
Transition metal-free, visible-light-mediated construction of α,β-diamino esters via decarboxylative radical addition at room temperature†
Guibing
Wu
a,
Jingwen
Wang
a,
Chengyu
Liu
a,
Maolin
Sun
a,
Lei
Zhang
a,
Yueyue
Ma
a,
Ruihua
Cheng
b and
Jinxing
Ye
 *a
*a
aEngineering Research Center of Pharmaceutical Process Chemistry, Ministry of Education, Shanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: yejx@ecust.edu.cn
bSchool of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 1st May 2019
Abstract
Transition-metal-free visible-light photoredox catalyzed decarboxylative radical addition has been developed for the construction of α,β-diamino esters from amino acids with glyoxylic oxime ethers under mild conditions. Cyclic and chain amino acids, dipeptides, and simple aliphatic acids are amenable to this addition as the alkyl radical source. A variety of α,β-diamino esters with broad functional groups were synthesized in satisfactory yields at room temperature. The employment of acridinium as the photocatalyst and its potential scalability also demonstrate the synthetic value in practice.
α,β-Diamino acids are essential building blocks and abundant in many bioactive compounds,1 such as peptides, antibiotics, natural products and pharmaceuticals. Accordingly, various methods have been developed to synthesize the compounds.2 As a powerful and efficient protocol, C–C coupling reactions are widely applied for the construction of α,β-diamino acids from simple nitrogen-containing blocks, and the typical representative is the Mannich reaction.3 Due to its high reactivity and great tolerance of functional groups, the radical mediated Mannich reaction is drawing increasing attention. Meanwhile, stoichiometric metals and dangerous agents such as triethyl boron, rare metal indium and antimony are required for the generation of radicals.4 Thus, a mild and environmentally friendly strategy for the generation of alkyl radicals is in urgent need for the synthesis of α,β-diamino acid derivatives.
As widely accessible and inexpensive substrates, carboxylic acids have been extensively studied as an alkyl radical source in the radical coupling reactions. Hatanaka and co-workers disclosed an efficient radical addition reaction between carboxylic acids and oxime ethers, using stoichiometric phenanthrene and 1,4-dicyanobenzene as a photosensitizer and an electron-acceptor.5 In 2015, Inoue and co-workers developed a novel and valuable decarbonylative radical coupling for the synthesis of δ-amino and α,β-diamino acids (Scheme 1).6 In this work, 3.0 equiv. of Et3B were required to activate the decarboxylation of aminoacyl tellurides and form alkyl radicals. Very recently, Baran et al. have reported a Ni-catalyzed radical reaction between glyoxylate-derived sulfinimines and N-hydroxytetrachlorophthalimide-esters in situ generated from alkyl carboxylic acids, which paved a way for the preparation of enantiopure α-amino acids.7
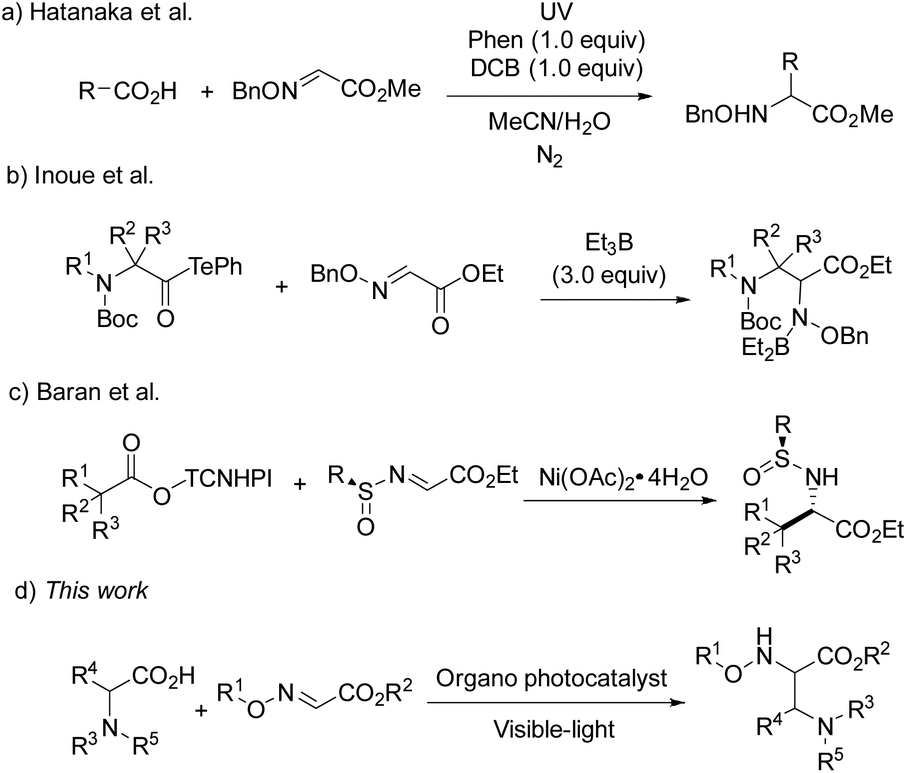 | ||
| Scheme 1 Decarboxylative radical Mannich reaction. | ||
Transition metal photoredox catalysis is a powerful tool for the decarboxylation and many excellent studies were pioneered in the radical coupling reaction.8 The pre-functionalization of carboxylic acids was not involved in these transformations. Meanwhile, as the widely used organic photocatalyst, 9-mesityl-10-methylacridinium perchlorate (Acr+ClO4−II) is also applied in the radical reaction gradually.9,10 However, the negative ground state reduction potential (E1/2(C/C−) = −0.57 V vs. SCE) is weak. Furthermore, its strongly positive excited state oxidation potential (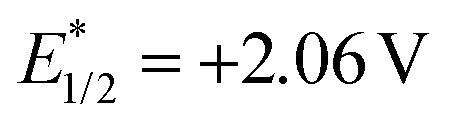 vs. SCE) can lead to substrate decomposition through unselective oxidation processes. In addition, DiRocco et al. designed several novel acridinium-based photocatalysts, which have been demonstrated to have excellent photoredox capability.11 These achievements have expanded the scope of photocatalysts with highly potential, applicable value in the organic synthesis field. Herein, we envisioned that α,β-diamino esters could be built via the decarbonylative radical Mannich reaction between amino acids and glyoxylic oxime ethers using an acridinium-based photocatalyst as the photosensitizer.
vs. SCE) can lead to substrate decomposition through unselective oxidation processes. In addition, DiRocco et al. designed several novel acridinium-based photocatalysts, which have been demonstrated to have excellent photoredox capability.11 These achievements have expanded the scope of photocatalysts with highly potential, applicable value in the organic synthesis field. Herein, we envisioned that α,β-diamino esters could be built via the decarbonylative radical Mannich reaction between amino acids and glyoxylic oxime ethers using an acridinium-based photocatalyst as the photosensitizer.
Results and discussion
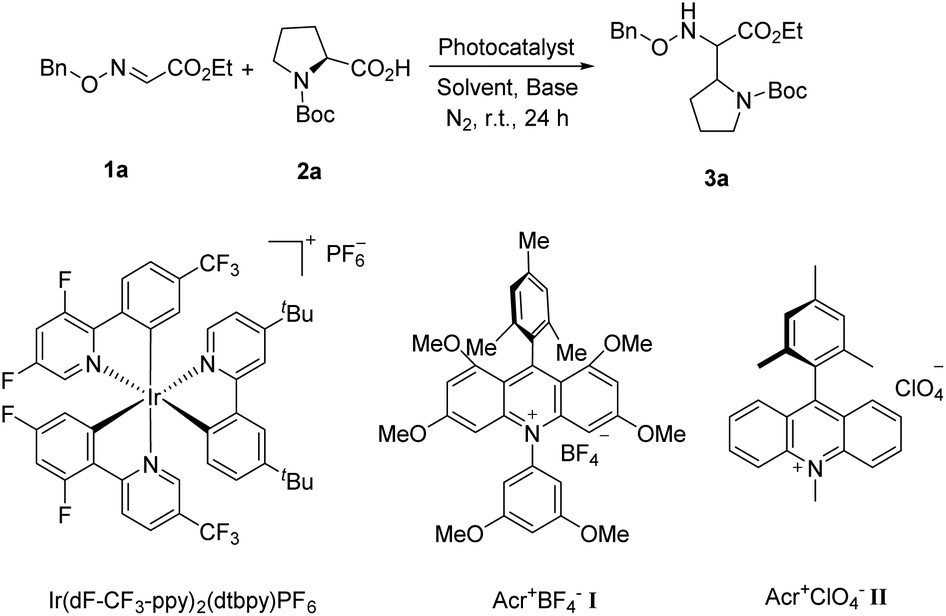
Due to the excellent reactivity toward nucleophilic carbon radicals,12 glyoxylic oxime ether (1a) was chosen as a radical acceptor to screen the optimal conditions with N-Boc-L-proline (2a). A set of solvents were evaluated in the presence of 2.0 mol% of Acr+BF4−I, and 1.5 equiv. of K2HPO4·3H2O, irradiated with blue LEDs at room temperature for 24 h (Table 1, entries 1–4). The results illustrated that non-polar solvent CH2Cl2 was the best, and toluene provided competitive yield as well (Table 1, entries 2 and 4, 73% and 72% yield, respectively). Impressively, we found that Acr+BF4−I presented appreciable catalytic efficacy in comparison with any other photocatalysts, such as Ir and Ru photocatalysts and other organic dyes, which proved to be the optimal photocatalyst (Table 1, entries 4–8). It is mainly due to the high oxidation potential value of the positive excited state of Acr+BF4−I (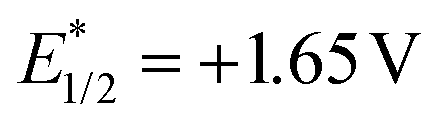 vs. SCE).11 Notably, the yield of this reaction irradiated with blue LEDs was much higher than that obtained with fluorescence light (Table 1, entries 4, 11). However, no desired product was detected in the absence of a light resource or photocatalyst, suggesting the important roles played by the light resource and photocatalyst in this conversion (Table 1, entries 9, 10 and 12). Furthermore, excellent yield was obtained when 2.5 equiv. of Cs2CO3 were utilized as the base instead of K2HPO4·3H2O (Table 1, entries 2 and 15). Only a trace of 3a was obtained without additive bases (Table 1, entries 13). We reasoned that the basicity of the reaction system is essential for the decarboxylation and the following formation of C–C bonds.
vs. SCE).11 Notably, the yield of this reaction irradiated with blue LEDs was much higher than that obtained with fluorescence light (Table 1, entries 4, 11). However, no desired product was detected in the absence of a light resource or photocatalyst, suggesting the important roles played by the light resource and photocatalyst in this conversion (Table 1, entries 9, 10 and 12). Furthermore, excellent yield was obtained when 2.5 equiv. of Cs2CO3 were utilized as the base instead of K2HPO4·3H2O (Table 1, entries 2 and 15). Only a trace of 3a was obtained without additive bases (Table 1, entries 13). We reasoned that the basicity of the reaction system is essential for the decarboxylation and the following formation of C–C bonds.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Light sources | Base | Solvent | Photocatalyst | Yieldb (%) |
| a Unless otherwise noted, all reactions were performed on a 0.2 mmol scale using 1 equiv. of 1a, 1 equiv. of 2a, 1.5 equiv. of base, 2 mol% photocatalyst, solvent (0.1 M). b Isolated yield. c Reaction performed with 3 mol% Ir(dF-CF3-ppy)2(dtbpy)PF6. d Reaction performed with 3 mol% Ru(bpy)3Cl2·6H2O. e Reaction performed in the absence of the photocatalyst. f Reaction performed in the absence of visible light. g Reaction performed in the absence of the base. h Reaction performed with 2.5 equiv. of Cs2CO3. | |||||
| 1 | Blue LEDs | K2HPO4·3H2O | CH3CN | Acr+BF4−I | 64 |
| 2 | Blue LEDs | K2HPO4·3H2O | Toluene | Acr+BF4−I | 72 |
| 3 | Blue LEDs | K2HPO4·3H2O | Acetone | Acr+BF4−I | 64 |
| 4 | Blue LEDs | K2HPO4·3H2O | CH2Cl2 | Acr+BF4−I | 73 |
| 5c | Blue LEDs | K2HPO4·3H2O | CH2Cl2 | Ir(dF-CF3-ppy)2(dtbpy)PF6 | 64 |
| 6 | Blue LEDs | K2HPO4·3H2O | CH2Cl2 | Acr+ClO4−II | 40 |
| 7d | Blue LEDs | K2HPO4·3H2O | CH2Cl2 | Ru(bpy)3Cl2·6H2O | Trace |
| 8 | Blue LEDs | K2HPO4·3H2O | CH2Cl2 | Eosin-Yellow | Trace |
| 9e | Blue LEDs | K2HPO4·3H2O | CH2Cl2 | — | 0 |
| 10e | Fluorescence | K2HPO4·3H2O | CH2Cl2 | — | 0 |
| 11 | Fluorescence | K2HPO4·3H2O | CH2Cl2 | Acr+BF4−I | 24 |
| 12f | — | K2HPO4·3H2O | CH2Cl2 | Acr+BF4−I | 0 |
| 13g | Blue LEDs | — | Toluene | Acr+BF4−I | 5 |
| 14 | Blue LEDs | Cs2CO3 | Toluene | Acr+BF4−I | 84 |
| 15h | Blue LEDs | Cs2CO3 | Toluene | Acr+BF4−I | 93 |
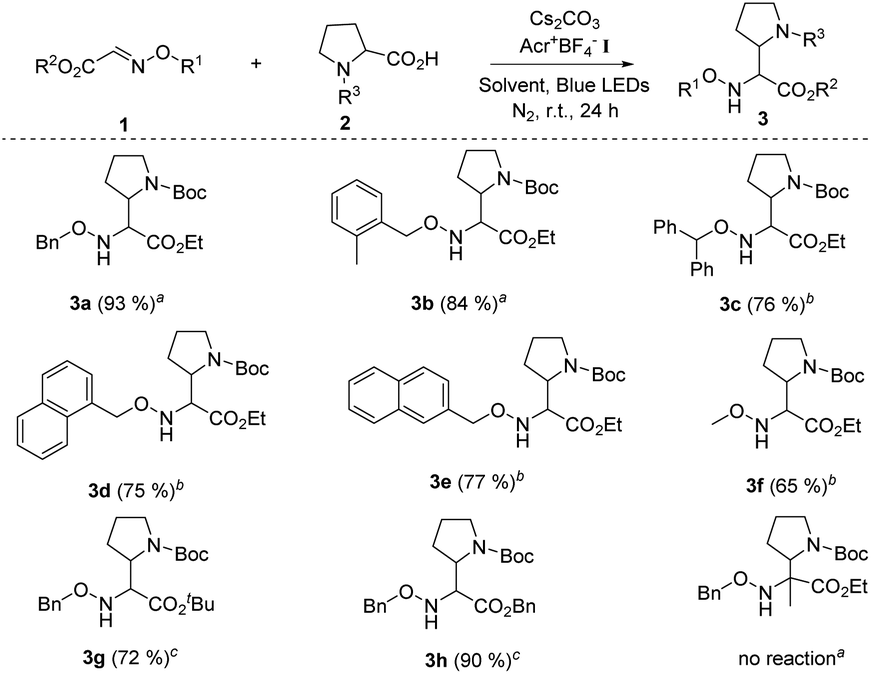
On the basis of the optimal reaction conditions, the scope of glyoxylic oxime ethers 1 was evaluated. As shown in Table 2, a variety of glyoxylic oxime ethers bearing different substitution patterns were well tolerated in the catalytic system (Table 2, 3a–h, 65–93%). When the phenyl group was replaced by other aromatic moieties, the process underwent efficiently and formed the corresponding α,β-diamino esters in good to excellent yields (Table 2, 3a–3e, 75–93%). Notably, O-methyl oxime ether 1f is also the suitable radical acceptor and provided 3f in slightly decreased yield. When the carboxyl group of glyoxylic oxime ether is protected by other groups, such as the tert-butyl group and benzyl group, 3g and 3h were synthesized in 72% and 90% yields, respectively. Unfortunately, when the ketone oxime ether substrate was involved in the reaction, no desired product was detected possibly due to the steric effect of ketone oxime ether.
a Unless otherwise noted, all reactions were carried out using 1.0 equiv. of 1 (0.2 mmol), 1.0 equiv. of 2 (0.2 mmol), 2.0 mol% Acr+BF4−I, toluene (0.1 M), 2.5 equiv. of Cs2CO3, under N2, at room temperature and irradiation with blue LEDs for 24 h.
b Using DCE (0.1 M) as the solvent while other conditions remained the same.
c The ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (0.2 mmol) is 1.5:1.0, and using DCE (0.1 M) as the solvent, while other conditions remained the same. :2 (0.2 mmol) is 1.5:1.0, and using DCE (0.1 M) as the solvent, while other conditions remained the same.
|
|---|
|
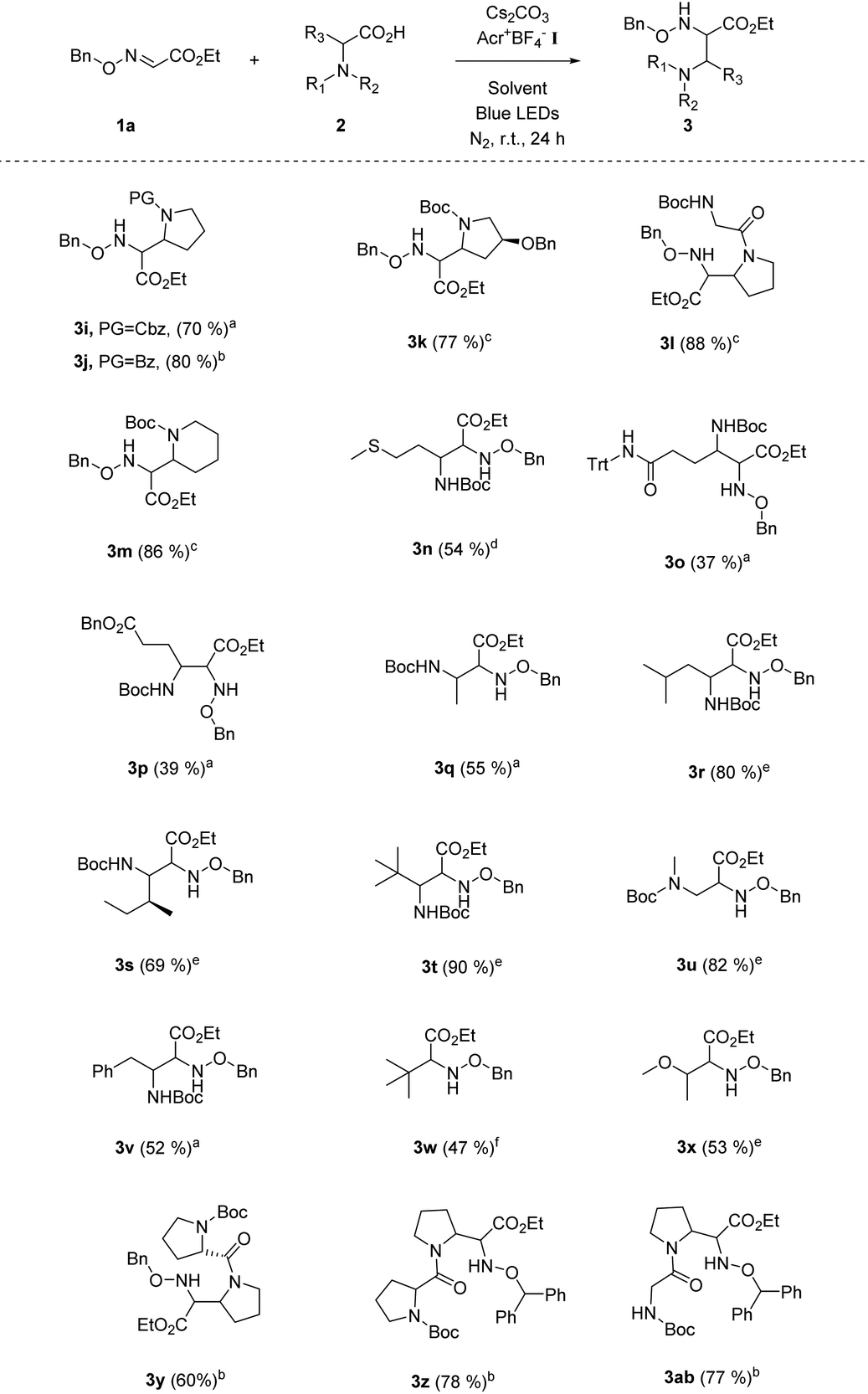
Our following task was to explore the substrate scope with respect to carboxyl acids. A number of N-protected five-membered cyclic amino acids (e.g. N-Cbz-proline, N-Bz-proline) readily participated in this radical coupling with glyoxylic oxime ether 1a, and provided the corresponding products in good yields (Table 3, 3i–l). The N-Boc protected six-membered cyclic amino acid is also the suitable alkyl radical source for this decarbonylative radical Mannich reaction (Table 3, 3m). Moreover, a series of chain amino acids with a variety of functional groups, such as the methylthio group, ester group and N-Trt protected amide group, could be tolerated in this radical coupling reaction (Table 3, 3n–p). Except for the N-Boc-alanine, other N-Boc protected natural chain amino acids (e.g., Boc-protected leucine, isoleucine, sarcosine, phenylalanine) could be smoothly transformed into the target products in moderate to good yields (Table 3, 3q–v). Intriguingly, the radical Mannich reaction was also amenable to various aliphatic acids, such as pivalic acid and 2-methoxypropionic acid (Table 3, 3w–x). We were delighted to find that dipeptides were successful substrates to obtain multi-amino esters in good yields (Table 3, 3y–ab). It provides a valuable and useful routine for the building of more complex α,β-diamino ester derivatives.
|
a Unless otherwise noted, all reactions were carried out using 1.0 equiv. of 1a (0.2 mmol), 1.0 equiv. of 2 (0.2 mmol), 2.0 mol% Acr+BF4−I, toluene (0.1 M), 2.5 equiv. of Cs2CO3, under N2, at room temperature and irradiation with blue LEDs for 24 h.
b Using DCE (0.1 M) as the solvent while other conditions remained the same.
c The ratio of 2:1a (0.2 mmol) is 1.5:1.0, and using DCE (0.1 M) as the solvent, while other conditions remained the same.
d The ratio of 2:1a (0.2 mmol) is 3.0:1.0, and using DCE (0.1 M) as the solvent, while other conditions remained the same.
e The ratio of 2:1a (0.2 mmol) is 2.0:1.0, and using DCE (0.1 M) as the solvent, while other conditions remained the same.
f Using DCE (0.1 M) as the solvent, pivalic acid as the alkyl carboxylic acid, while other conditions remained the same.
|
|---|
|
Finally, the reaction was carried out at the gram-scale. Under the standard conditions, the radical coupling between 3.37 mmol 1a and 2a was performed and 1.20 g 3a was obtained in excellent yield (Scheme 2, 92%). The scalability of this protocol demonstrates great value in the practical synthesis of α,β-diamino esters.
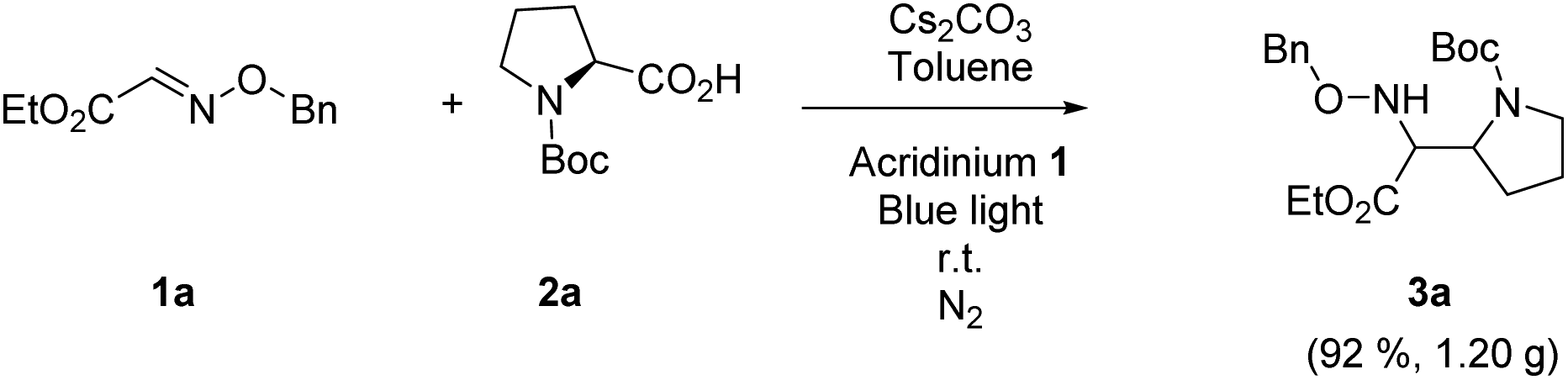 | ||
| Scheme 2 Gram-scale reaction of the radical Mannich reaction. | ||
Based on the previous reports, two plausible reaction mechanisms are shown in Scheme 3: the radical addition pathway5,13 and the radical–radical coupling pathway.14 Irradiation of Acr+BF4−I with visible light generates photoexcited state Acr*. Alkyl acid 2a can be deprotonated and then oxidized by Acr* (E1/2(C/C−) = −0.82 V vs. SCE in MeCN) which will be transformed into Acr*−, affording alkyl radical A following the decarboxylation process. On the one hand, the radical addition of A to Glyoxylic oxime ether 1a provides nitrogen radical B, which could be stabilized by the neighboring oxygen atom. Subsequently, the single electron transfers between B and Acr*˙−, generating glyoxylic oxime ether anion C and Acr. On the other hand, glyoxylic oxime ether 1a is reduced by Acr*˙− to produce glyoxylic oxime ether radical anion D. Then it undergoes radical–radical coupling with B to afford nitrogen anion C. The final product 3a is obtained via the following protonation of C.
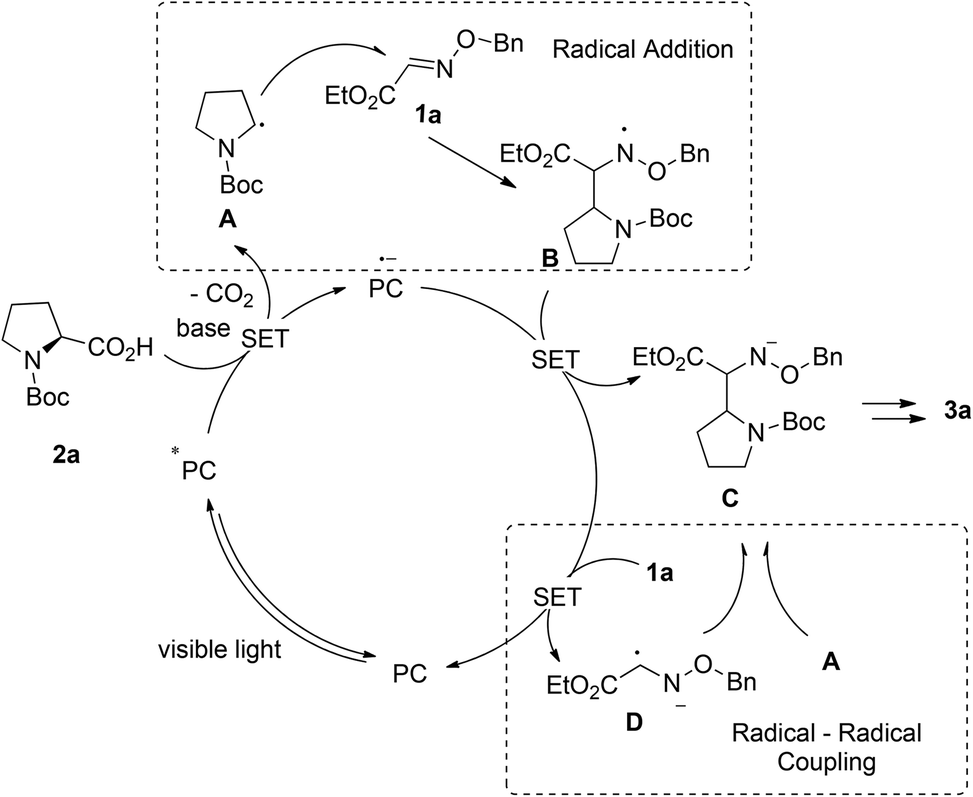 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel protocol to construct α,β-diamino esters via the radical Mannich reaction using alkyl carboxylic acids as radical precursors under mild photoredox conditions. This method is subjected to a wide range of substrates with broad functional group tolerance. Moreover, the gram scale reaction also maintains an excellent yield.Conflicts of interest
The authors declare no competing financial interest.Notes and references
- (a) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580 CrossRef CAS; (b) A. Viso, R. F. de la Pradilla, A. García and A. Flores, Chem. Rev., 2005, 105, 3167 CrossRef CAS PubMed; (c) K. Okamoto, M. Sakagami, F. Feng, H. Togame, H. Takemoto, S. Ichikawa and A. Matsuda, J. Org. Chem., 2012, 77, 1367 CrossRef CAS PubMed; (d) S. Izumi, Y. Kobayashi and Y. Takemoto, Org. Lett., 2016, 18, 696 CrossRef CAS PubMed; (e) H. Zhang, Z. Yang, B. N. Zhao and G. Li, J. Org. Chem., 2018, 83, 644 CrossRef CAS PubMed.
- (a) M. S. Valle, M. F. Saraiva, P. Retailleau, M. V. de Almeida and R. H. Dodd, J. Org. Chem., 2012, 77, 5592 CrossRef CAS PubMed; (b) C. Jing, D. Xing, Y. Qian and W. Hu, Synthesis, 2014, 46, 1348 CrossRef; (c) R. Lei, Y. Wu, S. Dong, K. Jia, S. Liu and W. Hu, J. Org. Chem., 2017, 82, 2862 CrossRef CAS PubMed.
- (a) C. Mannich and W. Krösche, Arch. Pharm., 1912, 250, 647 CrossRef CAS; (b) M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044 CrossRef; (c) H. Miyabe, R. Shibata, M. Sangawa, C. Ushiro and T. Naito, Tetrahedron, 1998, 54, 11431 CrossRef CAS; (d) L. Bernardi, A. S. Gothelf, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2003, 68, 2583 CrossRef CAS PubMed; (e) H. Miyabe, M. Ueda, A. Nishimura and T. Naito, Tetrahedron, 2004, 60, 4227 CrossRef CAS; (f) M. M. B. Marques, Angew. Chem., Int. Ed., 2006, 45, 348 CrossRef CAS PubMed; (g) R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940 RSC; (h) G. Callebaut, S. Mangelinckx, P. Van der Veken, K. W. Törnroos, K. Augustyns and N. De Kimpe, Beilstein J. Org. Chem., 2012, 8, 2124 CrossRef CAS PubMed; (i) G. Callebaut, T. Meiresonne, N. De Kimpe and S. Mangelinckx, Chem. Rev., 2014, 114, 7954 CrossRef CAS PubMed; (j) C. Huo, M. Wu, X. Jia, H. Xie, Y. Yuan and J. Tang, J. Org. Chem., 2014, 79, 9860 CrossRef CAS PubMed.
- (a) G. Callebaut, S. Mangelinckx, L. Kiss, R. Sillanpää, F. Fülöp and N. De Kimpe, Org. Biomol. Chem., 2012, 10, 2326 RSC; (b) H. Fujino, M. Nagatomo, A. Paudel, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, Angew. Chem., Int. Ed., 2017, 56, 11865 CrossRef CAS PubMed.
- Y. Yoshimi, K. Kobayashi, H. Kamakura, K. Nishikawa, Y. Haga, K. Maeda, T. Morita, T. Itou, Y. Okada and M. Hatanaka, Tetrahedron Lett., 2010, 51, 2332 CrossRef CAS.
- M. Nagatomo, H. Nishiyama, H. Fujino and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 1537 CrossRef CAS.
- S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 14560 CrossRef CAS.
- For selected examples, see: (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS; (b) A. Noble, S. J. Mccarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624 CrossRef CAS; (c) S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654 CrossRef CAS; (d) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS; (e) J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522 CrossRef CAS; (f) N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed.
- (a) C. Cassani, G. Bergonzini and C.-J. Wallentin, Org. Lett., 2014, 16, 4228 CrossRef CAS PubMed; (b) X. Wu, C. Meng, X. Yuan, X. Jia, X. Qian and J. Ye, Chem. Commun., 2015, 51, 11864 RSC; (c) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340 CrossRef CAS.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244 CrossRef CAS.
- (a) H. Miyabe, C. Ushiro and T. Naito, Chem. Commun., 1997, 1789 RSC; (b) H. Miyabe, C. Ushiro, M. Ueda, K. Yamakawa and T. Naito, J. Org. Chem., 2000, 65, 176 CrossRef CAS PubMed.
- Y. Yoshimi, S. Washida, Y. Okita, K. Nishikawa, K. Maeda, S. Hayashi and T. Morita, Tetrahedron Lett., 2013, 54, 4324 CrossRef CAS.
- E. Fava, A. Millet, M. Nakajima, S. Loescher and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6776 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and ESI spectra for compounds. See DOI: 10.1039/c9qo00407f |
| This journal is © the Partner Organisations 2019 |