
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible control of magnetism: on the conversion of hydrated FeF3 with Li to Fe and LiF†
Ruby
Singh
ab,
Ralf
Witte
a,
Xiaoke
Mu
a,
Torsten
Brezesinski
 a,
Horst
Hahn
ab,
Robert
Kruk
a and
Ben
Breitung
*ac
a,
Horst
Hahn
ab,
Robert
Kruk
a and
Ben
Breitung
*ac
aInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
bJoint Research Laboratory Nanomaterials, Technische Universität Darmstadt, Karlsruhe Institute of Technology (KIT), Otto-Berndt-Str. 3, 64287 Darmstadt, Germany
cKarlsruhe Nano-Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 30th August 2019
Abstract
Conversion-type electrode materials are prominent examples of potential Li-ion battery cathodes and/or anodes with large specific capacities. Conversion reactions often rely on complete reduction or oxidation of a transition metal, leading to multi-electron redox processes per formula unit, accompanied by changes of crystal structure and electronic configuration and, consequently, of materials properties such as magnetization. Here, we report on the utilization of the FeF3/Fe conversion reaction to reversibly control the magnetization. Hydrated FeF3 was chosen as electrode material and the changes in magnetization upon reversible redox reaction against Li counter electrode monitored by means of Superconducting Quantum Interference Device magnetometry. In situ measurements provide insights into the changes in magnetization occurring during the conversion reaction, specifically revealing the transition from paramagnetism to ferromagnetism.
1. Introduction
Materials with tailorable and tunable properties are receiving great interest in materials science. The possibility to tailor or tune properties is a desired feature for the development of functional materials for various applications. While tailoring means generating a specific structure or architecture of the material to achieve a certain property, the tuning concept addresses reversible changes of properties by means of an external control parameter. One possibility to reversibly change the characteristics of a material is to vary the magnetic interactions between electrons, thus leading to different types of magnetic responses in a magnetic field. This concept of magnetization tuning has been shown by, e.g., magnetoelectric coupling at ferromagnetic/ferroelectric interfaces.1–8 Additionally, electron- or hole-doping has been shown to modify the type of magnetism in La1−xSrxMnO3 (LSMO); doping can be achieved either by altering the elemental composition of LSMO (additional Sr leads to hole doping, resulting in a tailored structure) or by applying an electric field, which reversibly changes the carrier concentration in LSMO.9–14However, the tuning of LSMO using an external electric field constitutes only a surface effect depending on the affected magnetic depth.15 In order to address the bulk material, a different concept has been applied recently.16–19 For example, the magnetization of bulk γ-Fe2O3 could be tuned by reversible intercalation of Li+ into vacant sites of the lattice structure.20,21 This idea is derived from the concept of Li-ion batteries (LIBs), where active materials are reversibly de/lithiated over thousands of cycles and show distinct redox reactions depending on the applied potential. These redox reactions are often accompanied by changes in magnetization due to the transferred electrons. The most common electrode materials for LIBs are intercalation materials,22–26 where lithium is intercalated into a host structure but does not extensively change the crystal structure of the active material. Usually, only a fraction of the redox active ions in the active material is being reduced or oxidized (e.g., LiCo(III)O2/Co(IV)O2) to prevent material degradation due to changes in crystal structure. In contrast to intercalation materials, conversion materials are based on complete structural conversion. The reduction of, e.g., Fe(III)F3 to Fe(0) requires three electrons per formula unit and completely rebuilds the material. In the latter case, the conversion is accompanied by a transition of antiferromagnetic FeF3 to ferromagnetic Fe. Many different metal fluoride compounds are reported to be LIB cathode alternatives with high specific capacity but low reversibility resulting from the complex conversion reactions.27–30
In the present work, the concept of in situ monitoring the lithiation of an electrochemically active material is extended from the intercalation approach to the reversible conversion reaction of FeF3·3H2O (β-FeF3) (eqn (1)). The conversion of FeF3·3H2O to Fe delivers a specific capacity of 712 mA h g−1 owing to the three electron reduction.31–33 The conversion mechanism is a two-step process, comprising an insertion and a subsequent conversion reaction step.34,35
| Fe(III)F3·3H2O + Li → LiFe(II)F3·3H2O (insertion) | (1a) |
| LiFe(II)F3·3H2O + 2Li → 3LiF + Fe(0) + 3H2O (conversion) | (1b) |
These measurements were performed by coupling Superconducting Quantum Interference Device (SQUID) magnetometry with cyclic voltammetry (CV) measurements. A customized electrochemical cell using an ionic liquid as electrolyte was built to adapt to the conditions of the SQUID. The changes in magnetization during the reversible FeF3/Fe conversion were tracked and the features assigned to the respective increase and decrease of current during cycling operation. The magnetic data are supported by TEM, EELS and Mössbauer spectroscopy. Apart from the complete conversion, the insertion of Li into the FeF3·3H2O host structure has been studied as well.
2. Results and discussion
The changes in magnetization during the conversion reaction were monitored using combined CV/SQUID measurements. FeF3 and FeF3·3H2O are known conversion materials and subject of many studies (for application as potential cathodes in LIBs).36–40 The electrodes for the measurements were prepared using polyvinyl alcohol (PVA) as binder, since it is known to form a flexible polymer network. PVA is often used for conversion and alloying electrodes, as it can withstand volume expansions during the redox reactions. PVA is prepared as aqueous solution, therefore we chose the most hydrated FeF3 form as starting material to prevent changes during slurry preparation. Commercial FeF3·3H2O material was analyzed using Mössbauer spectroscopy and contains around 97% FeF3·3H2O and 3% FeF3 (Table 1).| Sample | IS (mm s−1) | QS (mm s−1) | B HF (T) | Γ (mm s−1) | Area (%) | |
|---|---|---|---|---|---|---|
| Pristine | Fe(III) doublet | 0.432 | 0.63 | — | 0.27 | 97 |
| Fe(III) sextet | 0.47 | 0.003 | 39.8 | 0.40 | 3 | |
| Discharged | Fe(III) | 0.47 | 0.54 | — | 0.47 | 23 |
| Fe(II) | 1.220 | 1.73 | — | 0.85 | 53 | |
| Fe(0) | 0* | — | — | 0.6 | 24 |
The sample for combined CV/SQUID measurements was prepared in a customized electrochemical cell using a mixture of FeF3·3H2O, conductive carbon black and PVA binder as electrode and Li foil as counter electrode. A detailed description of the slurry preparation can be found in the Experimental section. The measurement setup and cell used are described in more detail in Fig. S1.† CV measurements were performed while measuring the changes in magnetization at an applied field of 0.1 T. A considerably weak magnetic field was used to prevent side reactions that appeared when stronger fields were applied, most probably due to Fe(0) particles responding to the field and penetrating the separator, leading to short circuits. The redox reactions of FeF3·3H2O upon lithiation have been described in the literature (eqn (1)) and comprise insertion of Li into FeF3 and subsequent conversion to Fe.41 While the insertion reaction is reported to be reversible, the conversion reaction leads to accelerated degradation.42 These two mechanisms were independently monitored and Fig. 1 shows the evolution of the magnetic moment with cycling in the insertion regime between 2 and 4.5 V vs. Li+/Li.
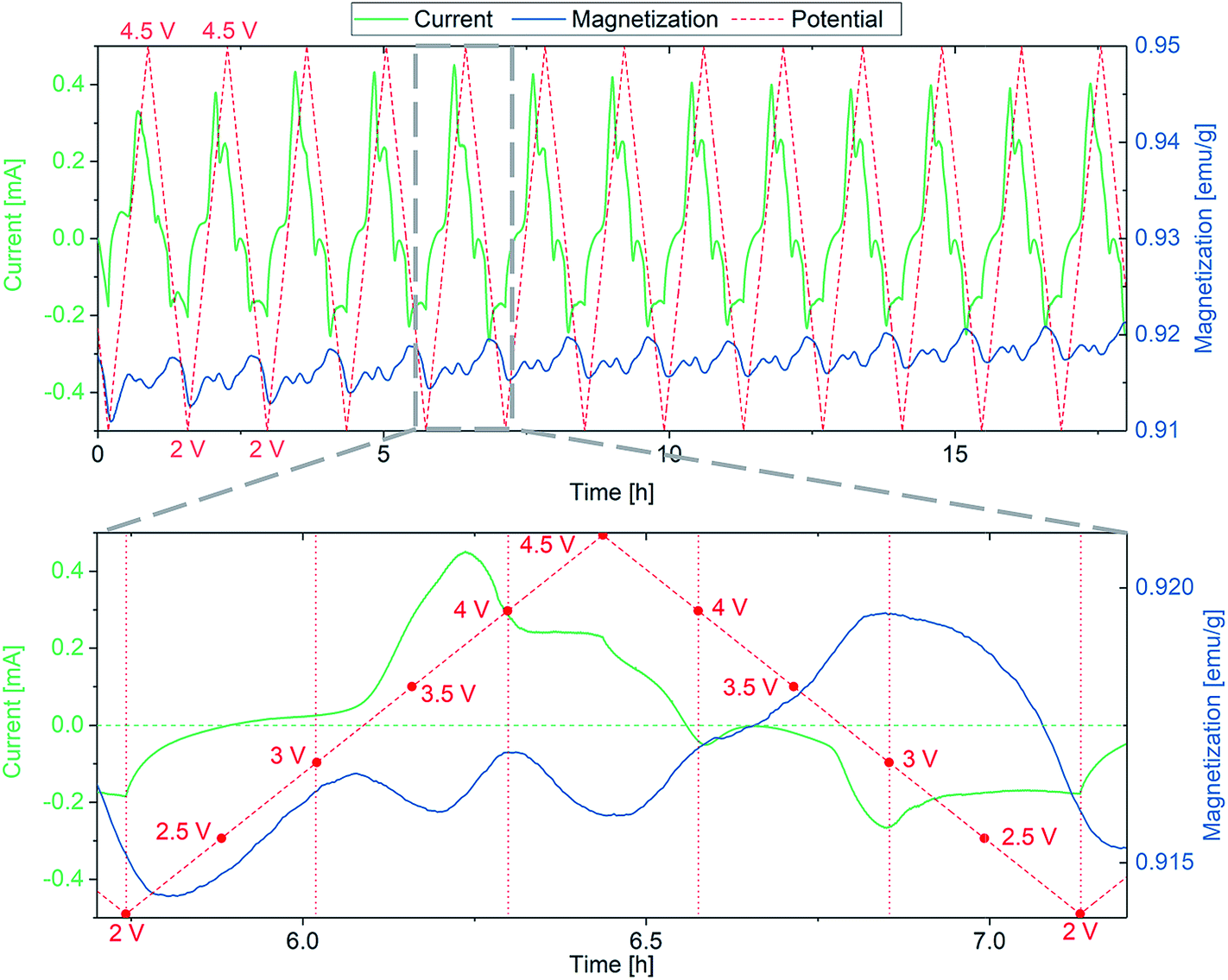 | ||
| Fig. 1 First 13 cycles of the FeF3·3H2O/Li insertion reaction (top) with a more detailed view of the 5th cycle (bottom). CV/SQUID measurements showing the evolution of magnetization with current and potential. The sweep rate was 1 mV s−1. The horizontal dashed green line in the bottom panel represents the zero line of current, while the vertical red dotted lines denote the potential at the respective time. Corresponding cyclic voltammograms can be found in the ESI (Fig. S2†). | ||
The insertion is described to be reversible, since, in contrast to a conversion reaction, the crystal structure undergoes minor changes.43 Therefore, the respective insertion reaction of FeF3·3H2O to LiFeF3·3H2O only results in a slight rearrangement of the structure.43 As discussed, the sample represents a mixture of about 3% FeF3 and 97% FeF3·3H2O, thus the changes in magnetic moment can be attributed to either of these species. FeF3 is known to exhibit antiferromagnetism at room temperature, whereas the zero-field-cooled/field-cooled (ZFC/FC) curves for FeF3·3H2O reveal a Néel temperature at around 140 K (Fig. S3†). Below this temperature, some antiferromagnetic orientations of spins are apparent (1-dimensional antiferromagnetism along the chains) and the behavior increases with decreasing temperature as the chains begin to couple.44 Density functional theory (DFT) calculations by Zheng et al. indicate an alternating change of the ground state magnetization between antiferromagnetism and ferrimagnetism depending on the lithiation level of FeF3.43 While LiFeF3, Li0.5FeF3 and FeF3 were found to be antiferromagnetic in nature, both Li0.75FeF3 and Li0.25FeF3 appear to show ferrimagnetic behavior. In our data, which were recorded at room temperature, we see a similar behavior, i.e., an alternating increase and decrease of magnetization depending on the lithiation degree. The insertion reactions seem to involve several steps, which are repeatedly changing the magnetization of the sample. The current progression and the respective changes in magnetization appear to be very reversible, therefore we conclude that the redox reactions do not result in major changes of the crystal structure (note that this would lead to material degradation). The magnetization after one complete cycle does not coincide with the magnetization before cycling; in fact, an increased value is found after each cycle. The increase is noticed in every cycle and may be the reason for the continuous growth of magnetization background with cycling operation. A possible explanation might be the slight change of structure when achieving a lithiation level of x > 0.5 in LixFeF3, which apparently is not be fully reversible. Yamakawa et al. reported a pronounced change in XRD pattern upon insertion of >0.5 Li in FeF3, resulting from the conversion of the FeF3 crystal structure (ReO3-type).45 Moreover, Doe et al. describe a possible precipitation of nanosized Fe when x > 0.5 (in LixFeF3), which may be the reason for the irreversible increase in magnetization.33,45 We assume that a combination of structural and electronic processes, appearing during the insertion processes, lead to the described increase in magnetization. Nevertheless, the clear reason for this result remains unclear at present.
Similar measurements have been performed between 1 and 4.5 V (vs. Li+/Li) to include the conversion reaction of FeF3·3H2O (eqn (1)). Fig. 2 shows the dependence of magnetization on the redox reactions occurring during this conversion, the change in magnetization being around 1000 times larger compared to the insertion regime. This is a clear indication for the conversion of the paramagnetic material to a ferromagnetic one.
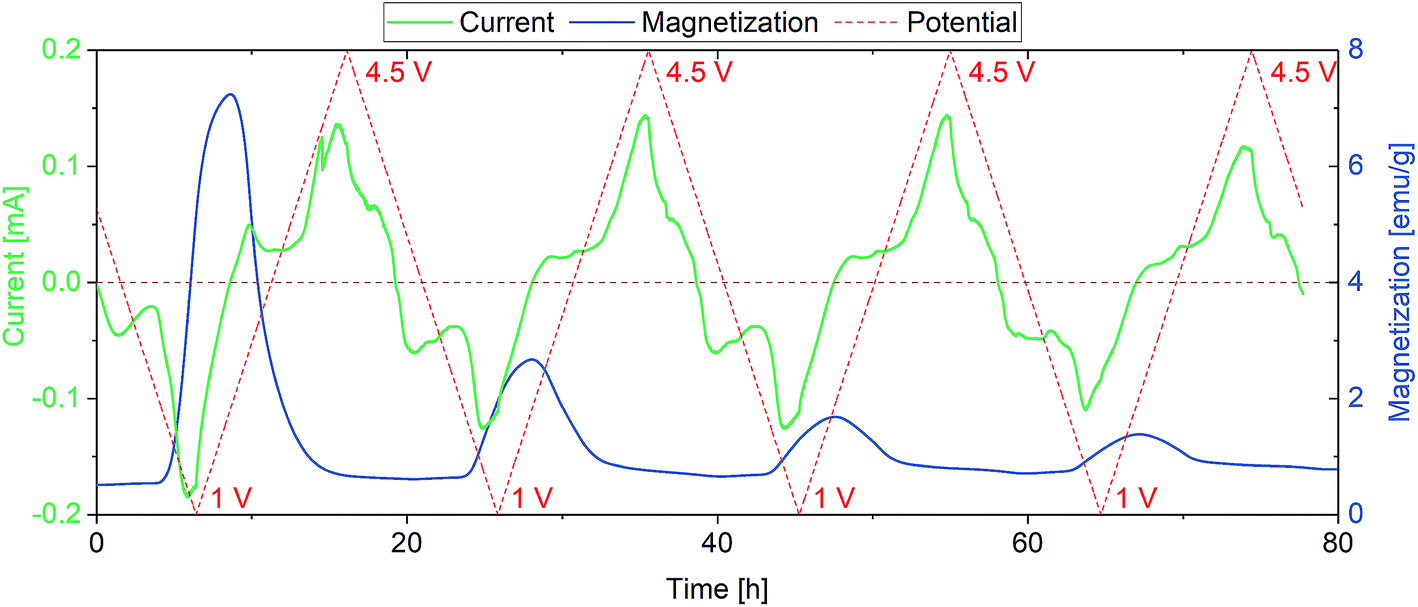 | ||
| Fig. 2 Conversion reaction of FeF3·3H2O. The sweep rate was set to 0.1 mV s−1 to account for the slow kinetics of conversion reactions in general. Magnetization degradation with cycling is observed, reflecting the decreasing appearance of Fe(0). | ||
As explained above, the overall reversibility of the FeF3·3H2O conversion reaction is reported to be relatively low due to structural changes. The magnetization changes during conversion are superimposing the weak insertion variations. The curves in Fig. 2 can be explained as follows: upon lithiation, FeF3·3H2O converts to Fe and LiF. The reduction reaction can be tracked by the increase in absolute current (green line, negative current values) and results in an increase in magnetization below the FeF3/Fe redox potential. The magnetization reaches its maximum when the potential is not sufficient anymore to reduce more FeF3 to Fe. With increasing potential, the current increases and when it becomes positive the reverse reaction (Fe + 3LiF → FeF3 + 3Li) takes place. This is accompanied by a decrease in magnetization. The highest slope of the magnetization increase is coinciding with the lowest potential, indicating the proceeding conversion reaction. The decreasing intensity of the magnetization change with cycling is related to the irreversibility of the conversion reaction. It appears that with every cycle less FeF3·3H2O can be converted to Fe, and therefore, the specific capacity decreases. This behavior is reflected as well by the current, showing decreasing values at low potential during cycling. This partial irreversibility of the FeF3 conversion reaction has been reported in several studies.35,36,46 Additionally, stepwise background increase of magnetization after each conversion reaction is observable. This indicates the presence of particles that remain in a ferri- or ferromagnetic state and do not participate in the reaction anymore. To further evaluate the magnetization, magnetization curves were recorded. The evolution of magnetization with increasing magnetic field is shown in Fig. 3, where the as-prepared material, the electrode at different lithiation states (2 V for insertion, 1 V for conversion) and at the higher cutoff potential (4.5 V) are compared with the empty electrochemical tuning cell.
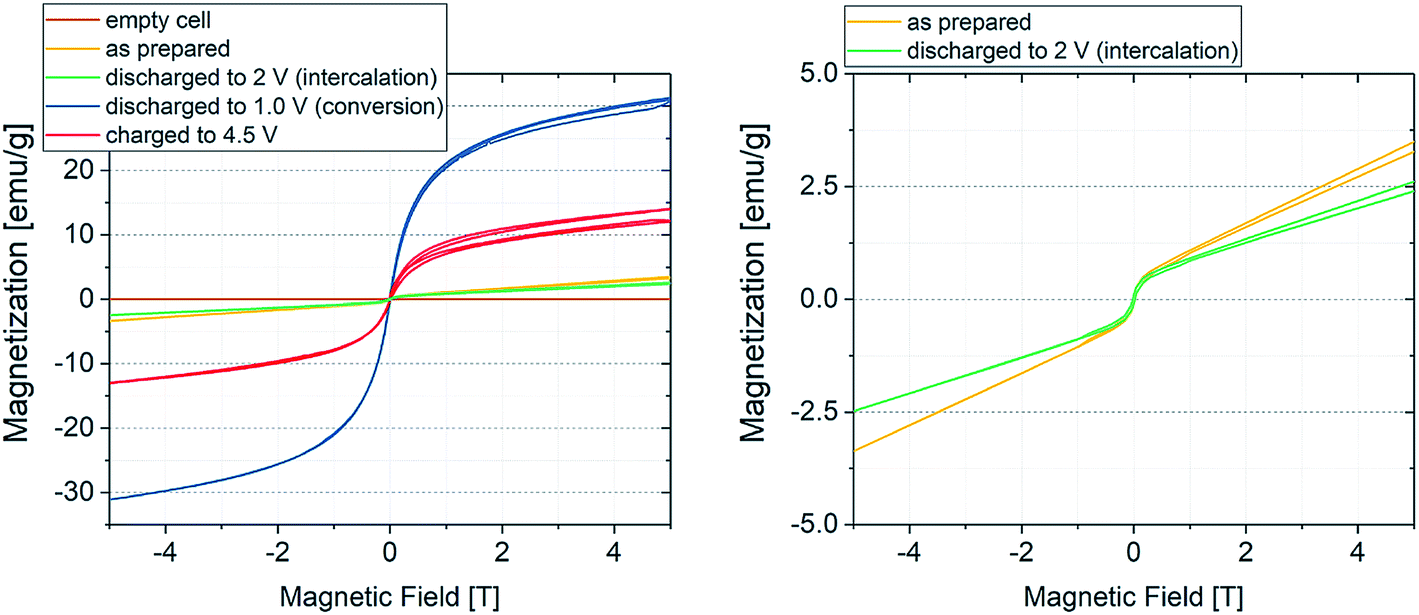 | ||
| Fig. 3 Room temperature magnetization curves of the sample at different potentials. The brown line represents the empty electrochemical tuning cell, while the blue and red curves display the magnetization of the material in its fully discharged (Fe + LiF) and fully charged (FeF3) state, respectively. The green curve denotes the material discharged to 2 V (insertion regime) and the yellow curve the as-prepared material. The cure splitting results from self-discharge during the measurement. Since the sample is not completely in equilibrium state, the open circuit potential slightly changes over time. This indicates that even small state of charge changes can be detected using coupled CV/SQUID measurements. | ||
The magnetization curves were measured to exclude a possible contribution of the electrochemical cell housing plus all integral parts such as wires, seals and connections to the measurement devices. Hereby, the as-prepared sample (yellow line) will first be lithiated (green curve, 2 V, insertion) and converted to the fully discharged state (blue curve, 1 V, conversion) with a subsequent delithiation to the charged state at 4.5 V (red curve). The brown line indicates that there is no significant contribution of the housing to the magnetization. The as-prepared material shows paramagnetic behavior, while the fully discharged (Fe + LiF) and fully charged (FeF3) states indicate cooperative effects, presumably ferromagnetic coupling. Note that the fully delithiated species can be also described as LiyFeFx, as many indications appear in the literature that FeF3 cannot be completely regained.33,41 Nevertheless, the weak ferromagnetic behavior is not expected in the fully charged state of the material. As mentioned, the evolution of magnetization in the CV/SQUID measurements shows a continuous increase in background with increasing cycle number. This may suggest that not every Fe particle formed during the lithiation process of the conversion reaction can be converted back during the delithiation process. These remaining Fe particles would show a ferromagnetic signature. Thus, the shape of curve for the fully charged sample likely results from an overlap of the ferromagnetic state of the remaining Fe particles and the state of the delithiated material.
Quantification of the Fe species (Fe(0), Fe(II), Fe(III)) present in the as-prepared and the fully lithiated species was performed using Mössbauer spectroscopy. Additionally, the magnetic states were investigated. The as-prepared sample shows a spectrum, which can be well represented with a doublet, with isomer shift (IS) of FeF3·3H2O (β-FeF3) and quadrupole splitting (QS) characteristic of Fe(III), being in good agreement with the literature (Fig. 4).47 An additional component with magnetic sextet can be attributed to antiferromagnetic FeF3, with a magnetic hyperfine field BHF of about 40 T. Table 1 summarizes the fitting results.48
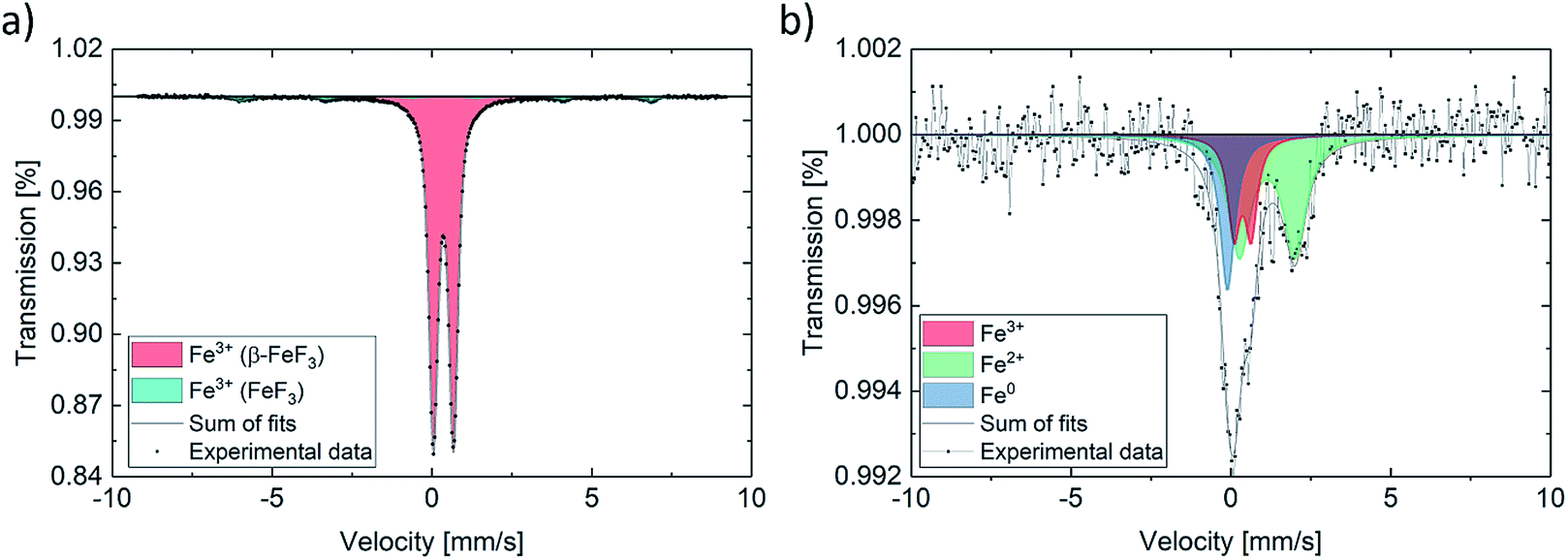 | ||
| Fig. 4 Mössbauer spectrum for (a) as-prepared powder and (b) fully lithiated material (initial discharge). | ||
In contrast, the discharged sample features a drastically altered spectrum, as expected based on the results of the changes observed in the magnetic properties during the electrochemical reduction process. The spectrum can be represented using three sub-spectra. A minor component with an IS and a QS typical of Fe(III), which is very similar to the pristine material, a distinctly split Fe(II) component and a singlet-like component with an IS being very close to what is expected for metallic Fe. The fit is a bit ambiguous due to the strong overlap of the sub-spectra, however, with additional evidence from TEM (next paragraph), it is reasonable to attribute the singlet component to reduced Fe(0). Fe(0) is present in a nanoparticulate form and therefore shows a singlet, characteristic of the superparamagnetic state. The latter interpretation is in agreement with the magnetometry data, which do not show the full magnetic moment of Fe(0). This can be well understood by the fact that the magnetization of Fe nanoparticles may not be fully saturated in the accessible magnetic field. The spectrum and its interpretation correspond well with what has been found by ex situ and operando Mössbauer experiments on a similar electrochemical cell.29,49 The quantification of Fe species in the discharged sample further shows that only some of the FeFx·3H2O can be converted to Fe(0) by using these measurement parameters. These results support the finding by other groups, describing the formation of differently structured FeF3 and LixFeF3 species instead of full conversion reaction to Fe(0) and FeF3.33 The magnetization further indicates that less of ferromagnetic Fe(0) is formed with increasing cycle number, which helps explain the low overall magnetization.
The expected saturation magnetization of Fe(0) amounts to 217.6 emu g−1.50,51 When compared to the measured values in Fig. 3, this value is not achieved. The sample shows a saturation magnetization of about 30 emu g−1 at 5 T. This difference can be explained by two effects. From the results of the Mössbauer measurements, the conversion reaction seems not to be complete, the amount of formed Fe(0) is smaller than the theoretical quantity calculated for a full conversion. This would lead to a lower absolute saturation magnetization. The second effect is related to the small particle size of the formed Fe(0). It is known that conversion reactions result in very small crystalline or amorphous nanoparticles, with sizes often being in the nm range.45 Particles of such small size typically only show a single magnetic domain and specific electron spin behavior, therefore they are superparamagnetic in nature. The superparamagnetic behavior has been described for Fe(0) nanoparticles of diameter around 10 nm.52–54
To analyze if the Fe(0) particles in the cycled sample have diameters in the lower nm region and therefore could show superparamagnetic behavior, TEM investigations have been performed. Fig. 5a shows a HAADF-STEM micrograph of the fully discharged sample, with the SAED indicating an fcc structure, most probably that of LiF (Fig. 5b).
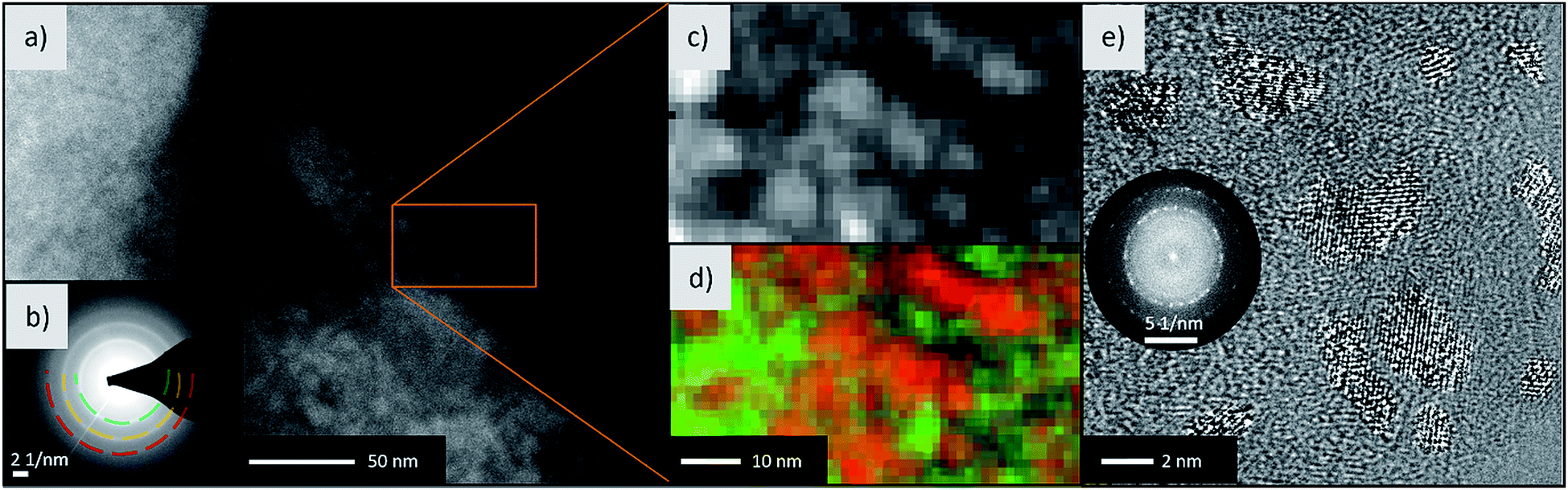 | ||
| Fig. 5 Electron microscopy of the discharged material. (a) STEM-HAADF micrograph with (b) the SAED pattern of the lithiated sample. The broad ring of d-spacing 2.34 Å can be attributed to the (111) plane of LiF. (c) Enlarged micrograph of the area denoted by the rectangular box and (d) the corresponding elemental map for Li+ (green) and Fe atoms (red) from EELS (Li–K and Fe–M edge). (e) HR-TEM micrograph and FFT of the shown crystallites (contrast enhanced to make the crystallites better visible). | ||
To identify the elements, EELS has been conducted on the same area (Fig. 5c and d), revealing anticorrelation of the spatial distribution of Li (green) and Fe (red). The Fe(0) size has been determined by HR-TEM (Fig. 5e). In fact, particles having diameters between 5 and 10 nm and a bcc structure were found. Consequently, the size dependent transition from ferromagnetic to superparamagnetic state during the conversion reaction is a possible scenario and helps explain the magnetization in Fig. 3. For identification of Li and Fe, the Li–K and Fe–M edges located between 55 to 75 eV were used. These edges were chosen to prevent beam damage on beam sensitive LiF and to still obtain good signal-to-noise ratios. Because the F–K edge is located at 685 eV and the EELS signal reduces exponentially with higher energies, a 50 times higher dose is required to map the F in LiF. During our experiments, this led to severe beam damage on the LiF particles. Thus, we determined LiF and Fe via SAED and showed the anticorrelation of Li and Fe with EELS. Taken together, this demonstrates the presence of Fe and LiF in the discharged state of the electrode.
3. Experimental
Sample preparation
The FeF3·3H2O/carbon C65 nanocomposite was obtained by ball-milling a FeF3·3H2O (Alfa Aesar)/carbon black (C65) (Timcal) mixture at 500 rpm for 3 h using a Retsch PM 100 planetary ball mill with WC vials and a ball-to-powder ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Afterwards, the slurry was prepared by mixing the ball-milled sample with carbon C65 additive and an aqueous Selvol 425 PVA binder solution (70:20:10 weight ratio). The resulting slurry was pasted onto Cu foil (Gould Electronics) by doctor-blading and then dried in a vacuum oven at 80 °C for 12 h. The active material loading varied from 0.35 to 2 mg cm−2, depending on the measurement type (M/H-curves, CV/SQUID measurements).
:1. Afterwards, the slurry was prepared by mixing the ball-milled sample with carbon C65 additive and an aqueous Selvol 425 PVA binder solution (70:20:10 weight ratio). The resulting slurry was pasted onto Cu foil (Gould Electronics) by doctor-blading and then dried in a vacuum oven at 80 °C for 12 h. The active material loading varied from 0.35 to 2 mg cm−2, depending on the measurement type (M/H-curves, CV/SQUID measurements).
Cell assembly
An electrochemical tuning cell (Fig. S1†) was used for combined CV/SQUID measurements. Therefore, the prepared electrode, glass fiber separator (GF/A, Whatman) and lithium foil (China Lithium Ltd) as counter electrode were stacked and assembled in the cell. LiTFSI (20 wt%, Sigma Aldrich) in EMIM-TFSI (Sigma Aldrich) was used as electrolyte to adapt to the SQUID measurement conditions (no organic solvent could be used, since accidental evaporation would lead to cell failure and contamination of the magnetometer).Instrumentation
Potentiostatic measurements were performed using a μAutolab Type 3 (PGSTAT12/30/302) coupled with an MPMS3 SQUID with vibrating sample magnetometer (VSM). S/TEM was performed on a Titan 80-300 (FEI) operated at 300 kV equipped with imaging aberration (Cs) corrector and Gatan GIF Tridium 863 energy filter. 57Fe Mössbauer spectra were recorded using a spectrometer in transmission geometry with a moving source of 57Co in a Rh matrix and a triangular velocity variation. Care was taken to seal the sensitive samples into a plastic bag inside an argon-filled glove box to avoid oxidation. The isomer shift is given relative to bcc-Fe at room temperature.4. Conclusions
The impact of redox reactions on the magnetization during conversion of FeF3·3H2O to Fe has been successfully studied. While the insertion reaction is reversible, the conversion reaction is found to trigger severe degradation processes and irreversible reactions. The insertion reaction leads to different magnetic states of differently lithiated LixFeF3 species. The conversion of paramagnetic FeF3·3H2O to ferromagnetic Fe(0), leading to major changes in magnetization, could be clearly tracked at low cell potentials. The saturation magnetization at larger magnetic fields can be explained by incomplete conversion reactions and by the small particle size (<10 nm), leading to superparamagnetism. The presence of crystalline Fe(0) particles of diameter around 10 nm was observed by means of HR-TEM, SAED and EELS. Furthermore, Mössbauer spectroscopy confirmed the formation of superparamagnetic Fe(0) particles during the conversion reaction. These combined CV/SQUID measurements demonstrate that the reversibility issues, which have been reported for conversion-based FeF3·xH2O materials in electrochemical energy storage applications, also appear in the magnetic tuning experiments and can be analyzed in detail by monitoring the magnetization changes with cycling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
One of the authors acknowledges the financial support by Deutsche Forschungsgemeinschaft under contract number HA 1344/34-1 and DA 1781/1-1. The authors express their special thanks to Prof. Subho Dasgupta, Indian Institute of Science, Bangalore for fruitful discussions.References
- P. M. Leufke, R. Kruk, R. A. Brand and H. Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 1–9 CrossRef.
- X. Chen, X. Zhu, W. Xiao, G. Liu, Y. P. Feng, J. Ding and R. W. Li, ACS Nano, 2015, 9, 4210–4218 CrossRef CAS.
- G. Wei, L. Wei, D. Wang, Y. Tian, Y. Chen, S. Yan, L. Mei and J. Jiao, RSC Adv., 2017, 7, 2644–2649 RSC.
- T. Yamada, K. Morita, K. Kume, H. Yoshikawa and K. Awaga, J. Mater. Chem. C, 2014, 2, 5183–5188 RSC.
- Y. Kasahara, T. Nishijima, T. Sato, Y. Takeuchi, J. Ye, H. Yuan, H. Shimotani and Y. Iwasa, J. Phys. Soc. Jpn., 2011, 80, 1–4 CrossRef.
- J. Carvell, E. Ayieta, A. Gavrin, R. Cheng, V. R. Shah and P. Sokol, J. Appl. Phys., 2010, 107, 103913 CrossRef.
- V. Sivakumar, S. Kumar, C. Ross and Y. Shao-Horn, ECS Trans., 2007, 2, 1–11 Search PubMed.
- N. A. Chernova, M. Ma, J. Xiao, M. S. Whittingham, J. Breger and C. P. Grey, Chem. Mater., 2007, 19, 4682–4693 CrossRef CAS.
- A. Molinari, P. M. Leufke, C. Reitz, S. Dasgupta, R. Witte, R. Kruk and H. Hahn, Nat. Commun., 2017, 8, 1–9 CrossRef.
- C. Reitz, D. Wang, D. Stoeckel, A. Beck, T. Leichtweiss, H. Hahn and T. Brezesinski, ACS Appl. Mater. Interfaces, 2017, 9, 22799–22807 CrossRef CAS.
- S. Dong, R. Yu, S. Yunoki, G. Alvarez, J. M. Liu and E. Dagotto, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 1–4 Search PubMed.
- F. Matsukura, Y. Tokura and H. Ohno, Nat. Nanotechnol., 2015, 10, 209–220 CrossRef CAS.
- A. Rocher, O. Durand, J. Maurice, F. Pailloux, A. Barthe, R. Lyonnet and J. Contour, Appl. Surf. Sci., 2002, 188, 176–181 CrossRef.
- X. Hong, A. Posadas, A. Lin and H. Ahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 1–5 Search PubMed.
- A. Molinari, H. Hahn and R. Kruk, Adv. Mater., 2018, 1703908 CrossRef.
- C. Reitz, C. Suchomski, D. Wang, H. Hahn and T. Brezesinski, J. Mater. Chem. C, 2016, 4, 8889–8896 RSC.
- L. A. Dubraja, C. Reitz, L. Velasco, R. Witte, R. Kruk, H. Hahn and T. Brezesinski, ACS Appl. Nano Mater., 2018, 1, 65–72 CrossRef CAS.
- C. Navarro-Senent, A. Quintana, E. Menéndez, E. Pellicer and J. Sort, APL Mater., 2019, 7, 030701 CrossRef.
- Q. Zhang, X. Luo, L. Wang, L. Zhang, B. Khalid, J. Gong and H. Wu, Nano Lett., 2016, 16, 583–587 CrossRef CAS.
- S. Dasgupta, B. Das, M. Knapp, R. A. Brand, H. Ehrenberg, R. Kruk and H. Hahn, Adv. Mater., 2014, 26, 4639–4644 CrossRef CAS.
- S. Dasgupta, B. Das, Q. Li, D. Wang, T. T. Baby, S. Indris, M. Knapp, H. Ehrenberg, K. Fink, R. Kruk and H. Hahn, Adv. Funct. Mater., 2016, 26, 7507–7515 CrossRef CAS.
- L. de Biasi, B. Schwarz, T. Brezesinski, P. Hartmann, J. Janek and H. Ehrenberg, Adv. Mater., 2019, 31, 1900985 CrossRef.
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 CrossRef CAS.
- X. Lou, R. Li, X. Zhu, L. Luo, Y. Chen, C. Lin, H. Li and X. S. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 6089–6096 CrossRef CAS.
- Q. Fu, R. Li, X. Zhu, G. Liang and L. Luo, J. Mater. Chem. A, 2019, 19862–19871 RSC.
- X. Zhu, J. Xu, Y. Luo, Q. Fu, G. Liang, L. Luo, Y. Chen, C. Lin and X. S. Zhao, J. Mater. Chem. A, 2019, 7, 6522–6532 RSC.
- M. Nishijima, I. D. Gocheva, S. Okada, T. Doi, J. Ichi Yamaki and T. Nishida, J. Power Sources, 2009, 190, 558–562 CrossRef CAS.
- F. Badway, N. Pereira, F. Cosandey and G. G. Amatucci, J. Electrochem. Soc., 2003, 150, A1209 CrossRef CAS.
- A. Pohl, M. Faraz, A. Schröder, M. Baunach, W. Schabel, A. Guda, V. Shapovalov, A. Soldatov, V. S. K. Chakravadhanula, C. Kübel, R. Witte, H. Hahn, T. Diemant, R. J. Behm, H. Emerich and M. Fichtner, J. Power Sources, 2016, 313, 213–222 CrossRef CAS.
- X. Xu, S. Chen, M. Shui, L. Xu, W. Zheng, J. Shu, L. Cheng, L. Feng and Y. Ren, Ionics, 2015, 21, 1003–1010 CrossRef CAS.
- T. Li, L. Li, Y. L. Cao, X. P. Ai and H. X. Yang, J. Phys. Chem. C, 2010, 114, 3190–3195 CrossRef CAS.
- X. Fan, Y. Zhu, C. Luo, T. Gao, L. Suo, S. C. Liou, K. Xu and C. Wang, J. Power Sources, 2016, 307, 435–442 CrossRef CAS.
- R. E. Doe, K. A. Persson, Y. S. Meng and G. Ceder, Chem. Mater., 2008, 20, 5274–5283 CrossRef CAS.
- Y. L. Shi, N. Wu, M. F. Shen, Y. L. Cui, L. Jiang, Y. H. Qiang and Q. C. Zhuang, ChemElectroChem, 2014, 1, 645–654 CrossRef.
- T. Takami, K. Matsui, H. Senoh, N. Taguchi, M. Shikano, H. Sakaebe and T. Fukunaga, J. Alloys Compd., 2018, 769, 539–544 CrossRef CAS.
- L. Liu, H. Guo, M. Zhou, Q. Wei, Z. Yang, H. Shu, X. Yang, J. Tan, Z. Yan and X. Wang, J. Power Sources, 2013, 238, 501–515 CrossRef CAS.
- L. Li, F. Meng and S. Jin, Nano Lett., 2012, 12, 6030–6037 CrossRef CAS.
- G. Ali, J. Lee, W. Chang, B.-W. Cho, H.-G. Jung, K.-W. Nam and K. Y. Chung, Sci. Rep., 2017, 7, 42237 CrossRef CAS.
- B. Breitung, M. A. Reddy, V. S. K. Chakravadhanula, M. Engel, C. Kübel, A. K. Powell, H. Hahn and M. Fichtner, Beilstein J. Nanotechnol., 2013, 4, 705–713 CrossRef CAS.
- M. A. Reddy, B. Breitung, V. S. K. Chakravadhanula, C. Wall, M. Engel, C. Kübel, A. K. Powell, H. Hahn and M. Fichtner, Adv. Energy Mater., 2013, 3, 308–313 CrossRef CAS.
- F. Cosandey, J. F. Al-Sharab, F. Badway, G. G. Amatucci and P. Stadelmann, Microsc. Microanal., 2007, 13, 87–95 CrossRef CAS.
- J. Lee and B. Kang, Chem. Commun., 2016, 52, 9414–9417 RSC.
- Y. Zheng, R.-F. Li, S.-Q. Wu, Y.-H. Wen, Z.-Z. Zhu and Y. Yang, Electrochemistry, 2013, 81, 12–15 CrossRef CAS.
- G. Nénert, O. Fabelo, K. Forsberg, C. V. Colin and J. Rodríguez-Carvajal, Dalton Trans., 2015, 44, 14130–14138 RSC.
- N. Yamakawa, M. Jiang, B. Key and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 10525–10536 CrossRef CAS.
- D. L. Ma, Z. Y. Cao, H. G. Wang, X. L. Huang, L. M. Wang and X. B. Zhang, Energy Environ. Sci., 2012, 5, 8538–8542 RSC.
- M. Burbano, M. Duttine, O. Borkiewicz, A. Wattiaux, A. Demourgues, M. Salanne, H. Groult and D. Dambournet, Inorg. Chem., 2015, 54, 9619–9625 CrossRef CAS.
- G. K. Wertheim, H. J. Guggenheim and D. N. E. Buchanan, Phys. Rev., 1968, 169, 465–470 CrossRef CAS.
- D. E. Conte, L. Di Carlo, M. T. Sougrati, B. Fraisse, L. Stievano and N. Pinna, J. Phys. Chem. C, 2016, 120, 23933–23943 CrossRef CAS.
- A. J. McGrath, S. Cheong, A. M. Henning, J. J. Gooding and R. D. Tilley, Chem. Commun., 2017, 53, 11548–11551 RSC.
- J. Crangle and G. M. Goodman, Proc. R. Soc. A, 2006, 321, 477–491 CrossRef.
- G. C. Papaefthymiou, Nano Today, 2009, 4, 438–447 CrossRef CAS.
- R.-P. Methling, V. Senz, E.-D. Klinkenberg, T. Diederich, J. Tiggesbäumker, G. Holzhüter, J. Bansmann and K. H. Meiwes-Broer, Eur. Phys. J. D, 2001, 16, 173–176 CrossRef CAS.
- D. Kumar, J. Narayan, A. Kvit, A. Sharma and J. Sankar, J. Magn. Magn. Mater., 2001, 232, 161–167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta08928d |
| This journal is © The Royal Society of Chemistry 2019 |