
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sulfonium tethered peptide ligand rapidly and selectively modifies protein cysteine in vicinity†
Dongyuan
Wang‡
a,
Mengying
Yu‡
a,
Na
Liu
a,
Chenshan
Lian
a,
Zhanfeng
Hou
a,
Rui
Wang
b,
Rongtong
Zhao
a,
Wenjun
Li
a,
Yixiang
Jiang
a,
Xiaodong
Shi
a,
Shuiming
Li
*c,
Feng
Yin
 *a and
Zigang
Li
*a
*a and
Zigang
Li
*a
aState Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, China. E-mail: lizg@pkusz.edu.cn; yinfeng@pkusz.edu.cn
bDepartment of Biomedical Sciences, City University of Hong Kong, Kowloon, Hong Kong. E-mail: rwang46@cityu.edu.hk
cCollege of Life Sciences and Oceanography, Shenzhen University, Shenzhen, 518055, China. E-mail: shuimingli@szu.edu.cn
First published on 25th March 2019
Abstract
Significant efforts have been invested to develop site-specific protein modification methodologies in the past two decades. In most cases, a reactive moiety was installed onto ligands with the sole purpose of reacting with specific residues in proteins. Herein, we report a unique peptide macrocyclization method via the bis-alkylation between methionine and cysteine to generate cyclic peptides with significantly enhanced stability and cellular uptake. Notably, when the cyclized peptide ligand selectively recognizes its protein target with a proximate cysteine, a rapid nucleophilic substitution could occur between the protein Cys and the sulfonium center on the peptide to form a conjugate. The conjugation reaction is rapid, facile and selective, triggered solely by proximity. The high target specificity is further proved in cell lysate and hints at its further application in activity based protein profiling. This method enhances the peptide's biophysical properties and generates a selective ligand-directed reactive site for protein modification and fulfills multiple purposes by one modification. This proof-of-concept study reveals its potential for further broad biological applications.
Introduction
Site selective protein conjugation provides a controllable tool to directly and precisely analyze protein functions in important cellular processes.1 Thus, multiple approaches have been developed to selectively modify endogenously reactive amino acid residues in proteins.2–5 Chemo-selective reactions on particular residues in proteins are broadly utilized, including cysteine,6–9 lysine,10–15 tyrosine,16 tryptophan,17 arginine18 and methionine.19,20 The development of chemical tools for site-selective protein labeling is in high demand due to its high precision and versatility. These tools include the genetic incorporation of unnatural amino acids within proteins equipped with “bioorthogonal” reactivity,21,22 genetic incorporation of peptide sequences (tag) for spatial recognition23 and the utilization of N-terminal/C-terminal sites for protein labeling.24Another alternative approach for achieving regio-selectivity is the ligand-induced protein conjugation which is mainly based on a precise spatial positioning between a functional group of a ligand and a reactive residue in protein. Meares et al. pioneered this concept in developing antibody conjugation with infinite affinity.25 Recently, Hamachi et al. established ligand-directed (LD) chemistry to specifically label a protein of interest (POI) in living cells.26,27 Howarth et al. reported the Spytag-Spycatcher system in which the 13-residue Spytag peptide spontaneously and selectively forms an isopeptide bond with the Spycatcher-tagged proteins.28 These ligand-directed approaches provide promising opportunities to balance the reactivity and selectivity for protein modification.
Regarding the selection of amino acid residues for ligand-induced protein conjugation, the high nucleophilicity and low abundance of cysteine in proteins make it a prime residue for selective protein conjugation. In general, cysteine residues were allowed to (i) react with electrophilic moieties such as haloalkyl or alkenyl groups; (ii) undergo metal assisted reactions and (iii) be converted into dehydroalanine for further modifications. The diverse methods for selective protein labeling have been widely used in the study of post-translational modifications (PTMs), cellular imaging, activity based protein profiling (ABPP) or covalent drug discovery.2,29–31 Notably, covalent inhibitors of key kinases, such as ibrutinib and rociletinib, were recently approved by the FDA as efficient cancer therapeutics.32–34
Peptides have been recently utilized as promising ligands for covalent protein modification due to their high binding affinity, selectivity and biocompatibility. For example, Xia et al. installed a mildly electrophilic α-chloroacetyl moiety onto peptide ligands for covalent conjugation with cysteine near protein–peptide interaction sites.35 Walensky and Fairlie et al. independently developed covalent BFL-1 inhibiting stapled peptides with an additional electrophilic warhead acrylamide.36–38 Wang et al. incorporated aryl sulfonyl fluoride (Ar-SO2F) in the SAHp53-8 peptide which interrupted p53–Mdm2/4 interactions.39 To the best of our knowledge, all reported methods require a pre-arranged reactive moiety solely for the purpose of conjugation. However, the reactive group either requires special steps to prepare it or may undergo non-specific reactions. For peptides such as the BFL-1 ligands, an additional stapling step is also necessary for enhanced stability and cellular uptake. Thus, we are seeking to develop a facile peptide cyclization method which could enhance the peptides' stability and cellular uptake, meanwhile the modification also generates a highly selective reaction site for reactive amino acid residues in protein.
Based on Deming's pioneering work of selective methionine alkylation, we recently developed a bisalkylation modification of Met to generate cyclic peptides with better cellular uptake and stability.40,41 Taking the current protein labelling demands into consideration, we envisioned that this method could be further developed into a novel and facile methodology to fulfil the multiple purposes mentioned above. As shown in Scheme 1, the bis-alkylation between Cys and Met could generate a cyclic peptide with a tuneable tether and an on-tether sulfonium center, which could help in improving the peptide's stability and cellular uptake. The labile sulfonium center may further undergo proximity promoted Cys substitution with the available Cys in the vicinity of the POI's binding pocket. Based on previous reports and our own findings, the sulfonium centers are stable in the presence of thiols for a reasonable time, indicating their potential cellular application.20,42
 | ||
| Scheme 1 Schematic representation of Cys–Met bis-alkylation and proximity-promoted protein labeling with a sulfonium tethered peptide. (A) Deming et al.'s report of selective methionine alkylation and our previous report of chemoselective methionine bis-alkylation. (B) Peptide macrocyclization through cysteine and methionine bis-alkylation. (C) Ligand induced protein conjugation using the Cys–Met macrocyclization method. | ||
Results and discussion
To meet the demands for both peptide stapling and protein modification, we first used a model hexapeptide (1) to assess the efficiency of peptide cyclization as shown in Fig. 1A. The peptides were constructed via conventional Fmoc-based solid phase peptide synthesis. The cyclization involved two steps: (1) the deprotection of Trt-protected cysteine and alkylation with di-halogenated linkers on resin; and (2) the cleavage of the peptide from the resin to give the resulting bis-alkylated cyclic peptides. The alkylation of Met occurred spontaneously during resin cleavage to generate the cyclic peptide 1-I with an 86% conversion based on HPLC integration. The formation of the cyclization product was further supported by LC-MS and 1H-NMR with a clear shift of the phenyl proton shown in Fig. S1.† We further confirmed that the cyclization process occurred during the resin cleavage step instead of the DIPE/DMF step shown in Fig. S2.† Notably, as the Met alkylation will generate a new on-tether chiral center, we successfully isolated two epimers as indicated in Fig. S3.† Then we tested the reaction efficiency of different di-alkylating linkers and found that the peptide (1) reacted smoothly with different linkers to provide products with high conversions as indicated in Fig. 1A and S3.† The epimer ratios of different linkers were generally ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and in some cases, the peptide epimers were not separable under our HPLC conditions (Fig. S3.†). Notably, the sulfonium chiral center of the purified epimer is not very stable and would slowly racemize into the initial epimer mixtures. In addition, the separable epimers showed similar secondary structures in CD spectroscopy measurements, suggesting that the chiral center had a limited effect on the peptide’s secondary structure (Fig. S4†). To assess the functional group tolerance of our method, ten peptides with different sequences were prepared as summarized in Fig. 1B. All peptides efficiently generated the corresponding cyclic peptides with high conversion rates.
:1 and in some cases, the peptide epimers were not separable under our HPLC conditions (Fig. S3.†). Notably, the sulfonium chiral center of the purified epimer is not very stable and would slowly racemize into the initial epimer mixtures. In addition, the separable epimers showed similar secondary structures in CD spectroscopy measurements, suggesting that the chiral center had a limited effect on the peptide’s secondary structure (Fig. S4†). To assess the functional group tolerance of our method, ten peptides with different sequences were prepared as summarized in Fig. 1B. All peptides efficiently generated the corresponding cyclic peptides with high conversion rates.
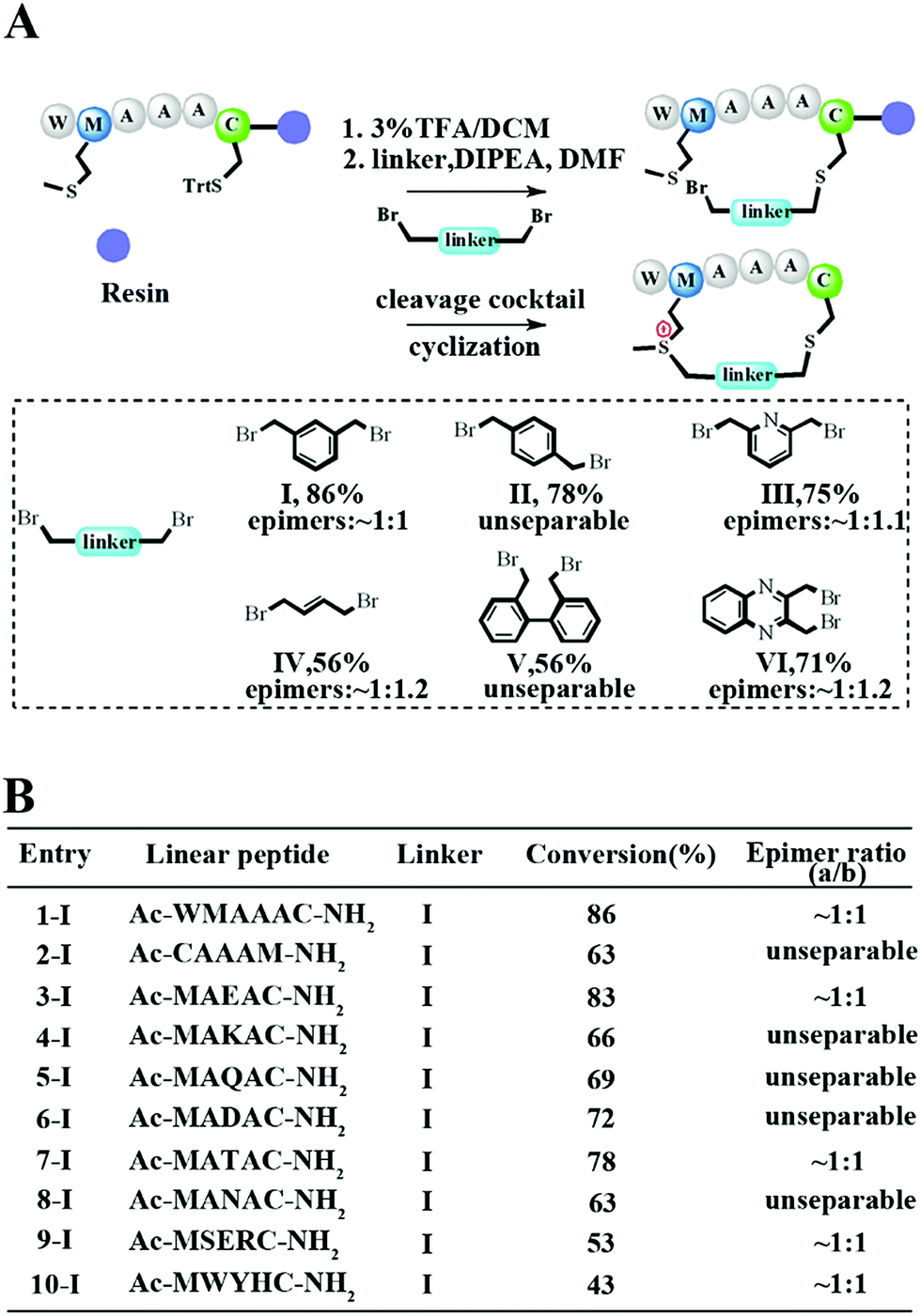 | ||
| Fig. 1 Facile construction of stabilized peptides by bisalkylation between Cys and Met. (A) Constrained peptide preparation and different linkers tested in this study. The Trt-protected cysteine was deprotected with 3%TFA in DCM until the yellow colour was no longer present. Then a di-halogenated linker (2.0 equiv.) was added with DIPEA (4.0 equiv.) in DMF and was left to react for 3 hours. Methionine alkylation and peptide cyclization were completed when the resin was cleaved in a TFA mixture (TFA:TIS:H2O = 95:2.5:2.5). The epimer ratio was calculated according to the HPLC traces. (B) Ten different peptides were tested for functional residue tolerance. | ||
The Met alkylation was reported to be reversible in the presence of appropriate reductants under mild conditions (pH 7.4 PBS, 37 °C).40 Among the reductants and nucleophiles tested in previous studies, GSH was reported to be the least reactive.40 We first tested the reaction rate of sulfonium tethered peptide 1-Ia (1 mM) with 2-mercaptopyridine (10 mM) in PBS buffer (pH 7.4) as shown in Fig. 2A. The LC-MS analysis clearly showed the time-dependent reduction of peptide 1-Ia (1 mM) with 2-mercaptopyridine (10 mM) in PBS (pH 7.4). We then tested the reaction rate of sulfonium tethered peptides 1-Ia and 1-Ib (1 mM) in the presence of different reductants (10 mM, 2-mercaptopyridine, thiourea or GSH) in PBS buffer (pH = 7.4) as shown in Fig. 2B and S5.† The reduction experiments were tracked by LC-MS at different time points and GSH was confirmed to be the weakest reductant among the tested reagents. The dealkylation efficiencies of different linkers and their epimers were further tested and are summarized in Fig. S6.† The peptide epimers showed briefly similar kinetics. Notably, peptide 1 equipped with linker V showed the quickest dealkylation rate (Fig. S6†). The sulfonium center on Met is stable in 20 mM thiourea as reported by Gaunt et al. The on-tether sulfonium centers shown in Fig. 2B showed similar stability.20 Serum stability of peptide 1 analogues was tested and the cyclic ones were found to have significantly enhanced serum stability (Fig. S7†). To test whether our tethering strategy could improve cellular uptake, peptide 11 with three arginine residues was prepared and reactions were performed with different linkers I–VI. The reactions went smoothly with high conversions and the epimers were separated if possible. A2780 cells (Fig. 2C) and 293 T cells (Fig. S8†) were treated with 10 μM FAM-labeled peptides for 4 hours and then incubated with 0.05% trypan blue before FACS analysis.43 In both A2780 and 293 T cell lines, the constrained peptides showed significantly increased cellular uptake compared to linear peptide 11. Confocal microscopy images of A2780 cells indicated the different cellular distributions of peptides with different linkers (Fig. S9†). To sum up, the model peptides constructed with this method showed enhanced stability and cellular uptake and could conjugate with physiologically relevant nucleophiles. Notably, the sulfonium center showed limited conjugation with GSH after 48 hours at physiologically relevant concentrations.
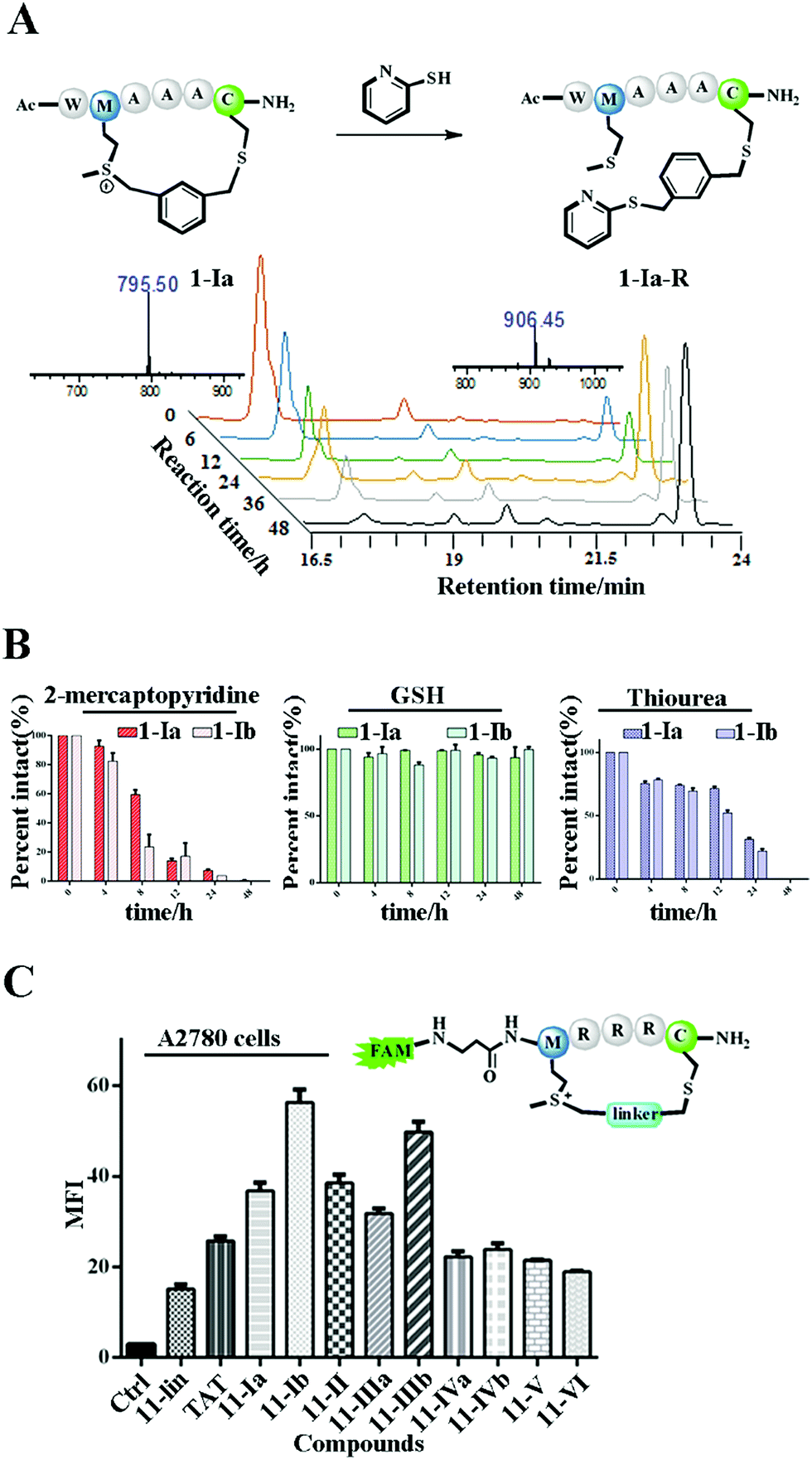 | ||
| Fig. 2 The biochemical properties of the constrained peptides. (A) Dealkylation of model peptide 1-Ia (1 mM) in the presence of PyS (10 mM) in PBS (pH = 7.4) at 37 °C for 48 hours. HPLC traces of the time-dependent conversion between peptide 1-Ia and its conjugated product 1-Ia-R. (B) The kinetics of peptide dealkylation reactions with different reductants under the same reaction conditions as indicated in (A). (C) Cellular uptake of the cyclic and linear peptides in A2780 cells treated with 10 μM FAM-labeled peptides for 4 hours. All cells were incubated with 0.05% trypan blue for 3 minutes before further analysis. | ||
To further prove the potential of this methodology for proximity induced cysteine modification, we constructed peptide ligands for PDZΔRGS3 as shown in Fig. 3A. PDZΔRGS3 (PDZΔRGS3 denotes the PDZ domain of PDZ-RGS3, and the protein sequence is shown in Fig. S10†) plays an important role in ephrin-B reverse signaling which is associated with SDF-1 (stromal derived factor 1) neuronal chemotaxis.44 PDZΔRGS3 was reported to be covalently labeled at a cysteine by a peptide ligand bearing a chloroacetamide moiety and we envisioned it as an ideal showcase for this proof-of-concept study.35 There are three Cys residues (C33, C34, and C73) in the vicinity of the peptide ligand binding site of PDZΔRGS3.35 To study the peptide selectivity and site selective cysteine conjugation of targets, a series of peptide ligands with different cyclization sites and different PDZΔRGS3 mutants were then prepared including PDZΔRGS3 C33SC34S and PDZΔRGS3 C73S (Fig. 3A and S10†). We first tested the peptides' binding affinity to PDZΔRGS3 and their mutants by fluorescence polarization assays shown in Fig. 3B and S11.† The peptides with different stapling sites showed quite different binding affinities, while the Cys–Ser mutations (PDZΔRGS3 C33S/C34S, PDZΔRGS3 C73S) appeared to have no obvious detrimental effects on peptide binding.
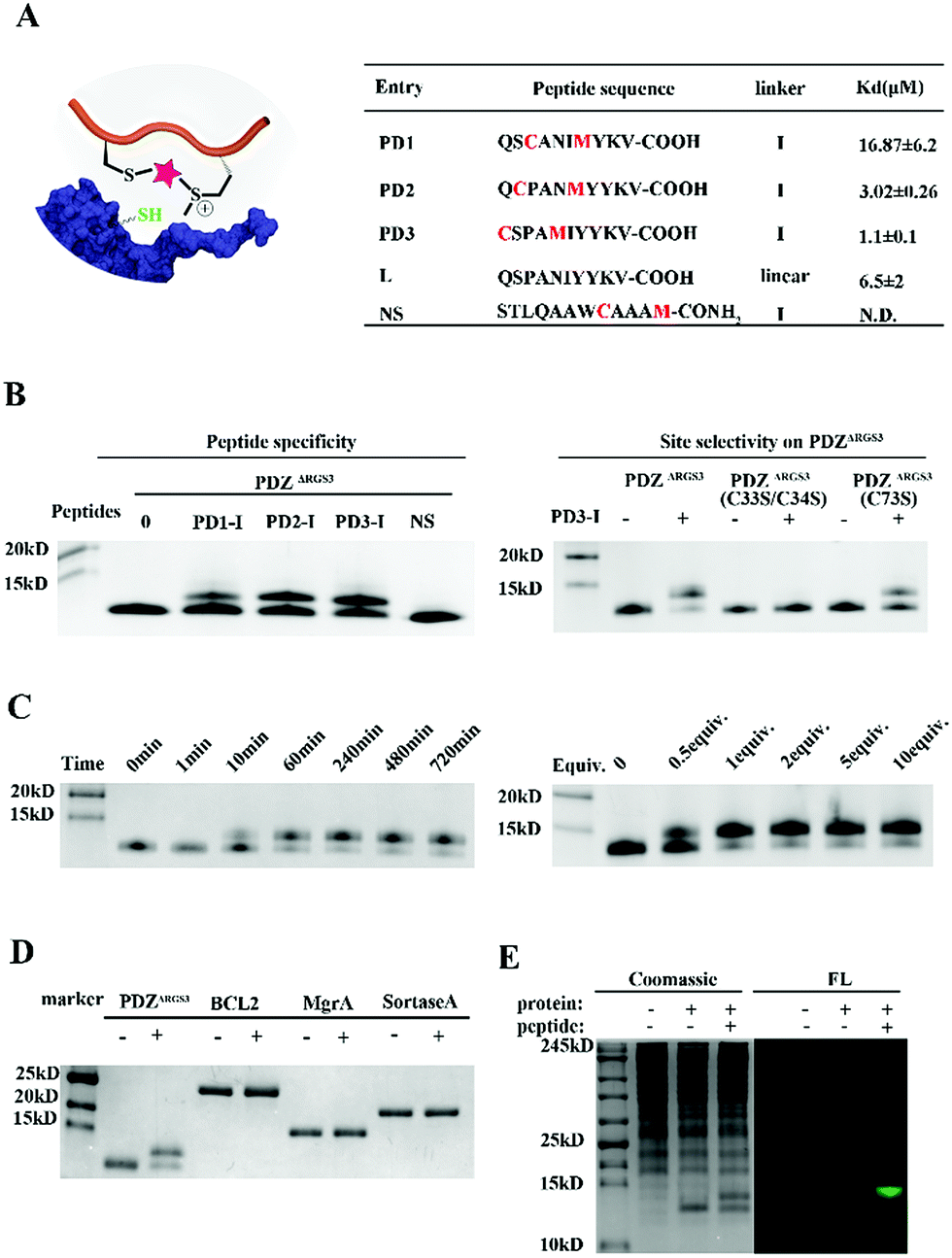 | ||
| Fig. 3 Design and synthesis of reactive peptide ligands for covalent PDZΔRGS3 conjugation (PDZΔRGS3 denotes the PDZ domain of PDZ-RGS3). (A) Peptide sequences designed for protein conjugation. (B) Peptide specificity and site selective cysteine conjugation of PDZΔRGS3. PDZΔRGS3 was reacted with different peptides (protein/peptide 20/100 μM, pH 7.4, 1 hour). PDZ mutants were incubated with peptide PD3-I (protein/peptide 20/100 μM, pH 7.4, 12 hours). (C) Reaction kinetics study of peptide PD3-I (protein/peptide 15/75 μM, pH 7.4) for 0 min, 1 min, 10 min, 60 min, 240 min, 480 min and 720 min, respectively. Stoichiometric study of peptide PD3-I from 0.5 equiv. to 10.0 equiv. by incubating with PDZΔRGS3 for 4 hours. (D) Ligand induced protein conjugation. Other proteins containing free Cys would not react with peptide PD3-I (protein/peptide 15/75 μM, pH 7.4, 12 hours). (E) FAM-labelled peptides (PD3-I, 50 μM) and PDZΔRGS3 (10 μg) were incubated with 293 T cell lysates (300 μg) for 24 hours. FL, in-gel fluorescence scanning. | ||
Then peptides PD1-I to PD3-I were reacted with PDZΔRGS3 in pH 7.4 PBS (protein/peptide 20/100 μM, 37 °C, 1 hour). As shown in Fig. 3B, peptide PD1-I with a binding affinity of 16.87 μM showed weak conjugation, while peptides PD2-I and PD3-I with a Kd of 3.02 μM and 1.1 μM showed rapid covalent conjugation. Rationally, the scrambled peptide NS with no binding affinity for PDZΔRGS3 couldn't conjugate with PDZΔRGS3, which further confirmed the concept of ligand-induced protein conjugation. The protein conjugates still retained their secondary structure as confirmed by circular dichroism (CD) and thermal shift assays as shown in Fig. S12.† We also found that the reaction efficiency was reduced with an excess of competitive linear peptides confirming the ligand-induced conjugation (Fig. S13†). We then tested the site selectivity of peptide PD3-I with wild type PDZΔRGS3 and its mutants. Peptide PD3-I showed the most efficient reaction with wild PDZΔRGS3, and a little weaker reaction with PDZΔRGS3 C73S but negligible reaction with PDZΔRGS3 C33SC34S (Fig. 3B). The MS data further confirmed the satisfactory covalent conjugation of PDZΔRGS3 C73S and poor covalent conjugation of the mutant PDZΔRGS3 C33SC34S (Fig. S14–S17†), indicating that the cysteine in the vicinity of the protein–peptide interaction site was essential for ligand conjugation. The reaction kinetics and stoichiometric study between PDZΔRGS3 and peptide PD3-I was then performed as shown in Fig. 3C. The reaction started within 10 minutes and went to an ∼80% conjugation within 60 minutes. One equiv. of peptide was briefly enough to complete the conjugation. The kinetics and stoichiometric study clearly showed the efficient conjugation. We also found that the reaction efficiency was reduced in acidic buffer but went more smoothly under basic conditions (Fig. S18†). The resulting protein–peptide conjugate was stable in 10 mM GSH for at least 12 hours at 37 °C with no leaks being detected (Fig. S18†), which hinted at its potential biological application in antibody–drug conjugates or protein post-translational modifications.
Then protein selectivity was carefully examined by using other proteins containing free Cys residues such as BCL2, MgrA and SrtA (Table S3†). No conjugation was observed as shown in Fig. 3D which indicates the site-selective cysteine modification of our designed peptides. To assess the ability of peptide PD3-I to label PDZΔRGS3 in a complex proteome environment, 293 T cell lysates (300 μg) were spiked with PDZΔRGS3 (10 μg), and then treated with 50 μM FAM labeled peptide PD3-I as shown in Fig. 3E, referring to the work of Sun et al.45 The gel data showed a clear single fluorescence band with the right molecular weight indicating a clean and selective conjugation of peptide PD3-I to PDZΔRGS3. The successful conjugation was further confirmed by a pull down assay using Ni-NTA agarose beads as shown in Fig. S19.†
The peptide was then tested for conjugation efficiency in HA-PDZΔRGS3 transfected cell lysates as shown in Fig. 4. Anti-HA western analyses revealed that peptide PD3-I could conjugate with PDZΔRGS3 in a concentration-dependent manner while the scrambled peptide NS couldn't label this target even at 100 μM with incubation for 8 hours, as shown in Fig. 4A. The reaction in cell lysates could be started in 30 min and reached high conjugation within 8 hours at room temperature with 50 μM PD3-1, as shown in Fig. 4B. The reaction efficiency would be decreased when competed with the excess of linear peptide ligand L, indicating that the reaction in cell lysates occurs by the ligand-induced protein conjugation (Fig. 4C). The successful conjugation in cell lysates showed its potential for future cellular applications, such as cell imaging, the study of protein–protein interaction, the discovery of covalent inhibitors and activity based protein profiling.
 | ||
| Fig. 4 The covalent reaction in HA-PDZΔRGS3 transfected cell lysates. (A) Covalent bonding of peptide PD3-1 or peptide NS to HA-PDZΔRGS3 after 8 h incubation at room temperature with HA-PDZΔRGS3 transfected cell lysates. (B) Reaction kinetics study of peptide PD3-I and HA-PDZΔRGS3 conjugation in HA-PDZΔRGS3 transfected cell lysates for 0 min, 30 min, 1 h, 2 h, 4 h and 8 h respectively. (C) The high concentration of linear peptide competitively blocked the labeling of peptide PD3-1 (30 μM) to HA-PDZΔRGS3 in cell lysates in a dose-dependent manner. | ||
As different linkers demonstrated different reaction rates (Fig. S6†), we then tested the conjugation efficiency of peptide PD3 equipped with different linkers as shown in Fig. 5A. The linker IV showed the slowest cysteine conjugation rate. The successful conjugation was further confirmed by ESI-MS as shown in Fig. 5B, S17 and S20–S22† which suggested that only one cysteine residue in PDZΔRGS3 could be conjugated with the peptide. To further identify the modification sites, trypsin digestion of a single reaction band cut from the gel was sent for MS/MS analysis as shown in Fig. 5C and S23–S25.† The MS/MS results suggested an expected peptide fragmentation containing both peptide PD3-I and a peptide segment containing Cys33, Cys34 or Cys73 of PDZΔRGS3 indicating that our peptide can selectively label PDZ in the vicinity of ligand binding sites.
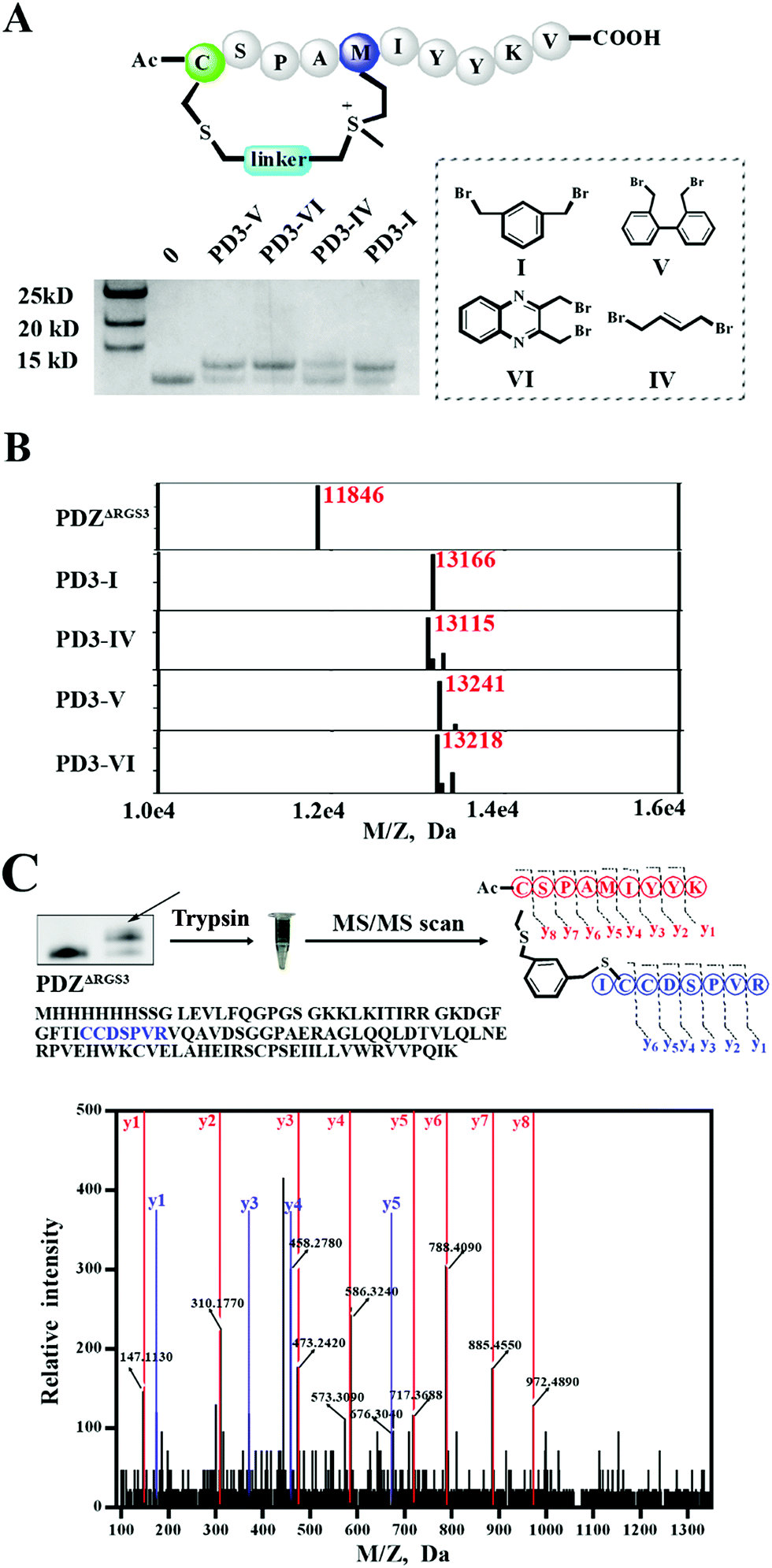 | ||
| Fig. 5 ESI MS analysis of the PDZ–peptide covalent conjugates. (A) The proteins were incubated with peptides equipped with different linkers (protein/peptide 15/75 μM, pH 7.4, 12 hours). (B) The mass analysis of peptide–PDZΔRGS3 conjugation, figures prepared using Prism based on original MS data shown in Fig. S14–S18.† (C) Mass/mass spectrometry analysis of trypsin-digested PDZ–peptide conjugates indicating that peptide PD3-I binds covalently to C33 or C34 in PDZΔRGS3. | ||
Conclusions
In summary, we designed a facile macrocyclization method via bis-alkylation between cysteine and methionine with improved serum stability and cellular uptake. Additionally, the tethered sulfonium could be a novel warhead for site selective protein modification under biocompatible conditions. To the best of our knowledge, this is the first attempt to combine peptide stapling and the installation of a selectively reacting moiety in one simple design. To demonstrate the principle, we constructed peptide ligands for PDZΔRGS3 and clearly showed that the reaction is rapid, efficient, highly selective and solely proximity-prompted. The reaction is viable in PBS, doesn't require any further special conditions and is selective in cell lysates. This method could have great potential in affinity-based protein profiling, target identification, and novel PPI modulator development.Author contributions
Z. L., F. Y. and D. W. prepared the manuscript and designed the research presented. D. W., M. Y. and Z. H. designed and performed the synthetic and biological experiments. N. L. and C. L. prepared various proteins. R. Z. and W. L. provided HPLC, MS and CD data. R. W and S. L. helped analyze the MS data. Y. J. and X. S. helped prepare the cellular experiments.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China grants 21778009, 81701818 and 81572198; the Shenzhen Science and Technology Innovation Committee, JCYJ20170412150609690, KQJSCX20170728101942700 and JCYJ20170807144449135. This work is supported by the High-Performance Computing Platform of Peking University and Beijing National Laboratory for Molecular Sciences (BNLMS). The PDZ-RGS3 plasmid is a kind gift from Prof Jiang Xia's lab at The Chinese University of Hong Kong.Notes and references
- R. Rakhit, R. Navarro and T. J. Wandless, Chem. Biol., 2014, 21, 1238–1252 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- E. Basle, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213–227 CrossRef CAS PubMed.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- C. Zhang, P. Dai, A. A. Vinogradov, Z. P. Gates and B. L. Pentelute, Angew. Chem., Int. Ed., 2018, 57, 6459–6463 CrossRef CAS PubMed.
- A. M. Embaby, S. Schoffelen, C. Kofoed, M. Meldal and F. Diness, Angew. Chem., Int. Ed., 2018, 57, 8022–8026 CrossRef CAS PubMed.
- Y. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart and S. Weiss, Bioconjugate Chem., 2008, 19, 786–791 CrossRef CAS PubMed.
- J. Yu, X. Yang, Y. Sun and Z. Yin, Angew. Chem., Int. Ed., 2018, 57, 11598–11602 CrossRef CAS PubMed.
- S. Choi, S. Connelly, N. Reixach, I. A. Wilson and J. W. Kelly, Nat. Chem. Biol., 2010, 6, 133–139 CrossRef CAS PubMed.
- J. M. Hooker, A. P. Esser-Kahn and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 15558–15559 CrossRef CAS PubMed.
- Y. Takaoka, H. Tsutsumi, N. Kasagi, E. Nakata and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 3273–3280 CrossRef CAS PubMed.
- J. C. Gildersleeve, O. Oyelaran, J. T. Simpson and B. Allred, Bioconjugate Chem., 2008, 19, 1485–1490 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martinez-Saez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jimenez-Oses and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- S. R. Adusumalli, T. G. Rawale, U. Singh, P. Tripathi, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301–6308 CrossRef CAS PubMed.
- T. Oya, N. Hattori, Y. Mizuno, S. Miyata, S. Maeda, T. Osawa and K. Uchida, J. Biol. Chem., 1999, 274, 18492–18502 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- T. A. Cropp and P. G. Schultz, Trends Genet., 2004, 20, 625–630 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. M. Chalker, G. J. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- A. J. Chmura, M. S. Orton and C. F. Meares, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8480–8484 CrossRef CAS PubMed.
- H. Nonaka, S. Tsukiji, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2007, 129, 15777–15779 CrossRef CAS PubMed.
- H. Nonaka, S. H. Fujishima, S. H. Uchinomiya, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 9301–9309 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS.
- P. V. Robinson, G. de Almeida-Escobedo, A. E. de Groot, J. L. McKechnie and C. R. Bertozzi, J. Am. Chem. Soc., 2015, 137, 10452–10455 CrossRef CAS PubMed.
- J. C. Henise and J. Taunton, J. Med. Chem., 2011, 54, 4133–4146 CrossRef CAS PubMed.
- E. D. Deeks and G. M. Keating, Drugs Ther. Perspect., 2018, 34, 89–98 CrossRef PubMed.
- J. R. Brown, Current Hematologic Malignancy Reports, 2013, 8, 1–6 CrossRef PubMed.
- A. O. Walter, R. T. Sjin, H. J. Haringsma, K. Ohashi, J. Sun, K. Lee, A. Dubrovskiy, M. Labenski, Z. Zhu, Z. Wang, M. Sheets, T. St Martin, R. Karp, D. van Kalken, P. Chaturvedi, D. Niu, M. Nacht, R. C. Petter, W. Westlin, K. Lin, S. Jaw-Tsai, M. Raponi, T. Van Dyke, J. Etter, Z. Weaver, W. Pao, J. Singh, A. D. Simmons, T. C. Harding and A. Allen, Cancer Discovery, 2013, 3, 1404–1415 CrossRef CAS PubMed.
- Y. Yu, M. Liu, T. T. Ng, F. Huang, Y. Nie, R. Wang, Z. P. Yao, Z. Li and J. Xia, ACS Chem. Biol., 2016, 11, 149–158 CrossRef CAS PubMed.
- A. D. de Araujo, J. Lim, A. C. Good, R. T. Skerlj and D. P. Fairlie, ACS Med. Chem. Lett., 2017, 8, 22–26 CrossRef CAS PubMed.
- A. J. Huhn, R. M. Guerra, E. P. Harvey, G. H. Bird and L. D. Walensky, Cell Chem. Biol., 2016, 23, 1123–1134 CrossRef CAS PubMed.
- A. D. de Araujo, J. Lim, K. C. Wu, Y. Xiang, A. C. Good, R. Skerlj and D. P. Fairlie, J. Med. Chem., 2018, 61, 2962–2972 CrossRef PubMed.
- C. Hoppmann and L. Wang, Chem. Commun., 2016, 52, 5140–5143 RSC.
- J. R. Kramer and T. J. Deming, Chem. Commun., 2013, 49, 5144–5146 RSC.
- X. Shi, R. Zhao, Y. Jiang, H. Zhao, Y. Tian, J. Li, W. Qin, F. Yin and Z. Li, Chem. Sci., 2018, 9, 3227–3232 RSC.
- J. R. Kramer and T. J. Deming, Chem. Commun., 2013, 49, 5144–5146 RSC.
- Y. Tian, J. Li, H. Zhao, X. Zeng, D. Wang, Q. Liu, X. Niu, X. Huang, N. Xu and Z. Li, Chem. Sci., 2016, 7, 3325–3330 RSC.
- Q. Lu, E. E. Sun, R. S. Klein and J. G. Flanagan, Cell, 2001, 105, 69–79 CrossRef CAS.
- Y. Xie, J. Ge, H. Lei, B. Peng, H. Zhang, D. Wang, S. Pan, G. Chen, L. Chen, Y. Wang, Q. Hao, S. Q. Yao and H. Sun, J. Am. Chem. Soc., 2016, 138, 15596–15604 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supporting tables and figures. See DOI: 10.1039/c9sc00034h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |