
Redox-switchable olefin cross metathesis (CM) reactions and acyclic diene metathesis (ADMET) polymerizations†
Yeonkyeong
Ryu
 ab,
Huiling
Shao
c,
Guillermo
Ahumada
ab,
Peng
Liu
cd and
Christopher W.
Bielawski
*abe
ab,
Huiling
Shao
c,
Guillermo
Ahumada
ab,
Peng
Liu
cd and
Christopher W.
Bielawski
*abe
aCenter for Multidimensional Carbon Materials (CMCM), Institute for Basic Science (IBS), Ulsan 44919, Republic of Korea
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
cDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA
dDepartment of Chemical and Petroleum Engineering, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, USA
eDepartment of Energy Engineering, UNIST, Ulsan 44919, Republic of Korea
First published on 12th August 2019
Abstract
We show that redox-switchable catalysis may be used to control acyclic diene metathesis (ADMET) polymerizations and related reactions. A Ru(II) complex was found to display catalytic activities that were dependent on the oxidation state of a quinone-containing ligand. While the neutral form of the complex was found to catalyze ADMET polymerizations at rates that were commensurate with a commercially-available catalyst, significantly lower activities were observed when the complex was reduced. Using the rate differential, a series of ADMET polymerizations were modulated by alternately reducing and oxidizing the catalyst over time. A similar approach was also used to regulate the molecular weights of the polymers produced. Cross metathesis reactions and computational studies were performed in parallel to gain a deeper understanding of the underlying redox-switchable chemistry.
Introduction
Olefin metathesis is a widely-implemented transformation used to synthesize small molecules and macromolecular materials.1–6 Acyclic diene metathesis (ADMET) polymerization7,8 is a variation of olefin metathesis that affords unsaturated polymers in a step-growth manner through cross metathesis (CM), a process wherein terminal olefins are coupled concomitantly with the formation of ethylene.9 ADMET has been used to prepare a broad range of polymers, including those that feature pendant groups in precisely defined positions along their corresponding backbones.10,11 Similar to other step-growth techniques, the average molecular weights of the polymers produced using ADMET are dependent on the extent of the corresponding polymerization reaction. As such, the synthesis of polymers with specific molecular weights often involves adding chain transfer agents or quenching the polymerization reactions at pre-determined conversions.12,13We hypothesized that the use of a catalyst that can be rapidly deactivated and subsequently re-started may bestow control over ADMET polymerizations, including the molecular weights of the polymers produced. Such a goal is rooted in the burgeoning field of switchable catalysis,14 which seeks to modulate the intrinsic activities and selectivities displayed by catalysts through the introduction of thermal,15 chemical,16 photochemical,17 redox18–22 or mechanical23 stimuli. Redox approaches are particularly attractive in part because a broad range of chemical oxidants and reductants are currently available.24,25
The first example of a redox-controlled transformation was disclosed by Wrighton.26 The Rh complex I shown in Scheme 1 was found to promote hydrogenation or isomerization reactions when its cobaltocene-based ligand was in a neutral form; however, upon oxidation, the catalyst facilitated hydrosilylations. Gibson and Long subsequently showed that the ring-opening polymerization (ROP) of lactide may be modulated by changing the oxidation state of the ferrocenyl units attached to a Ti-based catalyst (II).21 Control over ROPs using other organometallic catalysts were reported by Diaconescu, Byers and Long (III–V).27–29 Redox-controlled olefin polymerizations have also been demonstrated by Chen30–32 as well as Long and Tennyson33 (VI–IX).
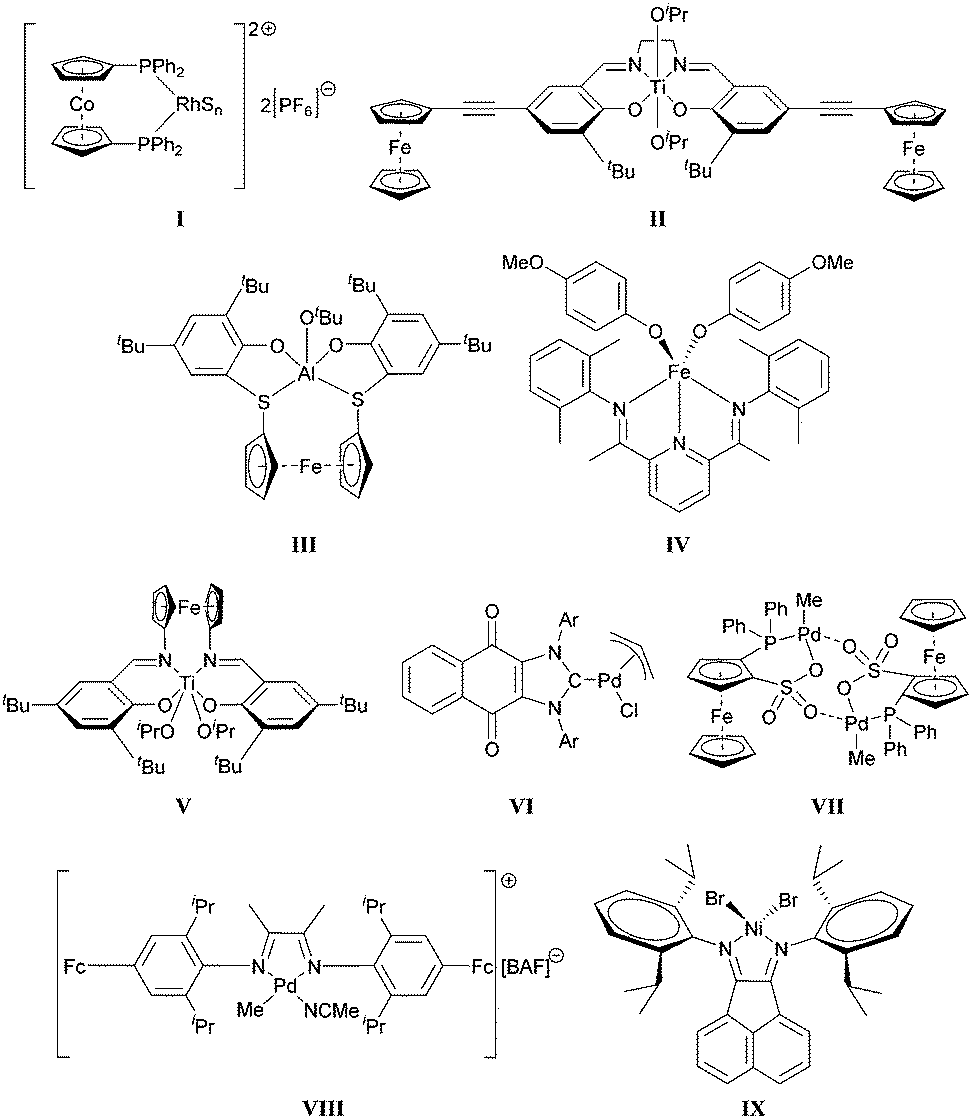 | ||
| Scheme 1 Examples of redox-switchable catalysts. S = (CH3)2CO, BAF = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate, Ar = 2,6-dimethylphenyl or 2,6-diisopropylphenyl. | ||
Our contributions to the field of redox-switchable catalysis34 have included the design and deployment of Ru-based complexes for controlling olefin metathesis reactions. As summarized in Scheme 2, we demonstrated that catalysts equipped with naphthoquinone-containing N-heterocyclic carbenes (e.g., 1) undergo reduction upon exposure to a reductant (e.g., cobaltocene; CoCp2); subsequent exposure of a reduced catalyst to an oxidant (e.g., 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DDQ) restored its neutral form. The activity of the redox-switchable catalyst was found to be dependent on the oxidation state of the quinone group. For example, the rate constants measured for ring-closing metathesis (RCM)35 and ring-opening metathesis polymerization (ROMP) reactions36 were significantly attenuated upon reduction.‡ Moreover, the intrinsic monomer selectivities displayed by some catalysts were found to depend on the oxidation states of the redox active ligands, features that permitted control over copolymerization reactions.36
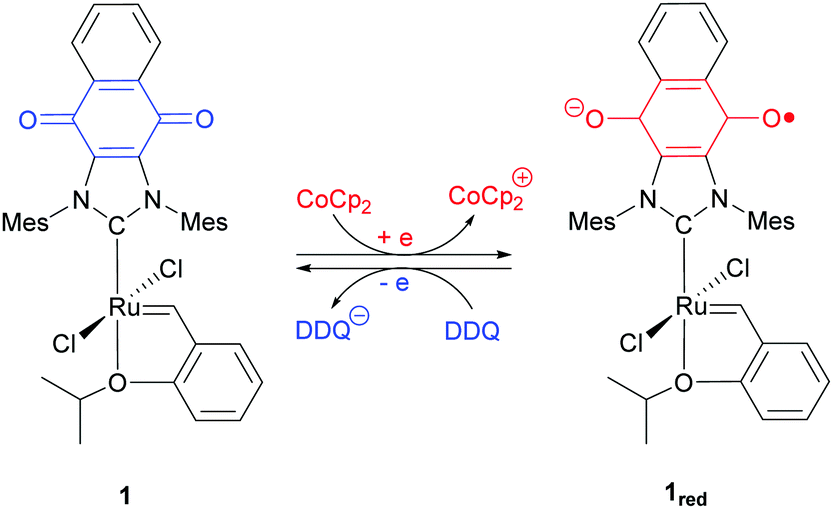 | ||
| Scheme 2 Addition of CoCp2 reduces 1 to 1red; subsequent exposure to DDQ reverses the reaction. | ||
Building on these results, we describe herein a series of redox-controlled ADMET polymerizations. Since such polymerizations are predicated on cross metathesis (CM), a summary of efforts to control such reactions is also described as the results obtained therefrom were used to guide the polymerization chemistry. We also show that the molecular weights of the polymers produced using ADMET can be modulated using redox-switchable catalysts. Finally, a mechanism derived from a series of calculations is proposed.
Experimental
General considerations
All procedures were performed in a nitrogen-filled glove box or using standard air-free techniques unless otherwise noted. Solvents were dried and degassed using a Vacuum Atmospheres Company solvent purification system and stored over 4 Å molecular sieves in a nitrogen-filled glove box. Compounds 2, 4 and 6 were purchased from commercial sources. Monomer 8 was synthesized by following a procedure reported in the literature.37 Substrates and monomers were distilled from CaH2 under reduced pressure in a Straus flask equipped with a Teflon valve, degassed by three freeze–pump–thaw cycles, and stored in a nitrogen-filled glove box. Complex 1 was synthesized according to previously reported procedures.35 NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts (δ) are given in ppm and are referenced to the residual solvent (1H: CDCl3, 7.26 ppm). Gel permeation chromatography (GPC) was performed on a Malvern GPCmax system that featured a series of two fluorinated polystyrene columns maintained at 35 °C and employed THF stabilized with BHT as the mobile phase at a flow rate of 0.8 mL min−1. Detection was performed using a Viscotek VE 3580 RI detector. Molecular weight data are reported as their polystyrene equivalents.Kinetics measurements
Reaction kinetics were monitored by measuring the quantity of gaseous byproducts (e.g., ethylene) that were produced over time using a modified setup reported in the literature.38 A flame-dried, 50 mL, 2-neck Schlenk flask equipped with a 14/20 ground-glass joint was connected via Tygon tubing and a Schlenk adapter to a Teflon valve. The end of the tubing was connected to a 24/40 ground-glass joint which was capped with a rubber septum and pierced with a 60 cm cannula (18 gauge). The other side of the cannula was placed in a water-filled burette that was partially submerged in a pool of water (Fig. S1, ESI†). The ideal gas equation in conjunction with the change in volume over time were used to calculate the quantity of gas produced and thus the extent of the reaction.39 Additional details may be found in the ESI.†General procedure used to monitor cross metathesis reactions
A flame-dried, 50 mL, 2-neck Schlenk flask equipped with 14/20 ground-glass joint and a magnetic stir bar was cooled to room temperature under vacuum. Afterward, the flask was purged with nitrogen, charged with 1 (5.0 μmol), and then connected to the apparatus described above. For experiments that used 1red as the initiator, 0.05 mL of a 0.1 M stock solution of CoCp2 (1.0 equiv. rel. to 1) dissolved in 1,2-dichlorobenzene was added. Reducing the pressure inside of the burette through the application of vacuum from the top of the system caused the water level to rise. Before the water reached the tip of the cannula, the vacuum was halted. Next, the Teflon valve that connects the reaction flask to the burette was opened. After waiting for 5 min for the system to equilibrate, 2.25 mmol of the substrate was injected into the reaction flask (time = 0). To generate 1redin situ, 0.05 mL of a 0.1 M stock solution of CoCp2 (1.0 equiv. rel. to 1) dissolved in 1,2-dichlorobenzene was added to 1. Conversely, the reaction mixture was charged with 0.15 mL of a 0.05 M stock solution of DDQ (1.5 equiv. rel. to 1) in 1,2-dichlorobenzene to oxidize the reduced complex. The reactions were quenched by adding 1.0 mL of a chloroform solution containing 10 mM of ethyl vinyl ether and 4.3 mM of 2,6-di-tert-butyl-4-methylphenol (BHT). The extent of reaction was calculated from the amount of ethylene that evolved over the course of the reaction as determined in part by the change in volume. The crude products were analyzed by 1H NMR spectroscopy.General procedure used to monitor ADMET polymerizations
A 7.50 mM of stock solution of 1 in 1,2-dichlorobenzene was prepared. A flame-dried Schlenk flask was charged with 0.4 mL of the stock solution of 1. For experiments that used 1red as the initiator, 0.03 mL of a 0.1 M stock solution of CoCp2 (3.0 μmol, 1.0 equiv. rel. to 1) dissolved in 1,2-dichlorobenzene was added to the solution of 1. Vacuum was then applied to the top of the system, which caused the water level to rise, and then closed before the water reached the tip of the cannula. Next, the Teflon valve that connects the reaction flask to the burette was opened. After waiting for 5 min for the system to equilibrate, 1.35 mmol of the monomer was injected into the reaction flask. To generate 1redin situ, 0.03 mL of a 0.1 M stock solution of CoCp2 (3.0 μmol, 1.0 equiv. rel. to 1) dissolved in 1,2-dichlorobenzene was added to the reaction mixture. Conversely, the reaction mixture was charged with 0.09 mL of a 0.05 M stock solution of DDQ (4.5 μmol, 1.5 equiv. rel. to 1) in 1,2-dichlorobenzene to oxidize the reduced complex. The reactions were quenched by adding 0.8 mL of a chloroform solution containing 10 mM of ethyl vinyl ether and 4.3 mM of BHT. For each reaction, the degree of polymerization was ascertained from the amount of ethylene that was generated and independently confirmed by analyzing the crude product mixtures using 1H NMR spectroscopy.General bulk polymerization procedure
Three oven-dried, 25 mL Schlenk tubes equipped with magnetic stir bars were taken into a nitrogen filled glove box. Each tube was independently charged with 1 (24.11 μmol) followed by appropriate quantities (i.e., 0.0, 0.5 or 1.0 equiv. rel. to 1) of a 50 mM of stock solution of CoCp2 in 1,2-dichlorobenzene. Additional 1,2-dichlorobenzene was added to each flask to maintain a constant initial concentration of 1 (i.e., 3.86 mL, 3.62 mL and 3.38 mL, respectively). After 5 min, 1,9-decadiene (6) (2.0 mL, 1.5 g, 10.85 mmol, 60/10 = 450) was added to each flask. The tubes were then sealed, removed from the glove box, and connected to a high vacuum manifold at 75 °C. Bubbles were observed upon the application of a static vacuum. Once the bubbling ceased, a full vacuum (ca. 0.1 mmHg) was applied to the reaction vessels for 5 days. Excess ethyl vinyl ether was added to quench the reaction followed by removal of residual solvent under high vacuum. The crude products were analyzed by GPC.Results and discussion
Initial efforts were directed toward the cross metathesis of 1-decene (2). Although CM and ADMET polymerizations are often conducted under vacuum to drive product formation,40,41 we adapted a system reported in the literature to monitor the evolution of ethylene and thus the reaction conversion over time.38 Adding 2 to catalyst 1 (20/10 = 450) at 65 °C initiated a reaction that proceeded with a second order rate constant (k) of 3.2 × 10−3 M−1 s−1. In a separate experiment it was discovered that charging a reaction vessel containing the catalyst with a concentrated solution of CoCp2 (1.0 equiv. rel. to 1) before adding the substrate resulted in a slower reaction (kred = 8.9 × 10−5 M−1 s−1). Under these conditions it was surmised that 1 underwent reduction in situ, in accord with previous results.35 For comparison, the rate constant for an analogous reaction performed with the commercially-available catalyst HG2 was measured to be 5.1 × 10−3 M−1 s−1.To determine if the aforementioned condensation can be controlled in a temporal manner, redox agents were added to the reaction mixture at various points in time. As summarized in Fig. 1, the CM of 2, as initiated with 1 (20/10 = 450), was measured to proceed with an initial rate constant (k) of 1.6 × 10−3 M−1 s−1 at 65 °C. After a 19% conversion of 2 to 3 was reached (30 s), CoCp2 (1.0 equiv. rel. to 1) was introduced to the reaction mixture. The addition resulted in a lower measured rate constant, kred = 2.0 × 10−4 M−1 s−1 (k/kred = 8.0). At a later point in time (120 s), a slight excess of DDQ (1.5 equiv. rel. to 1) was added which resulted in restoration of the initial catalytic activity, kred-ox = 1.3 × 10−3 M−1 s−1 (kred-ox/kred = 6.5).
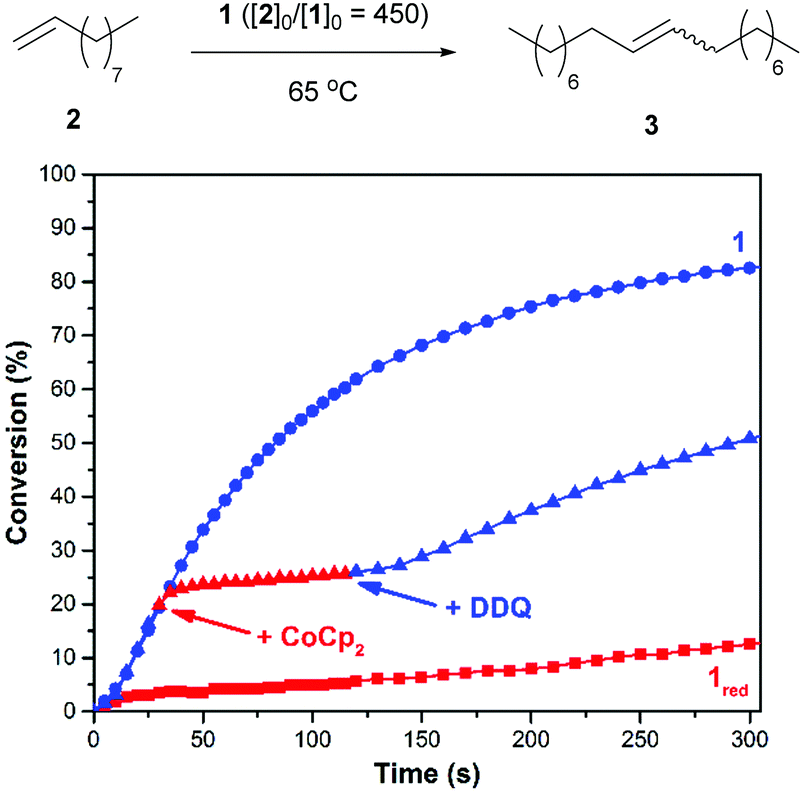 | ||
| Fig. 1 The cross metathesis of 2 (top) and corresponding plots of conversion vs. time (bottom). Conditions: 65 °C, 20 = 2.25 mmol, 10 = 5.0 μmol. The labels “+CoCp2” and “+DDQ” indicate when said reagent was added (see text). The label “1red” refers to an experiment where the catalyst was subjected to CoCp2 prior to the addition of 2. | ||
Next, efforts shifted toward exploring the scope of substrates that are amenable to control using a redox-switchable catalyst. Attention was directed toward the CM of an aromatic substrate, particularly allylbenzene (4), in part because related monomers (e.g., 1,4-diallylbenzene) can be expected to afford polymers that may be less prone to intramolecular cyclization and thus of relatively high molecular weight.37 The addition of 1 to neat 4 at 50 °C (40/10 = 450) resulted in the condensation of ethylene gas whose evolution was measured to proceed with a corresponding reaction rate constant (k) of 1.8 × 10−3 M−1 s−1. For comparison, the reduced catalyst, which was generated in situ with CoCp2 using the procedure described above, resulted in a significantly slower reaction (kred = 6.2 × 10−5 M−1 s−1). As summarized in Fig. 2, the CM of 4 was also controlled over time. The initial rate of the reaction (k = 1.1 × 10−3 M−1 s−1) was significantly reduced upon reduction of the catalyst (kred = 1.5 × 10−4 M−1 s−1; k/kred = 7.3); subsequent addition of DDQ enhanced the rate of the reaction (kred-ox = 5.7 × 10−4 M−1 s−1; kred-ox/kred = 3.8).
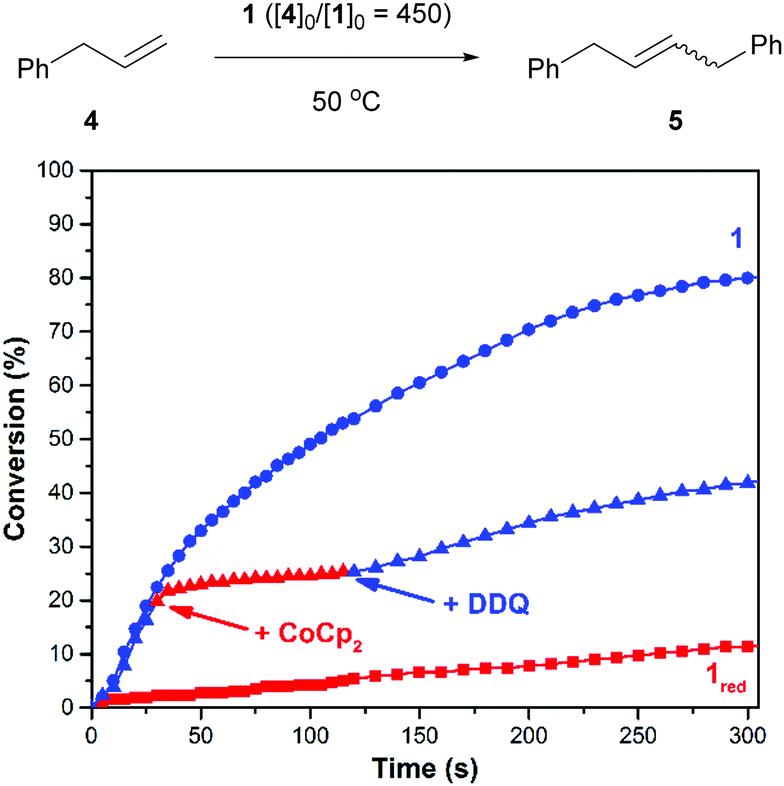 | ||
| Fig. 2 The cross metathesis of 4 (top) and corresponding plots of conversion vs. time (bottom). Conditions: 50 °C, 40 = 2.25 mmol, 10 = 5.0 μmol. The labels “+CoCp2” and “+DDQ” indicate when said reagent was added (see text). The label “1red” refers to an experiment where the catalyst was subjected to CoCp2 prior to the addition of 4. | ||
After demonstrating that the redox-active catalyst may be used to control CM reactions, efforts were directed toward polymerizing ditopic analogues. As summarized in Fig. 3, a suitable monomer, 1,9-decadiene (6), was added to a reaction vessel which contained a solution of 1 in 1,2-dichlorobenzene (60/10 = 450, 60 °C). The corresponding polymerization reaction proceeded with a rate constant (k) of 1.7 × 10−3 M−1 s−1. For comparison, an analogous reaction conducted with 1red, which was prepared by reducing the catalyst in situ prior to introduction to the monomer, was measured to proceed at a slower rate (kred = 1.2 × 10−4 M−1 s−1).
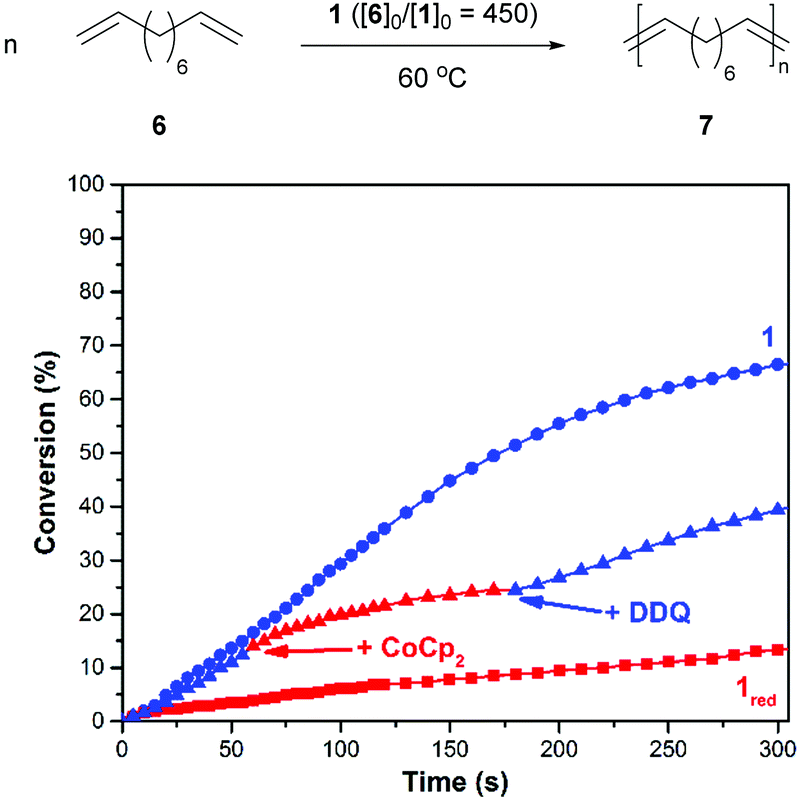 | ||
| Fig. 3 The ADMET polymerization of 6 (top) and corresponding plots of conversion vs. time (bottom). Conditions: 60 = 1.35 mmol, 10 = 3 μmol, 60 °C. The labels “+CoCp2” and “+DDQ” indicate when said reagent was added (see text). The label “1red” refers to an experiment where the catalyst was subjected to CoCp2 prior to the addition of 6. | ||
In parallel with the experiments described above, redox agents were added to a reaction mixture at pre-determined intervals and the corresponding changes were monitored to determine if the polymerization can be modulated over time. For example, the ADMET of 6, as initiated with 1 (60/10 = 450, 60 °C) was measured to proceed with an initial rate constant (k) of 6.5 × 10−4 M−1 s−1. After 14% of 6 converted to polymer, CoCp2 (1.0 equiv. rel. to 1) was introduced to the reaction mixture. The addition resulted in a lower measured reaction constant, kred = 3.6 × 10−4 M−1 s−1 (k/kred = 1.8). At a later point in time, a slight excess of DDQ (1.5 equiv. rel. to added 1) was introduced, which resulted in a restoration of the initial catalytic activity, kred-ox = 8.7 × 10−4 M−1 s−1 (kred-ox/kred = 2.4).
As shown in Fig. 4, the ADMET polymerization of p-diallylbenzene (8) was conducted under conditions that were similar to those described for 6. After charging a reaction vessel that contained a solution of 1 in 1,2-dichlorobenzene with monomer (80/10 = 450, 60 °C), the corresponding polymerization reaction proceeded with a rate constant (k) of 7.9 × 10−4 M−1 s−1. An analogous reaction that was conducted with 1red, which was prepared in situ prior to the addition of monomer, was measured to proceed at a relatively slow rate (kred = 1.9 × 10−5 M−1 s−1). Efforts to re-activate the reduced catalyst through the addition of DDQ were met with limited success. Isomerization products were observed upon analysis of the crude reaction mixtures using 1H NMR spectroscopy which may inhibit catalyst switching performance and/or facilitate premature catalyst decomposition.42
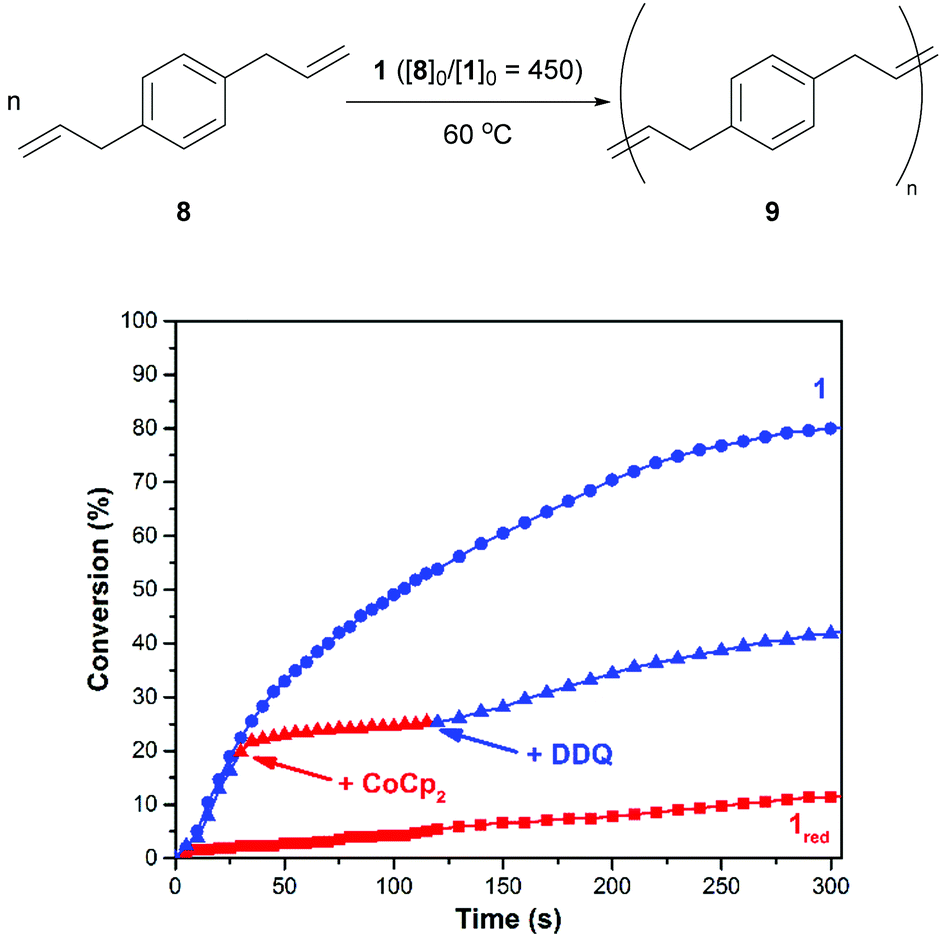 | ||
| Fig. 4 The ADMET polymerization of 8 (top) and corresponding plots of conversion vs. time (bottom). Conditions: 80 = 1.35 mmol, 10 = 3 μmol, 60 °C. The labels “+CoCp2” and “+DDQ” indicate when said reagent was added (see text). The label “1red” refers to an experiment where the catalyst was subjected to CoCp2 prior to the addition of 8. | ||
To fine tune the redox-switchable condensation chemistry underlying the reactions describe above, a series of experiments were conducted wherein the ratio of reductant to catalyst was varied. An ionic liquid, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMIM TFSI), was employed as a solvent to minimize volume changes, due its low vapor pressure, and to increase the accuracy of the measurements. As shown in Fig. 5 and Table S1 (ESI†), the quantity of added CoCp2 ([CoCp2]0 = 62.5 mM in a stock solution) inversely correlated with catalytic activity and overall substrate conversion. In other words, reaction performance was effectively adjusted by partitioning the catalyst between its neutral and reduced states.
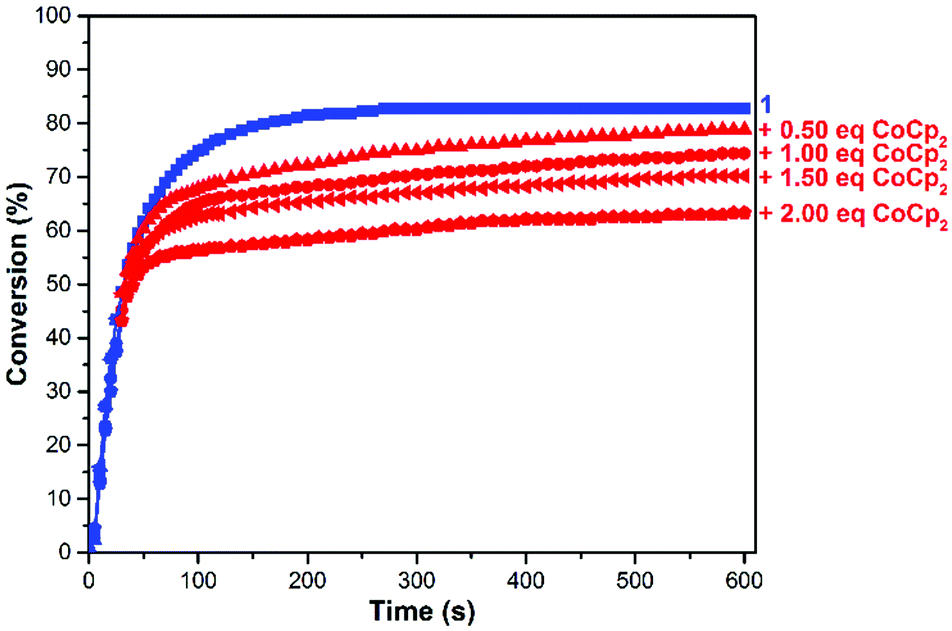 | ||
| Fig. 5 Plots of conversion of 2 to 3vs. time using 1 in conjunction with various quantities of CoCp2. Conditions: 20 = 2.25 mmol, 10 = 5.0 μmol, 65 °C, EMIM TFSI as solvent. The labels refer to the molar equivalents of CoCp2 (rel. to 1) that were added to the reaction mixture at 30 s. The label “1” refers to a control experiment wherein CoCp2 was not added to the reaction mixture. | ||
Finally, efforts were directed toward controlling the molecular weights of the ADMET polymers produced using a redox-switchable catalyst. Three separate experiments were conducted in parallel at 75 °C: one utilized the neutral form of the catalyst and the others used its reduced derivative as generated in situ from 1 and either 0.5 equiv. or 1.0 equiv. of CoCp2. After 5 days, the reactions were quenched, and then the crude product mixtures were directly analyzed to minimize fractionation that can accompany precipitation-based purification procedures. In accord with the kinetics measurements described above, the polymer synthesized with 1 was measured to exhibit a peak molecular weight of 6.0 kDa, which was the largest of the three samples analyzed. For comparison, the molecular weight the polymer prepared by reducing 1 with 0.5 equiv. of CoCp2 was 1.3 kDa and that with 1.0 equiv. of CoCp2 was 0.8 kDa.
Collectively, the results indicated that the reduced form of catalyst exhibited a lower catalytic activity in CM reactions and ADMET polymerizations than its neutral form. To gain a molecular level of understanding of the performance displayed by the redox-switchable catalyst, the propagation cycles for the ADMET of 6, as catalyzed by 1 or 1red, were calculated using density functional theory (DFT) (Fig. 6). The computed reaction energy profiles (Fig. S16, ESI†) revealed that the rate-determining transition state in the catalytic cycle was a retro-[2+2]-cycloaddition (TS4) that released the internal alkene product.42 For comparison, the monosubstituted ruthenacyclobutane intermediate 11 was calculated to be the resting state.§ The activation free energy of the retro-[2+2] cycloaddition using the reduced form of ligand (L1red) was calculated to be 0.7 kcal mol−1 higher than that of the neutral analogue (L1). Although the absolute values deviated, the change in the calculated rate constant ratio agreed with the value determined by experiment (k/kred = 3.6 vs. 14, respectively). The lower rate constant calculated for L1red may be attributed to the electronic stabilization of the Ru(IV) resting state 11 by the relatively strong electron donor ligand, consistent with previous computational studies of ROMP and RCM reactions which indicated that stronger donor ligands stabilize ruthenacyclobutane resting states and effectively suppress retro-[2+2] cycloadditions.35,36 Regardless of its oxidation state, the steric properties of the redox-switchable ligand remain largely unchanged and thus are not expected to affect the relative reactivities. The lower reactivity displayed by 1red in CM reactions can be rationalized by similar phenomena (see the ESI† for computational results that pertain to the CM of 1-pentene).
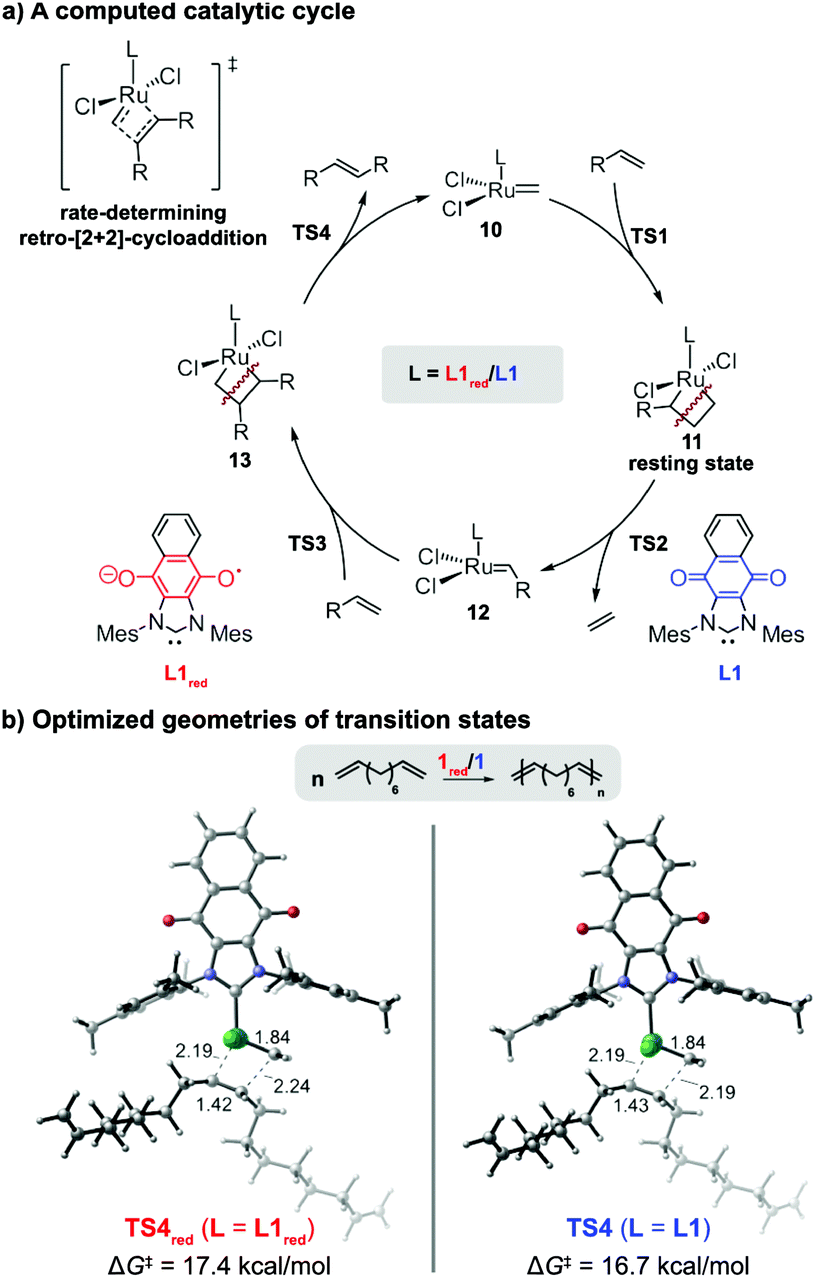 | ||
| Fig. 6 Computational studies on the origin of the ligand effects observed in the redox-switchable ADMET polymerization of 1,9-decadiene. The activation free energies of the retro-[2+2] cycloadditions (TS4red and TS4) are reported with respect the monosubstituted ruthenacyclobutane 11. All energies were calculated at the M06/SDD-6-311+G(d,p)/SMD(1,2-dichlorobenzene)//B3LYP/SDD-6-31G(d) level of theory (see text and ESI† for more details). | ||
Conclusions
A series of CM reactions and ADMET polymerizations were controlled using a redox-switchable Ru-based catalyst. The relatively high activities displayed by the catalyst in its neutral form were attenuated upon reduction and activity was restored upon subsequent oxidation. Likewise, reduced forms of the catalyst afforded polymers that exhibited lower molecular weights than analogous reactions performed using the neutral form of the catalyst. Catalytic activity was also tuned by varying the ratio of added reductant to catalyst. Based on a series of DFT calculations, a potential mechanism that explains how the catalyst enabled modulation of the olefin metathesis chemistry is proposed. In a broader context, these results represent the first examples of controlling step-growth polymerizations using a redox-switchable catalyst and scope the potential of using such types of catalysts to control a broad range of synthetic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Institute for Basic Science (IBS-R019), the Office of Naval Research (N00014-14-1-0650), and the BK21Plus Program as funded by the Ministry of Education and the National Research Foundation of Korea are acknowledged for support. We are grateful to Dr Tang Tang and Dr Songsu Kang for performing GPC analyses. DFT calculations were performed using supercomputer resources at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation.Notes and references
- A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef.
- R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef CAS.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS PubMed.
- C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed.
- O. M. Ogba, N. C. Warner, D. J. O’Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- R. H. Grubbs, A. G. Wenzel, D. J. O’Leary and E. Khosravi, Handbook of Metathesis, Wiley-VCH, Weinheim, Germany, vol. 1–3., 2015 Search PubMed.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- P. Atallah, K. B. Wagener and M. D. Schulz, Macromolecules, 2013, 46, 4735–4741 CrossRef CAS.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed.
- L. Caire da Silva, G. Rojas, M. D. Schulz and K. B. Wagener, Prog. Polym. Sci., 2017, 69, 79–107 CrossRef CAS.
- M. D. Schulz and K. B. Wagener, Macromol. Chem. Phys., 2014, 215, 1936–1945 CrossRef CAS.
- J. B. Matson, S. C. Virgil and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 3355–3362 CrossRef CAS PubMed.
- M. A. Hillmyer and R. H. Grubbs, Macromolecules, 1995, 28, 8662–8667 CrossRef CAS.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- O. Coulembier, A. P. Dove, R. C. Pratt, A. C. Sentman, D. A. Culkin, L. Mespouille, P. Dubois, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2005, 44, 4964–4968 CrossRef CAS PubMed.
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed.
- C. D. Varnado, Jr., E. L. Rosen, M. S. Collins, V. M. Lynch and C. W. Bielawski, Dalton Trans., 2013, 42, 13251–13264 RSC.
- E. M. Broderick, N. Guo, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS PubMed.
- K. Arumugam, C. D. Varnado, S. Sproules, V. M. Lynch and C. W. Bielawski, Chem. – Eur. J., 2013, 19, 10866–10875 CrossRef CAS PubMed.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed.
- C. Chen, ACS Catal., 2018, 8, 5506–5514 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- I. M. Lorkovic, R. R. Duff and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617–3618 CrossRef CAS.
- J. Wei, M. N. Riffel and P. L. Diaconescu, Macromolecules, 2017, 50, 1847–1861 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed.
- L. A. Brown, J. L. Rhinehart and B. K. Long, ACS Catal., 2015, 5, 6057–6060 CrossRef CAS.
- W. Zou, W. Pang and C. Chen, Inorg. Chem. Front., 2017, 4, 795–800 RSC.
- M. Chen, B. Yang and C. Chen, Angew. Chem., Int. Ed., 2015, 54, 15520–15524 CrossRef CAS PubMed.
- M. Zhao and C. Chen, ACS Catal., 2017, 7, 7490–7494 CrossRef CAS.
- W. C. Anderson, J. L. Rhinehart, A. G. Tennyson and B. K. Long, J. Am. Chem. Soc., 2016, 138, 774–777 CrossRef CAS PubMed.
- Y. Ryu, G. Ahumada and C. W. Bielawski, Chem. Commun., 2019, 55, 4451–4466 RSC.
- D. N. Lastovickova, A. J. Teator, H. Shao, P. Liu and C. W. Bielawski, Inorg. Chem. Front., 2017, 4, 1525–1532 RSC.
- D. N. Lastovickova, H. Shao, G. Lu, P. Liu and C. W. Bielawski, Chem. – Eur. J., 2017, 23, 5994–6000 CrossRef CAS PubMed.
- D. Steiger, C. Weder and P. Smith, Macromolecules, 1999, 32, 5391–5398 CrossRef CAS.
- S. E. Lehman and K. B. Wagener, Macromolecules, 2002, 35, 48–53 CrossRef CAS.
- K. B. Wagener, K. Brzezinska, J. D. Anderson, T. R. Younkin, K. Steppe and W. DeBoer, Macromolecules, 1997, 30, 7363–7369 CrossRef CAS.
- K. Brzezinska, P. S. Wolfe, M. D. Watson and K. B. Wagener, Macromol. Chem. Phys., 1996, 197, 2065–2074 CrossRef CAS.
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2017, 66, 7–12 CrossRef CAS.
- P. A. Fokou and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 368–373 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Kinetics data, NMR spectra, GPC chromatograms, and calculation data. See DOI: 10.1039/c9qm00391f |
| ‡ Ru catalysts equipped redox-active ligands that adopt a positive charge upon oxidation were also developed, see: ref. 18 and 22. |
| § The monodentate ruthenacyclobutane 11 is the lowest energy intermediate in the computed energy profile. However, under vacuum, the disubstituted ruthenacyclobutane 13 may also act as a catalyst resting state. Regardless, L1red was calculated to suppress reactivity due to stabilization of the ruthenacyclobutane. See ESI† for more details. |
| This journal is © the Partner Organisations 2019 |