
“Rod–coil” copolymers get self-assembled in solution
Fugui
Xu†
,
Jiacheng
Zhang†
 ,
Pengfei
Zhang
,
Xiangfeng
Luan
and
Yiyong
Mai
*
,
Pengfei
Zhang
,
Xiangfeng
Luan
and
Yiyong
Mai
*
School of Chemistry and Chemical Engineering, Shanghai Key Laboratory of Electrical Insulation and Thermal Ageing, Shanghai Jiao Tong University, 800 Dongchuan Road, 200240, Shanghai, P. R. China. E-mail: mai@sjtu.edu.cn
First published on 27th August 2019
Abstract
Supramolecular self-assembly of amphiphilic “rod–coil” copolymers in solution has attracted tremendous interest, as they exhibit distinct self-assembly behaviors profiting from the introduction of rigid segments, compared with coil–coil copolymers. The unique interplay between microphase separation of the rod and coil blocks with great geometric disparities leads to the formation of various ordered nanostructures including some unusual morphologies, such as ribbons, helices, nanosheets, and toroids, which are difficult to observe in the self-assembly of coil–coil copolymers. This article overviews the recent advances in tunable solution self-assembly of several common types of amphiphilic rod–coil copolymers, including block, alternating, graft, star, and hyperbranched/dendritic copolymers, which contain functional rod segments such as conjugated polymers, liquid crystalline polymers, polypeptides, and helical polymers. The discussions focus on the morphological control, distinctive optical/electronic properties, and stimuli-responsiveness of the assemblies, which make them potential functional materials particularly for optical, optoelectronic and biological applications.
 Yiyong Mai | Yiyong Mai received his PhD from Shanghai Jiao Tong University in April 2007 under the supervision of Professor Deyue Yan. He joined the group of Professor Adi Eisenberg at McGill University as a Postdoctoral Fellow in September 2008, where he was promoted as a Research Associate in March 2012. He has been a Professor in School of Chemistry and Chemical Engineering at Shanghai Jiao Tong University since March 2013. He visited the group of Professor Klaus Müllen at Max Plank Institute for Polymer Research (MPIP) as a visiting scholar from May to October 2013. He was selected as Shanghai Eastern Scholar in 2014/2018 and Shanghai Academic Research Leader in 2019, respectively. His present research interest involves polymer self-assembly and functional materials for optoelectronic and energy-related applications. |
Introduction
The self-assembly of amphiphilic copolymers in solution has proven to be a powerful bottom-up strategy for the preparation of diverse ordered structures, including spheres, cylinders, bicontinuous structures, lamellae, and vesicles, among others.1–6 The resultant superstructures exhibit great potential applications in many fields, such as biomedicine, microelectronics, energy storage and conversion, and catalysts.1–6 In recent years, thanks to the development of a wide range of organic synthesis and polymerization techniques, various types of amphiphilic copolymers have been successfully synthesized, including block copolymers (BCPs), alternating copolymers, graft copolymers (or polymer brushes), star-like polymers, and hyperbranched or dendritic polymers, among others.7–11 Depending on the flexibility of the building blocks, these copolymers can generally be classified into three categories: “coil–coil”, “rod–coil” and “rod–rod” copolymers, in which “rod–rod” type is not frequent to see in solution self-assembly. Rod–coil copolymers consist of rigid polymer segments, such as π-conjugated polymers, liquid crystalline (LC) polymers, polypeptides, and helical polymers, which are covalently bonded with flexible blocks.10,11 In general, the rigid polymers exhibit rod-like anisotropic conformation with a high bending energy, meaning that the bending of rigid polymers is constrained, while the coil polymers are flexible and behave like random coils.11,12 In theoretical simulations such as molecular dynamics (MD) simulation, the rod polymer is usually modeled as a linear chain consisting of several beads connected through bond stretching potential, and the rigidity of the rod block is introduced by the angle bend potential; without the angle bending potential constraint, the coil block is represented by a flexible chain.12 The anisotropic feature and rigid conformation of the rod segments result in a tendency for orientational ordered packing during aggregation, which affords rod–coil copolymer self-assembly behaviors different from those of the coil–coil type copolymers.11 Theoretical simulations have demonstrated the effect of chain rigidity on the self-assembled structures from rod–coil diblock copolymers.12,13 For instance, Lin and coworkers constructed a series of rod–coil copolymers with various rigid conformation fractions and rigid portion locations, through Brownian dynamics (BD) simulation. With a decrease in the fraction of the rigid conformation, the assemblies transit from cylindrical to spherical micelles.14,15 In experiments, apart from the common assemblies formed by coil–coil copolymers, such as spherical micelles, cylindrical micelles, lamellae and vesicles,1,4 unusual superstructures such as helices, ribbons, sheets, and toroids can also been frequently observed in the self-assembly of rod–coil copolymers due to the interplay between microphase separation of the rod and coil blocks with great geometric disparities.10–18The self-assembly of amphiphilic rod–coil copolymers in solution has been studied actively since the end of the 1990s. As one of the pioneer works, Jenekhe and coworkers reported the tunable self-assembly of poly(phenylquinoline)-block-polystyrene (PPQ-b-PS) rod–coil diblock copolymers in 1998, which produced robust, micrometer-scale, spherical, vesicular, cylindrical, and lamellar aggregates depending on the copolymer composition.20 Thereafter, remarkable achievements have been made in this field in the past two decades. Numerous crucial scientific issues have been explored, including self-assembly techniques, morphological control principles, theory and simulation, physical properties and potential applications of the assemblies.10–19,21,22 In light of the great interest and high level of activity of this area, this paper overviews its progress in the last 10 years, with an emphasis on five common types of rod–coil copolymers of different architectures, including block, alternating, graft, star-like, and hyperbranched copolymer systems (Fig. 1). Neither the synthesis of the rod–coil copolymers nor their self-assembly theory and simulation are covered in this paper; several review articles are recommended for these topics (ref. 8, 10–13, 16–19, 21 and 22). The following discussions concentrate on a number of crucial aspects including morphological control strategy, aggregation mechanism, physical properties and stimuli-responsiveness of rod–coil copolymer aggregates. Potential applications are briefly introduced along with the corresponding examples, which highlight case-by-case the promising perspective of rod–coil copolymer self-assembly especially in optical, optoelectronic, energy and biological applications.
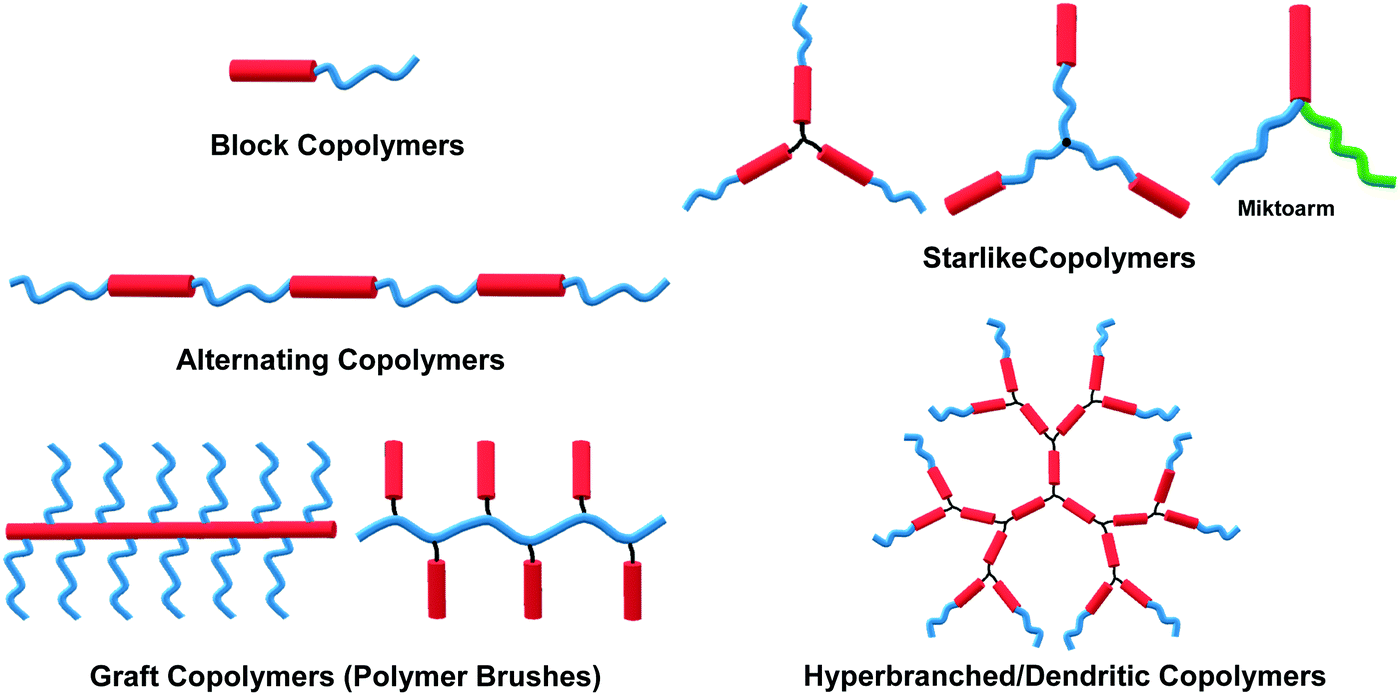 | ||
| Fig. 1 Molecular structures of various types of rod–coil copolymers. | ||
Self-assembly of rod–coil block copolymers
Rod–coil block copolymers based on conjugated polymers
π-Conjugated polymers, including polythiophene, polyphenylene, polyfluorene, polyacetylene, poly(p-phenylenevinylene), polyquinoline, polycarbazole, etc., have appealing optical and electronic properties.10,11,23–64 For example, polythiophene and their derivatives are an important type of conducting polymer and have been widely investigated as an active component of semiconducting devices such as organic field-effect transistors (FETs), solar cells, and light-emitting diodes (LEDs). Self-assembly of rod–coil copolymers containing conjugated blocks provides a powerful route for precisely tuning the ordered molecular organization mode as well as the resulting optical and electronic properties, which generally cannot be achieved from disordered aggregation of conjugated polymers.10,11 Among the different rod–coil copolymer systems, linear BCPs represent the most extensively studied family, in which the widely-used rigid and flexible blocks are summarized in Fig. 2.8,10,11,20–22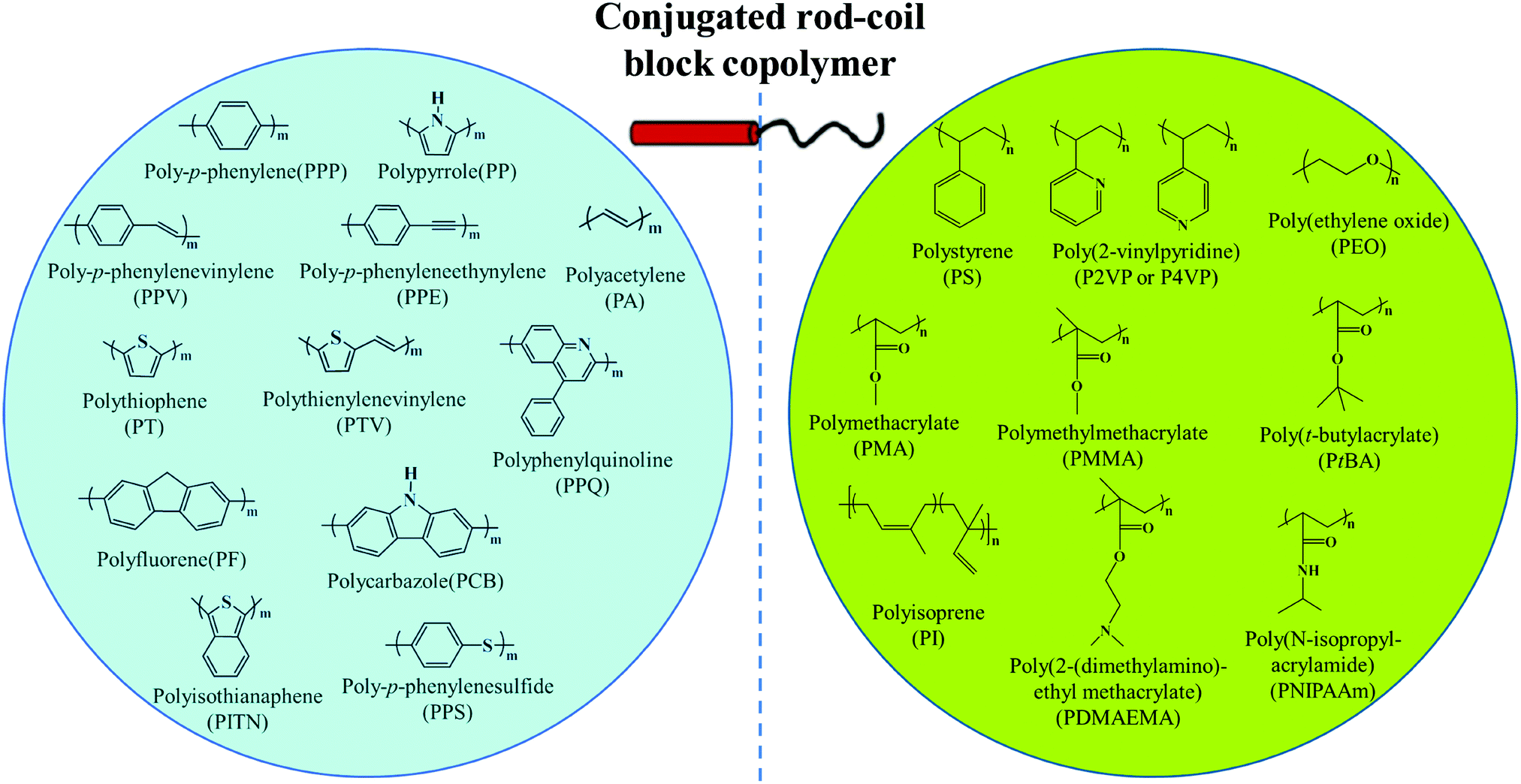 | ||
| Fig. 2 General rod and coil blocks of conjugated rod–coil block copolymers. Reproduced with permission from ref. 10. Copyright 2011, Elsevier. | ||
Polythiophene and their derivatives represent the most widely studied π-conjugated polymer systems, and the self-assembly of amphiphilic rod–coil BCPs containing polythiophene type blocks has attracted tremendous interest.26–47 This type of BCP can easily form low-dimensional (1D and 2D) structures as the ordered arrangement of the rigid and crystalline polythiophene type blocks inclines to follow a 1D or 2D direction. As a typical example, Winnik, Manners and coworkers reported the formation of cylindrical micelles with a crystalline poly(3-hexylthiophene) (P3HT) core by the crystallization-driven self-assembly of P3HT-block-poly(dimethylsiloxane) (P3HT-b-PDMS) copolymers in Et2O/toluene mixed solvents, using seed micelles as initiators (Fig. 3).26 The length of the cylinders was readily controlled in the range of 40 to 320 nm by adjusting the unimer-to-seed ratio, which is a typical characteristic of “living” self-assembly. In another case, they prepared stable nanofibers with precisely controllable length by living crystallization-driven self-assembly of conjugated diblock copolymers containing a crystalline regioregular P3HT core and a soluble, amorphous regiosymmetric P3HT corona.27 The nanofibers are solution-processible and electroactive, and thus are employed as the active layer in FETs. Interestingly, the resulting charge carrier mobility strongly depends on both the length of the core-forming block and the fiber length, but is independent of corona composition. This study highlights the significant potential of colloidally stable electroactive fibre-like micelles from common conjugated block copolymers in fundamental study of charge carrier processes in devices and in future electronic applications.
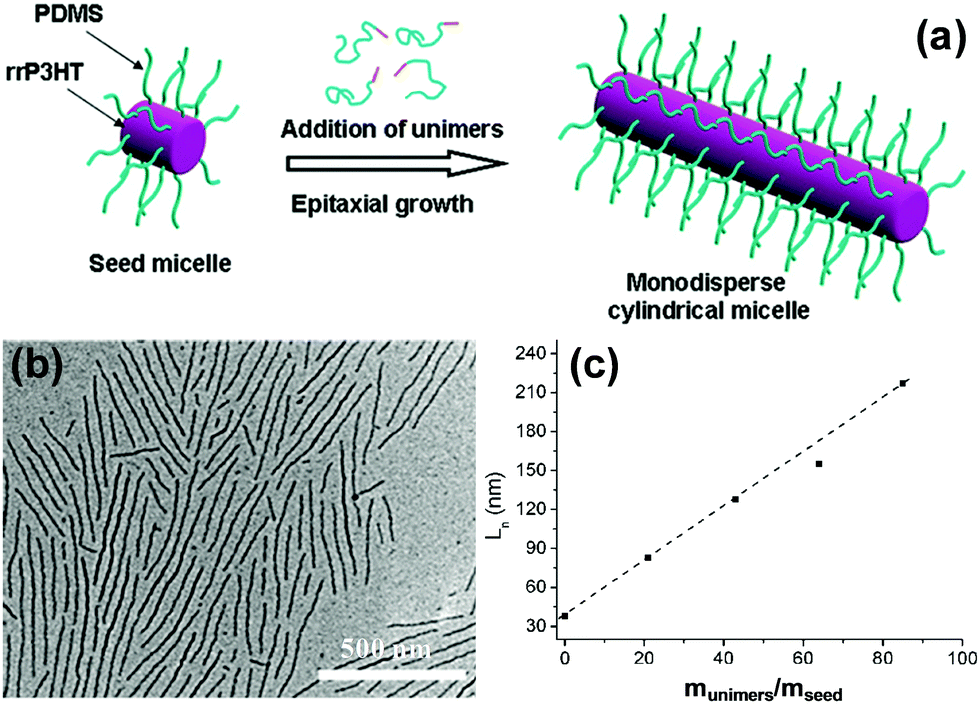 | ||
| Fig. 3 (a) Schematic representation of the formation of monodisperse P3HT-b-PDMS cylindrical micelles via crystallization-driven self-assembly using small seed micelles as initiators. (b) A typical TEM image of P3HT-b-PDMS cylindrical micelles. (c) Linear dependence of the micelle contour length on the unimer-to-seed ratio. Reproduced with permission from ref. 26. Copyright 2011, American Chemical Society. | ||
Park's group also achieved 1D nanocylinders of tunable lengths by the self-assembly of P3HT-b-poly(ethylene glycol) (P3HT-b-PEG) in chloroform/water solvent mixture. Rather than the crystallization-driven case, the control of the cylinder length was realized through varying the length of the PEG coils.32 The longer PEG chains had larger corona dimensions, which prevented the packing of more P3HT segments in the cylindrical core and thus yielded shorter nanofibers. By tuning the interactions among the flexible blocks, other interesting 1D superstructures have also been observed. Hayward, Emrick, and colleagues obtained superhelices by the self-assembly of P3HT-b-poly(3-triethylene glycol thiophene) (P3HT-b-P3(TEG)T) diblocks in solution.31 This copolymer first self-assembled into nanowires upon the addition of methanol to their chloroform solutions; the wire lengths were controlled by altering the block ratio or solvent nature. At suitable block ratios, the addition of K+ ions, which induced an increase in the steric hindrance among the P3(TEG)T coils during self-assembly, drove the formation of helical nanowires, which further bundled into double- or multi-stranded superhelices. The superstructures in solution exhibited a red-shift in spectral absorption compared with that of the copolymer.
2D self-assembly of π-conjugated polymers has attracted great interest very recently, in light of their advantages in planar electron transfer and potential applications for optoelectronic nanodevices.33 2D superstructures can be formed by finely adjusting the interplay between the interactions of the polythiophene-type blocks and those among the flexible blocks. For example, Park's group employed a conjugated diblock copolymer of a polythiophene derivative, poly[3-(2,5,8,11-tetraoxatridecanyl)thiophene]-b-PEG (PTOTT-b-PEG) as the precursor, which self-assembled into well-defined nanoribbons and nanosheets in water/methanol mixture, both of which have an average thickness of 7.5 ± 0.8 nm (Fig. 4).33 These superstructures were a result of a delicate interplay of the hydrogen bonding between the diblock copolymer and the solvent with the π–π interaction of the PTOTT blocks. Comparison of the thicknesses with the average dimension of the copolymer molecules coupled with the blue-shifted spectral absorption suggested that the PTOTT blocks probably adopted a twisted conformation in the 2D superstructures.
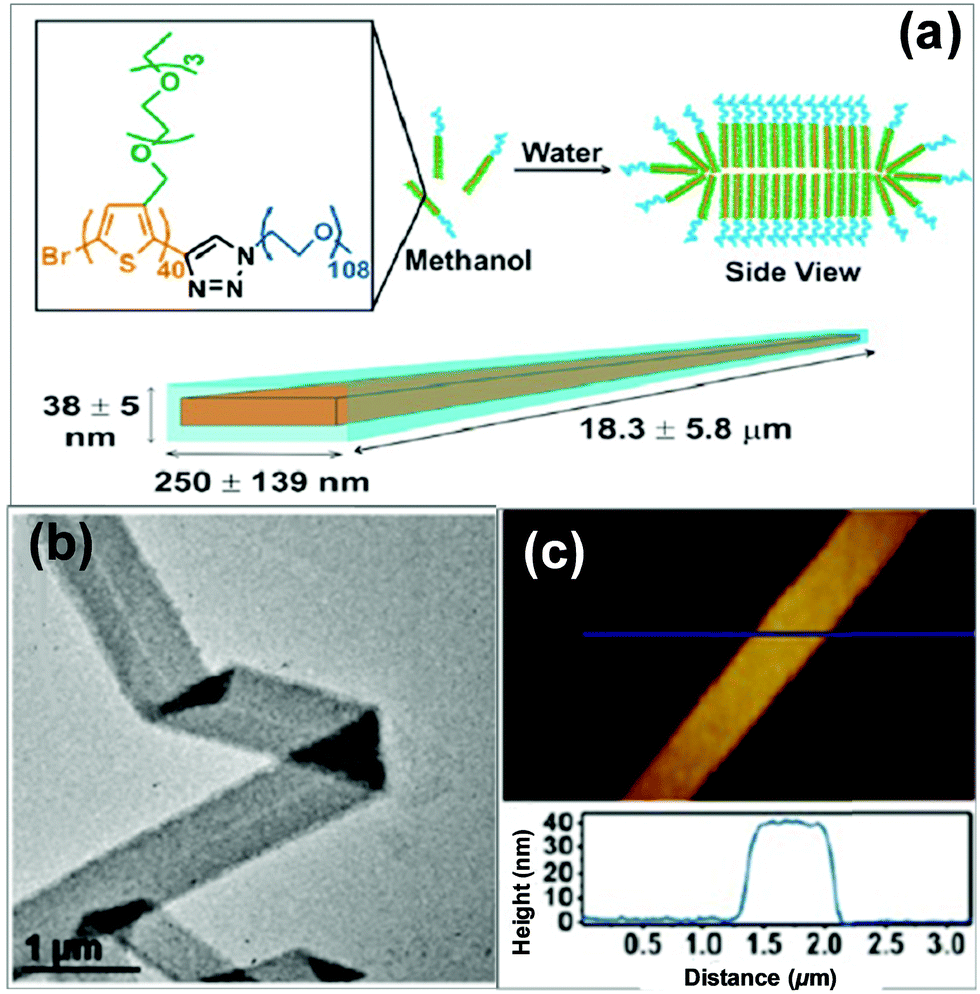 | ||
| Fig. 4 Supramolecular nanoribbons formed by the self-assembly of PTOTT-b-PEG in methanol/water mixed solvents. Reproduced with permission from ref. 33. Copyright 2014, American Chemical Society. | ||
Interestingly, this group also synthesized a conjugated diblock copolymer with DNA blocks, PTOTT-b-DNA (Fig. 5).34 This copolymer self-assembled into vesicles, a 3D nanostructure, with a PTOTT wall and DNA coronae in water; the average size of the vesicles was controlled over a broad length scale from a couple of hundred nanometers to over a micrometer by varying the concentration of the copolymers. Moreover, PTOTT-b-DNA showed a salt-induced reversible morphology transition from vesicles to lamellae due to the screening of negative charges on the phosphate backbone of DNA. In another case, Wu and colleagues prepared inverse vesicles by the self-assembly of poly(phenyl isocyanide)-b-(P3(TEG)T) (PPI-b-P3(TEG)T) in tetrahydrofuran (THF)/methanol mixed solvent. The vesicles had the P3(TEG)T blocks as the coronae and can be transformed to spherical micelles by varying the solvent composition.41 The light emissions of the aggregates were reversible upon their morphological transitions.
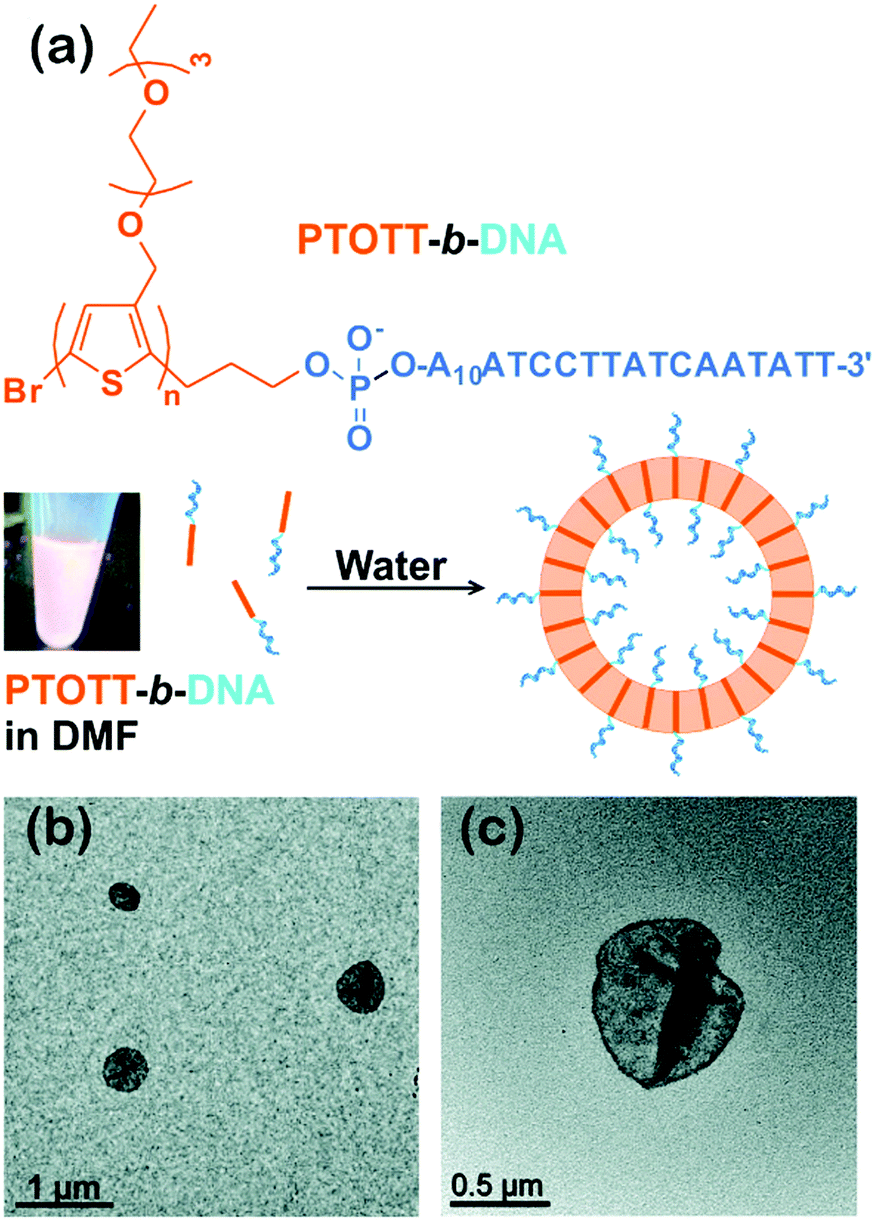 | ||
| Fig. 5 (a) Chemical structure of PTOTT-b-DNA and schematic depiction of the self-assembly of PTOTT-b-DNA into vesicles. (b) TEM images of PTOTT-b-DNA vesicles and (c) PTOTT-b-DNA vesicles stained with uranyl acetate. Reproduced with permission from ref. 34. Copyright 2014, American Chemical Society. | ||
Choi and colleagues reported the formation of 3D branched semiconducting superstructures including nanostars and nanonetworks via in situ nanoparticlization of conjugated polymers (INCP), which utilized diblock copolymers (P3EHT-b-PT) with a soluble poly(3-(2-ethylhexyl)thiophene) (P3EHT) block and an insoluble polythiophene block as the precursor.42 The INCP strategy employs insoluble conjugated polymers as the core-forming blocks, and their solvophobic and strong π–π interactions provide the driving force for in situ self-assembly of the conjugated copolymers during polymerization.42,43 By this method, the resultant nanostars and nanonetworks are kinetically trapped nanostructures, which are spontaneously and irreversibly formed, but are highly stable toward external stimuli such as heat and mechanical stress.
Apart from the above-discussed BCP systems, other recently investigated polythiophene-based rod–coil BCPs include P3HT-b-poly(4-vinylpyridine) (P3HT-b-P4VP),36 P3HT-b-poly(2-vinylpyridine) (P3HT-b-P2VP),38 and P3HT-b-poly(N-isopropylacrylamide) (P3HT-b-PNIPAM),45 although there have been only a small number of publications covering these systems. These BCPs can also be tunably self-assembled into cylinders, lamellae, and vesicles, among other nanostructures, depending on the copolymer composition and environmental factors.
Following similar self-assembly principles, a number of rod–coil BCP systems containing other types of conjugated polymers, such as polyfluorene,48–51 polyphenylene,52,53 polyacetylene,54 poly(p-phenylenevinylene) (PPV),43,55–61 and polyaniline,62 also exhibit controllable multi-dimensional self-assembly behavior in solution. Polyfluorene-based rod–coil BCPs possess high thermal/chemical stability and good fluorescence quantum yields, and thus their self-assembly in solution is of increasing interest.48,49 In 2018, Manners and coworkers prepared nanofibers of controlled lengths (1–5 μm) with a poly(di-n-hexylfluorene) (PDHF) core and a segmented corona consisting of PEG in the center and a polythiophene corona at each terminus, by seeded living crystallization-driven self-assembly of PDHF-b-PEG diblocks with a crystallizable π-conjugated PDHF block (Fig. 6).50 These nanofibers exhibit exciton transfer from the core to the lower-energy quaternized polythiophene (QPT) coronas at the end, which occurs in the direction of the interchain π–π stacking with the longest diffusion lengths (>200 nanometers) reported thus far and a high diffusion coefficient (0.5 square centimeters per second). Such diffusion lengths could enable light-harvesting devices using these polymer structures as antennae coupled to photodetector materials of limited absorption, and would also enable much simpler bilayer design of organic photovoltaics relative to those based on the bulk heterojunction.
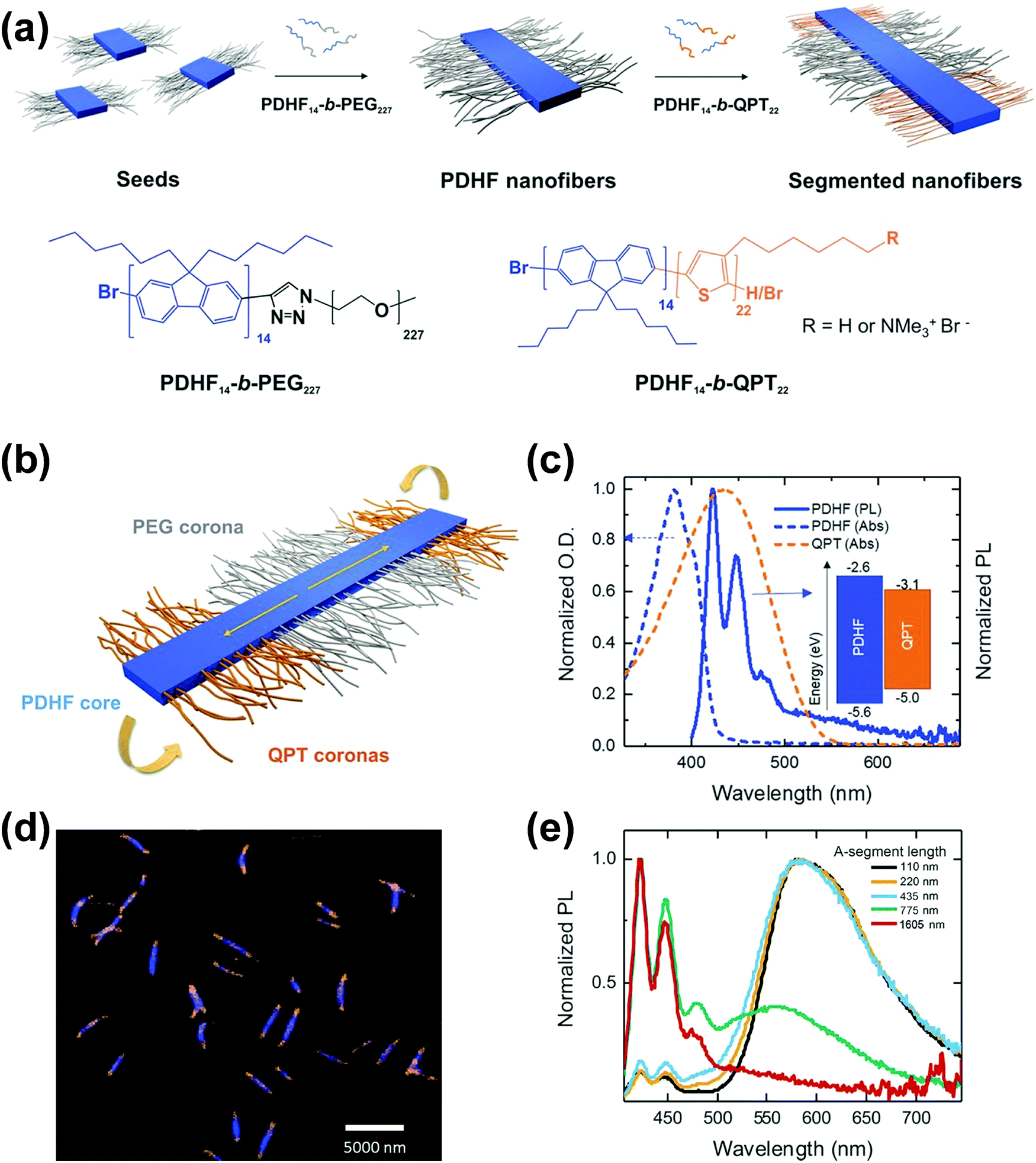 | ||
Fig. 6 (a) Schematic illustration of the seeded growth process and the molecular structures of PDHF14-b-PEG227 and PDHF14-b-QPT22. (b) The segmented B–A–B nanofiber structure with separate donor and acceptor domains. (c) Normalized absorption of QPT homopolymer in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (1:1) (orange dashes), and unsegmented PDHF nanofibers (blue dashes), and photoluminescence (PL) emission of unsegmented PDHF nanofibers in the same solution (solid blue line). The inset shows the energy levels of the PDHF and QPT. (d) Laser scanning confocal microscopy (LSCM) image of the uniform segmented PDHF nanofibers with a crystalline PDHF core (blue emission) and two terminal blocks with QPT coronas (orange emission). (e) Normalized PL spectra of segmented PDHF nanofibers with different A-segment lengths. Emission arising from direct excitation of the QPT in the 1605 nm sample was unresolved. Reproduced with permission from ref. 50. Copyright 2018, American Association for the Advancement of Science. :MeOH (1:1) (orange dashes), and unsegmented PDHF nanofibers (blue dashes), and photoluminescence (PL) emission of unsegmented PDHF nanofibers in the same solution (solid blue line). The inset shows the energy levels of the PDHF and QPT. (d) Laser scanning confocal microscopy (LSCM) image of the uniform segmented PDHF nanofibers with a crystalline PDHF core (blue emission) and two terminal blocks with QPT coronas (orange emission). (e) Normalized PL spectra of segmented PDHF nanofibers with different A-segment lengths. Emission arising from direct excitation of the QPT in the 1605 nm sample was unresolved. Reproduced with permission from ref. 50. Copyright 2018, American Association for the Advancement of Science. | ||
Poly(p-phenylenevinylene) (PPV) or oligo(p-phenylenevinylene) (OPV) is also one of the well-known π-conjugated materials with intriguing optical and electronic properties.43,55–61 Based on the seeded growth strategy of living crystallization-driven self-assembly, Huang and coworkers prepared, in ethanol, 1D nanofibers of controlled lengths ranging from 50 to 870 nm with a uniform OPV core and a poly(N-isopropylacrylamide) (PNIPAM) corona (Fig. 7).57 The seeded growth protocol was also applied to the preparation of ABA triblock comicelles containing an OPV core by the addition of OPV-b-PNIPAM unimers to seed micelles of OPV-b-PDEAEMA.
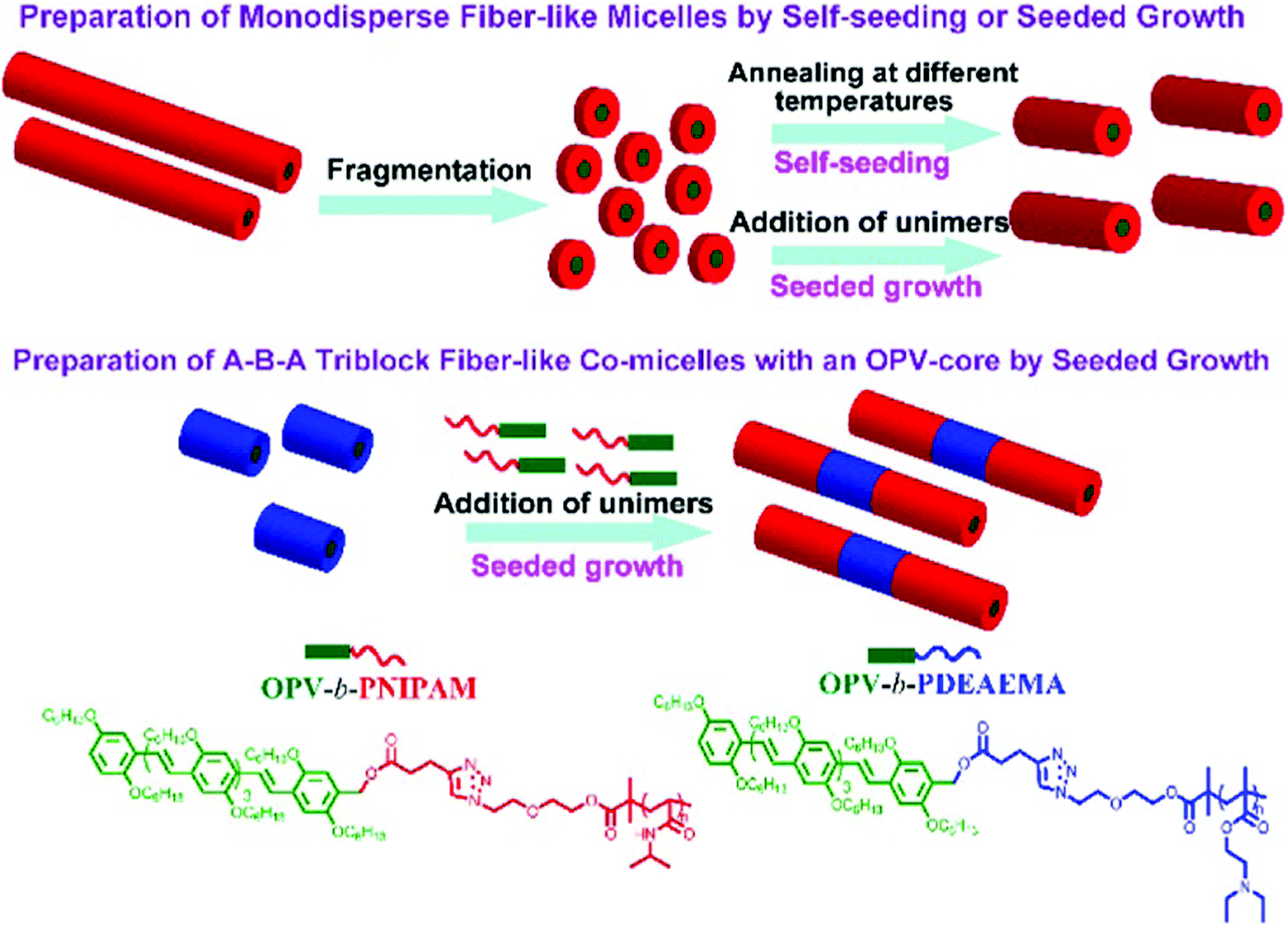 | ||
| Fig. 7 Illustration of the preparation of uniform nanocylinders and ABA triblock comicelles with an OPV core by living crystallization-driven self-assembly. Reproduced with permission from ref. 57. Copyright 2017, American Chemical Society. | ||
Aside from the above-mentioned crystallization ability, PPV polymers can also show interesting light-induced cis-to-trans isomerization.43,60 This photoisomerization can significantly reduce the solubility of PPV because cis-rich PPV has a twisted and coiled configuration, while the extended trans-rich PPV has a rod-like structure, which increases its π–π interactions and crystallinity, thereby facilitating its packing. Choi and coworkers reported the first light-induced crystallization-driven self-assembly (LI-CDSA) involving the initial synthesis of a soluble cis-rich polynorbornene-b-poly(p-phenylenevinylene-2,5-dimethoxy-p-phenylenevinylene) (PNB-b-MeO-PPV) copolymer, followed by its photoisomerization to more crystalline trans-rich PPV (Fig. 8).60 This stimulus triggered the self-assembly of the PNB-b-MeO-PPV copolymer into uniform nanofibers of controllable lengths by varying the unimer-to-seed ratio, as well as block or gradient comicelles by living seeded growth. Notably, LI-CDSA occurred rapidly within only a few minutes, and its living epitaxial growth was easily modulated by turning the light on and off. Since the PNB shell block can be easily modified, and the core contains fluorescent PPV, various functional polymeric materials with precise nanostructures can be designed for various applications.
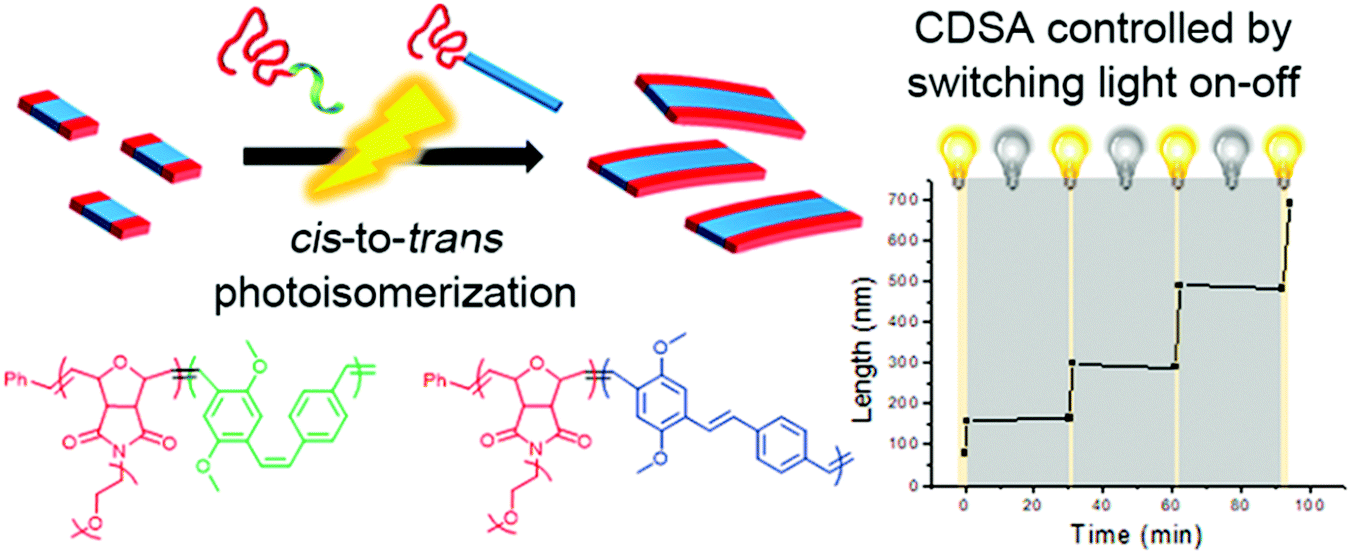 | ||
| Fig. 8 Schematic illustration of the formation of nanofibers through the LI-CDSA of PNB-b-MeO-PPV) block copolymer. Reproduced with permission from ref. 60. Copyright 2018, American Chemical Society. | ||
Highly regular and homogeneous 2D fluorescent square non-crystallization micelles were recently achieved by He's group,61 through the self-assembly of PPV-b-P2VP diblock copolymers in 2-PrOH solution (Fig. 9), which involved a dissolving-cooling-aging process. The dimension of the square micelles was controllable by varying the volume ratio of PPV/P2VP or the copolymer concentration. The mechanism study revealed that the PPV-b-P2VP diblocks first formed 1D structures, which further evolved into 2D structures in solution; the growth was driven by intermolecular π–π interaction between the PPV blocks. The square micelles were probably formed by the herringbone arrangement of the PPV segments, pertaining to the presence of the branched alkyl chains attached to the conjugated PPV cores.
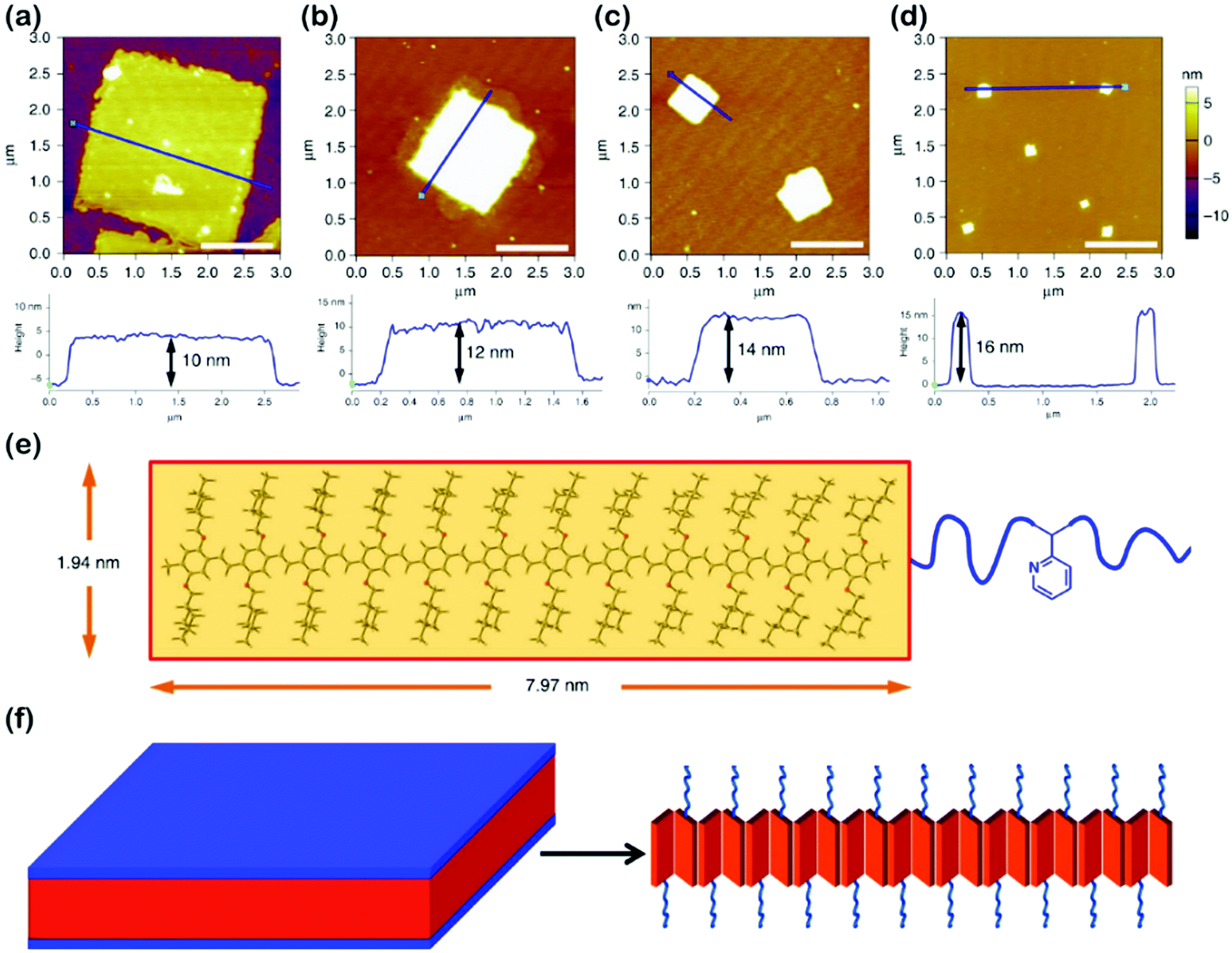 | ||
| Fig. 9 (a–d) AFM height images of 2D square micelles with different dimensions, obtained from 2-PrOH solution of PPV12-b-P2VP12 (a), PPV12-b-P2VP16 (b), PPV12-b-P2VP22 (c), and PPV12-b-P2VP36 (d). Scale bars represent 1 μm. (e) The optimized molecular structure and calculated scales of the PPV12 block by DFT calculations. (f) Schematic illustration and possible molecular packing of the square micelles. Reproduced with permission from ref. 61. Copyright 2018, Springer Nature. | ||
Rod–coil block copolymers containing liquid crystalline polymers
Amphiphilic rod–coil BCPs containing LC polymers as hydrophobic blocks are capable of self-assembling into well-defined nanostructures with ordered LC domains.10,11,63–65 The switchable order–disorder or order–order transition of the LC blocks induced by varying environmental parameters may lead to the variation of aggregate morphology. Therefore, LC BCPs can also be considered as ideal candidates to study the effect of additional order in the rod-like block on the morphology of assemblies. In the last ten years, most studies focused on the rod–coil BCP systems containing side-chain liquid crystalline polymers (SCLCPs), which show extraordinary self-assembly behavior in solution.66–93 Furthermore, stimuli-responsive SCLCPs give their BCP assemblies potential applications in controllable drug release, optical devices, intelligent displays, and so on.Azobenzene is a frequently-used LC mesogen in rod–coil BCPs, as its trans–cis isomerization upon alternative UV/vis light irradiation may result in interesting and reversible morphological changes of the aggregates.66,69,70,74,77,85–88 For instance, Yuan's group prepared a series of azo-containing copolymeric assemblies based on poly(N,N-dimethylaminoethyl methacrylate)-b-poly[(benzyl methacrylate)-co-(4-phenylazophenyl methacrylate)] [PDMA-b-P(BzMA-co-AzoMA)] by reversible addition–fragmentation chain transfer (RAFT) polymerization induced self-assembly (PISA) at high solid contents (Fig. 10).85 By adjusting the chain length of the P(BzMA-co-AzoMA) block, spheres, worms, and vesicles were readily obtained. In particular, the azo-containing wormlike micelles underwent reversible worm-to-vesicle transformation upon alternative UV/vis light irradiation. During the morphology evolution, a series of intermediates, including coalesced worms, “octopus”-like and “jellyfish”-like structures, were observed. The morphology transformation was rationalized by the volume change of the P(BzMA-co-AzoMA) block caused by the trans–cis conformation alteration of the azobenzene groups. Another interesting example of BCPs containing azobenzene LC mesogens involves the formation of snowman-like Janus nanoparticles (NPs) by emulsion solvent evaporation (Fig. 11).87 The azobenzene-containing poly(methacrylate) (PMAAz) head of the Janus NPs is in the smectic LC phase with ordered stripes, which becomes amorphous and enlarged due to trans–cis transformation under UV irradiation. The expanded PMAAz can consequently engulf the other head. The self-engulfed NPs can recover to their original state in both shape and LC state via visible-light irradiation. This strategy is promising for programmable load and release of different payloads by remote light trigger.
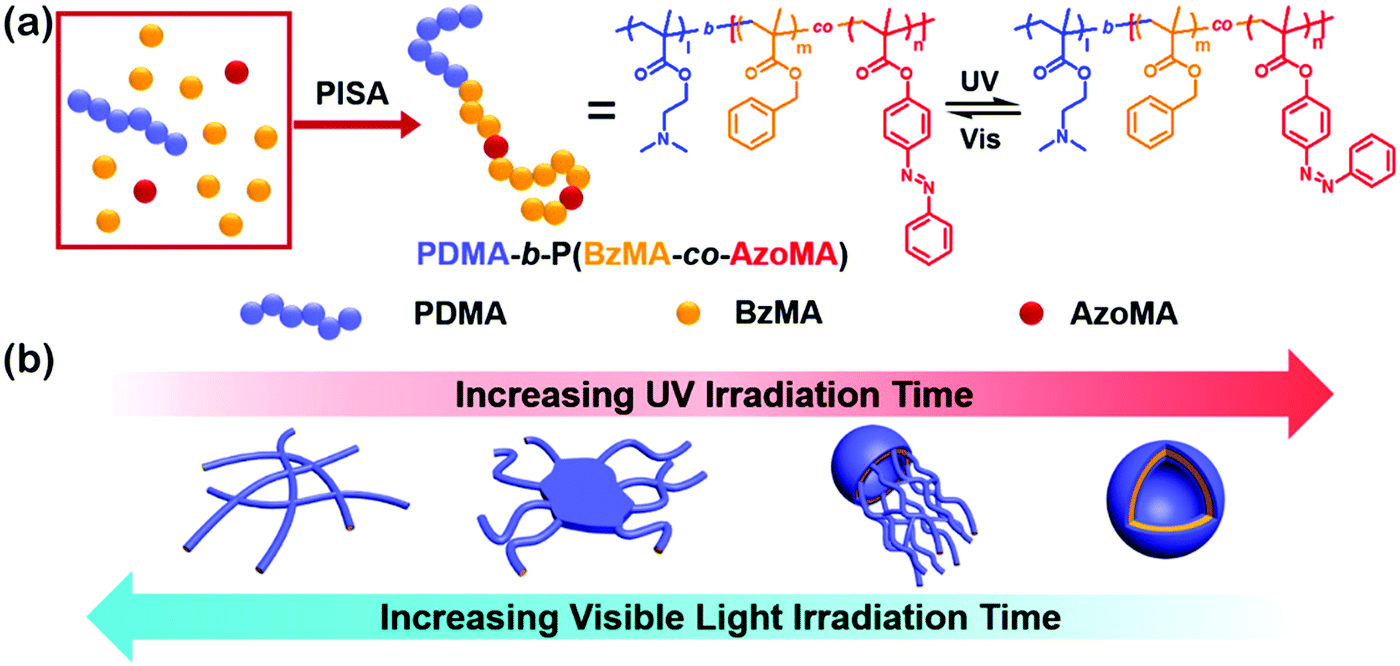 | ||
| Fig. 10 (a) Synthesis and isomerization of photoresponsive copolymers; (b) mechanism insights into the photoinduced reversible worm-to-vesicle transformation. Reproduced with permission from ref. 85. Copyright 2018, American Chemical Society. | ||
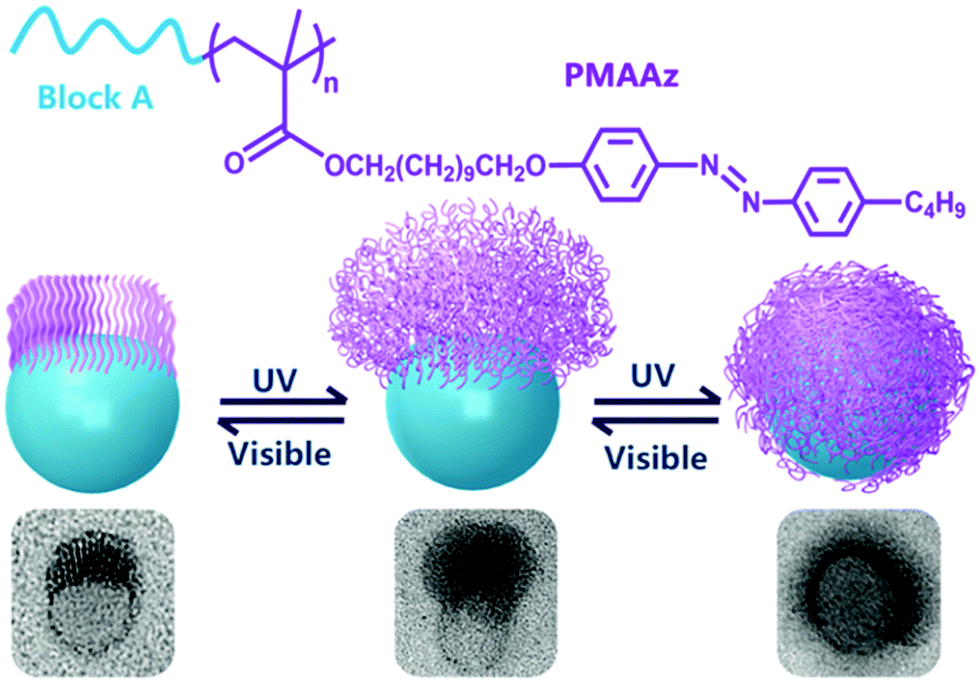 | ||
| Fig. 11 (a) Chemical structures of the PMAAz-based amphiphilic block copolymers and (b) light-triggered reversible self-engulfing of one example Janus nanoparticle. Reproduced with permission from ref. 87. Copyright 2018, American Chemical Society. | ||
Very recently, Li, Luo and colleagues reported an intriguing supramolecular self-assembly of an LC polymer-based diblock copolymer, P2VP-b-poly(2-(perfluorooctyl)ethyl methacrylate) (P2VP-b-PFMA) in solution.90,91 This diblock copolymer formed cylindrical micelles with a liquid crystalline (LC) micellar core from the PFMA block.90 The formation of the cylindrical micelles with a polydisperse length distribution is quick, suggesting a considerably low energy barrier for the nucleation and fast growth process. They discovered that through the addition of a small amount of initiators including macromolecules or nano-objects, which may interact with the copolymer, the subsequent growth driven by the LC ordering effect of the copolymer yields linear, branched, segmented, hairy plate-like, or star-like nanostructures in a one-pot manner (Fig. 12).91 In particular, depending on the interactions between the initiator and the copolymer, the self-assembly process in some cases can be reversible, and thus different micelle lengths are interconvertible by simply manipulating the initiator-to-polymer ratio and polymer concentration; and in other cases, the initiation sections can be fully crosslinked to enable a thermo-assisted seeded growth process.91 This in situ initiated self-assembly strategy represents an efficient and universal one-pot method to produce uniform hierarchical nanostructures.
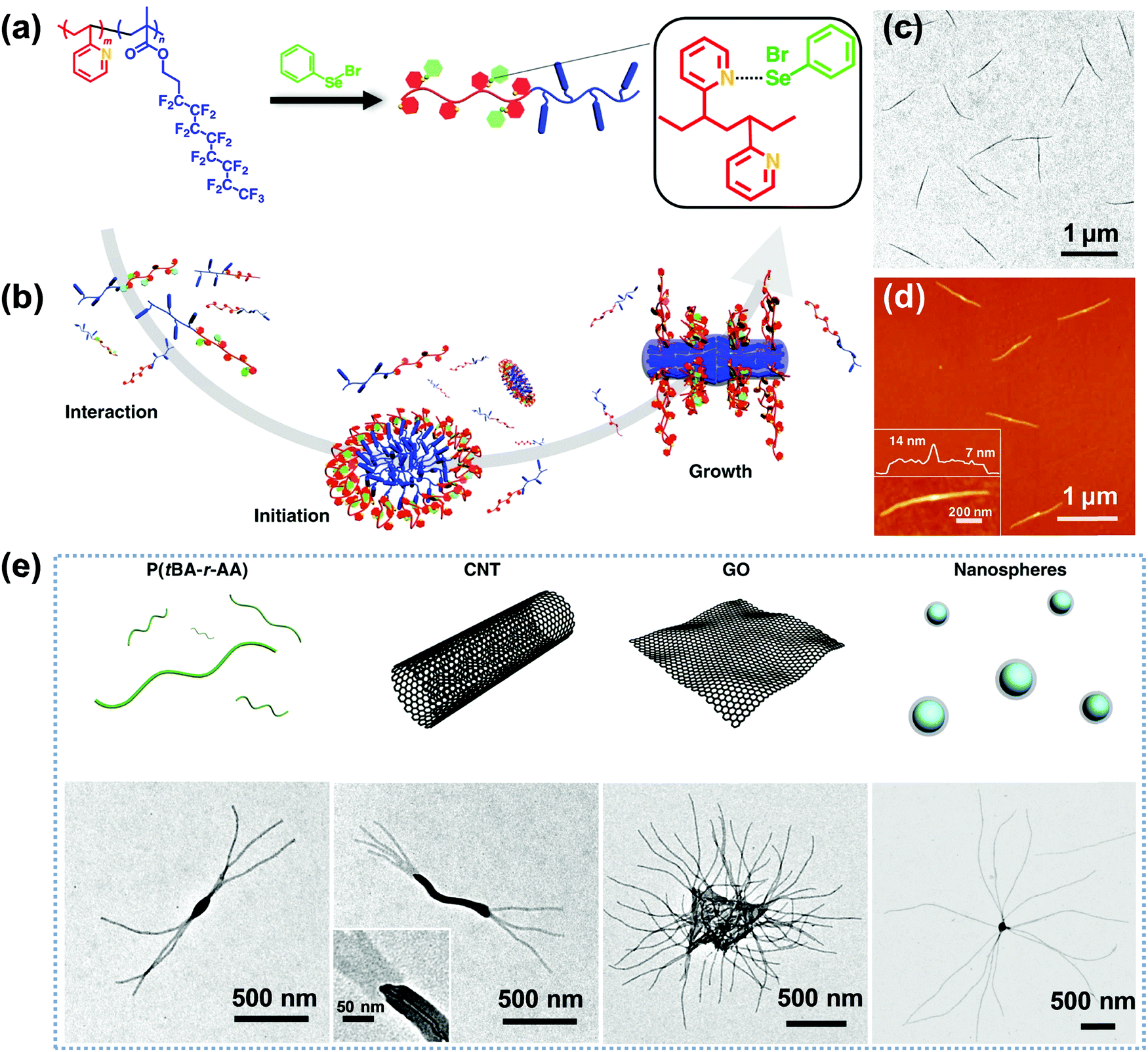 | ||
| Fig. 12 (a) Chemical structure of P2VP-b-PFMA diblock copolymer and the formation of dynamic covalent bonds between PhSeBr and P2VP units; (b) schematic illustration of the three steps of the one-pot synthesis of uniform micelles via the initiated assembly process; (c) TEM and (d) AFM height images of the cylindrical micelles from the initiation–growth process; (e) schematic cartoons and the corresponding TEM images of the initiated assembly by different large initiators, including P(tBA-r-AA), CNTs, GO, and PS nanospheres. Reproduced with permission from ref. 91. Copyright 2019, Springer Nature. | ||
Polymer cubosomes or hexasomes represent two very important categories of inverse self-assembled structures, which are porous superstructures and can be applied as soft templates for the synthesis of diverse porous materials with potential applications in energy storage and conversion, catalysis, gas or chemical separation, drug delivery and release, among others.92–97 Polymer cubosomes are actually inverse bicontinuous structures, which generally include three types of lattice structures, namely Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d, Pnm and Imm while hexasomes possess a typical p6mm structure.92–97 These aggregates with ordered pore structures are quite difficult to obtain due to the very narrow areas in the morphological phase diagram of block copolymers.4 In 2017, Mai and coworkers reported the first preparation of Pnm and Imm BCP cubosomes as well as p6mm hexasomes by tunable self-assembly of simple and commercial PS-b-PEO coil–coil diblock copolymers in dioxane/water mixed solvents.97 Later, Shen and colleagues studied the self-assembly of a rod–coil diblock copolymer, poly(ethylene oxide)-b-poly{2,5-bis[(4-methoxyphenyl)-oxycarbonyl]styrene} (PEO-b-PMPCS) in THF/water solvents (Fig. 13).93 In this copolymer, the PMPCS block is a typical mesogen-jacketed LC polymer that usually adopts an extended-chain conformation. By varying the copolymer composition, initial polymer concentration, or the solvent nature, which led to suitable head–tail asymmetry of the rod–coil copolymer, Pnm and Imm cubosomes along with p6mm hexasomes were also achieved (Fig. 13).93
d, Pnm and Imm while hexasomes possess a typical p6mm structure.92–97 These aggregates with ordered pore structures are quite difficult to obtain due to the very narrow areas in the morphological phase diagram of block copolymers.4 In 2017, Mai and coworkers reported the first preparation of Pnm and Imm BCP cubosomes as well as p6mm hexasomes by tunable self-assembly of simple and commercial PS-b-PEO coil–coil diblock copolymers in dioxane/water mixed solvents.97 Later, Shen and colleagues studied the self-assembly of a rod–coil diblock copolymer, poly(ethylene oxide)-b-poly{2,5-bis[(4-methoxyphenyl)-oxycarbonyl]styrene} (PEO-b-PMPCS) in THF/water solvents (Fig. 13).93 In this copolymer, the PMPCS block is a typical mesogen-jacketed LC polymer that usually adopts an extended-chain conformation. By varying the copolymer composition, initial polymer concentration, or the solvent nature, which led to suitable head–tail asymmetry of the rod–coil copolymer, Pnm and Imm cubosomes along with p6mm hexasomes were also achieved (Fig. 13).93
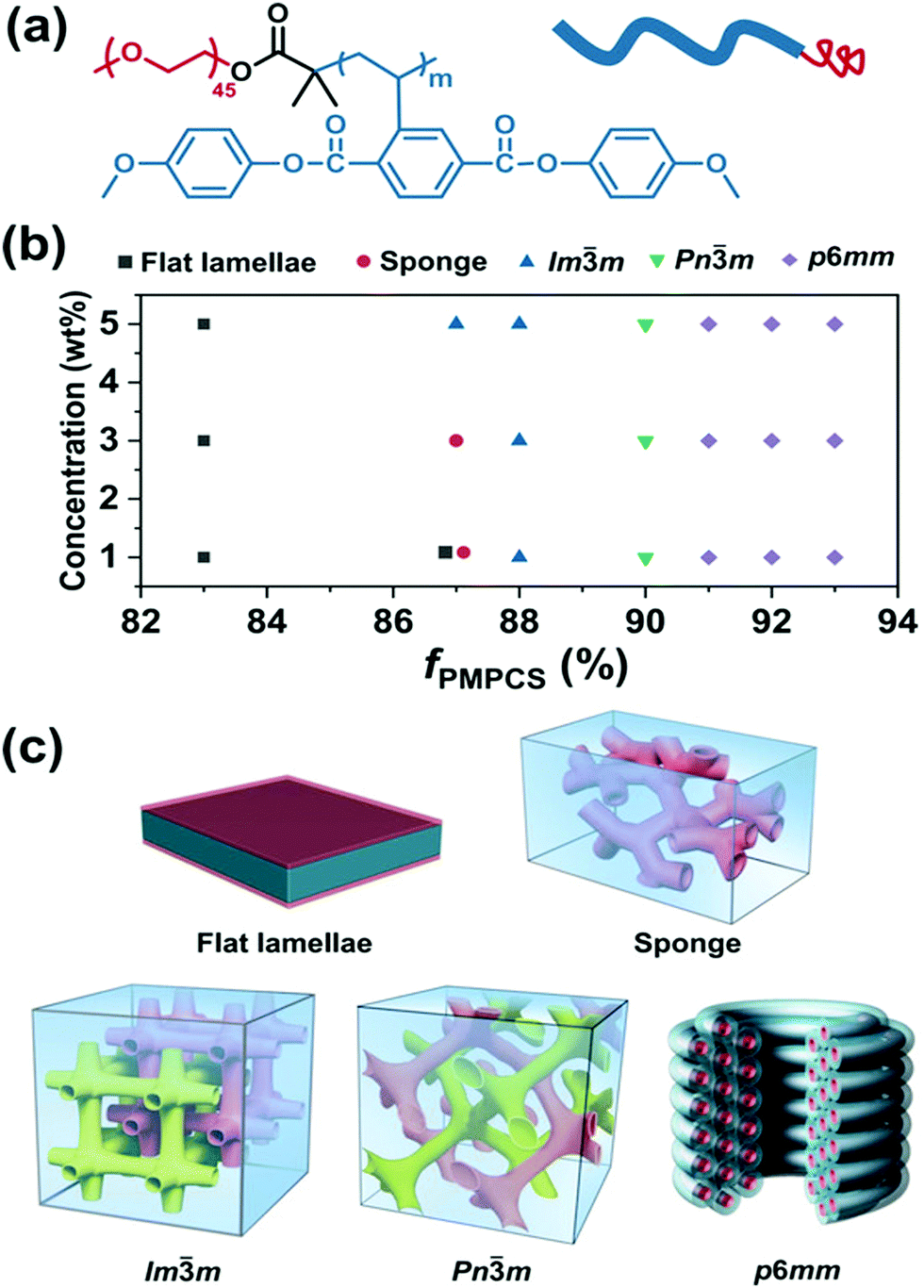 | ||
| Fig. 13 (a) Chemical structure of PEO-b-PMPCS and representation of the rod–coil copolymer; (b) morphological phase diagram of the copolymer as functions of initial concentration and volume fraction of the PMPCS block; (c) the corresponding self-assembled structures. Reproduced with permission from ref. 93. Copyright 2018 Wiley-VCH. | ||
Polypeptide-based rod–coil block copolymers
Polypeptide-based rod–coil copolymers are of particular interest for their biocompatibilities and biofunctionalities.11,16,18 Great contributions to the studies of a variety of polypeptide-based copolymers with various amino acid moieties in the polypeptide blocks as well as synthetic polymer blocks has been made by research groups of Gallot, Stupp, Klok, Schlaad, Lecommandoux, Deming, and Ulijn, amongst others.98–110 To date, amino acid species employed in the synthesis of polypeptide-containing BCPs have included lysine, leucine, aspartic acid, glutamic acid, valine, alanine, proline, tryptophan, glycine, and phenylalanine, while the synthetic polymer blocks include PEO, PS, PAA, PI, polybutadiene (PB), and so on.11,16,18External stimuli such as pH and temperature may induce reversible conformation transitions of polypeptides between random coils, α-helices, and β-sheets.11,16,18 This feature has been widely used to control the structure of assemblies and thus produce smart biomaterials.11,16,18 Previous studies have prepared stimuli-responsive polymer aggregates in association with the helix–coil secondary structure transition within peptide chains.98–110 Peptide sequences with a β-strand conformation offer intermolecular hydrogen bonding as the additional driving forces of self-assembly to produce ordered superstructures. Polypeptide-based BCPs can also be utilized to mimic self-assembly behavior of natural proteins. The resultant bioactive and stimuli-responsive superstructures are of great interest for biological applications, including drug delivery, biomimetic materials, and tissue engineering.11,16,18,98–126
Lin's group did elegant work on the self-assembly of polypeptide-based rod–coil BCPs.108–113 With the assistance of computer simulation, they systematically investigated the self-assembly behavior of poly(γ-benzyl-glutamate)-b-poly(ethylene glycol) (PBLG-b-PEG)/PBLG binary systems in solution.120 Very interestingly, they found that in THF/DMF/H2O solvent mixture, PBLG31000-b-PEG5000/PBLG528000 (the subscripts denote the molar mass of each block) with a 4:1 weight ratio of copolymer to homopolymer self-assembled into nanofibers, which further transformed to 1D helices and eventually to abacus-like assemblies with an increase in the ambient temperature (Fig. 14).120 In terms of computer simulation (inset of Fig. 14d), the abacus-like structures were formed with the copolymers aggregating into separated beads around the homopolymer bundles. This study demonstrated the possibility to build up novel superstructures in a designed fashion through stimuli-responsive self-assembly of polypeptides under the assistance of computer simulation.
 | ||
| Fig. 14 SEM images of aggregates self-assembled from the PBLG-b-PEG/PBLG binary system at various temperatures: (a) 10 °C, (b) 5 °C, (c) 30 °C, and (d) 40 °C. Inset in (d): simulation prediction of the abacus-like structure. Scale bars: 200 nm. Samples were prepared from aqueous solution. Reproduced with permission from ref. 120. Copyright 2012 Wiley-VCH. | ||
Cheng and colleagues reported the preparation of polypeptide vesicles with a multilayer membrane structure by solution self-assembly of PEG-b-poly(γ-(4,5-dimethoxy-2-nitrobenzyl)-L-glutamate) (PEG-b-PL).124 The polypeptide rod segments underwent smectic ordering with PEG buried between the hydrophobic polypeptide layers (Fig. 15). The size of both blocks and the rigidity of the hydrophobic polypeptide block played a crucial role in determining the membrane structures. An increase in the PEG length of the diblocks resulted in the formation of bilayer nanosheets, while using a random-coil polypeptide block led to the formation of large compound micelles. UV irradiation caused ester bond cleavage of the polypeptide side chain, which induced helix-to-coil transition, change of copolymer amphiphilicity, and eventual disassembly of vesicles. This work provides an insight into the control of self-assembled structures by precisely tuning the composition and conformation of polypeptide-containing amphiphiles.
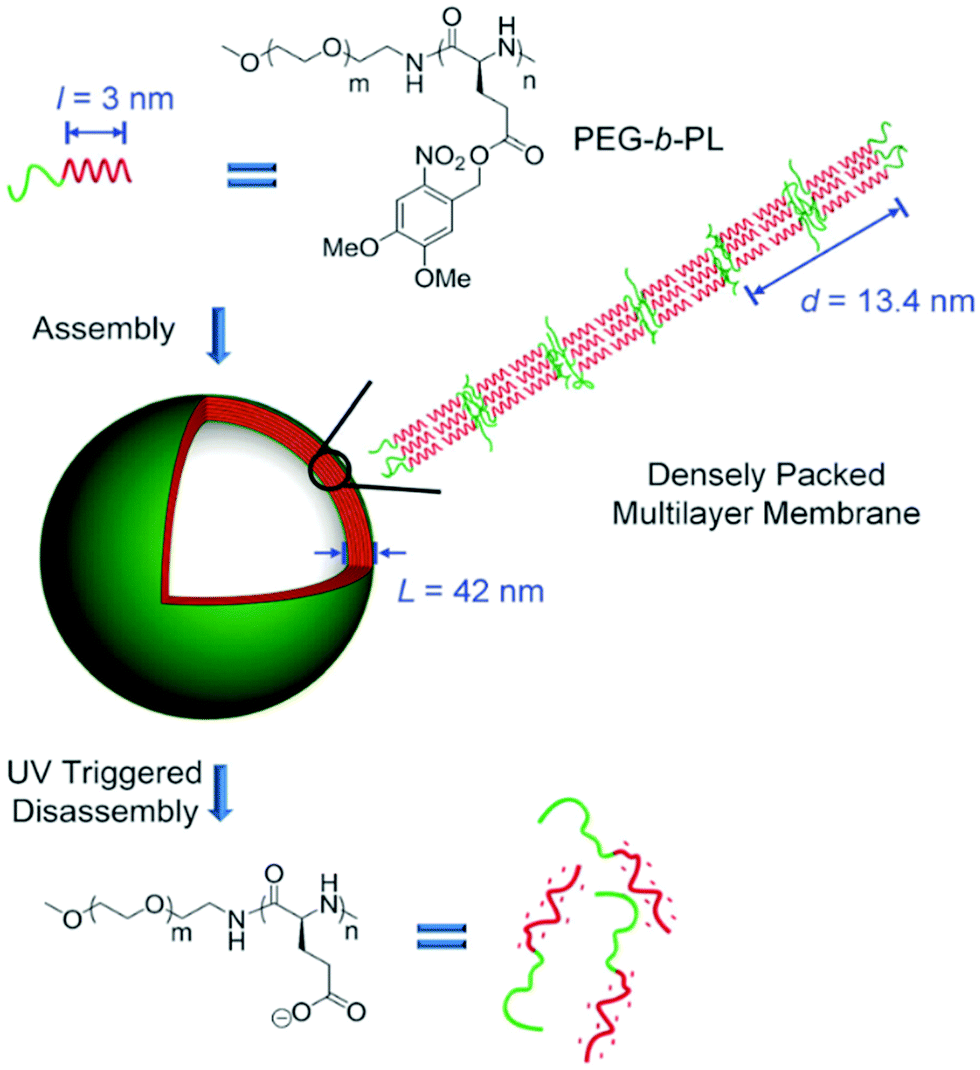 | ||
| Fig. 15 Schematic illustration of polypeptide vesicle formation with a densely packed multilayer membrane and UV-triggered disassembly process. Reproduced with permission from ref. 124. Copyright 2015 Wiley-VCH. | ||
The thermoreversible gel–sol behavior of polypeptide-based rod–coil BCPs has attracted considerable attention recently.125,126 It was found that PBLG-b-poly(dimethylsiloxane)-b-PBLG (PBLG-b-PDMS-b-PBLG) triblock copolymers formed thermoreversible gels in toluene, which correlated with the secondary structure of the peptidic block.121 As the volume fraction of the PBLG blocks increased, the content of α-helices increased while those of β-sheets and random coil dramatically decreased. The gels were formed by nanofibrils, which underwent gel–solution transition around 50 °C and reverted back to its original state when cooling down to room temperature. The gel strength enhanced with an increase in the molar mass of the PBLG blocks or the concentration of the copolymers.121
At this point, it should be raised that some rigid polymers, such as poly(carbodiimide)s,127,128 polyisocyanates,129–133 polyisocyanides,134–136 and polyacetylenes,54,137–139 possess a 1D helical structure similar to that of α-helical polypeptides.129,137 This type of polymer is called helical polymers, which exhibit unique optical activity solely due to their macromolecular helicity. Novak and colleagues prepared poly(carbodiimide)-based rod–coil copolymers with chiral poly(N-1-phenethyl-N′-methylcarbodiimide) (PPMC) rod segments and hydrophilic PEG coils.127,128 The self-assembly of such copolymers proved to be highly controllable.128 In dilute THF/H2O solutions, these interesting copolymers self-assembled into spherical micelles or vesicles depending on the relative amount of added H2O. Changing the selective solvent from H2O to MeOH resulted in the formation of interesting nanoworm and nanomaggot micelles. Blending PPMC homopolymer with the copolymers resulted in the formation of long, interconnected nanofibers, which were observed as parallel packing, perpendicular wrapping, and helical twisting of nanofibers. Finally, spin-coating copolymer/homopolymer blends from THF/ethanol induced the formation of long, bundled superhelical nanofibers with defined helical structures depending on the homopolymer–copolymer chiral pairing (i.e., (R)–(R) pairing formed P superhelical nanofibers and M superhelices for (S)–(S) pairing). The highly tunable nature of these polymeric nanostructures offers opportunities for the formation of nanoparticles of variable shapes and sizes with potential applications as biological mimics and drug delivery agents.
Wu's group synthesized a series of helical polyisocyanide-based rod–coil BCPs and studied their self-assembly in solution.134–136 For instance, they reported a class of pH and oxidation dual-responsive amphiphilic triblock copolymer, poly(L-lactic acid)(-IR780)-b-hydrophobic poly(phenyl isocyanide)-b-hydrophilic poly(phenyl isocyanide) (PLLA(-IR780)-HBPPI-HPPPI) (Fig. 16a).134 In neutral aqueous solution, the copolymers could form onion-like spherical micelles (∼84 nm diameter) consisting of PEGylated single left-handed helical PPI corona, endowing them rapid cell membrane permeability and internalization (10–20 min) with an analogous effect of cell penetrating peptides. Moreover, the phenylboronic pinacol ester embedded in the hydrophobic interlayer was stable under neutral and weak acid milieu and thus could minimize the premature drug leakage and systemic cytotoxicity. Upon exposure to H2O2, the interlayer was oxidized rapidly and accompanied by a hydrophobic–hydrophilic transition, leading to the release of encapsulated drugs and the creation of interconnected hydrophilic channels to the inner PLLA core. The degradation rates of micelles and the release rates of drugs could be easily tuned by adjusting the concentration of H2O2 and the acidity. On the other hand, the hyperthermia induced by the micelles could increase to ∼48 °C upon near-infrared (NIR) light irradiation due to the introduction of NIR absorptive IR780 dyes. Benefiting from the combined effect of chemotherapeutics, the onion-like micelles exhibited fatal and irreversible damage to cancer cells (Fig. 16c), thus being a promising multifunctional agent in the applications of disease diagnosis and therapy.
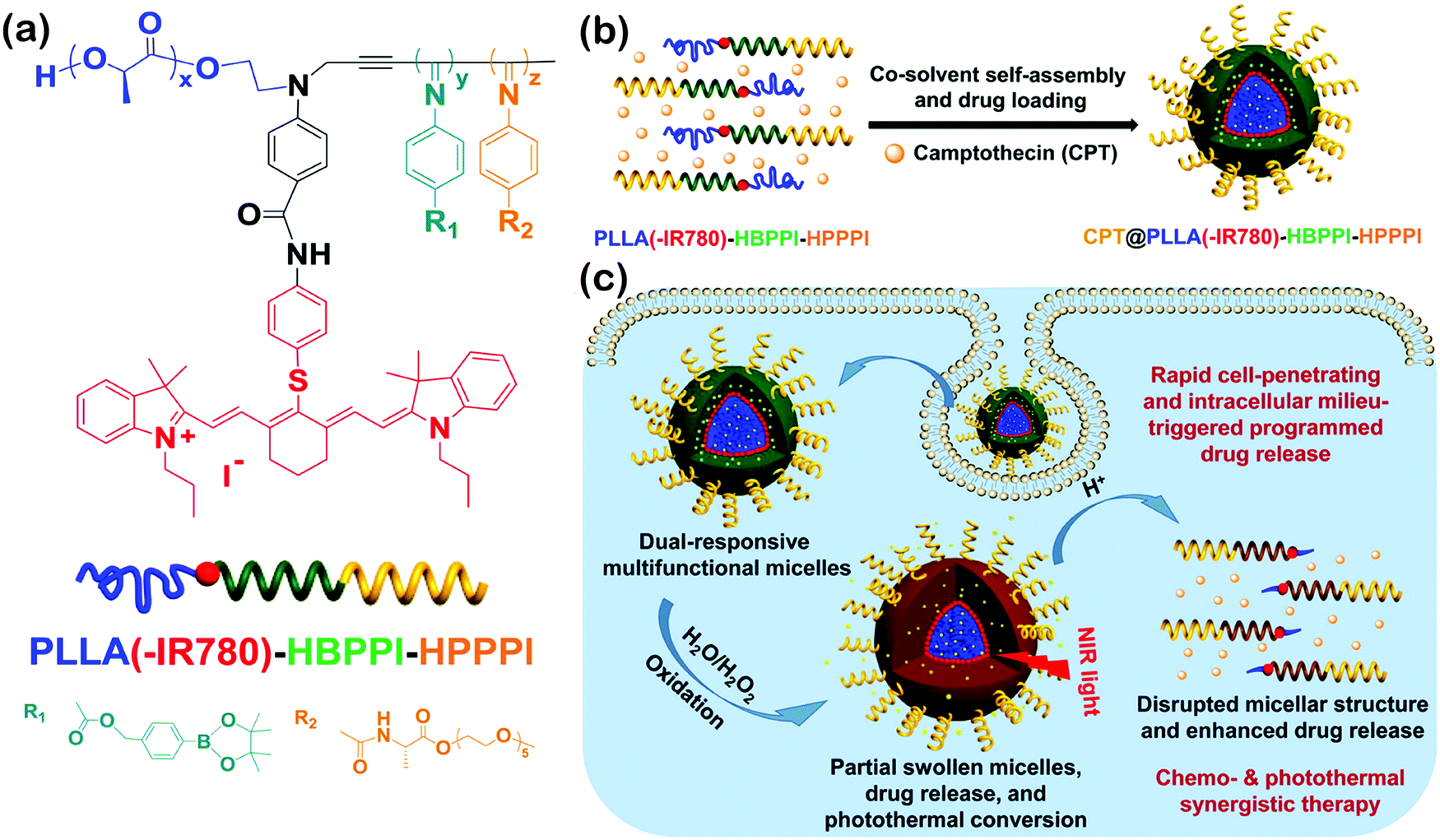 | ||
| Fig. 16 (a) Chemical structure of PLLA(-IR780)-HBPPI-HPPPI; (b) schematic cartoon for the fabrication of oxidation and pH dual-responsive multifunctional CPT@PLLA(-IR780)-HBPPI-HPPPI nanocarriers; (c) intracellular milieu-triggered programmed drug release as well as chemophotothermol synergistic therapy. Reproduced with permission from ref. 134. Copyright 2015, American Chemical Society. | ||
Self-assembly of rod–coil alternating copolymers
Alternating copolymers are a type of conventional polymers containing alternating repeating units (Fig. 1).140–149 Although the self-assembly of alternating copolymers remains much less explored compared with that of BCPs, it has attracted increasing attention in recent years due to their unique molecular architecture. For example, amphiphilic alternating copolymers generally have hydrophobic blocks of an equal length as well as the same length hydrophilic segments.144–149 These characteristics allow the self-assembly of alternating copolymers into nanostructures with hydrophobic or hydrophilic domains of uniform scale in spite of the molar mass dispersity of the copolymers. By taking advantage of this peculiarity, Ikeda and coworkers prepared 2D nanosheets with ultrathin and uniform thickness, which are generally hard to achieve by BCPs.142 They synthesized a polythiophene-containing alternating copolymer, poly(phenylcapped bithiophene-oligo(ethylene glycol)) (poly(Ph2TPh-OEG)), through “click chemistry” (Fig. 17).142 Supramolecular thiophene nanosheets were constructed by the hierarchical self-assembly of poly(Ph2TPh-OEG) in organic solvents including 1,2-dichlorobenzene (DCB), dimethylsulfoxide (DMSO), or DMF. The thickness of the resultant ultrathin nanosheets was about 3.5 nm, comparable to that of a thin lipid bilayer. It was found that two different processes, which involve disassembly of the nanosheets and unfolding of the copolymer, took place in solution with an increase in temperature. The disassembly temperature of the nanosheets (T1) was lower than the unfolding temperature of the copolymer (T2), suggesting that the folded conformation was more stable in solution. The two different temperatures also existed in the reverse direction, which confirmed that the self-organization of poly(Ph2TPh-OEG) involved two steps, i.e., the folding of the copolymers and the association of the folded copolymers into the nanosheets.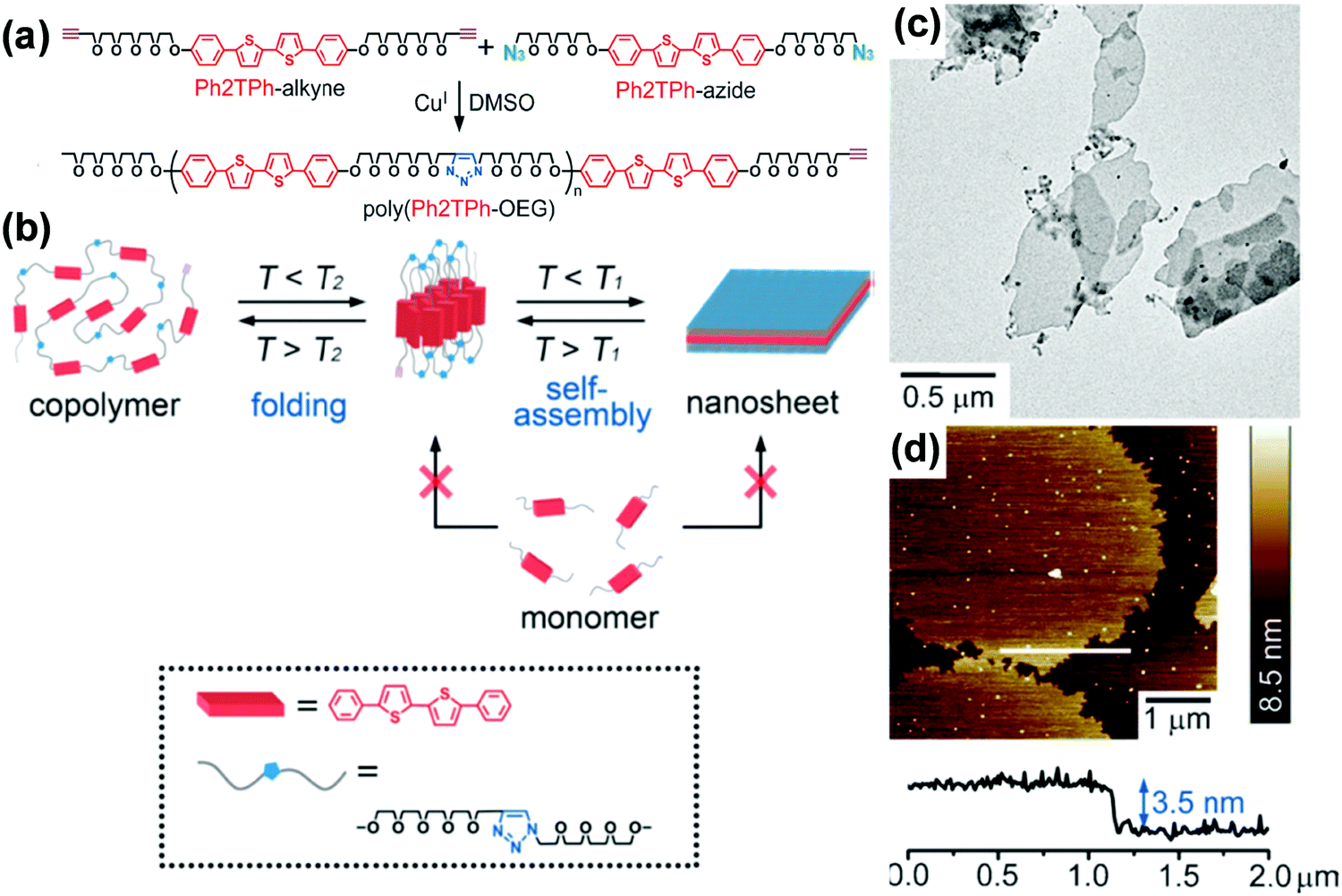 | ||
| Fig. 17 (a) Preparation of poly(Ph2TPh-OEG); (b) a proposed mechanism for the generation of supramolecular oligo-thiophene nanosheets (T1: dissociation temperature of the sheets, T2: unfolding temperature of the copolymer. T1 < T2); (c) TEM image of the poly(Ph2TPh-OEG) nanosheets; (d) AFM image and height profile of the poly(Ph2TPh-OEG) nanosheets (top left) on a silicon wafer; height scale bar: 8.5 nm. Reproduced with permission from ref. 142. Copyright 2013 Wiley-VCH. | ||
In another typical case, Mai, Zhou and coworkers synthesized an alternating copolymer, (poly(9,9′-bis(4-glycidyloxyphenyl)fluorene-alt-2,3-dihydroxy-butylene dithioether) (P(BGF-a-DHBDT)) with a broad molar mass dispersity, which contained carbon rich fluorene segments (Fig. 18).149 This alternating copolymer first self-assembled into hollow polymeric spheres (HPSs) in an N,N-dimethylformamide (DMF)/H2O solvent mixture. Then, the carbonization of the HPSs produced porous hollow carbon spheres (HCSs) with uniform micropores of very narrow size distribution (0.4–0.8 nm) in the wall, which benefited from the uniform DHBDT block length in the alternating copolymer (Fig. 18). Such a carbon structure is desirable as electrodes of energy storage devices, as the hollow cavity can reserve the electrolyte and shorten the ion diffusion distances from the exterior to the interior surfaces, while the uniform micropores (<2 nm) may render electrode materials with higher specific surface areas and also allow for easy control of their electrochemical performance. Indeed, when serving as electrode materials of supercapacitors, the activated HCSs exhibited high electrochemical performance, outperforming those of the commercial activated carbons and many well-documented porous carbon materials.
 | ||
| Fig. 18 Schematic illustration of the preparation of porous HCSs with uniform micropores through the self-assembly of amphiphilic alternating copolymers in solution. Reproduced with permission from ref. 149. Copyright 2019, American Chemical Society. | ||
Self-assembly of rod–coil graft copolymers
Rod–coil graft copolymers (also referred to as polymer brushes or molecular bottlebrushes) consist of a polymeric backbone with densely grafted side chains.150–154 In general, they are classed as two types, one consists of a rigid backbone grafted with flexible side chains and the other comprises a flexible polymer chain with rigid pendants (Fig. 1). The special 1D brush-like architecture of rod–coil graft copolymers leads to a number of novel and potentially useful properties. For example, unlike linear polymers, high molecular weight graft copolymers usually do not entangle and can self-assemble into structures with large domain sizes, even up to micrometers.10,11,19,150,151 Moreover, the microphase separation of rod–coil graft copolymers in their assemblies is different from that of linear BCPs or alternating copolymers.The introduction of pendant moieties onto conjugated polymers, forming graft copolymers, is an effective pathway to improve their solubility and tailor their optoelectronic properties, which also provides opportunities to study their self-assembly behavior and the physiochemical properties of their self-assembled structures in solution.155–161 Typically, our group synthesized a novel type of rod–coil graft copolymer containing an expanded poly-para-phenylene (PP) backbone grafted with PEO side chains (Fig. 19).159–161 Featuring a unique structure with a short average distance between neighboring PEO chains on the conjugated backbone, these graft copolymers exhibited temperature-dependent low-dimensional hierarchical self-assembly behavior in THF/H2O solution. At 25 °C, which is higher than the crystallization temperature (Tc) of the PEO coils, the achiral graft copolymers first organized into nanoribbons, which further bundled into ultralong helices with controlled pitch distance depending on the grafting percentage (GP) of the PEO chains (Fig. 19).159 Moreover, the 1D self-assembly is significantly affected by GP and the degree of polymerization (DP) of PEO.161 In the case of 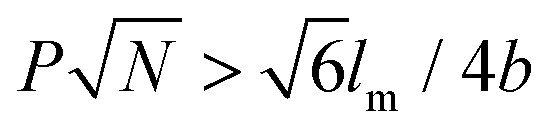 , where P represents GP, N denotes DP of PEO, lm expresses the length of a repeating unit in the poly-para-phenylene backbone and b is Kuhn monomer length of a free jointed PEO chain, the graft copolymers self-assembled into ultralong helices.161 However, when
, where P represents GP, N denotes DP of PEO, lm expresses the length of a repeating unit in the poly-para-phenylene backbone and b is Kuhn monomer length of a free jointed PEO chain, the graft copolymers self-assembled into ultralong helices.161 However, when 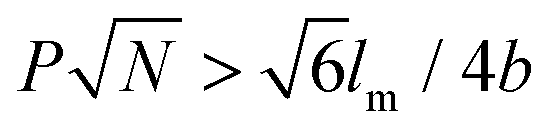 only nonhelical nanowires were observed. At 10 °C, which is below Tc of PEO, the copolymers self-assembled into polygonal multilayer nanosheets of micrometer-sized lateral dimensions when
only nonhelical nanowires were observed. At 10 °C, which is below Tc of PEO, the copolymers self-assembled into polygonal multilayer nanosheets of micrometer-sized lateral dimensions when 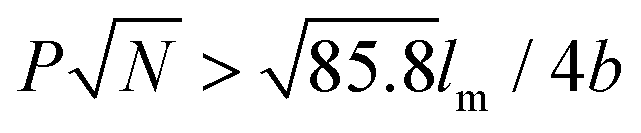 .151 Interestingly, the formation of the nanosheets experienced a morphological transition from ribbons to “raft-like” nanostructures and, eventually, to the sheets. While if
.151 Interestingly, the formation of the nanosheets experienced a morphological transition from ribbons to “raft-like” nanostructures and, eventually, to the sheets. While if  , helices or nanowires rather than nanosheets were formed.161 The maximum UV absorptions of both the helices and the nanosheets red-shifted to ∼290 nm, compared with 280 nm of the graft copolymers in THF, indicative of intermolecular π–π interactions associated with the aggregation.159
, helices or nanowires rather than nanosheets were formed.161 The maximum UV absorptions of both the helices and the nanosheets red-shifted to ∼290 nm, compared with 280 nm of the graft copolymers in THF, indicative of intermolecular π–π interactions associated with the aggregation.159
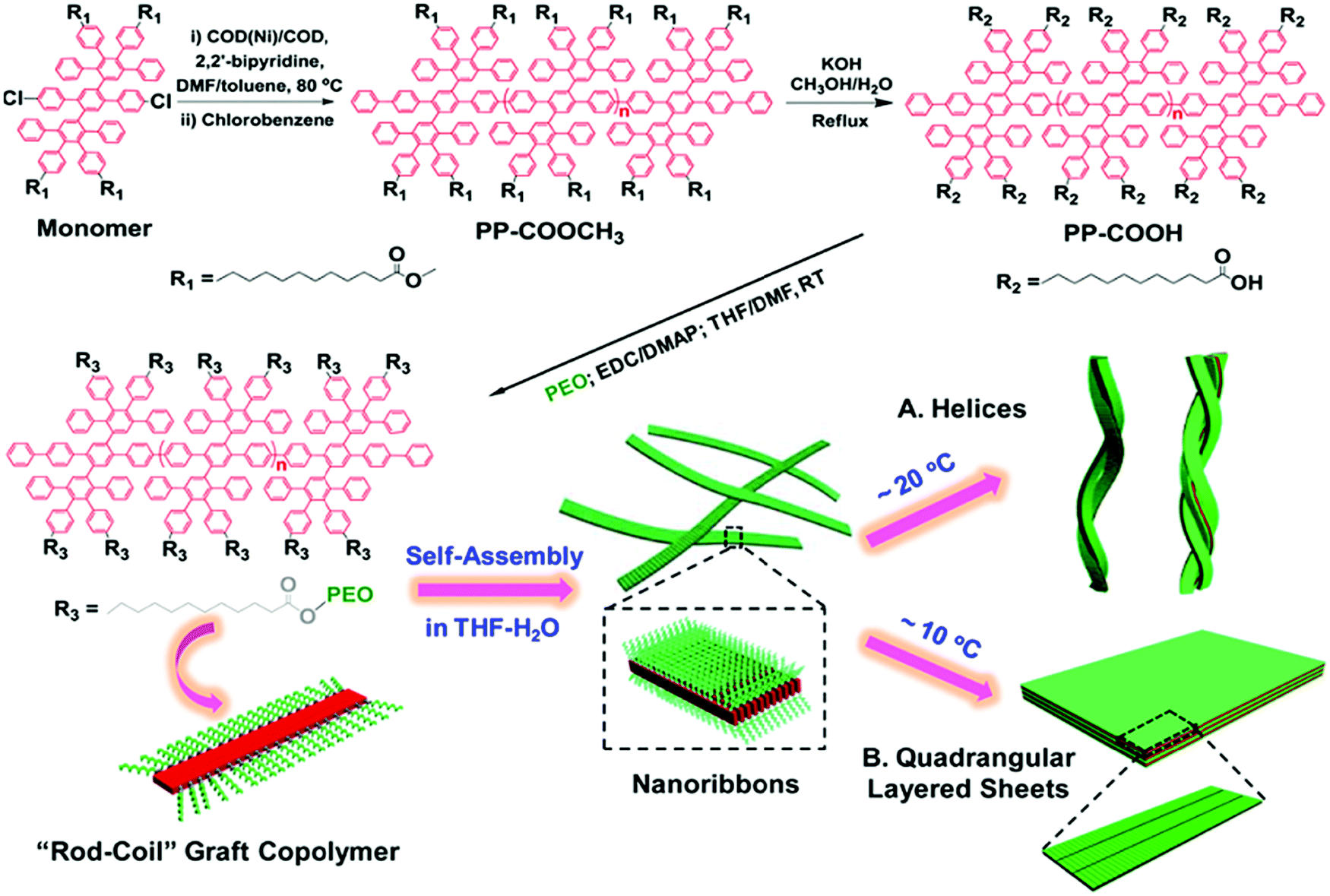 | ||
| Fig. 19 Schematic illustration of the synthesis of rod–coil graft copolymers containing a polyphenylene backbone grafted with PEO side chains as well as their temperature-dependent 1D and 2D hierarchical self-assembly process. Reproduced with permission from ref. 159. Copyright 2015, American Chemical Society. | ||
The poly-para-phenylene is an ideal precursor of graphene nanoribbons (GNRs), which hold great promise as next generation semiconductor nanomaterials.162–170 After the cyclodehydrogenation of the poly-para-phenylene, our group achieved structurally well-defined GNRs grafted with PEO chains (denoted as GNR-PEO, Fig. 20a).166 The PEO-modified GNRs show superb dispersibility in common organic solvents and in water (Fig. 21b). The excellent dispersibility offers opportunities for understanding physiochemical properties of GNRs by a wide range of solution-based physical characterizations. For instance, scanning probe microscopy revealed raft-like self-assembled monolayers of uniform GNRs on graphite substrates, in which individual GNRs were coaligned by a combination of side-by-side and end-to-end alignment (Fig. 20c). Self-assembled thin-film field effect transistors (FETs) of the GNRs exhibited a high carrier mobility of ∼0.3 cm2 V−1 s−1, manifesting a promising application of the polymer-functionalized GNRs in electronic devices.166
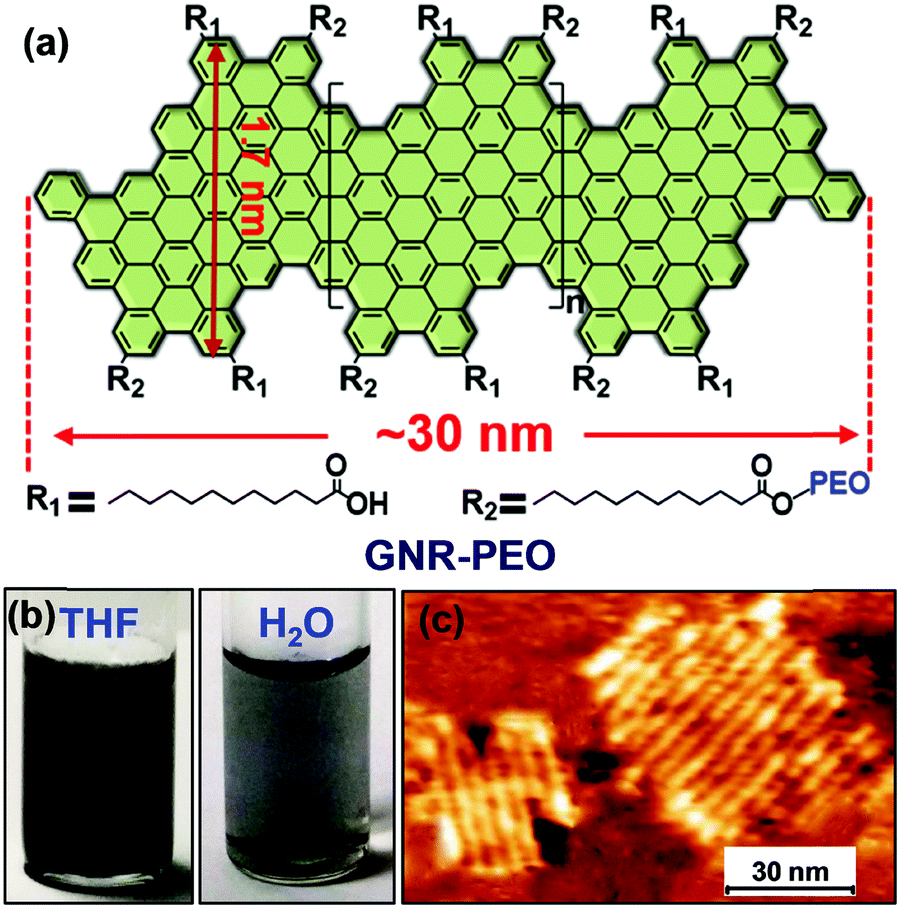 | ||
| Fig. 20 (a) Structurally well-defined GNRs grafted with flexible PEO chains. (b) Photos of the PEO-functionalized GNRs well dispersed in THF or water. (c) A typical atomic force microscopy image of self-assembled GNR monolayers on a highly oriented pyrolytic graphite substrate upon drying. Reproduced with permission from ref. 166. Copyright 2016, American Chemical Society. | ||
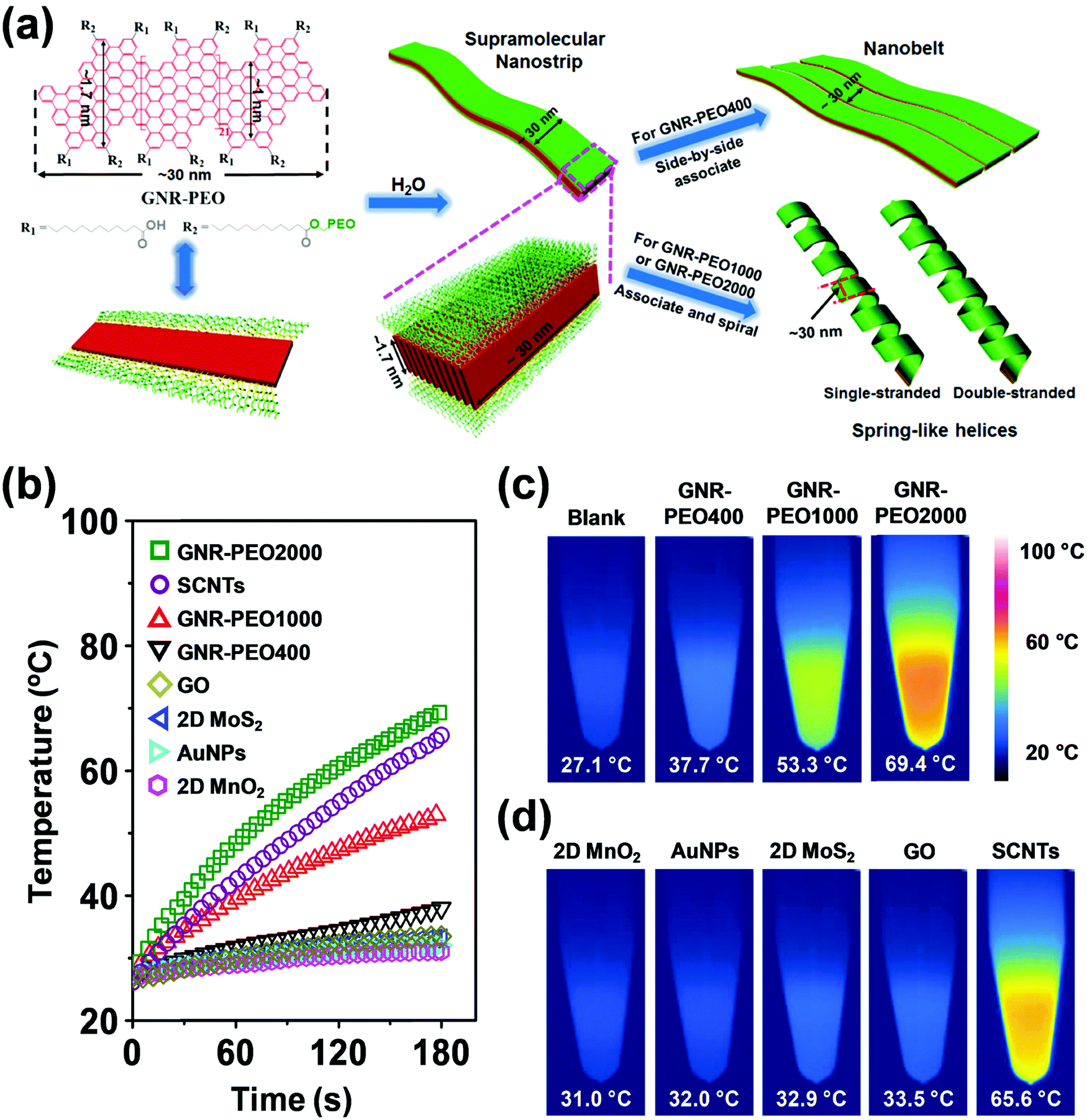 | ||
| Fig. 21 (a) Schematic diagram of the molecular structure of GNR-PEOs and their possible hierarchical self-assembly mechanism in H2O. The diagram of the molecular structure (left) expresses the GNR-COOH precursor when all R2 groups are replaced by R1. (b) Time-dependent temperature increase of aqueous dispersions of GNR-PEOs and different low-dimensional materials under NIR light irradiation (808 nm, 2.5 W cm−2). (c) Thermal images of the GNR-PEO aqueous dispersions. (d) Thermal images of the aqueous dispersions of different low-dimensional materials. Reproduced with permission from ref. 167. Copyright 2018 Wiley-VCH. | ||
Intriguingly, the GNR-PEO samples self-assembled into 1D hierarchical supramolecular nanostructures, including ultralong nanobelts and nanohelices (Fig. 21a), depending on the PEO chain length.167 A mechanistic study revealed that GNR-PEO400 (the number represents the molecular weight of the PEO chains), with the shortest PEO side chains, first aggregated into small nanostrips, and then the belt-like superstructures associated with each other by a side-by-side alignment due to the small interchain steric hindrance among the side PEO coils. In contrast, the longer PEO coils around the supramolecular nanostrips formed by GNR-PEO1000 or GNR-PEO2000 are much more crowded, generating large interchain steric hindrance and resulting in high system energy. To relieve the high energy, spring-like helices were formed, providing more peripheral space for the accommodation of the PEO side chains (Fig. 21a). The 1D superstructures of GNR-PEOs in the aqueous solutions showed strong near infrared (NIR) absorption, and exhibited high photothermal conversion performance with an efficiency of 31%, outperforming those of many conventional low-dimensional nanomaterials, including single-walled carbon nanotubes (SWCNTs), graphene oxide (GO), gold nanoparticles (AuNPs), 2D molybdenum disulfide (MoS2) and 2D manganese dioxide (MnO2) (Fig. 21b). The excellent photothermal conversion ability affords great chances for the applications of GNRs in photothermal tumor therapy, and antisepsis, among other applications.
By controlling over the interplay between the π–π interaction of GNR backbones and the interactions among the grafted polymers, our group also achieved the tunable self-assembly of structurally defined GNRs in common organic solvents. Dendronized GNRs were synthesized in a solution state, which consist of a well-defined GNR backbone grafted with benzyl ether-type dendrons of different generations (GNR-G1–G3, Fig. 22).169 The dendronized GNRs have grafting ratios of 0.59–0.68 for the dendrons of different generations, and show excellent dispersibility in common organic solvents such as THF. Thanks to the well-defined geometry of the grafted dendrons, the GNRs aggregate into different 1D superstructures including ultralong nanowires, helices, and short nanofibers in THF, depending on the dimension of the dendrons (Fig. 22). Ultrafast photoconductivity measurements reveal that GNR-G2 helices exhibit much longer free carrier (3.5 ps) and exciton lifetime (several hundred ps) than those of GNR-G1 nanowires and GNR-G3 nanofibers, and those of reported GNRs lack ordered superstructures.164 This discrepancy indicates the considerable effect of supramolecular structures on the optoelectronic properties of GNRs in the liquid phase, making them promising candidates for optoelectronic applications.
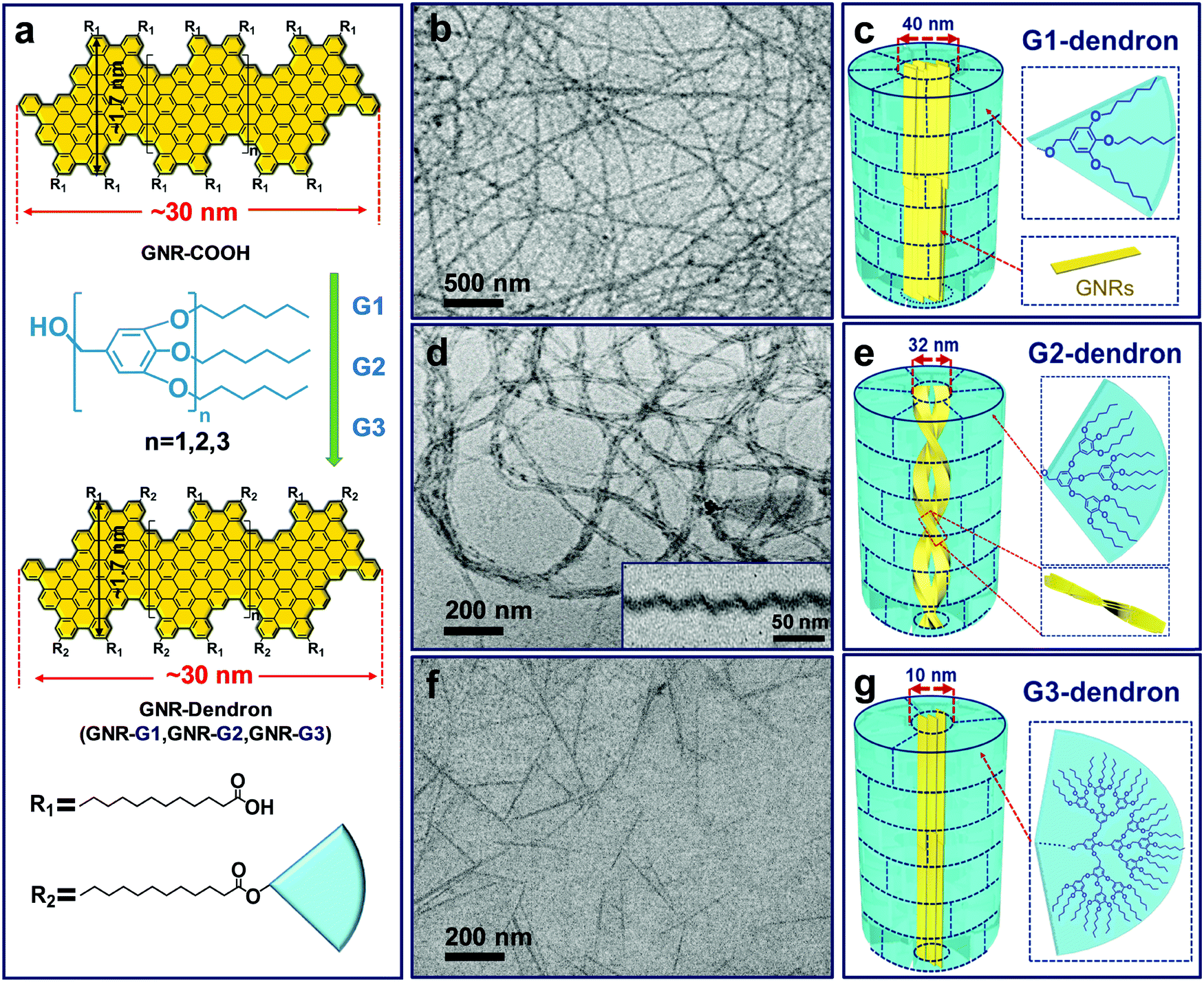 | ||
| Fig. 22 (a) Schematic diagram of the synthesis and molecular structure of GNR-Dendrons. (b–g) TEM images and the proposed aggregation models of the superstructures formed by the dendronized GNRs in THF. (b and c) GNR-G1 nanowires; (d and e) GNR-G2 helices, the inset in (d) shows a typical cryo-TEM image; (f and g) GNR-G3 nanofibers. For convenient illustration, in the schematic aggregation models the short alkyl chains between the GNR backbones and dendrons are omitted; the near-fan architectured dendrons are presented as “regular fans” separated by dashed lines, which are not the actual “borders” among the dendrons. Reproduced with permission from ref. 169. Copyright 2019, American Chemical Society. | ||
As an example of the self-assembly of rod–coil graft copolymers containing LC segments, Li et al. synthesized a series of brush-like amphiphilic liquid crystalline copolymers containing cholesteryl mesogens with different hydrophobic/hydrophilic block ratios.171 Governed by both amphiphilicity and smectic order, the self-assembly of the copolymers in different solvents, e.g. 1,4-dioxane/H2O, yielded micellar aggregates with well-organized architectures, including short cylindrical micelles, nanofibers, fringed platelets, and ellipsoidal vesicles with smectic micellar cores, with an increasing hydrophobic block ratio. The smectic order arising from strong in-plane interactions between cholesteryl mesogens played an important role in the micelle growth and their final morphological formation.
New rod–coil graft copolymers containing biomacromolecular blocks, such as polypeptide, polysaccharide, or alginate, have been recently synthesized for the construction of self-assembled structures in solution, which are particularly interesting for biological, medical, and biotechnological applications.172–182 Lee's group incorporated dendritic oligoether side groups into a peptide backbone, yielding peptide-based graft copolymers (Fig. 23).175 The reversible shielding of the peptide backbone from a water environment by the oligoether dendron side groups allowed conformational switching of the peptide from random coils to α-helices. Furthermore, the switchable α-helical peptides self-assembled into reversible membrane structures in which the rod-like α-helices were aligned parallel to each other. Peptide 2 based on two oligoether dendrons formed discrete nanodisks while peptide 3 based on three dendrons organized into vesicles (Fig. 23). The driving force for the formation of the switchable peptide nanostructures was the reversible stabilization of the α-helical conformation. Moreover, the vesicles could spontaneously capture a racemic mixture by the self-formation of the vesicular walls upon heating and selectively released the guest molecules through preferential diffusion across the walls. Such a unique peptide assembly–disassembly switching offers opportunities to explore biomedical applications for the controlled capture and release of proteins, genes, and drugs.175
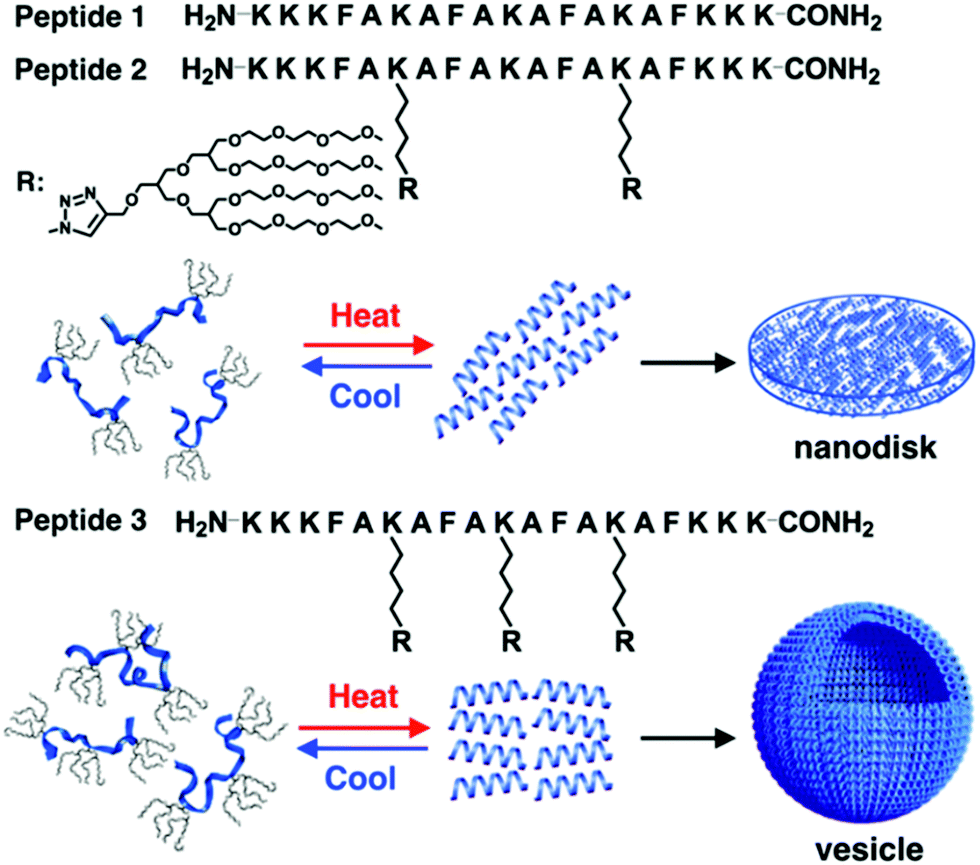 | ||
| Fig. 23 Schematic cartoon of reversible self-assembly of peptides 1, 2 and 3 and schematic illustrations of a nanodisk from peptide 2 and a vesicle from peptide 3. Reprinted with permission from ref. 175. Copyright 2016, American Chemical Society. | ||
Jiang, Chen and coworkers synthesized a novel type of alternating amphiphilic glycopolypeptide brushes (AAGBs) with pendants of glycodendrons and short peptides (Fig. 24).176 In such graft copoloymers, the interplay between the glycodendron attachment with a very high density of sugar units as well as the pendent β-sheet forming oligopeptide provided the driving force for the self-assembly resembling that occurs in nature. Remarkably, various self-assembled nanostructures were achieved, including nanowires, nanoribbons, and compound micelles in aqueous solution, depending on the number ratio of the sugar units to the amino acid species. Among these morphologies, the formation of the attractive nanowires was found to experience a hierarchical self-assembly process, in which the assemblies of the glycopolypeptides evolved from spherical micelles, to nanofilaments, branched filaments, and finally to nanowires. This study provides new opportunities to deeply understand the self-assembly mechanism of natural glyco-conjugates and expand the library of biomimetic materials.
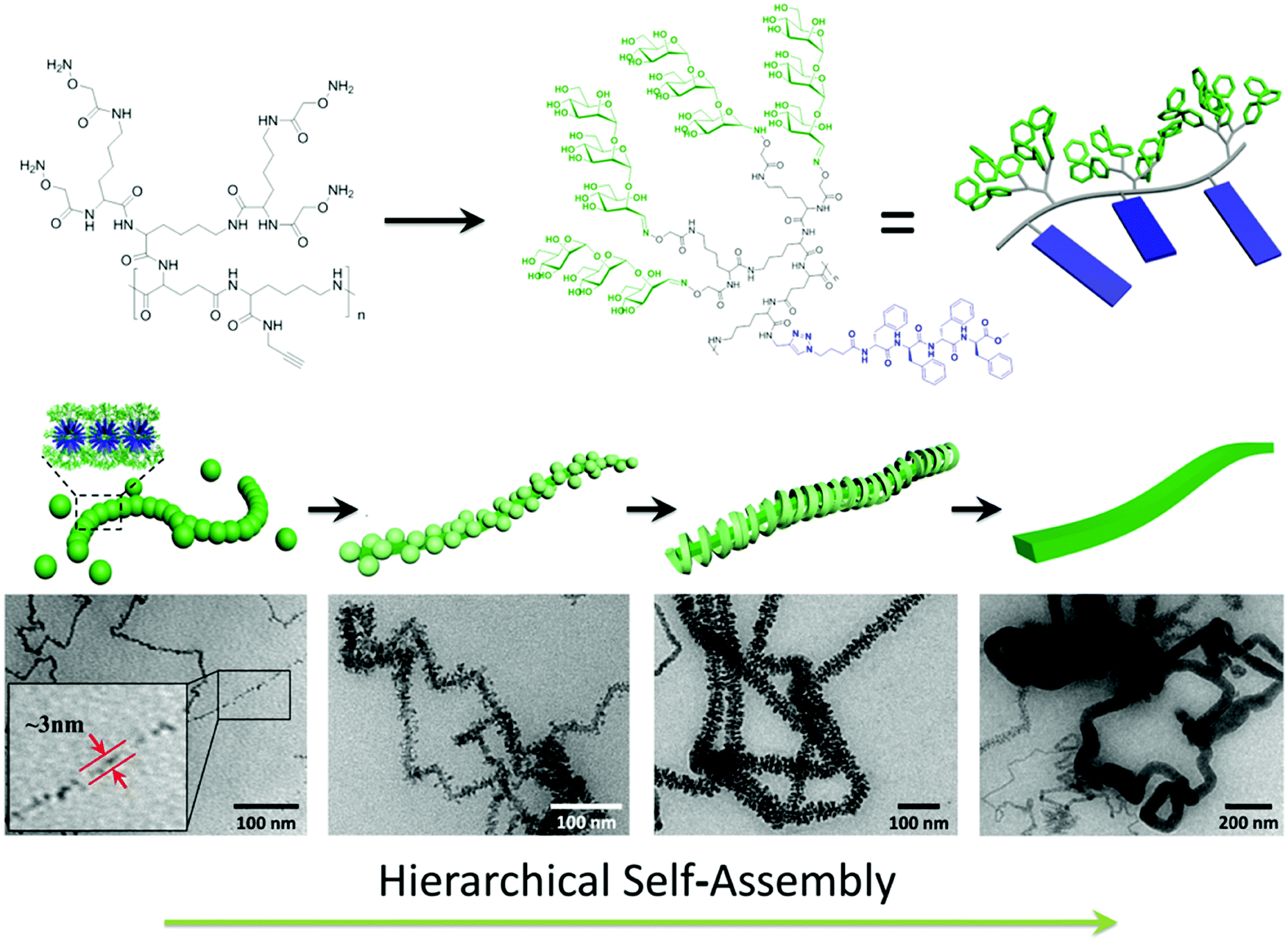 | ||
| Fig. 24 Hierarchical self-assembly of alternating amphiphilic glycopolypeptide brushes with pendants of high-mannose glycodendron and oligophenylalanine. Reprinted with permission from ref. 176. Copyright 2016, American Chemical Society. | ||
Ding et al. studied the potential applications of the assemblies of peptide-based rod–coil copolymers in drug delivery. They synthesized thermo-responsive rod–coil polypeptides by grafting of azideterminated (co)polymers of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) or 2-(2-(2-methoxyethoxy)ethoxy)ethyl methacrylate (MEO3MA) (i.e., PMEOiMA) onto poly(g-propargyl-L-glutamate) (PPLG).179 The modified polypeptides were employed to self-assemble into micelles for doxorubicin (DOX) loading and releasing. The micelles showed efficient internalization and intracellular DOX release towards HeLa cells. In vitro methyl thiazolyl tetrazolium assays revealed that the polypeptides were cytocompatible, and the DOX-loaded micelles showed efficient cellular proliferation inhibition. The micelles were hemocompatible, and the encapsulation with polypeptides significantly reduced the hemolysis ratio of DOX. Thereby, the thermo-responsive polypeptide micelles, which are stable under physiological conditions while releasing payloads in the acidic intracellular microenvironment, are promising for smart drug delivery.
Owing to the similar structure to that of α-helix polypeptides and chiral optical properties, some helical polymers have also been employed as the rigid backbone of rod–coil polymer brushes for preparing functional self-assembled nanostructures.183–185 Yang, Deng and coworkers reported the synthesis and self-assembly of amphiphilic rod–coil polymer brushes consisting of a hydrophobic optically active helical polyacetylene backbone and hydrophilic thermosensitive poly(N,N-dimethylamino-2-ethyl methacrylate) (PDMAEMA) side chains. The polymer brushes possessed optical activities, derived from the helical polyacetylene backbone.183 The copolymers could self-assemble into core/shell nanoparticles with optically active cores (helical polyacetylene) and thermoresponsive shells (PDMAEMA). Such nanoparticles showed considerable optical activity with intensive CD signals at ca. 350 nm, thus being expected to find potential applications in chiral materials, stimulus-responsive materials, etc.
A number of studies focused on the self-assembly of polysaccharide-based graft copolymers.186,187 Li et al. synthesized carboxymethyl konjac glucomannan-g-PEG (CKGM-g-PEG) and they obtained hollow nanospheres by co-assembly of CKGM-g-PEG and α-cyclodextrin (α-CD) in aqueous solution.186 In the spheres, the rod blocks formed by inclusion between grafted PEG coils and α-CD constructed the wall while the CKGM segments formed the coronae. The hollow nanospheres showed semi-permeability allowing enzyme substrates to pass through the surface while restricting the encapsulated enzyme (i.e. glucose oxidase) to the interior. These CKGM-g-PEG/α-CD nanospheres showed in vitro biocompatibility when exposed to L929 cells during the test using an MTT viability assay, suggesting that the self-assembly of CKGM-g-PEG and α-CD to form stable nanoparticles can be an effective strategy for enzyme encapsulation with potential biomedical applications. In another study, hollow spheres were also prepared by self-assembly of rod–coil alginate-g-PEG/α-CD complexes in water, in which the rod blocks were formed by inclusion between α-CDs and grafted PEG chains while the coil segments were protonated alginate backbones.187 The rod blocks formed the wall of the hollow spheres and the protonated alginate coils constructed the coronae. Because both α-CD and alginate-g-PEG are degradable, these hollow spheres exhibit good degradation in aqueous solution, indicating their great promise in the area of drug or gene delivery systems and artificial cells.
Polymerization induced self-assembly (PISA) has been predominantly undertaken using radical polymerization processes, most notably, reversible addition–fragmentation termination (RAFT) polymerization.188–190 Gianneschi and coworkers reported a new PISA process based on ring-opening metathesis polymerization (ROMP) of norbornenyl molecules.191,192 They utilized a peptide-based norbornenyl monomer as a hydrophobic unit to provide a range of nanostructures, including spheres, cylinders and vesicles, at room temperature yet at high solid concentrations of 20 wt% in combination with an oligoethylene glycol based norbornenyl monomer. The polymerizations occurred efficiently and maintained a living character. For the brush copolymers, the length of the grafted peptide block had a significant impact on the morphology control. With an increase in the peptide block, the aggregates of the graft copolymers transformed from spheres to cylinders and finally to vesicles. The demonstration broadens the scope of the PISA process to a new living polymerization methodology toward the development of easily accessible and highly functionalized nanostructures in situ.
Self-assembly of rod–coil star-like copolymers
Star-like polymers possess most of the properties of high molecular weight materials without the solution viscosity penalty of linear materials of similar molecular weight.193–212 Limited work was performed on the self-assembly of star-like rod–coil copolymers due to the difficulty in the synthesis. However, the limited studies have already revealed the specificity of the self-assembly of star-like rod–coil copolymers in solution, for example, forming distinct aggregates such as unimolecular micelles which are difficult or cannot be formed by other types of polymers. Lin and coworkers synthesized a series of novel amphiphilic 21-arm, star-like diblock copolymers, poly(acrylic acid)-b-poly(3-hexylthiophene) (PAA-b-P3HT), with well-defined molecular architectures.201 These star-like diblock copolymers possessed narrow molecular weight distribution with the molecular weight of both the core and shell blocks being well-controlled. They formed unimolecular micelles in solution as a result of the compact structure and reduced interchain entanglement.As another elegant example, Besenius and coworkers studied the stepwise self-assembly of AuI-metallopeptides in water.206 1D supramolecular polymer morphology was controlled using temperature-dependent self-assembly conditions (Fig. 25). At low temperatures, supramolecular polymerization of monomeric or small oligomeric disordered species (state 0) into nanorod-like morphologies of high molecular weight. At 5 °C, the lifetime of the disordered state 0 is very short, suggesting that the energetic barrier for the self-assembly of monomers into 1D supramolecular polymers is very low. At temperatures T < 20 °C, the 1D nanorod-like species (state I) are stable on the time-scale of several hours to days. Upon increasing the temperature T > 20 °C, the metastable state I is converted into thermodynamically stable helical fibrils, consisting of two intertwined nanorods (state II). The large activation energy required for this bundling most likely originates from the shielding effect of the flexible oligoethylene glycol chains that form a hydrated shell around the nanorods.
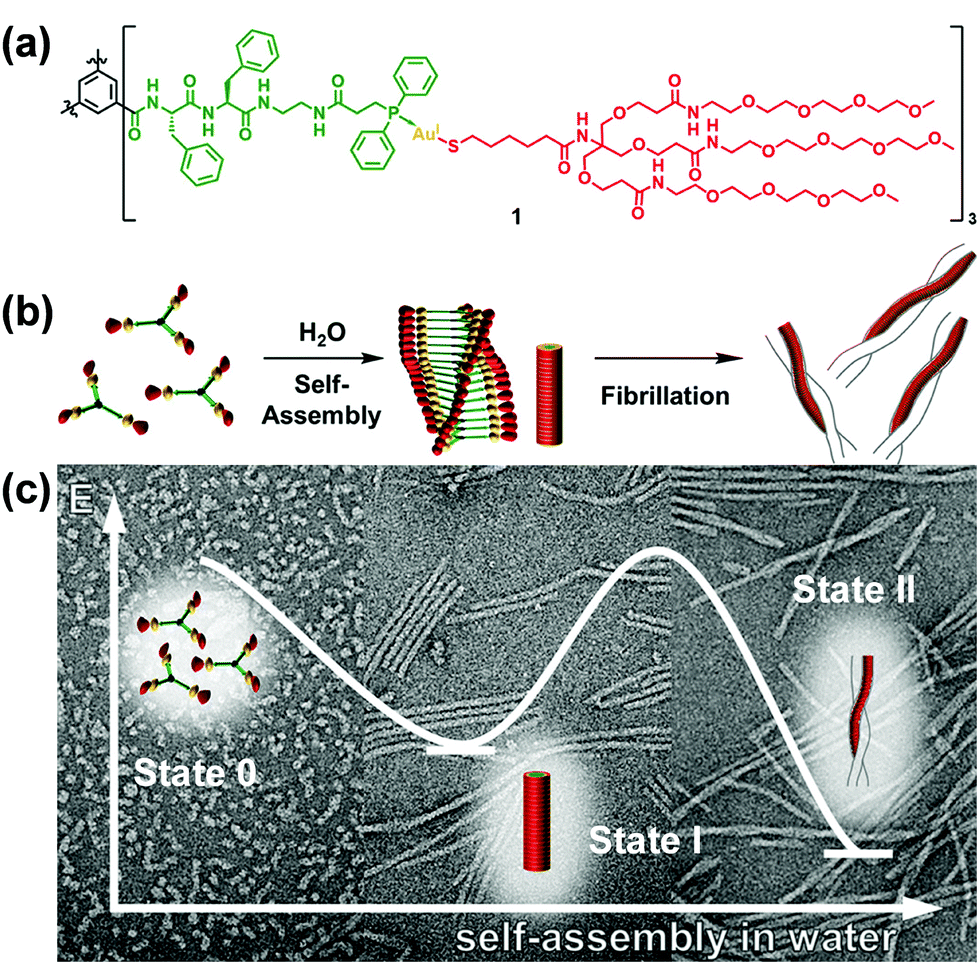 | ||
| Fig. 25 C 3-Symmetrical AuI-metalloamphiphile (a) and its self-assembly in water into 1D supramolecular nanorods and intertwined fibrils (b). (c) Negative stain TEM micrographs of the aggregates obtained at 6 °C, 20 °C and after heating to 60 °C, respectively. Reproduced with permission from ref. 206. Copyright 2018, American Chemical Society. | ||
ABC miktoarm star terpolymers contain three different blocks that are connected in a central organic group.193,194,198,207–212 The self-assembly of this type of copolymer has attracted considerable attention recently as such polymers show topographically constrained in the architecture, whereby the three blocks are interacted with one another. As this area is still in its infancy, there are only several papers related to this topic. He, Lin and coworkers synthesized photo-responsive ABC miktoarm star terpolymers consisting of hydrophilic poly(ethylene glycol)monomethyl ether (MPEG), hydrophobic PS and azobenzene containing poly[6-(4-methoxy-azobenzene-40-oxy)hexylmethacrylate] (PMMAZO) blocks by a combination of atom transfer radical polymerization (ATRP) and click chemistry.209 They found that these star terpolymers self-assembled into bowl-shaped or multibowl-shaped nanostructures depending on the lengths of the hydrophobic PS or PMMAZO chains. When the star terpolymers self-assembled into nanobowls in water, the azobenzene chromophores were arranged in the nanobowls in a orderly fashion and H/J-aggregates of azobenzene chromophores were formed during the self-assembly process. On the other hand, the H-aggregations were broken by the photoisomerization process and could not recover due to the absence of sufficient free volume in the aggregates. Wu's group synthesized ABC miktoarm terpolymers containing coil PCL, PS blocks, and rigid-rod helical poly(phenyl isocyanide) (PPI) through the combination of ring-opening polymerization (ROP), atom transfer radical polymerization (ATRP), and Pd(II)-initiated living polymerization. Such miktoarm star terpolymers were found to self assemble into well-defined spherical micelles.212
Self-assembly of rod–coil hyperbranched copolymers
To date, rod–coil hyperbranched copolymers employed as self-assembly precursors contain a dendritic core consisting of rigid segments and flexible polymer chains on the periphery of the core (Fig. 1).213–215 The unique structure makes the rod–coil hyperbranched copolymers form unimolecular micelles easily,216–218 which may further self-assemble into large micelles without phase separation through a “multi-micelle aggregate” (MMA) mechanism.219–221 This merit favors the fluorescence emission of aggregation-caused quenching molecules in their aggregates. Building on this concept, Zhu and colleagues synthesized two types of hyperbranched conjugated copolymers with different linking agents (e.g., pH-responsive acylhydrazone and stable ether) between a diformylcarbazole-based hyperbranched polymer (HCP) core and many linear PEG arms (Fig. 26a).214 Owing to the amphiphilic three-dimensional architecture, the hyperbranched copolymers self-assembled into multi-micelle aggregates from unimolecular micelles with excellent emission performance in the aqueous medium (Fig. 26b). The formation of MMAs efficiently restricted the concentration quenching effect and intermolecular interaction of conjugated polymer cores by many flexible arms. The length of the PEG arms showed great influence on the size and optical properties of the self-assembled micelles. Such a facile strategy for emission enhancement of conjugated polymers shows great potential in biological applications. For example, when doxorubicin (DOX) as a model drug was encapsulated into the copolymer micelles, the emission of the conjugated copolymer and DOX was quenched. In vitro biological studies revealed that fluorescent intensities of both the conjugated copolymer and DOX were activated when the drug was released from the micelles, leading to enhanced cellular proliferation inhibition against cancer cells. Importantly, the micelles formed by the pH-responsive HCP-N-PEG copolymer with an acylhydrazone linkage exhibited accelerated DOX release at a mildly acidic environment, due to the fast breakage of acylhydrazone in the endosomes or lysosomes of tumor cells.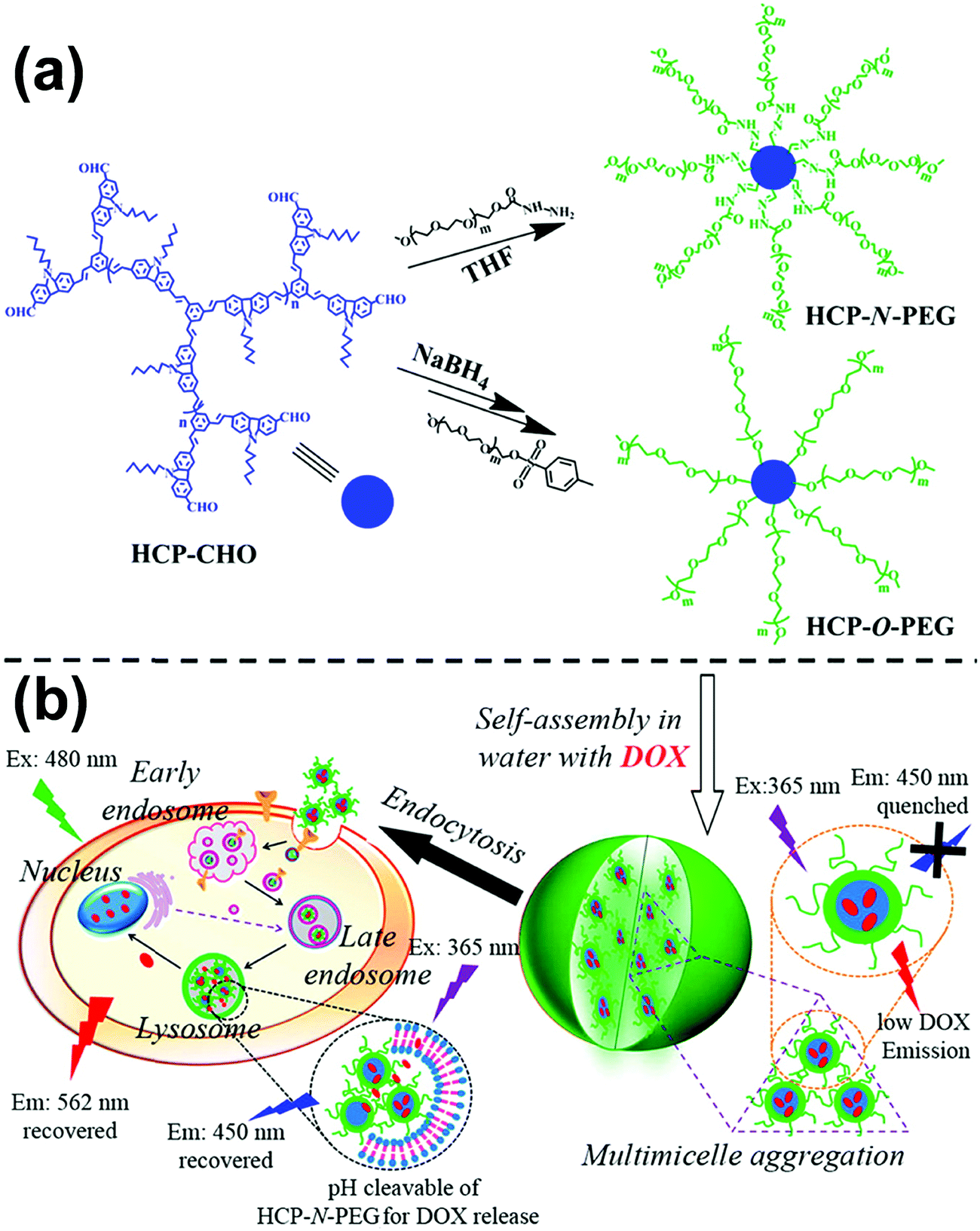 | ||
| Fig. 26 (a) Synthesis of HCP-N-PEG and HCP-O-PEG hyperbranched copolymers; (b) self-assembly of the copolymers and their endocytosis in the tumor cells. Reproduced with permission from ref. 214. Copyright 2014, American Chemical Society. | ||
Summary and outlook
This review paper describes the recent advances in the supramolecular self-assembly of amphiphilic rod–coil copolymers of various structures, including block, alternating, graft, star-like, and hyperbranched/dendritic copolymers in solution. The introduction concentrates on copolymer systems containing conjugated polymers, LC polymers, polypeptides, and helical polymers as the rod segments. Governed by microphase separation of the constituent blocks along with some additional interactions such as the π–π interaction and crystallization of the rod segments, these rod–coil copolymers can self-assemble into diverse ordered nanostructures, including a number of uncommon superstructures such as helices, ribbons, sheets and toroids, etc. By using representative examples, the principles underlying morphological control by regulating the conformation and aggregation mode of the rod segments are discussed case by case. The self-assembled structures of the rod–coil copolymers show appealing optical/electronic properties yet desirable stimuli-responsiveness and biocompatibility, thus holding great promise as functional materials particularly for optical, optoelectronic, energy, and biological applications.Despite the remarkable progress achieved in this area thus far, there are still a myriad of challenges and opportunities on the way. For example, (1) it is necessary to develop effective approaches for the synthesis of rod-like polymers with narrow molecular weight dispersity, which facilitates precise morphology control of their copolymer aggregates. (2) The study on the self-assembly of rod–coil alternating, graft, star-like, or hyperbranched copolymers is in its early infancy. Much effort needs to be devoted to these topics. The first priority is the development of more synthetic methods for controllable synthesis of these types of rod–coil copolymers. (3) Although chiral suprastructures have been observed in rod–coil copolymers in solution, the mystery of chiral expression from the molecular to the supramolecular level will be of great interest and challenge to study. (4) Compared with superstructures in nature, the complexity of the rod–coil copolymer assemblies drops much behind, which limits their multi-functionality and applications. To overcome this limitation, more powerful hierarchical self-assembly strategies should be developed for building up complex and multifunctional polymer superstructures. Meanwhile, deeper understanding of structure–property relationships is urgently required to allow prediction of the performance of a given structure. (5) It is highly important to formulate more systematic theories and simulations, which may greatly help to design experiment plans and explain results. With these objectives achieved, it is our hope that the supramolecular self-assembly of rod–coil copolymers will continue to blossom and the promise of their self-assembled structures as desirable materials with practical applications will be realized eventually.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (21774076 and 51573091), the Program of the Shanghai Committee of Science and Technology (17JC1403200), the Program of Shanghai Academic Research Leader (19XD1421700), and the Program of Shanghai Eastern Scholar.References
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS PubMed.
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567 CrossRef CAS PubMed.
- J. Du and R. K. O’Reilly, Chem. Soc. Rev., 2011, 40, 2402 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- Y. Mai and A. Eisenberg, Acc. Chem. Res., 2012, 45, 1657 CrossRef CAS PubMed.
- Y. Mai, F. Zhang and X. Feng, Nanoscale, 2014, 6, 106 RSC.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107 CrossRef CAS.
- K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424 RSC.
- F. Wurm and H. Frey, Prog. Polym. Sci., 2011, 36, 1 CrossRef CAS.
- C.-L. Liu, C.-H. Lin, C.-C. Kuo, S.-T. Lin and W.-C. Chen, Prog. Polym. Sci., 2011, 36, 603 CrossRef CAS.
- J. Zhang, X. Chen, H. Wei and X. Wan, Chem. Soc. Rev., 2013, 42, 9127 RSC.
- Q. Zhang, J. Lin, L. Wang and Z. Xu, Prog. Polym. Sci., 2017, 75, 1 CrossRef CAS.
- Q. Wang, Soft Matter, 2011, 7, 3711 RSC.
- W. Ding, S. Lin, J. Lin and L. Zhang, J. Phys. Chem. B, 2008, 112, 776 CrossRef CAS PubMed.
- S. Lin, X. He, Y. Li, J. Lin and T. Nose, J. Phys. Chem. B, 2009, 113, 13926 CrossRef CAS PubMed.
- L. Wang, J. Lin and X. Zhang, Polymer, 2013, 54, 3427 CrossRef CAS.
- W. Wu, C. Chen, W. Lee and W. Chen, Polymer, 2015, 65, A1 CrossRef CAS.
- C. Cai, J. Lin, Y. Lu, Q. Zhang and L. Wang, Chem. Soc. Rev., 2016, 45, 5985 RSC.
- D. Wu, Y. Huang, F. Xu, Y. Mai and D. Yan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1459 CrossRef CAS.
- S. A. Jenekhe and X. L. Chen, Science, 1998, 279, 1903 CrossRef CAS PubMed.
- L. A. McCullough and K. Matyjaszewski, Mol. Cryst. Liq. Cryst., 2010, 521, 1 CrossRef CAS.
- C. A. Machado, I. R. Smith and D. A. Savin, Macromolecules, 2019, 52, 1899 CrossRef CAS.
- E. E. Jelly, Nature, 1936, 138, 1009 CrossRef.
- L. G. S. Brooker, F. L. White, D. W. Heseltine, G. H. Keyes, S. G. Dent and E. J. Van Lare, J. Photogr. Sci., 1953, 1, 173 CrossRef CAS.
- M. Wang, G. L. Silva and B. A. Armitage, J. Am. Chem. Soc., 2000, 122, 9977 CrossRef CAS.
- S. K. Patra, R. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842 CrossRef CAS PubMed.
- X. Li, P. J. Wolanin, L. R. MacFarlane, R. L. Harniman, J. Qian, O. E. C. Gould, T. G. Dane, J. Rudin, M. J. Cryan, T. Schmaltz, H. Frauenrath, M. A. Winnik, C. F. J. Faul and I. Manners, Nat. Commun., 2017, 8, 15909 CrossRef CAS PubMed.
- D. W. Hayward, D. J. Lunn, A. Seddon, J. R. Finnegan, O. E. C. Gould, O. Magdysyuk, I. Manners, G. R. Whittell and R. M. Richardson, Macromolecules, 2018, 51, 3097 CrossRef CAS.
- E. L. Kynaston, A. Nazemi, L. R. MacFarlane, G. R. Whittell, C. F. J. Faul and I. Manners, Macromolecules, 2018, 51, 1002 CrossRef CAS.
- U. Tritschler, J. Gwyther, R. L. Harniman, G. R. Whittell, M. A. Winnik and I. Manners, Macromolecules, 2018, 51, 5101 CrossRef CAS.
- E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390 CrossRef CAS PubMed.
- A. C. Kamps, M. Fryd and S.-J. Park, ACS Nano, 2012, 6, 2844 CrossRef CAS PubMed.
- A. C. Kamps, M. H. M. Cativo, M. Fryd and S.-J. Park, Macromolecules, 2014, 47, 161 CrossRef CAS.
- A. C. Kamps, M. H. M. Cativo, X. Chen and S.-J. Park, Macromolecules, 2014, 47, 3720 CrossRef CAS.
- M. M. Cativo, D. K. Kim, R. A. Riggleman, K. G. Yager, S. S. Nonnenmann, H. Chao, D. A. Bonnell, C. T. Black, C. R. Kagan and S.-J. Park, ACS Nano, 2014, 8, 12755 CrossRef CAS PubMed.
- R. H. Lohwasser and M. Thelakkat, Macromolecules, 2012, 45, 3070 CrossRef CAS.
- P. M. Reichstein, J. C. Brendel, M. Drechsler and M. Thelakkat, ACS Appl. Nano Mater., 2019, 2, 2133 CrossRef CAS.
- Y.-J. Kim, C.-H. Cho, K. Paek, M. Jo, M.-K. Park, N.-E. Lee, Y.-J. Kim, B. J. Kim and E. Lee, J. Am. Chem. Soc., 2014, 136, 2767 CrossRef CAS PubMed.
- Y. Kim, H. J. Kim, J.-S. Kim, H. Yun, H. Park, J. Han and B. J. Kim, Chem. Mater., 2018, 30, 7912 CrossRef CAS.
- V. D. Deepak and P. R. Sundararajan, J. Phys. Chem. B, 2011, 115, 8458 CrossRef CAS PubMed.
- N. Liu, C.-G. Qi, Y. Wang, D.-F. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Macromolecules, 2013, 46, 7753 CrossRef CAS.
- I.-H. Lee, P. Amaladass, K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2013, 135, 17695 CrossRef CAS PubMed.
- S. Shin, M.-L. Gu, C.-Y. Yu, J. Jeon, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 475 CrossRef CAS PubMed.
- Y.-H. Lee, Y.-L. Yang, W.-C. Yen, W.-F. Su and C.-A. Dai, Nanoscale, 2014, 6, 2194 RSC.
- P. Kumari, M. K. Bera, S. Malik and B. K. Kuila, ACS Appl. Mater. Interfaces, 2015, 7, 12348 CrossRef CAS PubMed.
- J.-H. Li, Y. L. Li, J.-T. Xu and C. K. Luscombe, ACS Appl. Mater. Interfaces, 2017, 9, 17942 CrossRef CAS PubMed.
- Y. J. He, T.-H. Tu, M.-K. Su, C.-W. Yang, K. V. Kong and Y.-T. Chan, J. Am. Chem. Soc., 2017, 139, 4218 CrossRef CAS PubMed.
- J. Hu, G. Zhang, Y. Geng and S. Liu, Macromolecules, 2011, 44, 8207 CrossRef CAS.
- Y.-C. Chiu, C.-C. Shih and W.-C. Chen, J. Mater. Chem. C, 2015, 3, 551 RSC.
- X.-H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Science, 2018, 360, 897 CrossRef CAS PubMed.
- M. Zhou, J. Li, H. Zhang and K. Hong, Eur. Polym. J., 2018, 103, 304 CrossRef CAS.
- A. G. Dal Bó, V. Soldi, F. C. Giacomelli, C. Travelet, B. Jean, I. Pignot-Paintrand, R. Borsali and S. Fort, Langmuir, 2012, 28, 1418 CrossRef PubMed.
- S. K. Albert, I. Sivakumar, M. Golla, H. V. P. Thelu, N. Krishnan and R. Varghese, J. Am. Chem. Soc., 2017, 139, 17799 CrossRef CAS PubMed.
- S. Yang and J. He, Polym. Chem., 2016, 7, 4506 RSC.
- C. Zhou, N. Chen, J. Yang, H. Liu and Y. Li, Macromol. Rapid Commun., 2012, 33, 688 CrossRef CAS PubMed.
- L. Z. Borg, C. Schüll, H. Frey and R. Zentel, Macromol. Rapid Commun., 2013, 34, 1213 CrossRef PubMed.
- D. Tao, C. Feng, Y. Cui, X. Yang, I. Manners, M. A. Winnik and X. Huang, J. Am. Chem. Soc., 2017, 139, 7136 CrossRef CAS PubMed.
- D. Tao, C. Feng, Y. Lu, Y. Cui, X. Yang, I. Manners, M. A. Winnik and X. Huang, Macromolecules, 2018, 51, 2065 CrossRef CAS.
- Y. Cui, D. Tao, X. Huang, G. Lu and C. Feng, Langmuir, 2019, 35, 3134 CrossRef CAS PubMed.
- S. Shin, F. Menk, Y. Kim, J. Lim, K. Char, R. Zentel and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 6088 CrossRef CAS PubMed.
- L. Han, M. Wang, X. Jia, W. Chen, H. Qian and F. He, Nat. Commun., 2018, 9, 865 CrossRef PubMed.
- H. Kim, S.-M. Jeong and J.-W. Park, J. Am. Chem. Soc., 2011, 133, 5206 CrossRef CAS PubMed.
- M. Lee, B.-K. Cho and W.-C. Zin, Chem. Rev., 2001, 101, 3869 CrossRef CAS PubMed.
- H.-A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217 CrossRef CAS.
- C. Tschierske, Angew. Chem., Int. Ed., 2013, 52, 8828 CrossRef CAS PubMed.
- B. Yan, X. Tong, P. Ayotte and Y. Zhao, Soft Matter, 2011, 7, 10001 RSC.
- L. Jia, P.-A. Albouy, A. D. Cicco, A. Cao and M.-H. Li, Polymer, 2011, 52, 2565 CrossRef CAS.
- X. Xing, H. Shinb, M. J. Bowickc, Z. Yao, L. Jia and M.-H. Li, PNAS, 2012, 109, 5202 CrossRef CAS PubMed.
- S. Lin, Y. Wang, C. Cai, Y. Xing, J. Lin, T. Chen and X. He, Nanotechnology, 2013, 24, 085602 CrossRef PubMed.
- Y. Xing, S. Lin, J. Lin and X. He, Chin. J. Polym. Sci., 2013, 31, 833 CrossRef CAS.
- P. Liu, J. Liang, S. Chen and H. Zhang, RSC Adv., 2014, 4, 49028 RSC.
- K. Yuan, L. Chen and Y. Chen, J. Mater. Chem. C, 2014, 2, 3835 RSC.
- Y. Tu, Z. Ji, X. Yang, X. Wan and Q.-F. Zhou, Macromol. Rapid Commun., 2014, 35, 1795 CrossRef CAS PubMed.
- Z. Tong, Y. Li, H. Xu, H. Chen, W. Yu, W. Zhuo, R. Zhang and G. Jiang, ACS Macro Lett., 2016, 5, 867 CrossRef CAS.
- L. Zhou, D. Zhang, S. Hocine, A. Pilone, S. Trépout, S. Marco, C. Thomas, J. Guo and M. Li, Polym. Chem., 2017, 8, 4776 RSC.
- L. Shen, H. Guo, J. Zheng, X. Wang, Y. Yang and Z. An, ACS Macro Lett., 2018, 7, 287 CrossRef CAS.
- S. Li, J. Wang, J. Shen, B. Wu and Y. He, ACS Macro Lett., 2018, 7, 437 CrossRef CAS.
- J. Chen, B. Li, X. Li, J. Zhang and X. Wan, Polym. Chem., 2018, 9, 2002 RSC.
- Z. Yuan, W. Wei and H. Xiong, Polymer, 2018, 158, 65 CrossRef CAS.
- S. Dong, W. Sun, D. Wang, H. Zhao, G. Zu and Y. Zheng, Macromol. Rapid Commun., 2019, 40, 1900058 CrossRef PubMed.
- M. Huo, Y. Zhang, M. Zeng, L. Liu, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 8192 CrossRef CAS.
- M. Huo, G. Song, J. Zhang, Y. Wei and J. Yuan, ACS Macro Lett., 2018, 7, 956 CrossRef CAS.
- M. Huo, D. Li, G. Song, J. Zhang, D. Wu, Y. Wei and J. Yuan, Macromol. Rapid Commun., 2018, 39, 1700840 CrossRef PubMed.
- M. Huo, Z. Wan, M. Zeng, Y. Wei and J. Yuan, Polym. Chem., 2018, 9, 3944 RSC.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308 CrossRef CAS.
- S. Guan, C. Zhang, W. Wen, T. Qu, X. Zheng, Y. Zhao and A. Chen, ACS Macro Lett., 2018, 7, 358 CrossRef CAS.
- X. Hou, S. Guan, T. Qu, X. Wu, D. Wang, A. Chen and Z. Yang, ACS Macro Lett., 2018, 7, 1475 CrossRef CAS.
- S. Guan, Z. Deng, T. Huang, W. Wen, Y. Zhao and A. Chen, ACS Macro Lett., 2019, 8, 460 CrossRef CAS.
- W. Wen, T. Huang, S. Guan, Y. Zhao and A. Chen, Macromolecules, 2019, 52, 2956 CrossRef CAS.
- X. Li, B. Jin, Y. Gao, D. W. Hayward, M. A. Winnik, Y. Luo and I. Manners, Angew. Chem., Int. Ed., 2016, 55, 11392 CrossRef CAS PubMed.
- B. Jin, K. Sano, S. Aya, Y. Ishida, N. Gianneschi, Y. Luo and X. Li, Nat. Commun., 2019, 10, 2397 CrossRef PubMed.
- H. Cai, G. Jiang, C. Chen, Z. Li, Z. Shen and X.-H. Fan, Macromolecules, 2014, 47, 146 CrossRef CAS.
- X. Lyu, A. Xiao, W. Zhang, P. Hou, K. Gu, Z. Tang, H. Pan, F. Wu, Z. Shen and X.-H. Fan, Angew. Chem., Int. Ed., 2018, 57, 10132 CrossRef CAS PubMed.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534 CrossRef CAS PubMed.
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084 CrossRef CAS PubMed.
- H. He, K. Rahimi, M. Zhong, A. Mourran, D. R. Luebke, H. B. Nulwala, M. Möller and K. Matyjaszewski, Nat. Commun., 2017, 8, 14057 CrossRef PubMed.
- Z. Lin, S. Liu, W. Mao, H. Tian, N. Wang, N. Zhang, F. Tian, L. Han, X. Feng and Y. Mai, Angew. Chem., Int. Ed., 2017, 56, 7135 CrossRef CAS PubMed.
- A. Douy and B. Gallot, Polymer, 1987, 28, 147 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS PubMed.
- H. Kukula, H. Schlaad, M. Antonietti and S. Förster, J. Am. Chem. Soc., 2002, 124, 1658 CrossRef CAS PubMed.
- F. Chécot, S. Lecommandoux, Y. Gnanou and H. A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339 CrossRef.
- J. Babin, J. Rodriguez-Hernandez, S. Lecommandoux, H. A. Klok and M. F. Achard, Faraday Discuss., 2005, 128, 179 RSC.
- W. Agut, D. Taton, A. Brûlet, O. Sandre and S. Lecommandoux, Soft Matter, 2011, 7, 9744 RSC.
- A. Lübbert, V. Castelletto, I. W. Hamley, H. Nuhn, M. Scholl, L. Bourdillon, C. Wandrey and H. A. Klok, Langmuir, 2005, 21, 6582 CrossRef PubMed.
- H. A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1 CrossRef CAS.
- H. Schlaad, Adv. Polym. Sci., 2006, 202, 53 CrossRef CAS.
- T. Deming, Adv. Polym. Sci., 2006, 202, 1 CrossRef CAS.
- C. G. Pappas, R. Shafi, I. R. Sasselli, H. Siccardi, T. Wang, V. Narang, R. Abzalimov, N. Wijerathne and R. V. Ulijn, Nat. Nanotechnol., 2016, 11, 960 CrossRef CAS PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19 CrossRef CAS PubMed.
- T. O. McDonald, H. Qu, B. R. Saunders and R. V. Ulijn, Soft Matter, 2009, 5, 1728 RSC.
- Z. Song, Z. Han, S. Lv, C. Chen, L. Chen, L. Yin and J. Cheng, Chem. Soc. Rev., 2017, 46, 6570 RSC.
- Y. Lim, E. Lee and M. Lee, Macromol. Rapid Commun., 2011, 32, 191 CrossRef CAS PubMed.
- S. S. Naik, J. G. Ray and D. A. Savin, Langmuir, 2011, 27, 7231 CrossRef CAS PubMed.
- O. D. Krishna, K. T. Wiss, T. Luo, D. J. Pochan, P. Theato and K. L. Kiick, Soft Matter, 2012, 8, 3832 RSC.
- K. Klinker, O. Schfer, D. Huesmann, T. Bauer, L. Capelka, L. Braun, N. Stergiou, M. Schinnerer, A. Dirisala, K. Miyata, K. Osada, H. Cabral, K. Kataoka and M. Barz, Angew. Chem., Int. Ed., 2017, 56, 9608 CrossRef CAS PubMed.
- Z. Shi, Y. Wei, C. Zhu, J. Sun and Z. Li, Macromolecules, 2018, 51, 6344 CrossRef CAS.
- S. Ji, L. Xu, X. Fu, J. Sun and Z. Li, Macromolecules, 2019, 52, 4686 CrossRef CAS.
- W. Zhu, J. Lin and C. Cai, J. Mater. Chem., 2012, 22, 3939 RSC.
- C. Cai, L. Wang, J. Lin and X. Zhang, Langmuir, 2012, 28, 4515 CrossRef CAS PubMed.
- C. Cai, Y. Li, J. Lin, L. Wang, S. Lin, X.-S. Wang and T. Jiang, Angew. Chem., Int. Ed., 2013, 52, 7732 CrossRef CAS PubMed.
- Z. Zhuang, C. Cai, T. Jiang, J. Lin and C. Yang, Polymer, 2014, 55, 602 CrossRef CAS.
- C. Yang, Q. Li, C. Cai and J. Lin, Langmuir, 2016, 32, 6917 CrossRef CAS PubMed.
- J. Xue, Z. Guan, J. Lin, C. Cai, W. Zhang and X. Jiang, Small, 2017, 13, 1604214 CrossRef PubMed.
- Z. Song, H. Kim, X. Ba, R. Baumgartner, J. S. Lee, H. Tang, C. Leal and J. Cheng, Soft Matter, 2015, 11, 409 Search PubMed.
- V. K. Kotharangannagari, A. Sanchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2012, 45, 1982 CrossRef CAS.
- J. Huang, C. L. Hastings, G. P. Duffy, H. M. Kelly, J. Raeburn, D. J. Adams and A. Heise, Biomacromolecules, 2013, 14, 200 CrossRef CAS PubMed.
- J. F. Reuther, D. A. Siriwardane, O. V. Kulikov, B. L. Batchelor, R. Campos and B. M. Novak, Macromolecules, 2015, 48, 3207 CrossRef CAS.
- J. F. Reuther, D. A. Siriwardane, R. Campos and B. M. Novak, Macromolecules, 2015, 48, 6890 CrossRef CAS.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed.
- J. J. Cornelissen, M. Fischer, N. A. Sommerdijk and R. J. Nolte, Science, 1998, 280, 1427 CrossRef CAS PubMed.
- J. Min, P. N. Shah, J.-H. Ahn and J.-S. Lee, Macromolecules, 2011, 44, 3211 CrossRef CAS.
- C. G. Chae, P. N. Shah, J. Min, H. B. Seo and J. S. Lee, Macromolecules, 2014, 47, 1563 CrossRef CAS.
- N. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2014, 47, 1699 CrossRef CAS.
- Y. Chen, Z. H. Zhang, X. Han, J. Yin and Z.-Q. Wu, Macromolecules, 2016, 49, 7718 CrossRef CAS.
- Y. He, S. Shi, N. Liu, Y. Ding, J. Yin and Z.-Q. Wu, Macromolecules, 2016, 49, 48 CrossRef CAS.
- M. Su, N. Liu, Q. Wang, H. Wang, J. Yin and Z.-Q. Wu, Macromolecules, 2016, 49, 110 CrossRef CAS.
- F. Freire, E. Quiñoá and R. Riguera, Chem. Rev., 2016, 116, 1242 CrossRef CAS PubMed.
- K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, Macromol. Rapid Commun., 2015, 36, 1069 CrossRef CAS PubMed.
- B. Zhao, J. Deng and J. Deng, ACS Macro Lett., 2016, 6, 6 CrossRef.
- T. Muraoka, T. Shima, T. Hamada, M. Morita, M. Takagi and K. Kinbara, Chem. Commun., 2011, 47, 194 RSC.
- T. Muraoka, T. Shima, T. Hamada, M. Morita, M. Takagi, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2012, 134, 19788 CrossRef CAS PubMed.
- Y. Zheng, H. Zhou, D. Liu, G. Floudas, M. Wagner, K. Koynov, M. Mezger, H.-J. Butt and T. Ikeda, Angew. Chem., Int. Ed., 2013, 52, 4945 CrossRef.
- T.-G. Kim, C. Kim and J.-W. Park, Macromolecules, 2017, 50, 8185 CrossRef CAS.
- Z. Du, Y. Shan, J. Luo, N. Sun and B. Ren, ACS Macro Lett., 2019, 8, 279 CrossRef CAS.
- J. Chen, C. Yu, Z. Shi, S. Yu, Z. Lu, W. Jiang, M. Zhang, W. He, Y. Zhou and D. Yan, Angew. Chem., Int. Ed., 2015, 54, 3621 CrossRef CAS PubMed.
- Q. Xu, T. Huang, S. Li, K. Li, C. Li, Y. Liu, Y. Wang, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2018, 57, 8043 CrossRef CAS PubMed.
- Y. Zhang, C. Li, T. Rasheed, P. Huang and Y. Zhou, Chin. J. Polym. Sci., 2018, 36, 897 CrossRef CAS.
- T. Rasheed, C. Li, F. Nabeel, W. Huang and Y. Zhou, Chem. Eng. J., 2019, 358, 101 CrossRef CAS.
- C. Li, T. Rasheed, H. Tian, P. Huang, Y. Mai, W. Huang and Y. Zhou, ACS Macro Lett., 2019, 8, 331 CrossRef CAS.
- J. Yuan, Y. Xu and A. H. E. Müller, Chem. Soc. Rev., 2011, 40, 640 RSC.
- S. Ma, X. Zhang, B. Yu and F. Zhou, NPG Asia Mater., 2019, 11, 24 CrossRef.
- J. Ma, X. Ma, S. Deng, F. Li and A. Hu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1368 CrossRef CAS.
- J. Rzayev, ACS Macro Lett., 2012, 1, 1146 CrossRef CAS.
- H.-G. Jeong, B. Lim, D. Khim, M. Han, J. Lee, J. Kim, J.-M. Yun, K. Cho, J.-W. Park and D.-Y. Kim, Adv. Mater., 2013, 25, 6416 CrossRef CAS PubMed.
- M. G. Mohamed, C.-C. Cheng, Y.-C. Lin, C.-W. Huang, F.-H. Lu, F.-C. Chang and S.-W. Kuo, RSC Adv., 2014, 4, 21830 RSC.
- H. J. Kim, J.-H. Kim, J.-H. Ryu, Y. Kim, H. Kang, W. B. Lee, T.-S. Kim and B. J. Kim, ACS Nano, 2014, 8, 10461 CrossRef CAS PubMed.
- W. Lee, J.-S. Kim, H. J. Kim, J. M. Shin, K. H. Ku, H. Yang, J. Lee, J. G. Bae, W. B. Lee and B. J. Kim, Macromolecules, 2015, 48, 5563 CrossRef CAS.
- Y. Lyu, D. Cui, H. Sun, Y. Miao, H. Duan and K. Pu, Angew. Chem., Int. Ed., 2017, 56, 9155 CrossRef CAS PubMed.
- Y. Huang, Y. Mai, X. Yang, U. Beser, J. Liu, F. Zhang, D. Yan, K. Müllen and X. Feng, J. Am. Chem. Soc., 2015, 137, 11602 CrossRef CAS PubMed.
- Y. Huang, R. Yuan, F. Xu, Y. Mai, X. Feng and D. Yan, Polym. Chem., 2016, 7, 1234 RSC.
- D. Wu, F. Xu, Y. Huang, C. Chen, C. Yu, X. Feng, D. Yan and Y. Mai, Macromolecules, 2018, 51, 161 CrossRef CAS.
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC.
- W. Niu, J. Liu, Y. Mai, K. Müllen and X. Feng, Trends Chem., 2019, 1, 549 CrossRef.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. D. Feyter and K. Müllen, Nat. Chem., 2013, 6, 126 CrossRef.
- P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489 CrossRef CAS PubMed.
- Y. Huang, Y. Mai, U. Beser, J. Teyssandier, G. Velpula, H. van Gorp, L. A. Straasø, M. R. Hansen, D. Rizzo, C. Casiraghi, R. Yang, G. Zhang, D. Wu, F. Zhang, D. Yan, S. D. Feyter, K. Müllen and X. Feng, J. Am. Chem. Soc., 2016, 138, 10136 CrossRef CAS PubMed.
- Y. Huang, W. Dou, F. Xu, H. Ru, Q. Gong, D. Wu, D. Yan, H. Tian, X. He, Y. Mai and X. Feng, Angew. Chem., Int. Ed., 2018, 57, 3366 CrossRef CAS PubMed.
- Y. Huang, F. Xu, L. Ganzer, F. V. A. Camargo, T. Nagahara, J. Teyssandier, H. V. Gorp, K. Basse, L. A. Straasø, V. N. C. Casiraghi, M. R. Hansen, S. D. Feyter, D. Yan, K. Müllen, X. Feng, G. Cerullo and Y. Mai, J. Am. Chem. Soc., 2018, 140, 10416 CrossRef CAS PubMed.
- F. Xu, C. Yu, A. Tries, H. Zhang, M. Kläui, K. Basse, M. R. Hansen, N. Bilbao, M. Bonn, H. I. Wang and Y. Mai, J. Am. Chem. Soc., 2019, 141, 10972 CrossRef CAS PubMed.
- Y. Yano, N. Mitoma, K. Matsushima, F. Wang, K. Matsui, A. Takakura, Y. Miyauchi, H. Ito and K. Itami, Nature, 2019, 571, 387 CrossRef CAS PubMed.
- L. Li, F. Zhou, Y. Li, X. Chen, Z. Zhang, N. Zhou and X. Zhu, Langmuir, 2018, 34, 11034 CrossRef CAS.
- I. Park, Y.-R. Yoon, M. Jung, K. Kim, S. Park, S. Shin, Y. Lim and M. Lee, Chem. – Asian J., 2011, 6, 452 CrossRef CAS.
- W. Li, I. Park, S.-K. Kang and M. Lee, Chem. Commun., 2012, 48, 8796 RSC.
- W. Li, J. Li and M. Lee, Chem. Commun., 2013, 49, 8238 RSC.
- X. Chen, Y. He, Y. Kim and M. Lee, J. Am. Chem. Soc., 2016, 138, 5773 CrossRef CAS.
- Y. Liu, Y. Zhang, Z. Wang, J. Wang, K. Wei, G. Chen and M. Jiang, J. Am. Chem. Soc., 2016, 138, 12387 CrossRef CAS PubMed.
- Z. Ma, C. Zheng, Z. Shen, D. Liang and X.-H. Fan, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 918 CrossRef CAS.
- H.-J. Tian, W. Qu, Y.-F. Zhu, Z. Shen and X.-H. Fan, Polym. Chem., 2014, 5, 4948 RSC.
- J. Ding, L. Zhao, D. Li, C. Xiao, X. Zhuang and X. Chen, Polym. Chem., 2013, 4, 3345 RSC.
- S. Zhai, X. Song, C. Feng, X. Jiang, Y. Li, G. Lu and X. Huang, Polym. Chem., 2013, 4, 4134 RSC.
- Z. Wei, S. Zhu and H. Zhao, Polym. Chem., 2015, 6, 1316 RSC.
- J. Yuan, Y. Zhang, Y. Sun, Z. Cai, L. Yang and H. Lu, Biomacromolecules, 2018, 19, 2089 CrossRef CAS PubMed.
- L. Ding, Y. Huang, Y. Zhang, J. Deng and W. Yang, Macromolecules, 2011, 44, 736 CrossRef CAS.
- H. Zhang, L. Ding, Y. Chen, W. Yang and J. Deng, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4415 CrossRef CAS.
- L. Ding, C. Chen, J. Deng and W. Yang, Polym. Bull., 2012, 69, 1023 CrossRef CAS.
- Q. Li, B. Xia, M. Branham, W. Ha, H. Wu, S.-L. Peng, L.-S. Ding, B.-J. Li and S. Zhang, Carbohydr. Polym., 2011, 86, 120 CrossRef CAS.
- W. Ha, X.-W. Meng, Q. Li, M.-M. Fan, S.-L. Peng, L.-S. Ding, X. Tian, S. Zhang and B.-J. Li, Soft Matter, 2011, 7, 1018 RSC.
- J. Sun, C. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174 CrossRef CAS.
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495 CrossRef CAS.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925 CrossRef CAS.
- D. B. Wright, M. A. Touve, M. P. Thompson and N. C. Gianneschi, ACS Macro Lett., 2018, 7, 40 Search PubMed.
- W. Wu, W. Wang and J. S. Li, Prog. Polym. Sci., 2015, 46, 55 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, J. Am. Chem. Soc., 2010, 132, 10078 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Macromolecules, 2011, 44, 3179 CrossRef CAS.
- Y. Mai, L. Xiao and A. Eisenberg, Macromolecules, 2013, 46, 3183 CrossRef CAS.
- F. Xu, D. Wu, Y. Huang, H. Wei, Y. Gao, X. Feng, D. Yan and Y. Mai, ACS Macro Lett., 2017, 6, 426 CrossRef CAS.
- F. Xu, P. Zhang, J. Zhang, C. Yu, D. Yan and Y. Mai, ACS Macro Lett., 2018, 7, 1062 CrossRef CAS.
- J. Park, M. Moon, M. Seo, H. Choi and S. Y. Kim, Macromolecules, 2010, 43, 8304 CrossRef CAS.
- X. Pang, L. Zhao, C. Feng and Z. Lin, Macromolecules, 2011, 44, 7176 CrossRef CAS.
- J. G. Ray, J. T. Ly and D. A. Savin, Polym. Chem., 2011, 2, 1536 RSC.
- K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto, Y. Anraku, A. Kishimura, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2012, 134, 13172 CrossRef CAS PubMed.
- E. Blasco, B. V. K. J. Schmidt, C. B. Kowollik, M. Piñol and L. Oriol, Polym. Chem., 2013, 4, 4506 RSC.
- P. Wang, S. Cao, Y. Zhao, T. Iyoda and A. Chen, Macromol. Chem. Phys., 2017, 218, 1700148 CrossRef.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534 CrossRef CAS PubMed.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS PubMed.
- A. Hanisch, A. H. Gröschel, M. Förtsch, M. Drechsler, H. Jinnai, T. M. Ruhland, F. H. Schacher and A. H. E. Müller, ACS Nano, 2013, 7, 4030 CrossRef CAS.
- W. Sun, X. He, C. Gao, X. Liao, M. Xie, S. Lin and D. Yan, Polym. Chem., 2013, 4, 1939 RSC.
- T. Liu, Y. Zhang and S. Liu, Chin. J. Polym. Sci., 2013, 31, 924 CrossRef CAS.
- J. A. Kalow and T. M. Swager, ACS Macro Lett., 2015, 4, 1229 CrossRef CAS.
- C. Liu, Y. Mi, R. Wang, Z. Jiang, X. Zhang, N. Liu, J. Yin and Z.-Q. Wu, Polym. Chem., 2016, 7, 2447 RSC.
- F. Qiu, C. Tu, R. Wang, L. Zhu, Y. Chen, G. Tong, B. Zhu, L. He, D. Yan and X. Zhu, Chem. Commun., 2011, 47, 9678 RSC.
- F. Qiu, D. Wang, Q. Zhu, L. Zhu, G. Tong, Y. Lu, D. Yan and X. Zhu, Biomacromolecules, 2014, 15, 1355 CrossRef CAS PubMed.
- J. Luan, C. Lu, Y. Guo, F. Li and G. Wang, High Perform. Polym., 2017, 29, 257 CrossRef CAS.
- R. Dong, Y. Zhou and X. Zhu, Acc. Chem. Res., 2014, 477, 2006 CrossRef PubMed.
- W. Jiang, Y. Zhou and D. Yan, Chem. Soc. Rev., 2015, 44, 3874 RSC.
- H. Li, A. Zhang, K. Li, W. Huang, Y. Mai, Y. Zhou and D. Yan, Mater. Chem. Front., 2018, 2, 1040 RSC.
- Y. Mai, Y. Zhou and D. Yan, Macromolecules, 2005, 38, 8679 CrossRef CAS.
- H. Hong, Y. Mai, Y. Zhou, D. Yan and J. Cui, Macromol. Rapid Commun., 2007, 28, 591–596 CrossRef CAS.
- H. Tian, Z. Lin, F. Xu, J. Zheng, X. Zhuang, Y. Mai and X. Feng, Small, 2016, 12, 3155 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © the Partner Organisations 2019 |