DOI:
10.1039/C8SC05510F
(Edge Article)
Chem. Sci., 2019,
10, 3616-3622
Single-site metal–organic framework catalysts for the oxidative coupling of arenes via C–H/C–H activation†
Received
10th December 2018
, Accepted 17th February 2019
First published on 18th February 2019
Abstract
C–H activation reactions are generally associated with relatively low turnover numbers (TONs) and high catalyst concentrations due to a combination of low catalyst stability and activity, highlighting the need for recyclable heterogeneous catalysts with stable single-atom active sites. In this work, several palladium loaded metal–organic frameworks (MOFs) were tested as single-site catalysts for the oxidative coupling of arenes (e.g. o-xylene) via C–H/C–H activation. Isolation of the palladium active sites on the MOF supports reduced Pd(0) aggregate formation and thus catalyst deactivation, resulting in higher turnover numbers (TONs) compared to the homogeneous benchmark reaction. Notably, a threefold higher TON could be achieved for palladium loaded MOF-808 due to increased catalyst stability and the heterogeneous catalyst could efficiently be reused, resulting in a cumulative TON of 1218 after three runs. Additionally, the palladium single-atom active sites on MOF-808 were successfully identified by Fourier transform infrared (FTIR) and extended X-ray absorption fine structure (EXAFS) spectroscopy.
Introduction
The synthesis of biaryls has attracted much attention over the past decades since these motifs are abundantly present in pharmaceuticals, natural products, agrochemicals, specialty monomers and other fine chemicals.1–4 Typically, the synthesis of the biaryl groups in these compounds involves conventional coupling reactions, such as the Suzuki reaction, which requires pre-functionalized substrates and produces stoichiometric amounts of salt waste.1 In recent years, formation of biaryls via palladium catalyzed cross-dehydrogenative coupling (CDC) reactions has been proposed as a more cost-efficient and environmentally benign alternative, since simple arenes can be used as substrate and water is the only byproduct if O2 is used as the oxidant.5–7
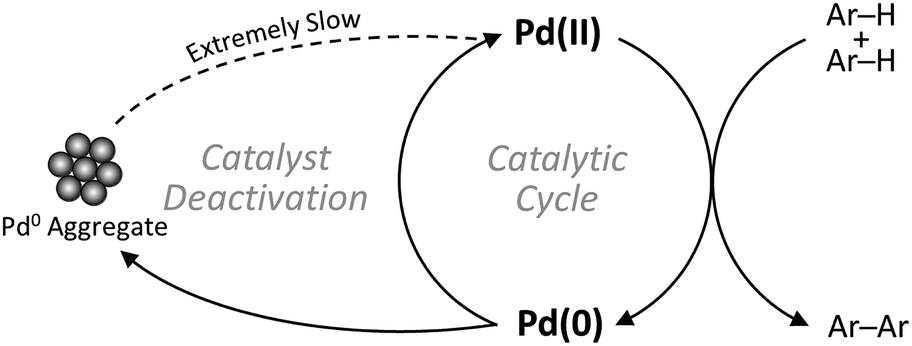
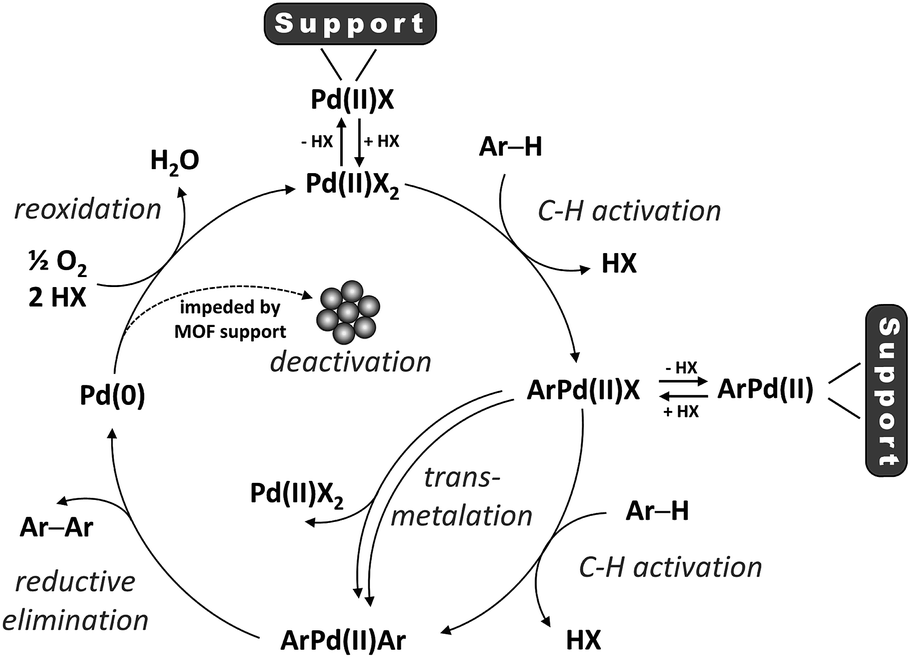
The primary focus of the scientific community regarding C–H activation of aromatic C(sp2)–H bonds has been so far on increasing the catalysts' activity and regioselectivity, besides expanding the substrate scope.8–13 However, given the relatively low turnover numbers (TONs) and high catalyst concentrations generally associated with CDC reactions compared to conventional coupling reactions, efficient recovery and recycling of the precious homogeneous palladium catalyst is a key aspect in the eventual implementation of this new synthetic strategy.14 Moreover, since the valence state of palladium changes between Pd(II) and Pd(0) in the catalytic cycle, formation of Pd(0) aggregates is commonly recognized as an important deactivation pathway (Scheme 1), highlighting the need for solid catalysts with stable single-atom active sites.15–17 Nevertheless, only very few heterogeneous catalysts for the oxidative coupling of arenes via C–H/C–H activation have been reported so far.18–22
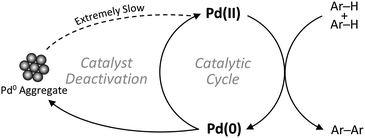 |
| | Scheme 1 Schematic representation of catalyst deactivation. | |
Metal–organic frameworks (MOFs), which are coordination polymers made up of inorganic secondary building units (SBUs; metal ions or clusters) and organic linkers, are an interesting group of porous, crystalline materials that can be transformed into heterogeneous single-site catalysts.23–26 Besides any catalytic activity inherent to MOFs, common strategies to imbue MOFs with well-defined and isolated active sites include anchoring active transition metals on the organic linkers27–31 or grafting them on open coordination sites of the SBUs.31–40 Due to their excellent stability, combined with high surface areas and tunable porosity, Zr-based MOFs have already been proven to be interesting heterogeneous scaffolds to anchor metals for several applications ranging from sensing to catalysis.41–45
Herein, we present the first heterogeneous MOF-based catalysts for the oxidative coupling of arenes via C–H/C–H activation, which exhibit superior TONs compared to their homogeneous analogues due to isolation of the Pd(II) centers on the MOF supports.
Results and discussion
Screening of MOF supports
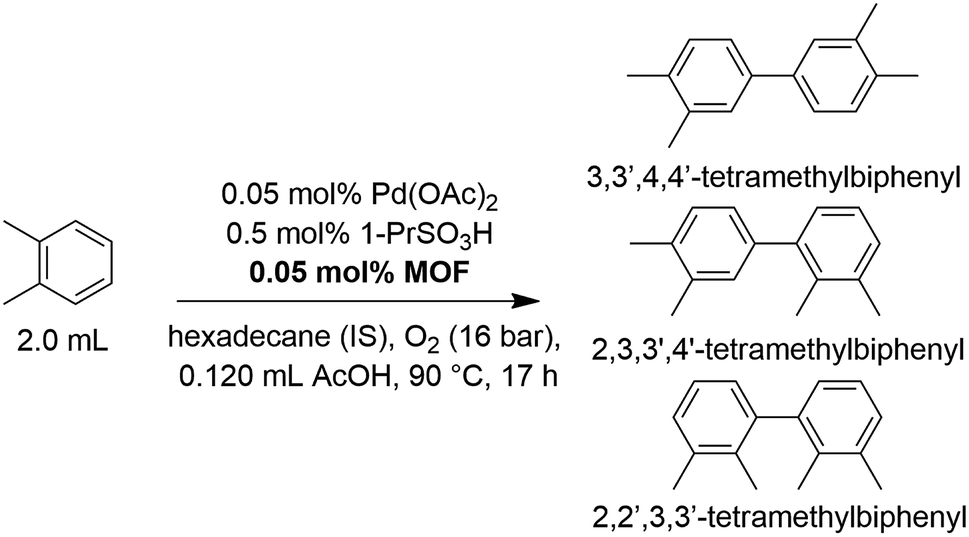
The oxidative homocoupling of o-xylene via C–H/C–H activation (Scheme 2) is considered to be a scientifically and industrially relevant CDC model reaction, since the 3,3′,4,4′-tetramethylbiphenyl product is an important intermediate in a more cost-efficient route to prepare the high-performance polyimide resin Upilex.7,14,46 Industrially, the reaction is stopped at low conversion to avoid oligomerization of the products and the starting feedstock is recycled. This justifies evaluating the reaction using a TON value instead of a single-pass yield.7 As a first step, several Zr-MOFs were evaluated for their potential as heterogeneous supports in the oxidative coupling of o-xylene (Table 1). Generally, an equimolar amount of MOF (based on its structural formula) with respect to Pd(OAc)2 was added, i.e. one Zr6-cluster of UiO-66 or MOF-808 per Pd atom. This ensures that a 4–6 fold excess of anchoring sites per Pd center are present in the frameworks (Table S1†). Additionally, 1-propanesulfonic acid was employed as strongly acidic additive, since recent findings by the group of Stahl reveal that strong acids or their corresponding salts dramatically increase the activity of Pd(II) for this reaction.12 Inspired by the excellent results obtained with 2,2′-bipyridine-grafted mesoporous silica for the related Pd(II)-catalyzed oxidative Heck coupling,17 a MOF containing 2,2′-bipyridine-5,5′-dicarboxylate (bpydc2−) linkers (UiO-67-bipy; [Zr6(μ3-O)4(μ3-OH)4(bpydc)6])27 was tested. However, the addition of this MOF resulted in a dramatic decrease in activity versus the homogeneous reaction (Table 1; entry 1 and 2). A similar deactivation effect was found for the analogous dissolved heterocyclic nitrogen-containing ligand, 4,4′-dimethyl-2,2′-bipyridine (Table S2†). Consequently, other MOFs featuring anchoring sites which correspond more closely to the carboxylates in the active Pd(OAc)2 complex were investigated. For instance, a UiO-66 analogue with pendent carboxylic acid groups as Pd(II) anchoring sites (UiO-66-COOH; [Zr6(μ3-O)4(μ3-OH)4(bdc-COOH)6], bdc2−-COOH = 1,2,4-benzenetricarboxylate)47 was synthesized and tested. In contrast to UiO-67-bipy, a significantly higher TON than in the homogeneous case was obtained after the same reaction time (Table 1; entry 3), highlighting the positive effect of active site isolation by anchoring the active Pd(II) centers on the pendent carboxylic acid groups. Inspired by previous research in which platinum-group metal complexes were grafted onto the inorganic SBUs of a MOF support,35,38,40 several Zr-MOFs with available coordination sites on the clusters (hydrogen-bonded OH/OH2 pairs)39 were synthesized (MOF-808, UMCM-309a and Zr-abtc). The hexanuclear Zr-clusters of MOF-808 ([Zr6(μ3-O)4(μ3-OH)4(btc)2(CH3COO)6], btc3− = 1,3,5-benzenetricarboxylate)48,49 are 6-fold coordinated by btc3− linkers, resulting in up to 6 open sites per cluster, after removing the acetate modulators using a simple acid treatment. A closely related MOF with 6-fold coordinated Zr-clusters is UMCM-309a ([Zr6(μ3-O)4(μ3-OH)4(btb)2(HCOO)6], btb3− = 1,3,5-(4-carboxylphenyl)benzene),50 which features a stable two-dimensional layered network instead of a three-dimensional framework. Recently, a new 8-connected Zr-MOF with 3,3′,5,5′-azobenzene-tetracarboxylate (abtc4−) linkers ([Zr6(μ3-O)4(μ3-OH)4(abtc)2(OH)4(H2O)4]) was reported,51 which contains 4 open sites per cluster and could be synthesized following a newly developed water-based green synthesis procedure (Fig. S2†). In line with the increase in activity for UiO-66-COOH, the addition of MOFs with open sites on the Zr-clusters resulted in significantly higher TONs compared to the homogeneous reference case (Table 1; entries 4–6). In addition, UiO-66 ([Zr6(μ3-O)4(μ3-OH)4(bdc)6], bdc2− = 1,4-benzenedicarboxylate),52 a well-known non-functionalized Zr-MOF, was tested as a reference, since it does not contain significant amounts of pendent carboxylic acid groups or open sites on the Zr-clusters to which Pd(II) could coordinate. The TON observed in the presence of UiO-66 was similar to that observed for homogeneous Pd(OAc)2 (Table 1; entry 7). This confirms that the increase in TON observed for other MOFs like MOF-808 is due to active site isolation of the Pd on the anchoring sites of the MOFs. Besides MOFs with anchoring sites, a moderate increase in TON could also be achieved by the addition of high-surface area zirconium oxide. Finally, higher chemoselectivities were obtained in the presence of MOFs with open sites on the Zr-clusters or pendent carboxylic acid groups since less triaryl side products were formed. The formation of these bulky triaryls may be suppressed in the presence of MOFs due to pore confinement.
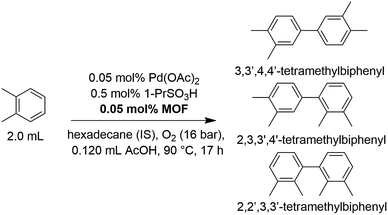 |
| | Scheme 2 The oxidative coupling of o-xylene under the standard reaction conditions: o-xylene (16.58 mmol), Pd(OAc)2 (8.29 μmol), MOF support (8.29 μmol), 1-propanesulfonic acid (82.85 μmol), acetic acid co-solvent (2.07 mmol), 90 °C, 16 bar O2, 17 h. | |
Table 1 Screening of the different MOF supports under the standard reaction conditions (cfr. Scheme 2)
| Entry |
MOF support |
Chemo-selectivitya (%) |
Regio-selectivityb (%) |
Yieldc (%) |
TONd |
TOFe (h−1) |
|
Chemoselectivity is defined as the percentage of biaryls relative to all formed products (oxidation products, biaryls and triaryls).
Regioselectivity is defined as the percentage of 3,3′,4,4′-tetramethylbiphenyl relative to all three biaryls.
Yield was determined by GC-FID with hexadecane as internal standard.
TON is defined as TON = 2 × mole (biaryl)/mole (Pd).
TOF was determined after 4 h.
10 mg of zirconium oxide was added.
|
| 1 |
— |
88 |
71 |
4.9 |
97 |
8.1 |
| 2 |
UiO-67-bipy |
74 |
66 |
0.3 |
6 |
1.1 |
| 3 |
UiO-66-COOH |
92 |
73 |
7.4 |
149 |
10.6 |
| 4 |
MOF-808 |
92 |
74 |
9.2 |
183 |
11.2 |
| 5 |
UMCM-309a |
93 |
74 |
8.4 |
168 |
11.1 |
| 6 |
Zr-abtc |
93 |
73 |
7.1 |
142 |
11.0 |
| 7 |
UiO-66 |
90 |
75 |
5.1 |
103 |
8.3 |
| 8 |
ZrO2f |
89 |
75 |
6.5 |
130 |
10.5 |
Heterogeneity of the single-site MOF catalysts
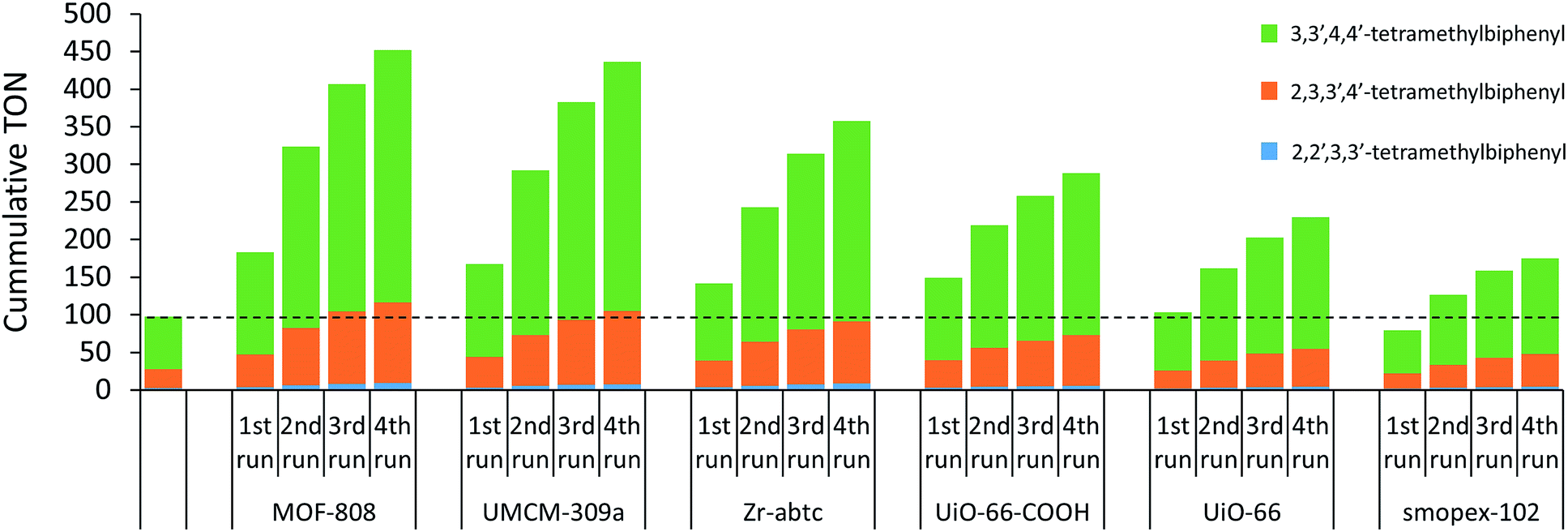
The heterogeneity of the single-site MOF catalysts was studied by recycling tests and metals analysis of the reaction solution. After the reaction, the sample was centrifuged and the MOF crystals were separated from the reaction solution. Fresh reactant (e.g. o-xylene), co-solvent (e.g. acetic acid), internal standard and strongly acidic additive (e.g. 1-propanesulfonic acid) were added to the recycled MOF and the reaction was resumed for a second run. Materials with open sites on the Zr-clusters (MOF-808, UMCM-309a and Zr-abtc) retained their activity better than UiO-66-COOH with pendent carboxylic acid groups, resulting in higher cumulative TONs after 4 runs (Fig. 1). In line with these results, a lower cumulative TON was noticed for UiO-66, which does not contain anchoring sites. In addition, the palladium and zirconium contents of the reaction solution were measured after the first run by inductively coupled plasma optical emission spectrometry (ICP-OES) (Table S3†). Generally, palladium leaching could be minimized to approximately 5% for MOFs with open sites on the Zr-cluster (MOF-808 and Zr-abtc) and no significant zirconium leaching was detected. However, more leaching was observed for UMCM-309a, presumably due to its two-dimensional layered structure. Higher leaching values were also obtained for UiO-66-COOH and UiO-66, indicating that palladium is best retained on MOFs with open sites on the Zr-clusters and with a three-dimensional pore structure, like MOF-808. Furthermore, an acrylic acid grafted polyolefin fiber (Smopex-102), one of the very few already reported heterogeneous supports for the oxidative coupling of arenes,22 was tested using a similar ratio of 1 Pd(II) center per 6 pendent carboxylic acid groups. The cumulative TON of this Pd-loaded polymer with pendent carboxylic acid groups is substantially lower than the TON of its MOF analogue (UiO-66-COOH), which might be due to inferior accessibility of the carboxylic acid groups. In addition, the stability of the MOFs was evaluated by comparing the powder X-ray diffraction patterns before and after the reaction (Fig. S5–S10†). No significant decrease in crystallinity was observed for MOF-808, UMCM-309a, Zr-abtc, UiO-66-COOH and UiO-66 after four runs at 90 °C. In contrast, UiO-67-bipy largely lost its long-range order after exposure to the reaction solvent and conditions.
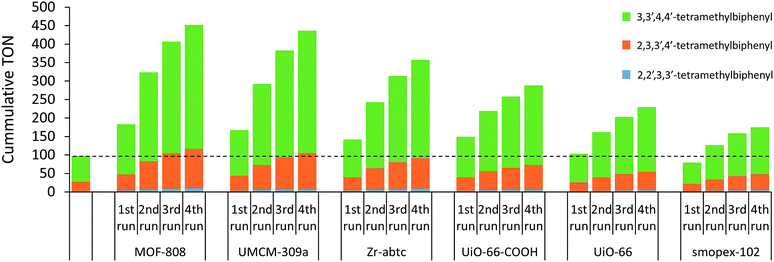 |
| | Fig. 1 Recycling experiments of several heterogeneous supports for the Pd-catalyzed oxidative coupling of o-xylene performed under the standard reaction conditions. | |
Substrate scope
In view of its superior activity, heterogeneity and facile synthesis procedure, MOF-808 was selected as model support to expand the substrate scope (Fig. S11†). High TONs were obtained for the homocoupling of toluene and tert-butylbenzene, while the activity was significantly lower for the coupling of p-xylene and 1,2-difluorobenzene due to steric hindrance and electron-withdrawing effects, respectively.
Optimizing the reaction conditions
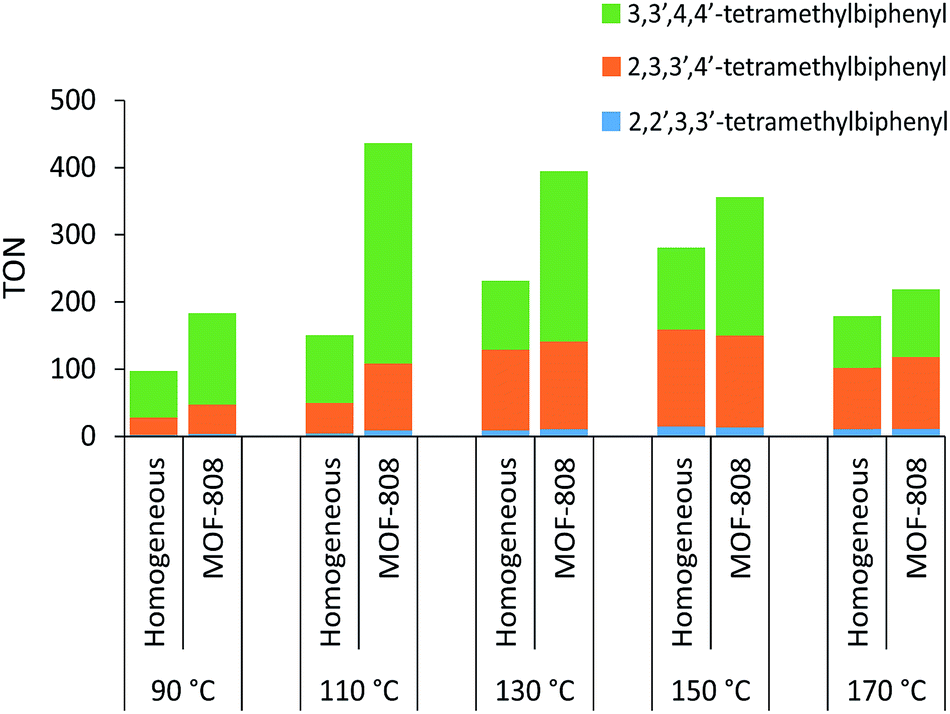
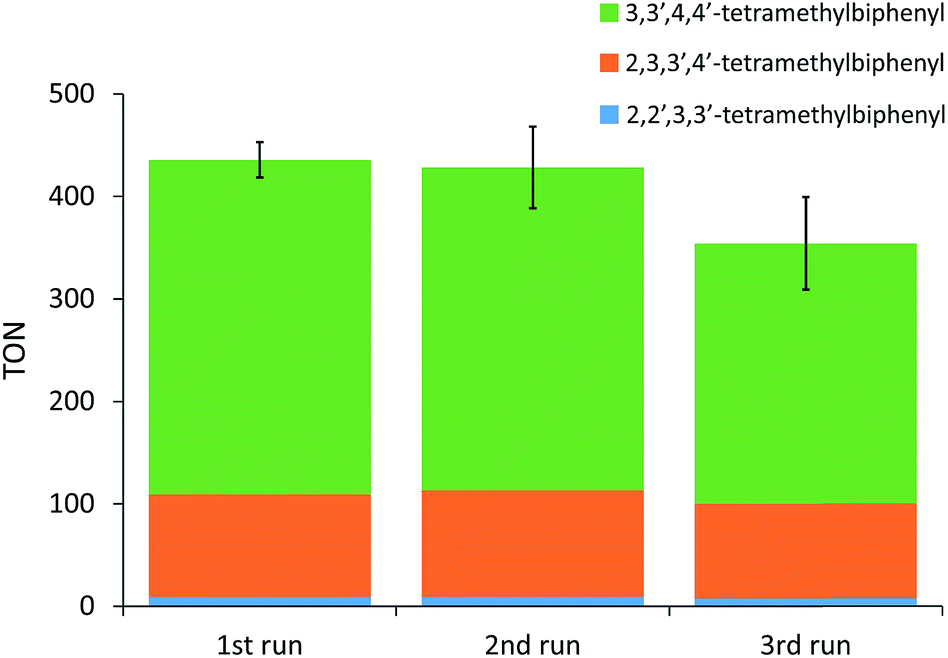
Significantly higher TONs for the oxidative coupling of o-xylene could be obtained for the heterogeneous system with MOF-808 by increasing the reaction temperature (Fig. 2). Notably, at 110 °C, the TON is 2.9 times higher for the MOF-808 system than for the analogous homogeneous system, which is a clear benefit of active site isolation enabled by the MOF. At even higher temperatures, the MOF support started to decompose (Fig. S12†), resulting in lower TONs, which were more comparable to those of the homogeneous system. After further optimizing the palladium loading of the MOF and the amount of 1-propanesulfonic acid and co-solvent (Table S2†), a TON of 436 ± 17 could be reached after the first run (Fig. 3) with only 2% Pd leaching (Table S4†). Since oxygen is used as terminal oxidant, a stoichiometric amount of water is produced and partially absorbed by the hydrophilic MOF material. Product inhibition experiments revealed that the formation of water did not lead to a significant decrease in activity at short reaction times (Fig. S13†). However, after many catalytic cycles (TONs > 400), a large amount of water is formed and product inhibition due to water formation is observed (Table S2†). Nevertheless, the activity of the solid MOF-based catalyst could be restored by reactivating the MOF materials after each run under vacuum (1 mbar) at room temperature for 24 h (Fig. 3). A cumulative TON of 1218 ± 36 with an overall regioselectivity of 73% for 3,3′,4,4′-tetramethylbiphenyl was obtained after three runs, which is well beyond the state-of-the-art.12,22 Moreover, only 5% of Pd was leached after three runs (Table S4†).
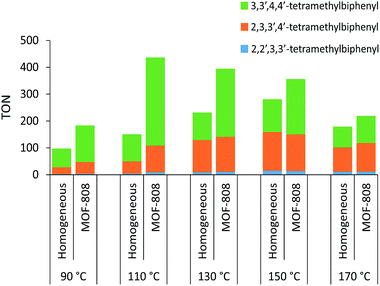 |
| | Fig. 2 Effect of temperature on the TON of the oxidative coupling of o-xylene with and without MOF-808 performed under the standard reaction conditions. | |
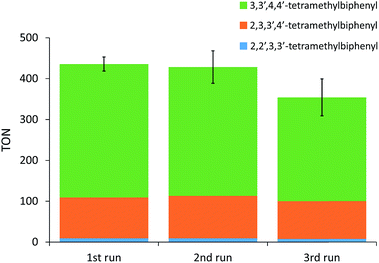 |
| | Fig. 3 Oxidative coupling of o-xylene performed under optimized reaction conditions and with reactivation of the Pd loaded MOF support under vacuum (1 mbar) at room temperature for 24 h in between consecutive runs. Conditions: o-xylene (16.58 mmol), Pd(OAc)2 (8.29 μmol), MOF-808 (41.43 μmol), 1-propanesulfonic acid (331.40 μmol), acetic acid (2.07 mmol), 110 °C, 20 bar O2, 17 h. The TONs are the average of three experiments. | |
Active site isolation
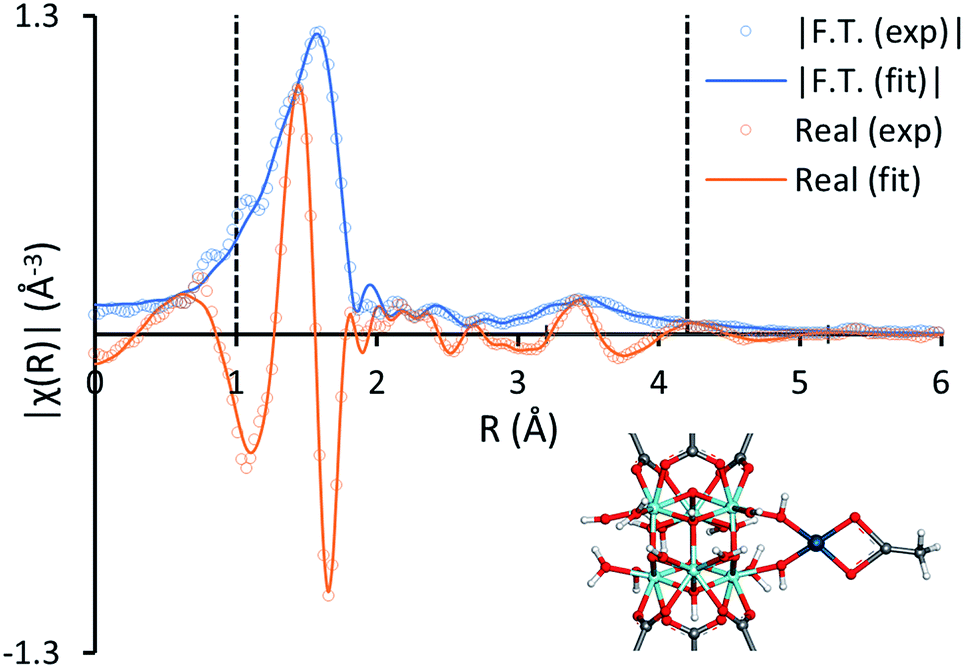
The binding mode of Pd(II) to the zirconium clusters of MOF-808 was studied by Fourier transform infrared spectroscopy (FTIR). Upon removal of a capping acetate from the cluster, a hydrogen-bonded OH/OH2 pair occupies the place of the missing carboxylate. This OH/OH2 pair displays a characteristic IR band at 2744 cm−1, while the sharp IR band at 3672 cm−1 results from a combination of non-hydrogen-bonded OH groups and μ3-OH groups inside the cluster (Fig. S15†).53,54 In agreement with previous research,33,35,36,38,55 the characteristic IR band at 2744 cm−1 disappears after chemisorption of the transition metal complex and the sharp IR band at 3672 cm−1 broadens, indicating that the palladium species interact with these open sites on the Zr-clusters. Moreover, the local environment of the MOF-supported Pd(II) was studied by X-ray absorption spectroscopy (XAS). In line with the FTIR data, the fit between the experimental and simulated extended X-ray absorption fine structure (EXAFS) data is excellent for the structure in which Pd(II) is anchored on the open site of the Zr-cluster after exchanging with the proton of the –OH2 group and liberating acetic acid (Fig. 4). Furthermore, the absence of a clear peak around 2.6 Å confirms that the trimeric Pd(OAc)2 complexes are converted into monomeric, MOF-supported Pd(II) species (Fig. S16†).17,22 Hence, the Pd(II) species formed after the reoxidation step and possibly also the Pd(II) complex formed after the first C–H activation step, can be anchored on the MOF support (Scheme 3). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of MOF-808 and UiO-66 after reaction in combination with energy-dispersive X-ray spectroscopy (EDX) showed that palladium anchoring reduces Pd nanoparticle aggregation and thus deactivation after the reductive elimination step (Fig. S19–S22†).
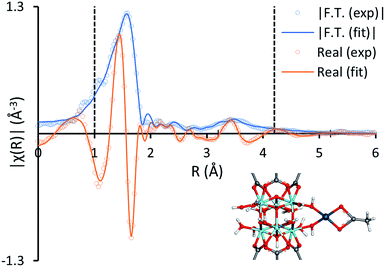 |
| | Fig. 4 The magnitude (blue) and real component (orange) of the Fourier transform of the experimental k2-weighted Pd K-edge EXAFS spectra of preloaded MOF-808 (1 Pd per Zr-cluster in o-xylene) (hollow circles) and the corresponding fits of the structure model (solid lines) in R-space. The vertical dashed lines indicate the fitting range. The Zr, O, C, H and Pd atoms are represented in the structure model by turquoise, red, gray, white and dark blue spheres, respectively. | |
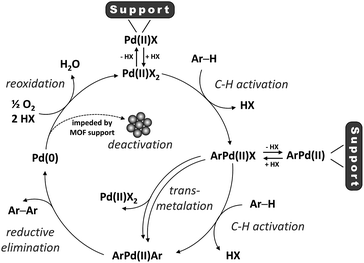 |
| | Scheme 3 The proposed catalytic cycle (X = acetate or 1-propanesulfonate). | |
Reaction mechanism
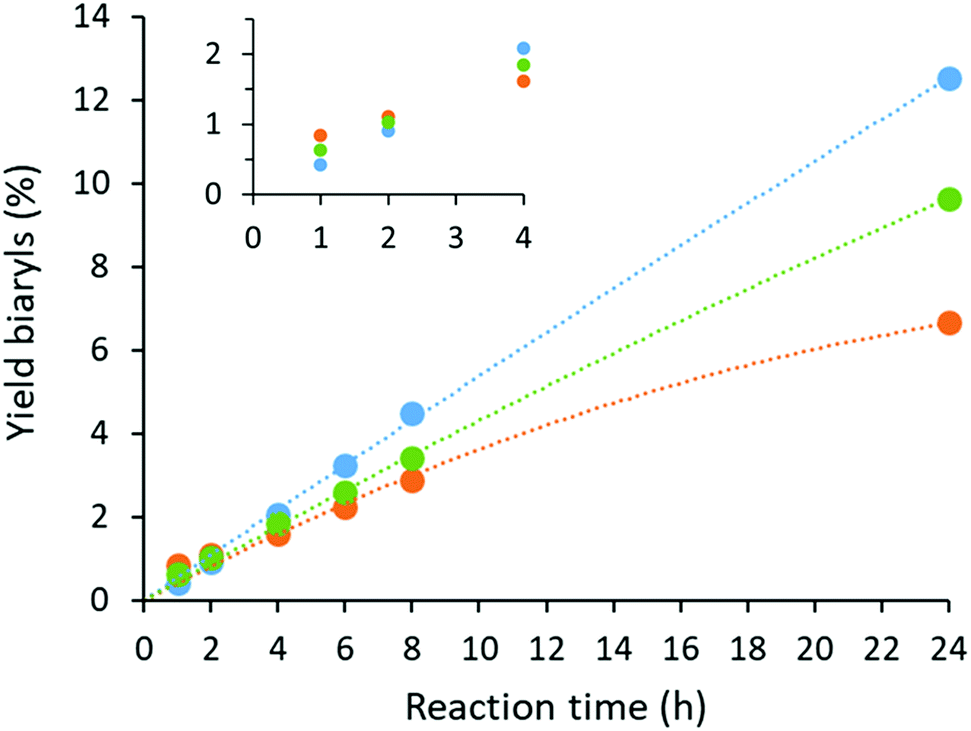
Catalyst deactivation was studied by analyzing the reaction kinetics (Fig. 5). Although the homogeneous system was found to be the most active at short reaction times, only moderate yields were obtained at longer reaction times due to catalyst deactivation. On the contrary, almost no deactivation was observed when MOF-808 was added, with an essentially constant rate up to 24 h reaction time. This implies that catalyst lifetime can be considerably prolonged by isolating the active sites on the MOF material. Moreover, a maximum TON was achieved for the heterogeneous system if more than 8 bar O2 was applied, indicating that deactivation is negligible under these conditions. In contrast, a clear dependence of the TON on the oxygen pressure could be seen in the homogeneous system for a wide range of oxygen pressures (Fig. S23†). To gather further insights into the catalytic mechanism, the kinetic isotope effect (KIE) was evaluated by comparing the conversion of o-xylene with o-xylene-d10 at short reaction times. The measured kH/kD values were 1.4 and 1.3 for the homogeneous and heterogeneous case, respectively, indicating a similar rate-limiting step for both systems. These modest KIE values are in accordance to the literature for the oxidative coupling of o-xylene under neat conditions.14
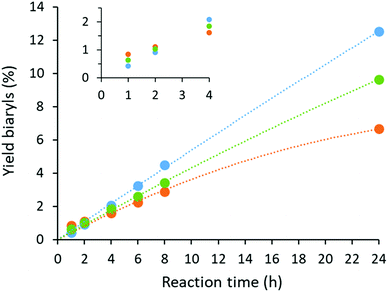 |
| | Fig. 5 Kinetic profiles of the oxidative coupling of o-xylene performed under the standard reaction conditions with MOF-808 (blue), UiO-66-COOH (green) and the homogeneous reaction without MOF support (orange). Lines were added as a guide to the eye. | |
Conclusions
In this work, we have shown for the first time that palladium loaded Zr-MOFs can be efficient single-site solid hybrid catalysts for the oxidative coupling of arenes via C–H/C–H activation. MOFs with various anchoring sites were screened first and the heterogeneity of these solid catalysts was studied by recycling tests and metals analysis of the reaction solution, which indicated that the heterogeneous catalysts could efficiently be reused. After optimization of the reaction conditions, a threefold higher TON could be achieved for the MOF-808 system compared to the analogous homogeneous system and Pd leaching was minimized to 2%. The activity of the solid MOF-based catalyst could be retained by reactivating the MOF materials by drying in between consecutive runs, resulting in a cumulative TON of 1218 after three runs. Finally, analysis of the reaction kinetics revealed that the superior TONs result from isolation of the active sites on the MOF material, which prolongs catalyst lifetime, and the isolated palladium single-atom active sites on MOF-808 were successfully identified by FTIR and EXAFS spectroscopy. These results show that the TON of palladium in C–H activation reactions can be significantly increased by developing stable heterogeneous single-site catalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research leading to these results has received funding from the NMBP-01-2016 Program of the European Union's Horizon 2020 Framework Program H2020/2014-2020/under grant agreement no. [720996]. N. V. V., S. S., J. V., B. B. and D. E. D. V. thank the FWO for funding (SB, Aspirant and postdoctoral grants). The electron microscopy work was supported by FWO funding G038116. D. E. D. V. is grateful for KU Leuven support in the frame of the CASAS Metusalem project and a C3 type project. The XAS experiments were performed on beamline BM26A at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to D. Banerjee at the ESRF for providing assistance in using beamline BM26A. Johnson Matthey and S. Bennett are gratefully acknowledged for providing Smopex-102.
Notes and references
- T. J. Colacot and J. Matthey, Platinum Met. Rev., 2011, 55, 84–90 CrossRef.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem.–Asian J., 2014, 9, 26–47 CrossRef CAS PubMed.
- Y. Izawa and S. S. Stahl, Adv. Synth. Catal., 2010, 352, 3223–3229 CrossRef CAS PubMed.
- T. W. Lyons, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 4455–4464 CrossRef CAS PubMed.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- K. M. Engle, D. Wang and J. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS PubMed.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 5704–5707 CrossRef CAS PubMed.
- B. J. Gorsline, L. Wang, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2017, 139, 9605–9614 CrossRef CAS PubMed.
- C. A. M. R. Van Slagmaat, G. K. M. Verzijl, L. Lefort, P. L. Alsters and M. Fern, ChemCatChem, 2018, 10, 2620–2626 CrossRef.
- E. M. Ferreira, H. Zhang and B. M. Stoltz, Tetrahedron, 2008, 64, 5987–6001 CrossRef CAS PubMed.
- Y. Liu, X. Wang, X. Cai, G. Chen, J. Li, Y. Zhou and J. Wang, ChemCatChem, 2016, 8, 448–454 CrossRef CAS.
- H. Duan, M. Li, G. Zhang, J. R. Gallagher, Z. Huang, Y. Sun, Z. Luo, H. Chen, J. T. Miller, R. Zou, A. Lei, Y. Zhao, T. Miller, R. Zou, A. Lei, Y. Zhao, J. T. Miller, R. Zou, A. Lei and Y. Zhao, ACS Catal., 2015, 5, 3752–3759 CrossRef CAS.
- T. Ishida, S. Aikawa, Y. Mise, R. Akebi, A. Hamasaki, T. Honma, H. Ohashi, T. Tsuji, Y. Yamamoto, M. Miyasaka, T. Yokoyama and M. Tokunaga, ChemSusChem, 2015, 8, 695–701 CrossRef CAS PubMed.
- T. Ishida, R. Tsunoda, Z. Zhang, A. Hamasaki, T. Honma, H. Ohashi, T. Yokoyama and M. Tokunaga, Appl. Catal., B, 2014, 150, 523–531 CrossRef.
- G. Mitran, O. D. Pavel, M. Florea and V. I. Pârvulescu, Appl. Catal., A, 2016, 514, 71–82 CrossRef CAS.
- L. M. Neal and H. E. Hagelin-Weaver, J. Mol. Catal. A: Chem., 2008, 284, 141–148 CrossRef CAS.
- Y. Liu, Y. Zhou, J. Li, Q. Wang, Q. Qin, W. Zhang, H. Asakura, N. Yan and J. Wang, Appl. Catal., B, 2017, 209, 679–688 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés I Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- S. Øien, G. Agostini, S. Svelle, E. Borfecchia, K. A. Lomachenko, L. Mino, E. Gallo, S. Bordiga, U. Olsbye, K. P. Lillerud and C. Lamberti, Chem. Mater., 2015, 27, 1042–1056 CrossRef.
- X. Li, R. Van Zeeland, R. V. Maligal-Ganesh, Y. Pei, G. Power, L. Stanley and W. Huang, ACS Catal., 2016, 6, 6324–6328 CrossRef CAS.
- R. Van Zeeland, X. Li, W. Huang and L. M. Stanley, RSC Adv., 2016, 6, 56330–56334 RSC.
- L. Chen, S. Rangan, J. Li, H. Jiang and Y. Li, Green Chem., 2014, 16, 3978–3985 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. I. Llabrés Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- R. J. Drout, A. J. Howarth, K. Otake, T. Islamoglu and O. K. Farha, CrystEngComm, 2018, 20, 6140–6145 RSC.
- K. Otake, Y. Cui, C. T. Buru, Z. Li, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 8652–8656 CrossRef CAS PubMed.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. Destefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- V. Bernales, D. Yang, J. Yu, G. Gümüşlu, C. J. Cramer, B. C. Gates and L. Gagliardi, ACS Appl. Mater. Interfaces, 2017, 9, 33511–33520 CrossRef CAS PubMed.
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- D. Yang, S. O. Odoh, J. Borycz, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, ACS Catal., 2016, 6, 235–247 CrossRef CAS.
- T. F. Liu, N. A. Vermeulen, A. J. Howarth, P. Li, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Eur. J. Inorg. Chem., 2016, 4349–4352 CrossRef CAS.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS PubMed.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- M. Zhao, S. Ou and C. De Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127–4170 RSC.
- S. Yuan, J. S. Qin, C. T. Lollar and H. C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed.
- J. S. Qin, S. Yuan, C. Lollar, J. Pang, A. Alsalme and H. C. Zhou, Chem. Commun., 2018, 54, 4231–4249 RSC.
- N. Erdmann, Y. Su, B. Bosmans, V. Hessel and T. Noël, Org. Process Res. Dev., 2016, 20, 831–835 CrossRef CAS.
- F. Ragon, B. Campo, Q. Yang, C. Martineau, A. D. Wiersum, A. Lago, V. Guillerm, C. Hemsley, J. F. Eubank, M. Vishnuvarthan, F. Taulelle, P. Horcajada, A. Vimont, P. L. Llewellyn, M. Daturi, S. Devautour-Vinot, G. Maurin, C. Serre, T. Devic and G. Clet, J. Mater. Chem. A, 2015, 3, 3294–3309 RSC.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- W. Liang, H. Chevreau, F. Ragon, P. D. Southon, V. K. Peterson and D. M. D'Alessandro, CrystEngComm, 2014, 16, 6530–6533 RSC.
- J. Ma, A. G. Wong-Foy and A. J. Matzger, Inorg. Chem., 2015, 54, 4591–4593 CrossRef CAS PubMed.
- H. Wang, X. Dong, J. Lin, S. J. Teat, S. Jensen, J. Cure, E. V. Alexandrov, Q. Xia, K. Tan, Q. Wang, D. H. Olson, D. M. Proserpio, Y. J. Chabal, T. Thonhauser, J. Sun, Y. Han and J. Li, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 6, 13850–13851 CrossRef PubMed.
- N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef CAS PubMed.
- D. Yang, V. Bernales, T. Islamoglu, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2016, 138, 15189–15196 CrossRef CAS PubMed.
- I. S. Kim, J. Borycz, A. E. Platero-Prats, S. Tussupbayev, T. C. Wang, O. K. Farha, J. T. Hupp, L. Gagliardi, K. W. Chapman, C. J. Cramer and A. B. F. Martinson, Chem. Mater., 2015, 27, 4772–4778 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, optimization of the reaction conditions, additional characterization data, substrate scope. See DOI: 10.1039/c8sc05510f |
|
| This journal is © The Royal Society of Chemistry 2019 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Steve
Waitschat
b,
Sachin M.
Chavan
a,
Steve
Waitschat
b,
Sachin M.
Chavan