and random copolymers of POx showing a broad range of cloud points between 5 °C and 75 °C have been synthesized.10 This thermo-responsive behavior was exploited, for example, to create a thermo-gelling physical hydrogel for bioprinting based on a block copolymer of 2-methyl-2-oxazoline and 2-n-propyl-2-oxazine by Lorson et al.11
A variety of chemically cross-linked POx hydrogels, based on multivalent monomers, macro-crosslinkers or side chain functionalized polymer precursors,12 has already been presented in the literature. The drawback of using multivalent monomers or macro-crosslinkers for hydrogel preparation is that they cannot be synthesized in situ as those hydrogels are usually formed in organic solvents such as methanol,8 ethanol13 and dichloromethane,14 which must be evaporated afterwards to ensure biocompatibility. Using side chain functionalized polymers in combination with a chemoselective reaction, like the UV initiated thiol–ene reaction, can therefore be advantageous for several reasons. It has been shown that the photoinitiator used for radical formation can be added at such low concentrations that no cytotoxic effects occur.15 Additionally, thiol–ene reactions mediated by radical formation are rather oxygen insensitive, which is favorable for present cells.16 In combination with a cytocompatible hydrophilic polymer and an appropriate light source, this route for hydrogel formation can be highly advantageous for in vivo applications allowing the preparation of complex shaped hydrogels under minimally invasive conditions inside a patient's body.17 POx copolymers of MeOx or EtOx in combination with unsaturated monomers (2-(dec-9-enyl)-2-oxazoline (DecEnOx) or 2-(3-butenyl)-2-oxazoline (ButEnOx)) have been synthesized and cross-linked with a variety of small molecule dithiols like dithiothreitol,18 2,2′-(ethylenedioxy)diethanol, glycol dimercaptoacetate19,20 and 1,3-propanethiol to 1,9-nonanedithiol,21 which always bear the risk of toxicity and have to be accurately weighed to minimize dangling cross-linking points. Hydrogels fabricated via the small molecule crosslinker route are usually pre-fabricated and then rehydrated before their intended biomedical application22 because of the poor solubility of the dithiol in water or because of the high concentration of dithiol needed, which exceeds the LC50 of the compound.21 One alternative to this approach is the replacement of the small dithiol molecule with a thiol side chain functionalized polymer as has been demonstrated by Stichler et al.,23 who were able to 3D print cytocompatible, mechanically strong and complex hydrogel structures via a rapid UV mediated thiol–ene reaction.
Here, we would like to present a hydrogel formulation based on the thiol–ene reaction of vinyl side chain functionalized POx with thiol side chain functionalized POx. We expect that the strategy of using two polymeric hydrogel precursors will significantly improve the mechanical stability due to the higher availability of reaction sites, resulting in denser networks. Our strategy should also lead to more homogenous networks as the chance of one functional group reacting with another of the same polymer chain is significantly reduced. Moreover, the chemical tunability of the POx hydrogel precursors, together with better control over the network density using thiol–ene chemistry, should be favorable for controlled release of guest molecules. While POx based micelles have been extensively examined for drug delivery and release applications,24,25 POx based hydrogels have so far not been the focus of research for drug loading and release.
We synthesized various random copolymers of MeOx or EtOx with ButEnOx, which were further functionalized with thiol or cysteine at the side chain with increasing molar percentage. Through the amine group, the cysteine side functions bring additional charges into the network, making the polymer sensitive to pH changes and allowing ionic interactions, for example, with negatively charged drugs. Based on the hydrophilicity of the monomer, the copolymers also exhibit different thermo-responsive behaviors. Therefore, we will investigate two systems, one being only sensitive to temperature change and the other being doubly stimuli responsive. The two systems will be examined for their swelling behavior and mechanical properties, and tested as potential drug delivery systems by loading the hydrogel network with two low molecular weight substances, methylene blue (positively charged) and sodium fluorescein (negatively charged), to study their release profiles depending on their different chemical environments.
2. Experimental
All materials used, the detailed synthesis protocol and the characterization of the polymers can be found in the ESI.†2.1 Polymer synthesis
2-Methyl-2-oxazoline (MeOx) and 2-ethyl-2-oxazoline (EtOx) were randomly copolymerized with 2-(3-butenyl)-2-oxazoline (ButEnOx) to yield poly(2-methyl-2-oxazoline-co-2-(3-butenyl)-2-oxazoline), abbreviated in the following as PMeOx-co-En, and poly(2-ethyl-2-oxazoline-co-2-(3-butenyl)-2-oxazoline), abbreviated in the following as PEtOx-co-En. The synthesis was carried out in a microwave synthesizer (Discover SP, CEM GmbH, Kamp-Lintfort, Germany) at 100 °C for several hours using methyl p-toluenesulfonate as initiator and acetonitrile as solvent.18,26 All monomers were added right at the beginning and the monomer molarity was kept at 4 M. All polymers were terminated with piperidine at room temperature. The MeOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ButEnOx and EtOx:ButEnOx monomer ratios were 0.9:0.1, 0.8:0.2 and 0.7:0.3 with a theoretical chain length of 50 repeating units (Fig. 1). After synthesis, the polymers were precipitated three times from methanol/chloroform or chloroform in ice cold diethyl ether.
:ButEnOx and EtOx:ButEnOx monomer ratios were 0.9:0.1, 0.8:0.2 and 0.7:0.3 with a theoretical chain length of 50 repeating units (Fig. 1). After synthesis, the polymers were precipitated three times from methanol/chloroform or chloroform in ice cold diethyl ether.
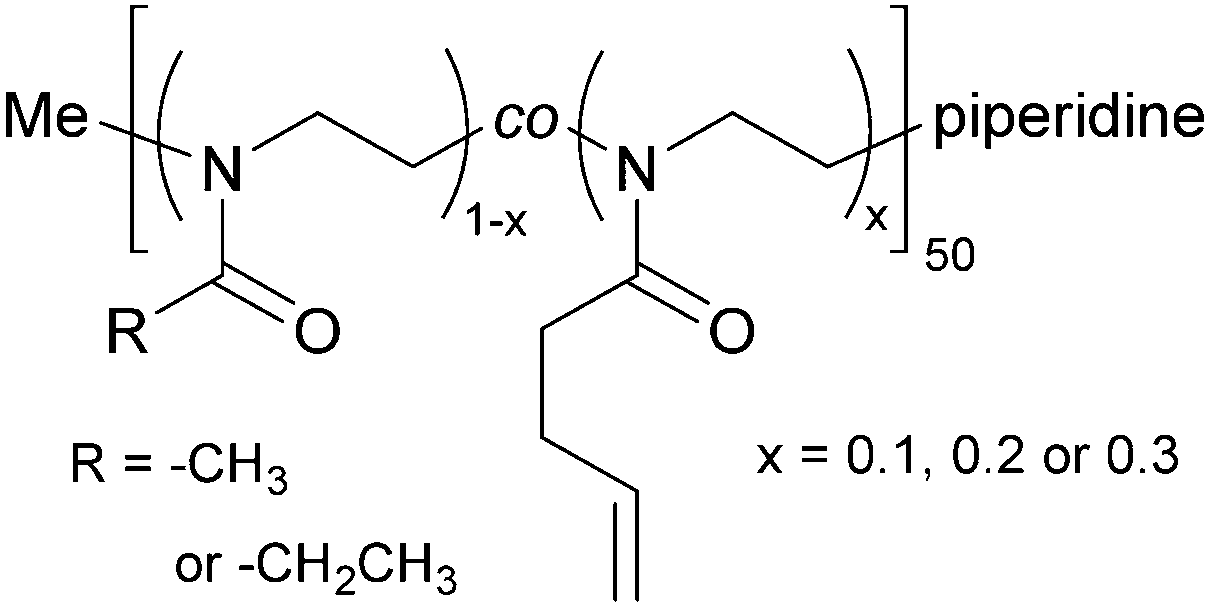 | ||
| Fig. 1 Chemical structure of the synthesized copolymers PMeOx-co-En and PEtOx-co-En. | ||
2.2 Post-polymerization functionalization with thiols
The copolymers were further functionalized at the side chain with thiols, yielding poly(2-methyl-2-oxazoline-co-2-thiobutyl-2-oxazoline) (PMeOx-co-SH) and poly(2-ethyl-2-oxazoline-co-2-thiobutyl-2-oxazoline) (PEtOx-co-SH), see Fig. 2. The thiol side chain functionality was introduced by creating a thioester through a thiol–ene reaction of thioacetic acid with the double bond of ButEnOx of both copolymer variations. The solvent was methanol and 2,2-dimethoxy-2-phenylacetophenone was used as a photo-initiator (λ = 365 nm). The polymer was precipitated from chloroform in cold diethyl ether after a reaction time of 0.5 h. The deprotection step was carried out at room temperature over 12 h in methanol using 1.5 eq. cysteine and 3 eq. NaBH4 under inert conditions. The thiol functionalized copolymer was obtained after precipitation in cold diethyl ether. The polymer was further dialyzed (MWCO = 3.5 kDa) against degassed water to remove the water soluble byproduct acetylcysteine.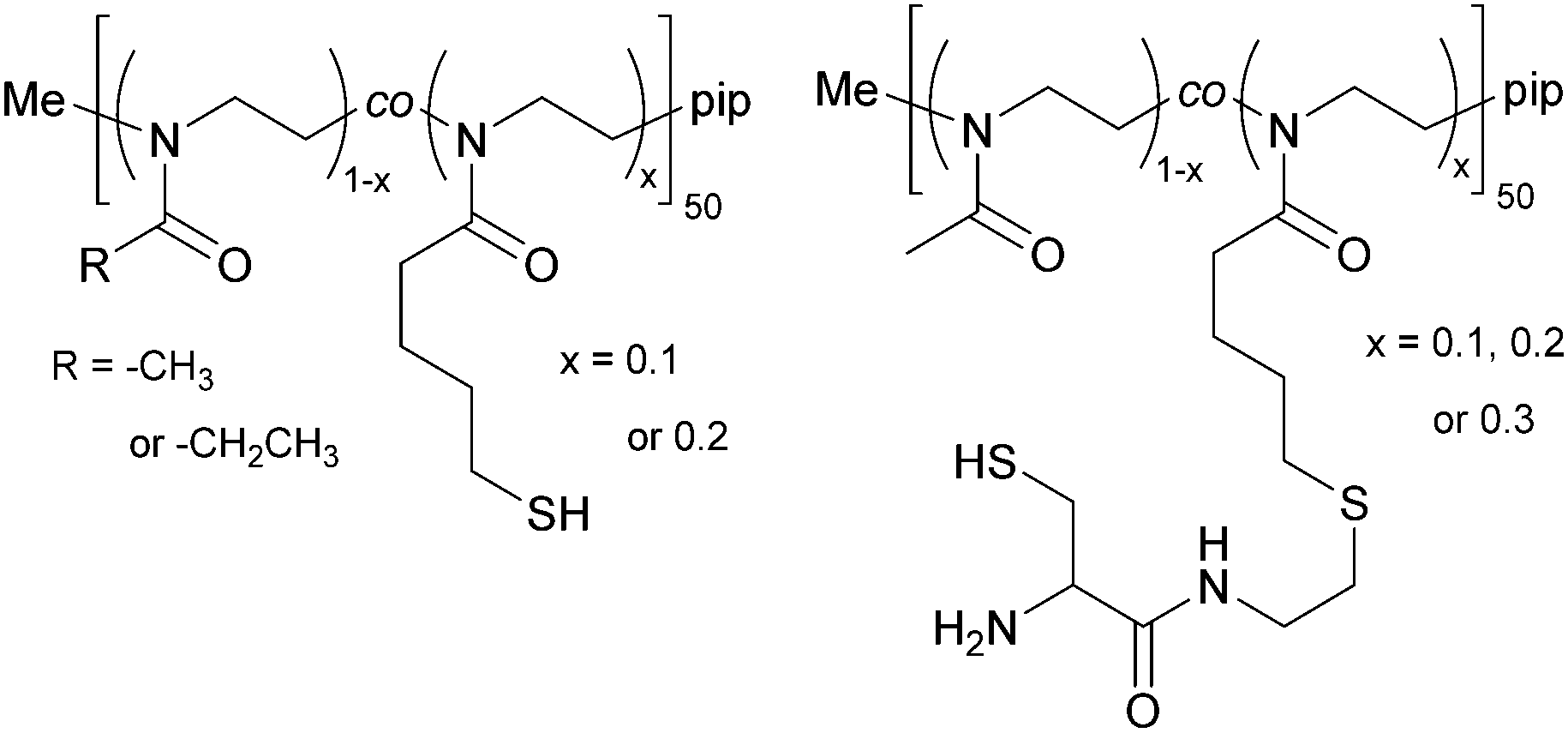 | ||
| Fig. 2 Chemical structure of the synthesized thiol or cysteine side chain functionalized POx. | ||
2.3 Post-polymerization functionalization with cysteine
The copolymer PMeOx-co-ButEnOx with 10, 20 and 30 mol% vinyl functionality was functionalized with 3-formyl-N-(2-mercaptoethyl)-2,2-dimethylthiazolidine-4-carboxamide, a protected cysteine molecule, as described elsewhere.27,28 The protected cysteine is deprotected at 70 °C in 0.1 M HCl for four days under reflux and dialyzed against degassed water for three days. Those polymers are denoted PMeOx-co-Cys. The deprotection step could not be performed with polymers based on EtOx with a functionality of 20 mol% or higher due to its cloud point below 60 °C and the occurring polymer precipitation.2.4 Hydrogel formation
All hydrogels were prepared in 1× phosphate buffered saline (PBS) to achieve an overall polymer content of 15 wt%. PMeOx-co-En with 10 and 20 mol% vinyl groups was combined with PMeOx-co-SH with 10 and 20 mol% thiol moieties, respectively, and PEtOx-co-En with 10 and 20 mol% vinyl groups was combined with PEtOx-co-SH with 10 and 20 mol% thiols, respectively, to obtain thiol cross-linked hydrogels. PMeOx-co-En with 10, 20 and 30 mol% vinyl groups was combined with PMeOx-co-Cys with 10, 20 and 30 mol% cysteine groups, respectively, to obtain cysteine cross-linked hydrogels, see Fig. 3. The vinyl:SH molar ratio was kept at 1:1 for all cases. A 9 wt% solution of the vinyl containing polymer in 1× PBS was used to dissolve the thiol/cysteine containing polymer so that an 18 wt% buffer solution was obtained. After complete dissolution, a defined amount of Irgacure (2.5 mg mL−1 in buffer) was added to reach the final polymer concentration of 15 wt%. The total concentration of Irgacure was always kept at 0.05 wt% as it has been shown that this amount is not toxic for various cell lines and for irradiation times up to 10 min at 8 mW cm−2 or 15 min at 1 mW cm−2.15,23,29 The mixed precursor solution (50 μL) was then pipetted into clear cylindrical silicon molds (d = 4 mm, h = 4 mm) and was irradiated for 15 min with a UV hand lamp at 365 nm with an intensity of ∼1 mW cm−2 (a UVL hand lamp with a filter, VL-4.LC, 8 W, A. Hartenstein, Wuerzburg, Germany). Solely the hydrogel precursor solution of PEtOx-co-En and PEtOx-co-SH with 20 mol% functionality had to be cooled with ice before and during hydrogel formation due to the LCST behavior of PEtOx-co-SH20.
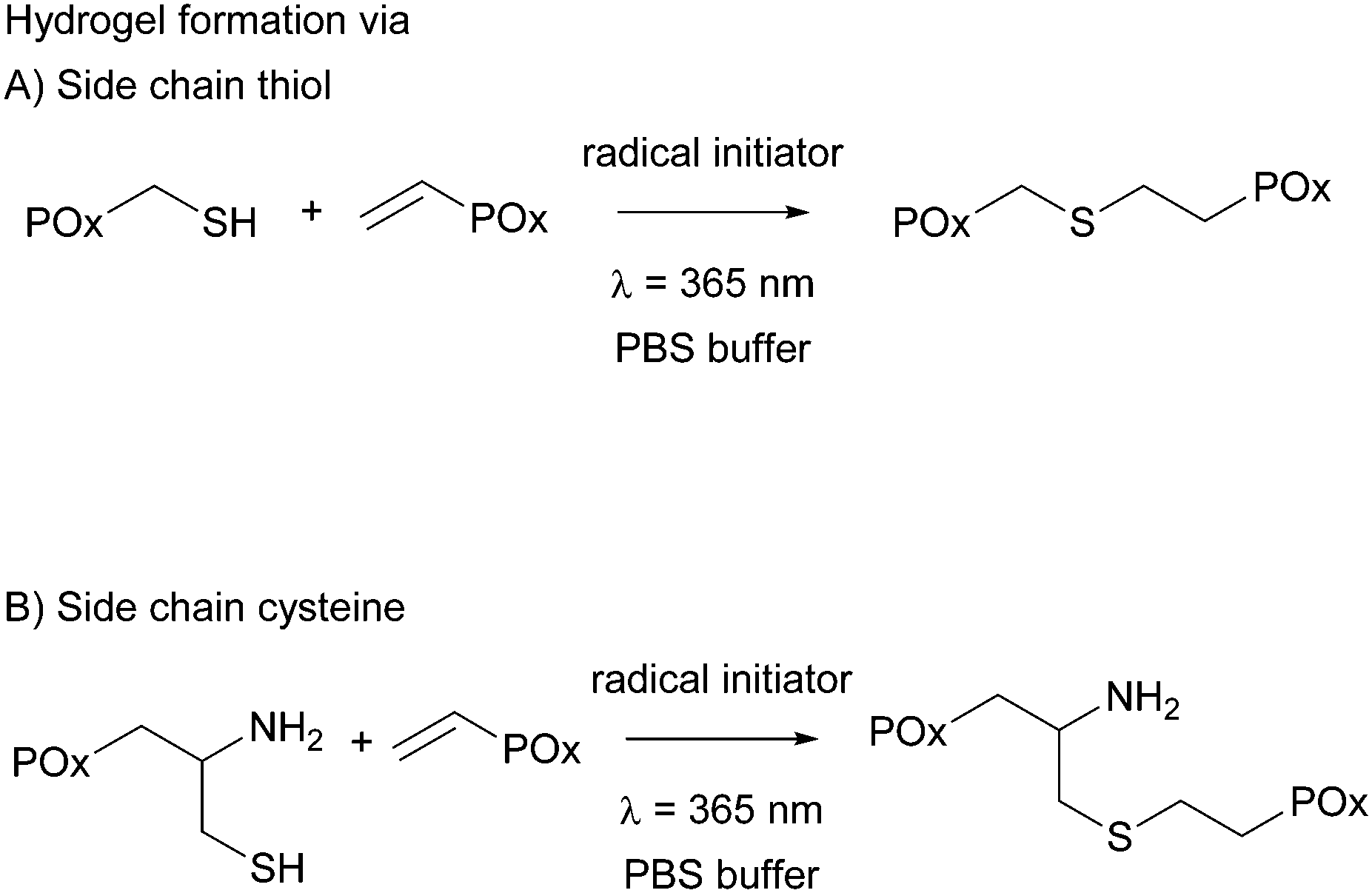 | ||
| Fig. 3 Hydrogel formation via thiol–ene chemistry. | ||
2.5 Cytocompatibility
A detailed description of the cell viability test can be found in the ESI.†Mouse fibroblasts (L 929 CC1) were cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies) containing 10% fetal bovine serum (Sigma-Aldrich) in the presence of 1% Penicillin–Streptomycin (Thermo Fisher Scientific).
Mouse fibroblasts in cell medium were plated in 48-well plates at a density of 50 000 cells per well and incubated for 48 h under standard cell culture conditions (37 °C, 5% CO2). All polymers were tested at concentrations of 15, 5, 1 and 0.1 mg mL−1 except for PEtOx-co-SH20, which would precipitate due to its low cloud point. Test results of 15 mg mL−1 of PMeOx-co-Cys20 and PMeOx-co-Cys30 could not be used as the polymer precipitated at this concentration and covered the cells.
Test samples were solubilized in medium to the desired treatment concentration and cells were treated with 0.5 mL of each solution in fourfold. As negative control, polystyrene with fresh medium was used. As positive control, the eluate of PVC plates (Vekaplan KT, König GmbH, Wendelstein, Germany) was used.
After 48 h incubation of the polymer eluates, the supernatant was carefully sucked away and 0.5 mL of the diluted assay reagent was added. The well plates were mixed on an orbital shaker for 2 min and afterwards incubated for 10 min to stabilize the luminescence signal. The luminescent signal was read out on a Tecan Spark® 20M multimode microplate reader (Tecan, Crailsheim, Germany).
000 cells per well (approximately 18000 cells per cm2) and incubated for 24 hours under standard cell culture conditions. The cell medium was removed, and freshly prepared hydrogels were placed on top of the cell layer. As negative control, 2 wt% agarose hydrogels were prepared, and as positive control, the eluate of PVC plates was used. New cell culture medium was added, and the hydrogels were incubated for 7 days, with the culture medium being changed after 3 days. After 7 days, the medium was removed from each well and 0.5 mL of diluted (1:10) WST reagent in fresh medium was added. After incubation at 37 °C for 30 min, duplicates of each well were pipetted into a 96-well plate, with the diluted WST reagent acting as the blank. The absorption was measured with a Tecan Spark® 20M at a wavelength of 450 nm.
2.6 Hydrogel characterization
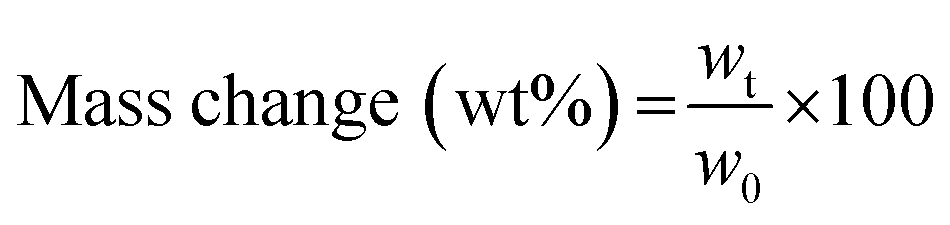 . Each timepoint was analysed in triplicate.
. Each timepoint was analysed in triplicate.
The swelling degree (SD) was determined by weighing the freshly prepared hydrogels after 1 d of incubation in 1× PBS (ws) at 37 °C and determining the weight after freeze-drying of the same sample (wd). The equilibrium swelling degree was determined by dividing the weight of the swollen hydrogel (ws) minus the weight of the freeze-dried sample (wd) by the weight of the freeze-dried sample and was calculated as follows: 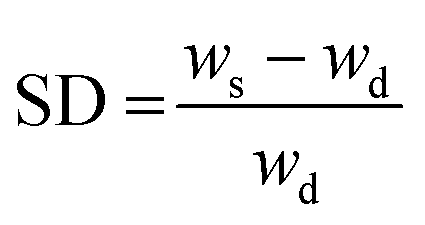 .
.
The gel fraction, i.e. the amount of polymer incorporated in the network, was determined by freeze-drying the hydrogel sample directly after preparation (wi) and swelling of the same dried hydrogel in deionized water for 3 days at 37 °C with frequent water changes. The residual hydrogel was again freeze-dried and weighed (wg). The gel fraction was then calculated as follows: 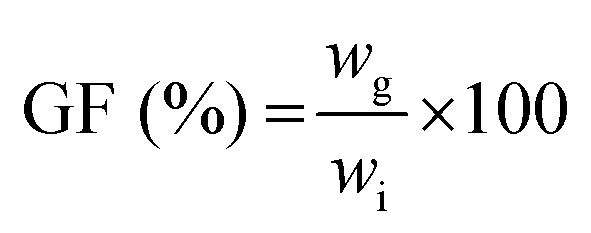 , with all measurements made in triplicate.
, with all measurements made in triplicate.
The Young's modulus was obtained by plotting the strain versus the true stress and applying a linear fit from 0.1 to 0.2 strain. The Young's modulus was calculated from the slope of the linear fit.
2.7 Flory–Rehner analysis
To estimate the mesh size of all hydrogels, which were used for the proof-of-principle study with low molecular weight substances, we applied the Flory–Rehner equation as described by Dargaville et al.19 for POx hydrogels. The molar mass between cross-links,![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) c, can be calculated as follows:
c, can be calculated as follows:This can further be used to determine the mesh size, ξ:
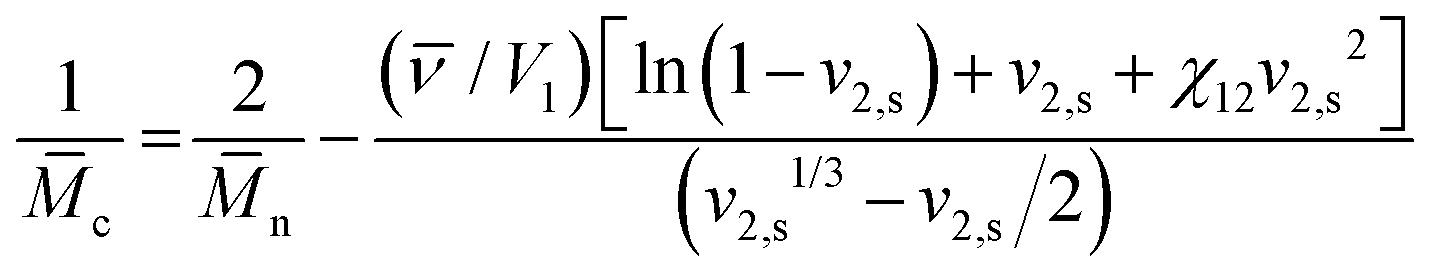
with the polymer volume fraction in the swollen state ν2,s, the specific volume of the polymer
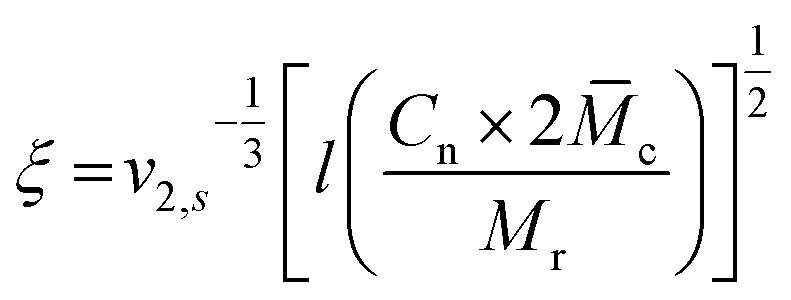
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) , the molar volume of water V1, the polymer–solvent interaction parameter χ12, the Flory characteristic ratio Cn and the molar mass of the monomer unit Mr. A detailed calculation with an example can be found in the ESI.†
, the molar volume of water V1, the polymer–solvent interaction parameter χ12, the Flory characteristic ratio Cn and the molar mass of the monomer unit Mr. A detailed calculation with an example can be found in the ESI.†
2.8 Loading and release of low molecular weight substances
All hydrogels were prepared as described above at 15 wt% in PBS. Triplicates of 50 μL polymer precursor solution were prepared for each possible combination with additional gels for all PEtOx-co-En/PEtOx-co-SH gels, where dye release was also performed at 4 °C. For dye loading, the hydrogels were immersed in either saturated buffered methylene blue solution (40 g L−1) or buffered fluorescein sodium salt solution (60 g L−1) directly after preparation. Hydrogels prepared with 30% cysteine were immersed in a fluorescein sodium salt solution with a higher concentration (80 g L−1). Fluorescein concentrations were chosen in a way that 50 μL of the dye solution would contain the calculated maximum amount of dye that could interact with the positive amino groups present in hydrogel specimens that had been cross-linked via cysteine. In order to compare non-charged and charged hydrogels, all hydrogels without cysteine were placed in the same dye solution as the hydrogel made with 20 mol% cysteine.For equilibration, all gels were incubated at room temperature overnight. Only hydrogels that were strongly thermo-responsive (PEtOx-co-En20/PEtOx-co-SH20) and the additional hydrogels of PEtOx-co-En10/PEtOx-co-SH10 for the cold release study were incubated at 4 °C overnight. After incubation, the hydrogels for the release study at 37 °C were placed at 37 °C for 1 h before they were taken from the highly concentrated dye solution, blotted to remove excess dye solution and then placed in clean vials.
For the release, the hydrogels were immersed in 9 mL of warm (37 °C) or cold (4 °C) PBS buffer. 1 mL of the solution was taken and replaced with 1 mL of fresh PBS buffer at the following time points: 0.5 h, 1 h, 2 h, 4 h, 8 h and 24 h, which describes phase one of the release. After one day the hydrogels were then taken out of the vials, blotted and placed in new clean vials to maintain the sink-conditions for the release. Phase two of the release was then started by adding fresh PBS buffer, 9 mL for fluorescein sodium and only 5 mL for methylene blue. Samples of 1 mL were taken and replaced with 1 mL of fresh buffer at time points 30 h, 48 h, 54 h, 72 h and 96 h. After 96 h the hydrogels were again taken out of the PBS buffer, blotted on tissue paper and placed in small Eppendorf tubes. To each hydrogel 0.5 mL of fresh tempered buffer was added and only 0.2 mL was taken out and replaced with fresh 0.2 mL of buffer at time points 5 d, 7 d, 9 d, 11 d and 14 d.
2.9 Loading and release with fluorescently labeled dextran
Hydrogels based on PEtOx-co-En and PEtOx-co-SH with 10 and 20 mol% were prepared as described before and fluorescently labeled dextran (FITC–dextran, purchased from Sigma-Aldrich) molecules with average molecular weights of 4, 40 and 500 kDa were mixed into the polymeric precursor solution before cross-linking with UV light. As the FITC–dextran with 40 kDa was substituted with only 0.001–0.008 mol FITC/mol glucose, the amount of FITC–dextran/hydrogel specimen was chosen twice as high with 0.016 ng/50 μL hydrogel solution compared to 0.008 ng/50 μL hydrogel for 4 and 500 kDa FITC–dextran, where 0.002–0.020 mol FITC mol−1 glucose were substituted. After cross-linking for 15 min, the hydrogels were incubated in 1 mL of 1× PBS at 37 °C and at 4 °C for hydrogels with 20 mol% functionality for 15 d. At each time-point, 0.8 mL of buffer was withdrawn for fluorescence spectroscopy (excitation wavelength = 485 nm, emission wavelength = 535 nm, Tecan Spark® 20M multimode microplate reader) and replaced with 0.8 mL of fresh buffer.2.10 Statistical analysis
Statistical evaluation with SigmaPlot12.5 (Systat Software, Erkrath, Germany) was performed using two way analysis of variance (two way ANOVA) or three way ANOVA for the time dependent swelling behavior and one way ANOVA for the comparison of the Young's moduli, swelling degrees and release of the model drug compounds. Significant differences were evaluated using post hoc Tukey's test using a significance level of p > 0.05.3. Results and discussion
3.1 Synthesis and characterization of poly(oxazoline) copolymers
Random poly(oxazoline) copolymers with varying hydrophilicity and functionality were synthesized. Through introduction of a statistical distribution of the functionalities along the polymer chain, we hoped to create a homogenous network later on during hydrogel preparation. However, it was reported by Dargaville et al. and Gress et al. that the two monomers MeOx and EtOx are polymerized slightly more rapidly than ButEnOx,18,30 leading to rather gradient copolymers, with the ButEnOx monomer being mostly located at the end of the polymer chain. Nevertheless, we expect that this gradient behavior is less pronounced for the combination of EtOx and ButEnOx as EtOx is polymerized more slowly than MeOx19,31 and that the effect is certainly weakened by higher mol% of ButEnOx added. 1H NMR end group analysis proved that the experimental values for the polymer composition and chain length were met with an error of 5–10% of the theoretical value. The dispersity was lower for PEtOx-co-En than for PMeOx-co-En copolymers, with values of 1.1 and 1.1–1.3 respectively; for details see Tables S1 and S2 (ESI†). GPC elugrams showed no shoulder for PEtOx-co-En copolymers but a shoulder at higher molecular weight for PMeOx-co-En, which also explains their higher dispersity (GPC elugrams shown in the ESI†). At this stage, we have no explanation for this phenomenon that only occurs when polymers contain MeOx and they are terminated with piperidine (data not shown).The proportion of functionalities was generally not increased above 30 mol% ButEnOx due to the steady decrease of the cloud point from 55.5 °C for PEtOx-co-En20 to 37 °C for PEtOx-co-En30 at 1 wt% in aqueous solution (Fig. 4), which hampers the processing of the polymers. In contrast, the more hydrophilic PMeOx-co-En copolymers did not display any cloud point.
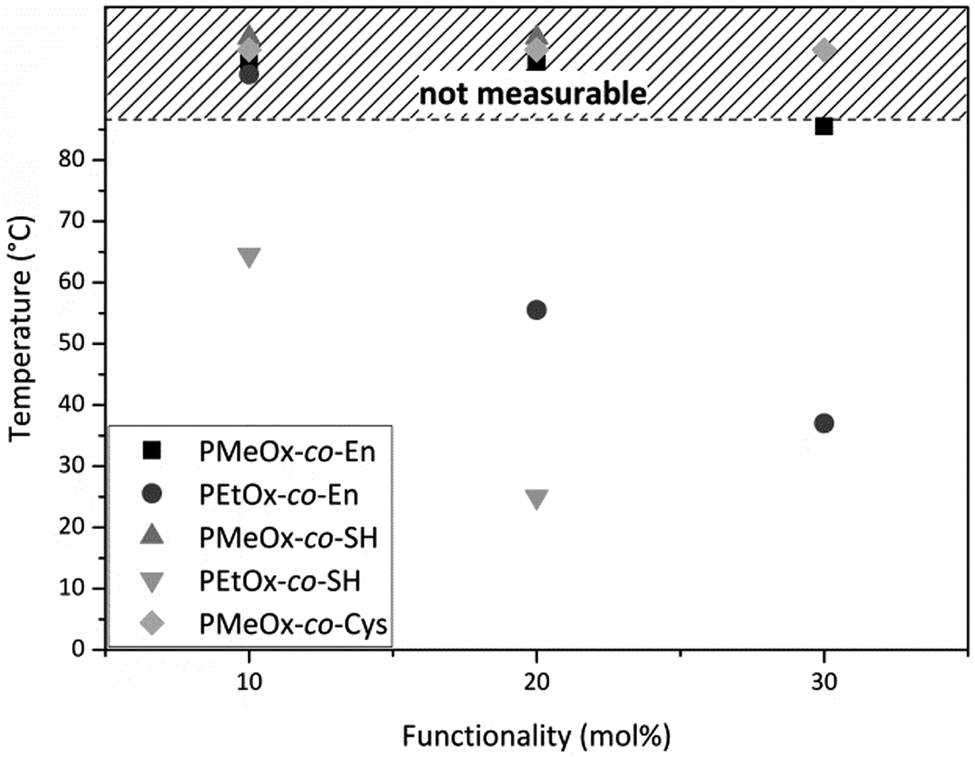 | ||
| Fig. 4 Cloud points of 1 wt% aqueous solutions of all synthesized poly(oxazoline) copolymers. All cloud points mapped in the shaded area were above the temperature limit of the device. | ||
For subsequent cross-linking, vinyl group containing copolymers were further functionalized with thiols at the side chain. The first POx with thiol side chain functionality was reported by Cesana et al.,32 who introduced thiols via a monomer with protected thiol side chains, but no further study or application was reported for this specific polymer. Usually, introducing functionalities through functional monomers is preferred, but depending on the specific functionality this can be rather difficult due to side reactions. Hence, post-polymerization functionalization can be preferable as not every nucleophilic group is compatible with the reaction conditions of the cationic ring opening polymerization.2,33 Accordingly, we chose the post-polymerization functionalization approach, as we intended to add two different functionalities to the same polymer backbone in order to compare their ability to cross-link hydrogels. To obtain a thiol functionality at the side chain, thioacetic acid was linked to the vinyl side chain via a radical thiol–ene reaction, which is a simple and very efficient method for post-polymerization functionalization.34 The prepared thioester can subsequently be deprotected under basic conditions at elevated temperatures to regain the thiol, which was already demonstrated recently.23 However, the deprotection step had to be slightly modified as polyoxazolines tend to degrade at the backbone under strongly basic conditions and high temperatures.35 Instead of working under aqueous conditions, we present an approach using cysteine to deprotect the thioester under reductive conditions in methanol at room temperature. The successful thiol functionalization was verified by 1H NMR, GPC and Ellman's assay (Tables S3 and S4, ESI†). The GPC curves of the modified polymers did not show any sign of degradation and only a slight peak broadening, which might be due to the increased functionality of the polymer and possible interactions with the column material. Ellman's assay confirmed the presence of thiols with values close to the theoretical mol-percentage. The successful functionalization of PMeOx-co-En with cysteine functions at the side chain was also confirmed by 1H NMR and Raman spectroscopy, which revealed full conversion and the presence of thiols.
After functionalization of PEtOx-co-En10 to PEtOx-co-SH10, the cloud point further decreased to 64.5 °C and even decreased to 25 °C for PEtOx-co-SH20 (Fig. 4), accordingly a thiol functionalization of PEtOx-co-En30 was not carried out as an even lower cloud point was expected, which would interfere with the biomedical application of these hydrogels. In contrast, PMeOx-co-SH and PMeOx-co-Cys did not display any cloud point due to the higher hydrophilicity of the co-monomer MeOx.
3.2 Cytocompatibility of POx copolymers and hydrogels
All polymers with vinyl, thiol and cysteine functionality were tested for cytotoxicity with the CellTiterGlo Assay, which assesses the cell viability with a luminescent reagent reacting with the adenosine triphosphate (ATP) generated and released by viable cells. A mouse fibroblast cell line (L 929) was used for culture and concentrations of 15, 5, 1 and 0.1 mg mL−1 polymer were tested.All tested polymers did not induce cytotoxic effects up to a concentration of 10 mg mL−1 with the exception of the thiol functionalized polymers. Here, weak cytotoxicity was observed at 5 mg mL−1 and no cytotoxicity was observed at 1 mg mL−1 and below. PEtOx-co-SH20 could not be tested due to its low cloud point at 23.5 °C. A detailed description and stereomicroscopy pictures can be found in the ESI† (Fig. S32).
Taken together, this proves that all tested polymers show an excellent cytocompatibility as non-crosslinked polymers, which is known from the recent literature on different poly(oxazoline)s.21,22
We further tested all hydrogel compositions in direct cell contact by placing the freshly prepared hydrogels on top of the cell layer (mouse fibroblasts, L 929) in a 48-well plate. To consider any mechanical influence of the hydrogel on top of the cell layer, we prepared 2 wt% agarose gels with the same dimensions and used them as an additional negative control to polystyrene. After incubation of the hydrogels for 7 days, the cell proliferation reagent WST-1 solution was added to the well and the absorbance measured. All hydrogels show very good cell compatibility after 7 d, see the ESI,† Fig. S33, indicating that the amount of initiator and uncross-linked polymer that might be released during incubation is not cell toxic. These results confirm several other studies with thiol–ene cross-linked hydrogels which showed no cytotoxic effects as well.21,36
3.3 Hydrogel swelling behavior
All hydrogels were prepared with an identical polymer content of 15 wt% as we aimed to compare how the cross-linking degree, hydrophilicity and side chain functionality influence the hydrogels’ properties. The swelling behavior of the synthesized hydrogels was measured gravimetrically in PBS buffer at 37 °C, and additionally at 4 °C for hydrogels that were composed of precursors exhibiting a LCST behavior (Fig. 5). The mass changes of all hydrogels were consistent with their volume changes (Fig. S36, ESI†), indicating that the increase or decrease in mass was solely generated by the ability of the hydrogel network to bind water.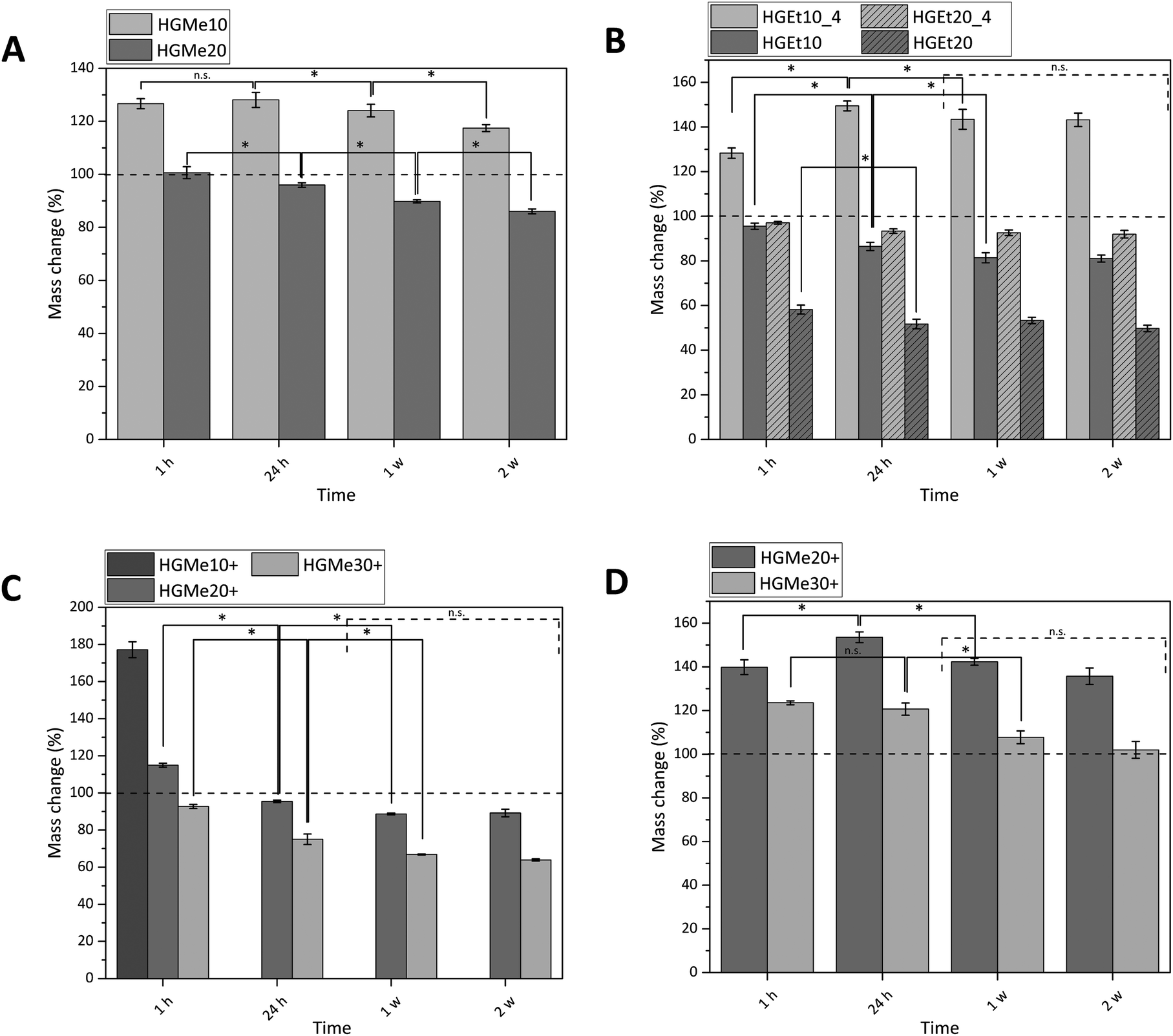 | ||
| Fig. 5 Swelling behavior of cross-linked hydrogels in PBS buffer: (A) thiol cross-linked HGMe with 10 and 20% functionality, (B) thiol cross-linked HGEt with 10 and 20% functionality at 4 °C and 37 °C, (C) cysteine cross-linked HGMe+ with 10, 20 and 30% functionality and (D) cysteine cross-linked HGMe+ with 20 and 30% functionality in deionized water. Means and standard deviations are shown (n = 3). Statistical significance values are shown with * (p < 0.050) and n.s. (no significant differences between the same types of hydrogels at consecutive time points). | ||
Fig. 5A shows that the swelling of the hydrogels based on the more hydrophilic monomer MeOx with 10 mol% functionality (HGMe10) was the strongest at 37 °C with a mass increase of about 130% after 24 h. In accordance with that, the SD was the highest for that hydrogel at 37 °C, with a value of 8.1 (see Table 1). As the cross-linking density was increased by increasing the molar percentage of the thiol, the hydrogels were less able to swell, which could be observed in terms of a mass change of ∼95% for HGMe20 and a lowered SD of 4.9. Over the course of two weeks, the hydrogels decreased significantly in mass even though no degradable covalent bonds were incorporated. This observation could be explained by the diffusion of non-cross-linked polymer chains, enhanced by the hydrophilicity of the polymer precursor.
| Hydrogel | Abbreviation | Temperature (°C) | SDa | Gel fractionb (%) |
|---|---|---|---|---|
| a SD = (ws − wd)/wd. b Gel fraction = wg/wi × 100. | ||||
| PMeOx-co-En10 + PMeOx-co-SH10 | HGMe10 | 37 | 8.1 ± 0.1 | 77.5 ± 1.9 |
| PMeOx-co-En20 + PMeOx-co-SH20 | HGMe20 | 37 | 4.9 ± 0.1 | 85.5 ± 0.5 |
| PMeOx-co-En10 + PMeOx-co-Cys10 | HGMe10+ | 37 | 18.5 ± 1.0 | n.d. |
| PMeOx-co-En20 + PMeOx-co-Cys20 | HGMe20+ | 37 | 5.7 ± 0.2 | 68.2 ± 2.6 |
| PMeOx-co-En30 + PMeOx-co-Cys30 | HGMe30+ | 37 | 4.5 ± 0.1 | 76.2 ± 0.4 |
| PEtOx-co-En10 + PEtOx-co-SH10 | HGEt10_4 | 4 | 9.4 ± 0.3 | n.d. |
| PEtOx-co-En10 + PEtOx-co-SH10 | HGEt10 | 37 | 4.8 ± 0.1 | 83.1 ± 0.3 |
| PEtOx-co-En20 + PEtOx-co-SH20 | HGEt20_4 | 4 | 5.8 ± 0.2 | n.d. |
| PEtOx-co-En20 + PEtOx-co-SH20 | HGEt20 | 37 | 2.6 ± 0.2 | 88.7 ± 2.3 |
Hydrogels formed with the more hydrophobic polymers PEtOx-co-En/PEtOx-co-SH did not swell at 37 °C; the mass even decreased to 86% after 24 h for 10 mol% of functionality (Fig. 5B). This effect became even more pronounced when the cross-linking density was increased. The mass of hydrogels formed with 20 mol% functionality decreased to ∼50% after 24 h at 37 °C. The SD decreased accordingly to 4.8 for 10 mol% and to 2.6 for 20 mol% thiol content. The collapse of the hydrogel network and hence the expulsion of water were expected, as the cloud point of one of the hydrogel components, PEtOx-co-SH20, was already at 23.5 °C and it is known from the literature that cross-links induce a further decrease of the cloud point.8
In consequence, hydrogels consisting of PEtOx-co-En/PEtOx-co-SH were also incubated at 4 °C to observe the swelling behavior at a temperature far below the cloud point of the polymer precursors. At this temperature, it was interesting to find that the hydrogel with 10 mol% thiol increased in mass by approx. 150%. The water loss was also less drastic for the hydrogel with 20 mol% thiol and the mass changed only to approx. 90%, instead of 50% at 37 °C. The SD also doubled for these hydrogels at 4 °C, with 9.4 for 10 mol% thiol and 5.8 for 20 mol% thiol. The masses of these hydrogels did not change significantly after one week, indicating that an equilibrium state was established. In addition, it might be more difficult for any non-cross-linked polymer precursor chain to diffuse due to the lower swelling of the hydrogel network and the less hydrophilic polymer precursors.
We then used the thiol group of the cysteine side chain functionalized copolymers to fabricate thiol–ene cross-linked hydrogels containing additional amine functions. For these gels, it was observed that no stable hydrogel formation was possible with only 10 mol% functionality. The hydrogels disintegrated completely in less than one hour in deionized water and in less than 24 h in PBS so that the mass change could only be measured at 1 h in PBS (Fig. 5C). These hydrogels showed a mass increase of approx. 180% and a SD of 18.5, which indicates a quite loose and incomplete network formation. By increasing the mol% of cysteine to 20, the mass change decreased to approx. 95% after 24 h and 90% after 1 w. The SD of these hydrogels was 5.7 after 24 h. A further increase to 30 mol% cysteine functionality caused a mass change of approx. 75% after 24 h and 65% after 1 w. The value of the SD also decreased to 4.5. No significant mass change was observed for these hydrogels after 1 week for the same possible reasons as already explained earlier for thiol cross-linked hydrogels based on EtOx.
It is interesting to note that the same swelling behavior with similar values for the SD could be observed for three completely different hydrogels HGMe20, HGEt10 and HGMe30+ at 37 °C. The influence of the hydrophilicity of MeOx allows the hydrogel network to swell to a larger extent even though more cross-links have been established and a much denser network was formed. The hydrogel with 30 mol% cysteine is more hydrophobic than the one with 20 mol% thiol as the amount of ButEnOx and the possible cross-linking degree are higher for the polymer precursors. However, two explanations for the similar SDs are possible. The first could be the additional charge of the amine functionality, which attracts more water molecules, and the second a lower amount of cross-links formed since the cysteine thiol–ene reaction is not as efficient and a looser network than expected was formed.
The swelling abilities of the hydrogels made with 20 mol% cysteine and 20 mol% thiol of the same copolymer backbone are very similar, although one would expect the hydrogel containing cysteine groups to swell to a larger degree due to the additional hydrophilic amino groups present in the network. This behavior seems to be suppressed by the large amounts of different ions present in the buffer. The cysteine containing hydrogel swells 1.5 times more in deionized water (Fig. 5D), where the positive charges of the amino groups repel each other, and the network is additionally widened, resulting in a higher water uptake. The stretching of the polymer chains, due to ionic repulsion, can also explain our observation that the thiol–ene reaction was less efficient in deionized water than in buffer for cysteine functionalized polymers resulting in faster dissolution of the polymer network. The effect of an increased swelling in pure water was also observed by Tan et al. who synthesized positively charged hydrogels by incorporating 2-(methacryloyloxy)ethyl-trimethylammonium chloride into PEG based hydrogels intended for bone tissue engineering.37 The swelling degree of the hydrogels containing tertiary amino groups was nearly doubled in deionized water compared to PBS.
As already observed by Dargaville et al. and Šrámková et al.,19,21 hydrogels made from the more hydrophobic 2-ethyl-2-oxazoline backbone are less able to swell at the same cross-linking degree as hydrogels made from 2-methyl-2-oxazoline. This fact was also confirmed in our case by a mass decrease to approx. 50% for hydrogels with 20 mol% thiol and the EtOx backbone compared to approx. 90–95% for hydrogels with 20 mol% thiol/cysteine and the MeOx backbone.
The fact that no stable hydrogels could be formed with 10 mol% cysteine and a relatively low gel fraction of the hydrogel formed with 20% cysteine (gel fraction = 68%) indicates that the close proximity of the primary amino group and the thiol group of the cysteine interferes with the thiol–ene reaction. The effect of different functional groups on the thiol–ene reaction under biologically relevant conditions has been extensively studied by Colak et al.38 A more detailed discussion on this effect can be found in the ESI.†
3.4 Solid-state NMR of thiol–ene cross-linked hydrogels
To further investigate the effect of the close proximity of the amino group on the efficiency of the thiol–ene reaction, we recorded 1H–13C cross polarization (CP)/Magic Angle Spinning (MAS) NMR spectra of the polymeric precursors as well as the thiol and cysteine cross-linked hydrogels. Fig. 6A shows the 13C NMR spectra of PEtOx-co-En10, PEtOx-co-SH10 and the cross-linked hydrogel. Overall, the polymers are largely comparable (backbone, amide unit, ethyl moiety) and even the amounts of impurities (grey boxes) are comparable. Furthermore, the representative CH2 groups of the specific sidechains can be assigned and distinguished. While the two CH2 units adjacent to the double bond show resonances at 32.3 and 29.8 ppm, two out of the four CH2-units in the side chain containing the SH group show signals at 32.9 ppm and the remaining two at lower ppm values overlaid by the main CH2 resonance. The major differences can be found in the area between 110 and 150 ppm, where resonances for the vinyl unit can be observed for the corresponding polymer. They are no longer present in the spectrum of the thiol cross-linked sample, which indicates that the majority of the double bonds has undergone a reaction when the thiol functionalized polymeric precursor is used for hydrogel formation. Fig. 6B shows stacked spectra of all hydrogels cross-linked via the cysteine functionality. The three 13C CP/MAS spectra were calibrated with respect to the carbonyl moiety at ∼170 ppm. The relative intensities of the signals in the region 100–150 ppm and that of the signal at ∼30 ppm can then be compared. While the relative amounts of impurities, e.g. signals at ∼130 ppm, are almost equal in all four samples, the double bond signals of the two carbon atoms increase in intensity with increasing content of cysteine. This confirms our assumption that the adjacent amino group interferes with the thiol–ene reaction.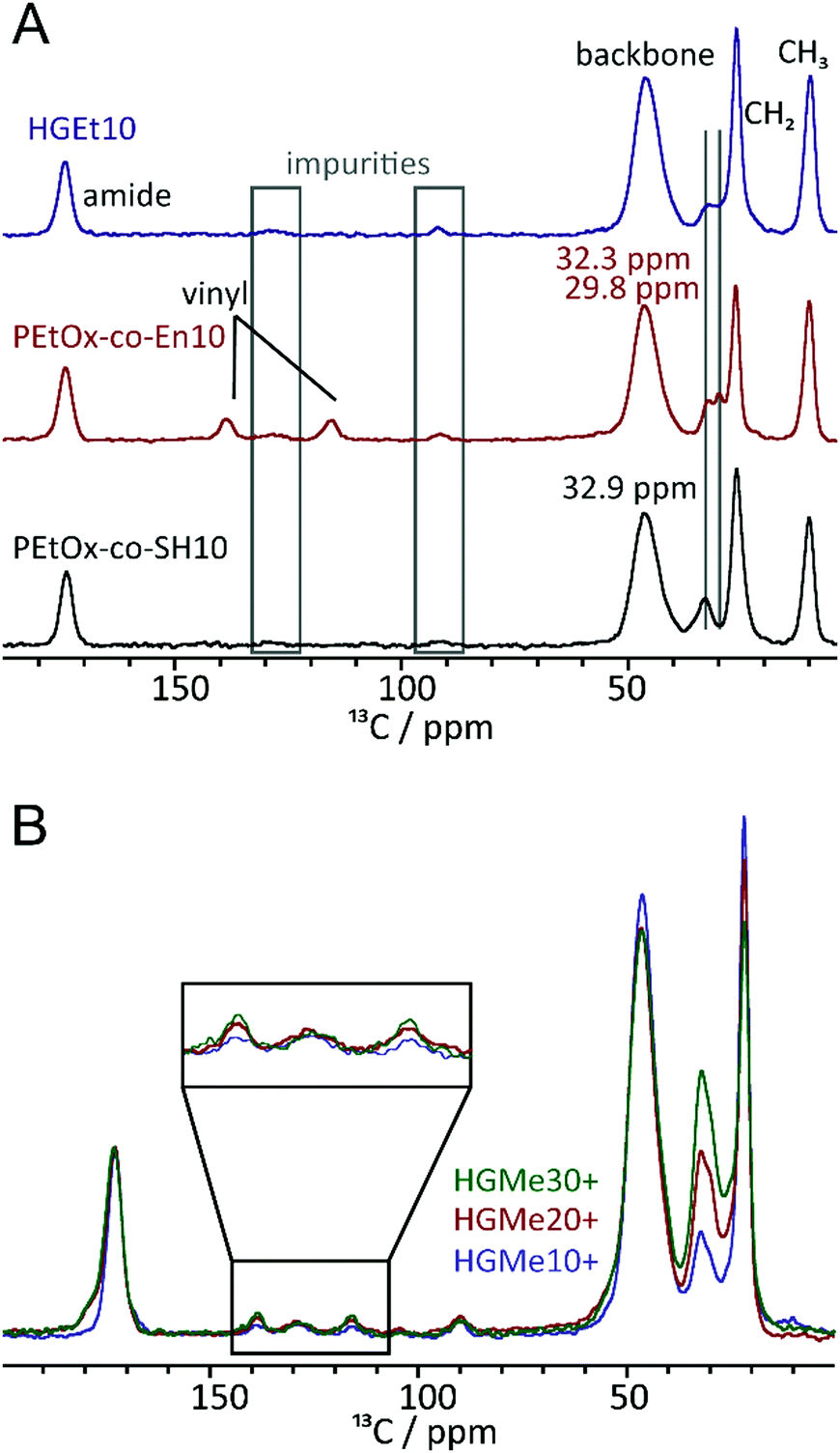 | ||
| Fig. 6 (A) 1H–13C CP/MAS solid-state NMR spectra of the cross-linked hydrogel HGEt10 (blue) as well as the two precursor polymers with thiol and vinyl units and (B) 13C CP/MAS solid-state NMR spectra of HGMe10+, HGMe20+ and HGMe30+. All experiments were recorded at 14.1 T and 12.5 kHz MAS. For the CP/MAS experiments, a contact time of 2 ms was used. | ||
3.5 Mechanical properties
As an additional characteristic of the formed hydrogel networks, the Young's modulus (also the compressive modulus) of all hydrogels was measured over the course of two weeks and was found to have reached its final value already after an equilibrium swelling time of 24 h due to the absence of degradable bonds. The values obtained after swelling in PBS for 24 h, which were determined by means of a mechanic testing device (22 N load, BOSE 5500, TA Instruments), are shown in Fig. 7. The arrows in Fig. 7 were added to illustrate the correlation between increasing cross-linking degree and decreasing swelling degree. It becomes clear that an increase in cross-linking density induces not only a decrease in swelling but also an increase in compressive strength and therefore mechanical stability. On the other hand, hydrogels based on the more hydrophilic backbone, i.e. MeOx, show a lower compressive modulus compared to hydrogels based on EtOx at the same cross-linking density. The Young's moduli for thiol–ene cross-linked hydrogels with the lowest cross-linking degree of 10 mol% were in the range of 100 kPa to 150 kPa, with the hydrogel HGEt10_4 exhibiting the lowest value as it also had the highest swelling degree. Hydrogels with the highest cross-linking degree of 20 mol%, HGMe20, HGEt20 and HGEt20_4, showed values between 240 kPa and 380 kPa, with HGMe20 having the lowest modulus, which is attributed to the fact the hydrophilic monomer MeOx strongly favors the binding of water to the polymer chains. The Young's modulus of HGEt20_4 did not change significantly compared to that of HGEt20 at 37 °C even though this hydrogel exhibited nearly a two times higher swelling degree. The same trend applies when comparing the mechanical strength of HGEt10_4 to that of HGEt10 at 37 °C. One explanation could be that at this point the hydrogel network is so dense that its mechanical properties are not much changed by an additional amount of water bound to the polymer chains. So far, POx hydrogels based on cross-linking via small dithiol molecules have been shown to exhibit compressive moduli between 6 and 130 kPa,18,21 with only one copolymer, PMeOx190-co-ButEnOx10 cross-linked with DTT, showing a compressive modulus of approximately 200 kPa in a 1:1 ratio of thiol:ene.18 Our findings show that the mechanical strength can be increased by employing side functionalized copolymers and can be finely tuned by varying the hydrophilicity and the functionalization degree.
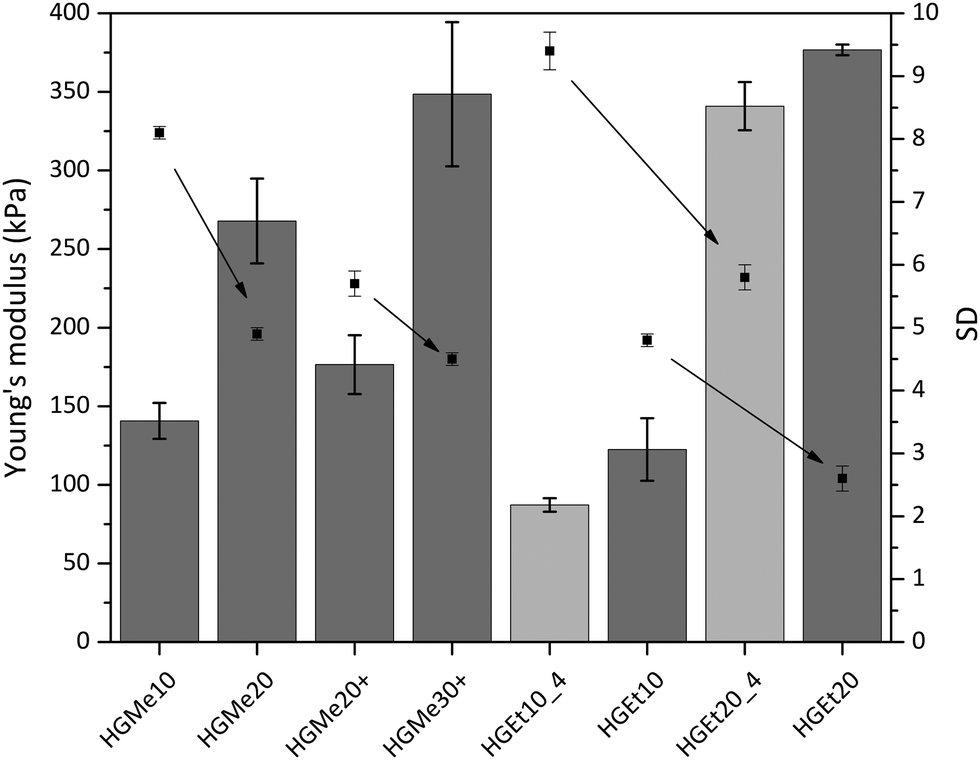 | ||
| Fig. 7 Young's moduli (columns) and swelling degrees (points) of thiol and cysteine (+) cross-linked hydrogels after 24 h incubation in PBS at 4 °C (light grey) or 37 °C (dark grey); arrows were added between the same hydrogel types with increasing cross-linking degree to show the correlation between the cross-linking degree and the swelling degree. | ||
3.6 Flory–Rehner analysis
The exact values of the mesh size determined by Flory–Rehner analysis can be found in Table S6, ESI.† The mesh size varies between 1 nm and 2.5 nm, with the highest values for hydrogels with the lowest degree of cross-linking and based on the more hydrophilic backbone. The degree of cross-linking follows the same trend ranging from 0.020 to 0.060 for HGMe10 to HGEt20. Dargaville et al. also compared copolymers based on MeOx and EtOx and observed the same trends that hydrogels based on MeOx displayed larger mesh sizes than hydrogels based on EtOx and that the mesh size was influenced by the sites of cross-linking available.19 Concerning the application of these hydrogels for the release of guest molecules, we expect that low molecular weight compounds should be able to diffuse into the network and be released. However, the hydrodynamic radius of the protein drug aprotinin with a molecular weight of 7 kDa has been reported to be 3.0 nm and bovine serum albumin (BSA) has a hydrodynamic radius of 7.2 nm with a molecular weight of 66 kDa.39,40 Hence, we expect that drug delivery of larger molecules such as proteins would be rather difficult with this system as these macromolecules will be entrapped in the network.3.7 Release of FITC–dextran from thermo-responsive hydrogels
Fluorescein isothiocyanate labeled dextran molecules with molecular weights of 4, 40 and 500 kDa were loaded into HGEt10 and HGEt20 hydrogels prior to cross-linking and their release was detected via fluorescence spectroscopy over the course of 15 d. The HGEt20 hydrogel specimens were additionally incubated at 4 °C to determine if the increased swelling had an effect on the release of the dextran macromolecules. The results are displayed in Fig. S38 (ESI†). It could be observed that dextran with a molecular weight of 4 kDa was released to the largest extent (22–25% of the total amount), except for the hydrogel HGEt10 where the dextran with a molecular weight of 40 kDa was released the most (40% of the total amount). We have no explanation for this behavior. Otherwise, the dextran with 40 kDa released is ∼17% for HGEt20 at 37 °C and ∼10% for HGEt20 at 4 °C. The same trend can be observed for the dextran with 500 kDa. Here the release at 4 °C is also ∼10% and at 37 °C ∼17% with a delayed release profile. One would expect that the release is greater for the hydrogels stored at 4 °C as the degree of swelling is nearly twice as high. We suspect that the dextran was pushed out of the network during shrinkage from cold to warm temperatures as the hydrogels had to be synthesized in the cold to keep the polymer from precipitating and was afterwards incubated at 37 °C.Sigma-Aldrich states that the Stokes’ radius of the FITC–dextran with 4 kDa is 1.4 nm and that of the FITC–dextran with 40 kDa is 4.5 nm. This is in accordance with the finding that the FITC–dextran with 4 kDa can be released to some extent from the hydrogel network in contrast to the larger FITC–dextran macromolecules which seem to be entrapped. However, none of the dextran molecules were fully released, which indicates that these hydrogel networks are not very well suited for the release of high molecular weight molecules as long as no degradation motif is incorporated, which could result in a controlled release of the entrapped macromolecules.
3.8 Interaction with low molecular weight substances
Fluorescein sodium salt and methylene blue were chosen as model drugs for this release study. Fluorescein sodium salt provides a negative charge for physical interaction with the positively charged cysteine cross-linked network and methylene blue offers a positive charge, which should be actively repelled by the positively charged amino groups of the cysteine moiety. The existence of a positively charged hydrogel network was also confirmed by zeta potential measurements of ground HGMe20+ and HGMe30+ particles in water. The zeta potential was 10.4 ± 0.5 mV for HGMe20+ and 16.6 ± 1.9 mV for HGMe30+, which exceeded the values measured by Tan et al.,37 where the highest zeta potential measured was 6.15 ± 0.40 mV.We chose to incubate the hydrogels in highly concentrated dye solutions directly after preparation instead of adding the dyes to the precursor solution before gel formation. By this approach, we intended to prevent any influence on the network formation to ensure the comparability of the results with the gels prepared for the testing described above.
To test how a positively charged, less hydrophilic drug would be incorporated into the hydrogel system, we incubated all hydrogels in a highly concentrated methylene blue (MB) solution. 50 μL of the highly concentrated solution contained 2 mg of MB, defining the maximum uptake by one hydrogel specimen (volume of the silicon mold = 50 μL).
Fig. 8A shows the amounts of MB that were released after 48 h and Fig. 8B displays the release profiles. HGMe10, HGMe20+ and HGMe30+ released the highest amounts of MB (∼1.5 to 1.8 mg). In contrast, HGMe20, HGEt10 and HGEt20 only released up to 0.2 to 0.3 mg of the maximum 2 mg that were calculated for one hydrogel specimen according to the concentration of the dye solution. The release profile shows a burst release during the first 10 hours for all hydrogel types. However, MB was able to diffuse to a larger extent into the hydrogels, which were based on the more hydrophilic MeOx as the main monomer of the polymeric precursor, as the photographs of the hydrogels after 72 h (see the ESI,† Figure S39A) show. HGMe10 and HGMe20 are of the darkest color, but HGMe20 only released 20% of the amount that was released by HGMe10, which could be caused by its higher cross-linking density. HGMe20+ and HGMe30+ are of a lighter color than hydrogels based on MeOx without amino groups, with HGMe30+ being darker than HGMe20+, which could again be caused by its higher cross-linking density. The lighter color in combination with the high amount of MB that was released during the first 24 h can be explained by the additional positive charge in the hydrogel, which would on the one hand hinder the diffusion and on the other hand lead to a quick repulsion of the dye. Hydrogel networks consisting of the copolymer with EtOx were not able to take up MB and only small quantities were released. This observation is in accordance with the intensity of coloring of the hydrogels, which was rather weak in comparison to the HGMe10 and HGMe20 hydrogels.
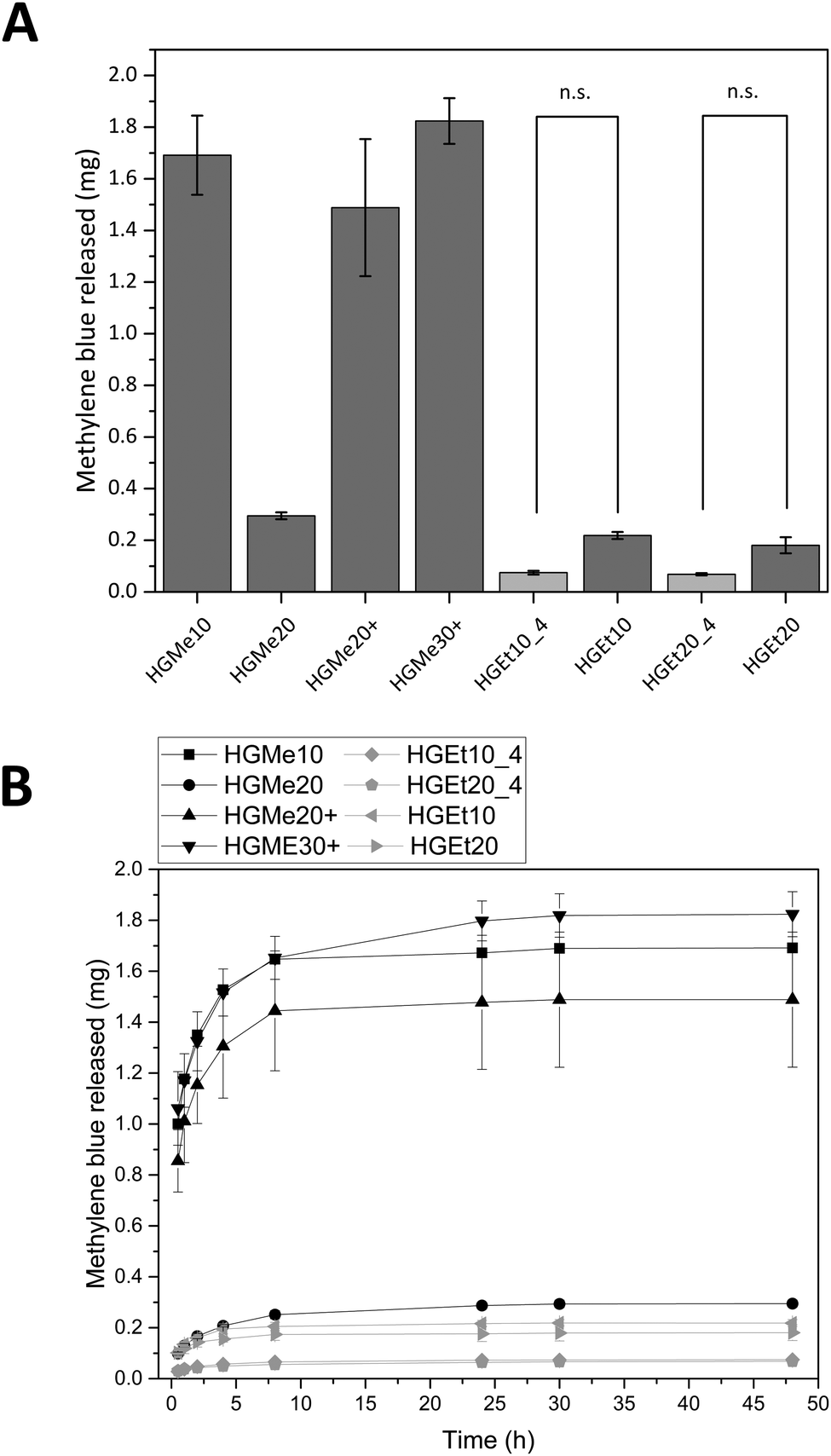 | ||
| Fig. 8 (A) Released masses of MB after 48 h and (B) release profiles during the first 48 h. There was always a significant difference between the hydrogel types (p < 0.05) unless otherwise indicated by n.s. (not significant). | ||
In order to trigger further release, the hydrogels were put into fresh PBS buffer after 24 h. A small increase in dye release was observed, which stopped after additional 24 h except for HGEt20_4 and HGMe20 hydrogels, which have the highest SD of all hydrogels with a cross-linking degree of 20 mol% and continued to release dye on the nanogram scale until day 14.
The general trend of this release study is that the cross-linking density plays a role in how rapidly MB is released and the hydrophilicity of the polymer precursor plays a major role in the uptake of MB. The additional positive charge in the hydrogel network results in a quick repulsion of the positively charged dye.
All hydrogels were also incubated in solutions of the negatively, hydrophilic model compound fluorescein (FSN) for 24 h. The concentration of FSN was chosen so that 50 μL would contain the maximum amount of drug that could interact with the amino groups available in one charged hydrogel specimen (volume of the silicon mold = 50 μL). This amount of FSN would be 3 mg for all hydrogel specimens except for HGMe30+, which had been incubated in a higher concentration of FSN as more amino groups are available. Here, the amount of FSN per HGMe30+ specimen would be 4 mg.
Fig. 9A shows the amounts of FSN that were released after 48 h and Fig. 9B shows the release profiles in the course of two weeks. All hydrogels based on the hydrophilic monomer MeOx release comparable amounts of FSN independent of the degree of cross-linking. The hydrogels based on the monomer EtOx show a temperature dependent release so that the hydrogels release more of FSN when they are incubated at 4 °C and the degree of swelling is greater. The lowest amount of FSN is released by HGEt20 at 37 °C, which can be explained by the dense network and the partial collapse of the network at elevated temperatures. The release profile shows that most of the dye is released during the first 24 h but a sustained release on the nanogram scale could be observed for all hydrogels until day 14. A photograph of the hydrogel specimens was again taken after 72 h, see the ESI,† Fig. S39B. Hydrogels with a positive charge, HGMe20+ and HGMe30+, show the most intense coloring, which can be explained by the additional ionic interaction and can lead to the assumption that more dye is bound to the hydrogel network.
 | ||
| Fig. 9 (A) Released masses of fluorescein sodium after 48 h, (B) release profiles during 14 d and (C) zoom in on the first 24 h. There was always a significant difference between the hydrogel types (p < 0.05) unless otherwise indicated by n.s. (not significant). | ||
As anticipated, the cross-linking density, which influences the degree of swelling, also influences the amount of FSN that is released, and in contrast to methylene blue, the hydrophilicity of the network seems to play a minor role for this highly water-soluble molecule. The high uptake of FSN is in accordance with the findings of Luef et al.,20 who loaded POx hydrogels with Eosin B, which is also a negatively charged drug with good water solubility, and observed a similar release behavior. Van der Heide et al.22 used the ability to stain POx hydrogels with Eosin Y to make hydrogel network structures visible under a fluorescence microscope.
4. Conclusions
In this study, we have presented hydrogels prepared from multifunctional POx-based building blocks via thiol–ene chemistry and their potential for loading and release of high and low molecular weight compounds. Copolymers PMeOx-co-En and PEtOx-co-En with increasing molar percentage of ButEnOx were synthesized and further functionalized with thiol groups or cysteine at the side chain after polymerization. UV-induced hydrogel formation in water revealed that the chemical environment of the thiol influences the conversion of the thiol–ene reaction and thus the extent of cross-linking. Hydrogels based on the same molarity of functional groups showed weaker cross-linking for cysteine functionalized than for thiol functionalized polymeric precursors. The hydrophilicity of the network as well as the cross-linking density strongly influenced the swelling behavior and the mechanical strength of the hydrogels. As anticipated, hydrogels based on the more hydrophilic monomer MeOx have the highest degree of swelling and hydrogels based on the less hydrophilic monomer EtOx show limited swelling at 37 °C due to their thermo-responsive behavior. The Young's moduli of the hydrogels fabricated were 100 to 380 kPa depending on the copolymer composition and degree of functionalization.All synthesized copolymers showed no cytotoxicity up to concentrations of 10 mg mL−1, except for thiol functionalized copolymers. However, all hydrogels displayed great cytocompatibility in direct cell contact up to seven days.
Flory–Rehner analysis and release experiments with fluorescently labeled dextran of different molecular weights showed that the formation of hydrogels using side chain functionalized POx as cross-linkers instead of small dithiol molecules creates dense networks, which are not suitable for the release of high molecular weight compounds as long as the network is not degradable. Nevertheless, the incorporation of degradable cross-links could combine the benefit of an effective entrapment with a controlled release of the high molecular weight compound.
Model drug release experiments with methylene blue (less hydrophilic and positively charged) and fluorescein (highly hydrophilic and negatively charged) showed that the dye diffusion into the hydrogel and the release strongly depend on the chemical environment of the hydrogel. Methylene blue was only taken up well by hydrogels based on MeOx and quickly released by the hydrogel containing positive charges due to the cysteine functionality. In contrast to this, fluorescein diffused well into all hydrogels, but was released to a larger extent by the hydrophilic hydrogel networks. A positive charge of the hydrogel resulted in a stronger adsorption of the negatively charged dye due to ionic interactions.
In summary, we demonstrated that mechanically strong hydrogels can be formed by using highly functional polymeric precursors made from poly(oxazoline)s and their covalent binding could be achieved via a very efficient radical thiol–ene reaction. The uptake and release properties of low molecular weight molecules can be fine-tuned in the following ways. The diffusion and release of the molecules can be influenced by the choice of monomer, leading to networks with different hydrophilicities and different thermo-responsive behaviors. In addition, increasing the cross-linking degree leads to a greater entrapment of the molecule keeping the possible drug at the desired site. In the future, the incorporation of degradable cross-links or functional side chains into polymers could lead to a more controlled and complete release of the hydrogel cargo.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council under grant agreement 617989 (Design2Heal) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 326998133 – TRR 225 (Sub Project A06).References
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, C. H. Abeln and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1895–1899 CrossRef CAS.
- A. Mero, G. Pasut, L. D. Via, M. W. Fijten, U. S. Schubert, R. Hoogenboom and F. M. Veronese, J. Controlled Release, 2008, 125, 87–95 CrossRef CAS PubMed.
- P. Goddard, L. E. Hutchinson, J. Brown and L. J. Brookman, J. Controlled Release, 1989, 10, 5–16 CrossRef CAS.
- F. C. Gaertner, R. Luxenhofer, B. Blechert, R. Jordan and M. Essler, J. Controlled Release, 2007, 119, 291–300 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- D. Christova, R. Velichkova, W. Loos, E. J. Goethals and F. D. Prez, Polymer, 2003, 44, 2255–2261 CrossRef CAS.
- P. Lin, C. Clash, E. M. Pearce, T. Kwei and M. Aponte, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 603–619 CrossRef CAS.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS PubMed.
- T. Lorson, S. Jaksch, M. M. Lübtow, T. Jüngst, J. R. Groll, T. Lühmann and R. Luxenhofer, Biomacromolecules, 2017, 18, 2161–2171 CrossRef CAS PubMed.
- M. Hartlieb, K. Kempe and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 526–538 RSC.
- Z. Zhu and X. Li, J. Appl. Polym. Sci., 2014, 131, 39867 Search PubMed.
- M. Hartlieb, S. Schubert, K. Kempe, N. Windhab and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 10–14 CrossRef CAS.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS PubMed.
- T. R. Dargaville, K. Lava, B. Verbraeken and R. Hoogenboom, Macromolecules, 2016, 49, 4774–4783 CrossRef CAS.
- T. R. Dargaville, R. Forster, B. L. Farrugia, K. Kempe, L. Voorhaar, U. S. Schubert and R. Hoogenboom, Macromol. Rapid Commun., 2012, 33, 1695–1700 CrossRef CAS PubMed.
- K. P. Luef, C. Petit, B. Ottersböck, G. Oreski, F. Ehrenfeld, B. Grassl, S. Reynaud and F. Wiesbrock, Eur. Polym. J., 2017, 88, 701–712 CrossRef CAS PubMed.
- P. Šrámková, A. Zahoranová, Z. Kroneková, A. Šišková and J. Kronek, J. Polym. Res., 2017, 24, 82 CrossRef.
- D. J. van der Heide, B. Verbraeken, R. Hoogenboom, T. R. Dargaville and D. K. Hickey, Biomaterials and Tissue Technology, 2017, 1, 1–5 Search PubMed.
- S. Stichler, T. Jungst, M. Schamel, I. Zilkowski, M. Kuhlmann, T. Bock, T. Blunk, J. Tessmar and J. Groll, Ann. Biomed. Eng., 2017, 45, 273–285 CrossRef PubMed.
- Z. He, A. Schulz, X. Wan, J. Seitz, H. Bludau, D. Y. Alakhova, D. B. Darr, C. M. Perou, R. Jordan and I. Ojima, J. Controlled Release, 2015, 208, 67–75 CrossRef CAS PubMed.
- Z. He, X. Wan, A. Schulz, H. Bludau, M. A. Dobrovolskaia, S. T. Stern, S. A. Montgomery, H. Yuan, Z. Li and D. Alakhova, Biomaterials, 2016, 101, 296–309 CrossRef CAS PubMed.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2012, 8, 659–671 CrossRef.
- M. Schmitz, M. Kuhlmann, O. Reimann, C. P. Hackenberger and J. Groll, Biomacromolecules, 2015, 16, 1088–1094 CrossRef CAS PubMed.
- M. Kuhlmann, O. Reimann, C. P. Hackenberger and J. Groll, Macromol. Rapid Commun., 2015, 36, 472–476 CrossRef CAS PubMed.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS PubMed.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. A. Leenen, M. A. Meier and U. S. Schubert, Macromolecules, 2005, 38, 5025–5034 CrossRef CAS.
- S. Cesana, A. Kurek, M. A. Baur, J. Auernheimer and O. Nuyken, Macromol. Rapid Commun., 2007, 28, 608–615 CrossRef CAS.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS.
- B. D. Fairbanks, D. M. Love and C. N. Bowman, Macromol. Chem. Phys., 2017, 218, 1700073 CrossRef.
- H. M. Lambermont-Thijs, J. P. Heuts, S. Hoeppener, R. Hoogenboom and U. S. Schubert, Polym. Chem., 2011, 2, 313–322 RSC.
- B. L. Farrugia, K. Kempe, U. S. Schubert, R. Hoogenboom and T. R. Dargaville, Biomacromolecules, 2013, 14, 2724–2732 CrossRef CAS PubMed.
- F. Tan, X. Xu, T. Deng, M. Yin, X. Zhang and J. Wang, Biomed. Mater., 2012, 7, 055009 CrossRef PubMed.
- B. Colak, J. C. Da Silva, T. A. Soares and J. E. Gautrot, Bioconjugate Chem., 2016, 27, 2111–2123 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2010, 31, 9527–9534 CrossRef CAS PubMed.
- D. Mohr, S. Frey, T. Fischer, T. Güttler and D. Görlich, EMBO J., 2009, 28, 2541–2553 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02575d |
| This journal is © The Royal Society of Chemistry 2019 |