
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitivity enhancement in lateral flow assays: a systems perspective
Joshua D.
Bishop
 a,
Helen V.
Hsieh
a,
David J.
Gasperino
a and
Bernhard H.
Weigl
*ab
a,
Helen V.
Hsieh
a,
David J.
Gasperino
a and
Bernhard H.
Weigl
*ab
aIntellectual Ventures Laboratory, Bellevue, 98007 WA, USA. E-mail: jbishop@intven.com
bDepartment of Bioengineering, University of Washington, Seattle, Washington 98195, USA. E-mail: bweigl@intven.com
First published on 28th June 2019
Abstract
Lateral flow assays (LFAs) are rapid, inexpensive, easy-to-manufacture and -use tests widely employed in medical and environmental applications, particularly in low resource settings. Historically, LFAs have been stigmatized as having limited sensitivity. However, as their global usage expands, extensive research has demonstrated that it is possible to substantially improve LFA sensitivity without sacrificing their advantages. In this critical review, we have compiled state-of-the-art approaches to LFA sensitivity enhancement. Moreover, we have organized and evaluated these approaches from a system-level perspective, as we have observed that the advantages and disadvantages of each approach have arisen from the integrated and tightly interconnected chemical, physical, and optical properties of LFAs.
 Joshua D. Bishop | Dr. Joshua D. Bishop is a Senior Scientist in the Biotechnology group at Intellectual Ventures Laboratory. His work focuses on the design, development, and construction of diagnostic technologies appropriate for use at the point-of-need in low-resource settings. Prior to joining Intellectual Ventures, he built portable, paper microfluidics-based, instrument-free, nucleic acid amplification test devices. He received his B.S. in Electrical and Computer Engineering from the University of Illinois at Urbana-Champaign, and his M.S. and Ph.D. in Electrical Engineering from the University of Washington, where he pursued interdisciplinary research the fields of control systems/robotics and synthetic biology. |
 Helen V. Hsieh | Dr. Helen V. Hsieh is a Senior Scientist at Intellectual Ventures Laboratory in Bellevue, WA. After earning her bachelor's degree in Chemistry at The Johns Hopkins University in Baltimore MD and doctorate in Chemistry at the University of North Carolina-Chapel Hill, she began developing medical diagnostic tests for Becton Dickinson. She joined Intellectual Ventures Laboratory in 2015 to develop innovative solutions for global health problems. Her research interests include point-of-care detection of viral, bacterial, and parasitic infectious diseases, as well as chronic diseases, and neglected tropical diseases. |
 David Gasperino | Dr. David Gasperino was previously a Principal Engineer at Intellectual Ventures/Global Good (IV/GG), and now works for Amazon in their Sustainability organization leading their computational modeling team. His work at IV/GG focused on building computational models to support low-resource diagnostic design and developing hardware/software systems to automate diagnostic optimization at the bench top. He received his B.S. in Chemical Engineering from the University of Washington, and his Ph.D. in Chemical Engineering from the University of Minnesota, where he focused on computational modeling of solution phase reactions and transport phenomena. |
 Bernhard H. Weigl | Dr. Bernhard H. Weigl is the Director of the Center for In-Vitro Diagnostics at Intellectual Ventures/Global Good (IV/GG) and Affiliate Professor at the University of Washington, Department of Bioengineering. His scientific interests include traditional and paper-based microfluidics as well as any assay platform that allows simplification and integration of previously complex assays. He received his M.Sc (Mag. rer. nat.) and Ph.D. (Dr. rer. nat.) from Karl-Franzens-University Graz and completed post-doctoral studies at the University of Southampton and the University of Washington. He has authored 130+ scientific publications and is an inventor on 80+ US patents and published patent applications. |
1. Introduction
Lateral flow assays (LFAs) have been developed for use in a range of fields, such as medical diagnostics, veterinary medicine, food safety, and environmental safety, to serve as analytical tests for a range of biochemical analytes, such as antigenic proteins, glycoproteins, glycolipids, lipids, antibodies, and nucleic acids.1 While these tests are highly valued as inexpensive, quick, and easy to use, they historically have lacked sensitivity compared to analytical reference methods.2An LFA is commonly constructed as a test strip (Fig. 1) that implements assay schemes wherein an analyte interacts with a detection reagent and an immobilized capture reagent. An LFA test strip is composed of several porous materials, onto which assay reagents are striped, sprayed, or spotted, then dried for storage in spatially distinct locations, and through which analyte, in a clinical or environmental sample, and assay reagents are transported by capillary action. A standard LFA test strip accepts sample (and a running buffer, if required) on a sample pad, stores detection reagents (i.e. conjugates) in a conjugate pad, arranges immobilized capture reagents in the form of test and control lines on an analytical membrane, and draws excess fluid into an absorbent pad. The test strip may be enclosed in a cassette or run as a dipstick in a test tube. The result of an LFA is almost always related to an optical signal generated at the test line. In a sandwich assay format, a signal is generated by the concentration of detection reagent bound to captured analyte. Alternatively, in a competitive assay format (commonly used for small target analytes such as drugs of abuse3), a signal at the test line is eliminated by the displacement of captured detection reagent with the analyte.
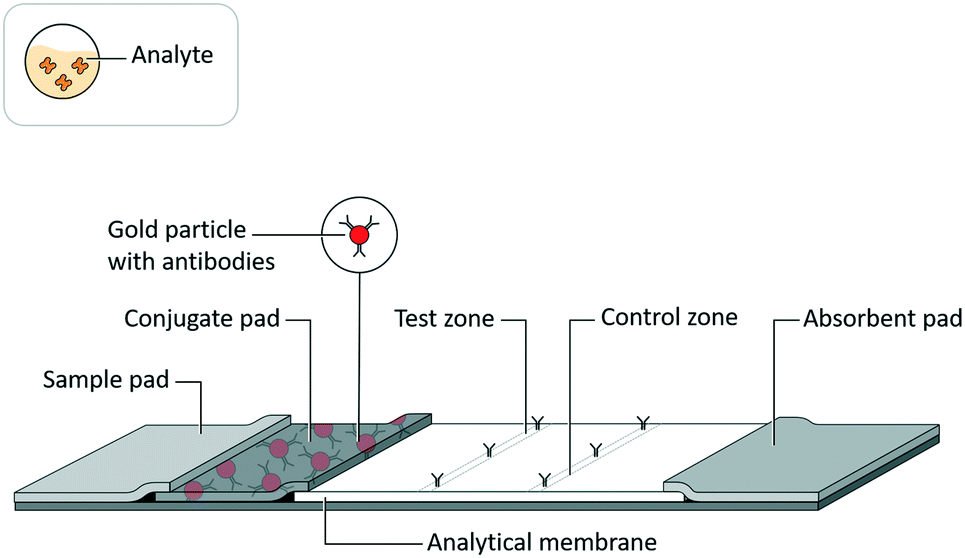 | ||
| Fig. 1 Lateral flow assay schematic. Adapted from Hsieh et al.105 (with permission under open license CC BY 4.0). | ||
The ideal LFA is sensitive, specific, stable, fast (<30 minutes), inexpensive, and user-friendly. It gives reproducible results and is robust to variation in patient samples and operating parameters such as sample and running buffer volumes. However, these performance characteristics arise from the integrated and interconnected chemical, physical, and optical properties of the LFA (Fig. 2). The complexity of these interconnections suggests that a system-level understanding will be critical to the realization of the ideal LFA and the delineation of its fundamental weaknesses.
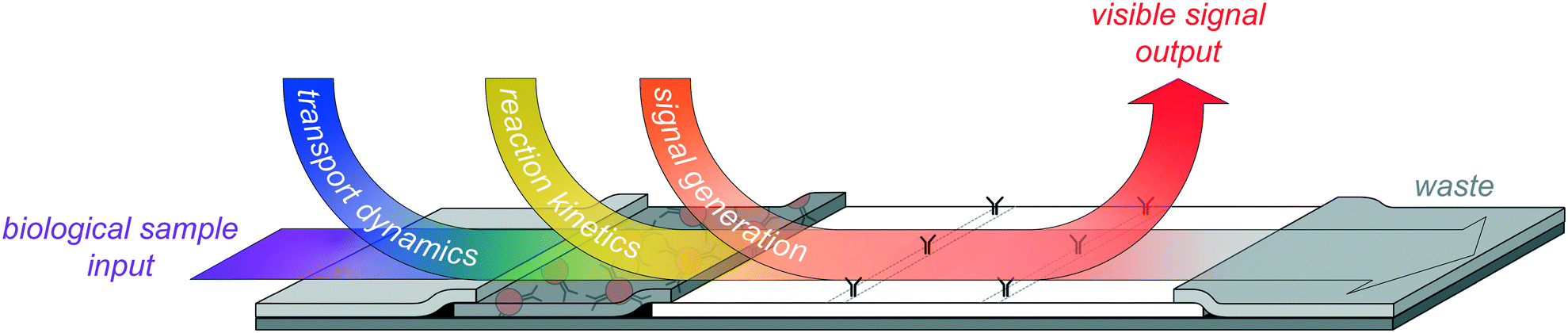 | ||
| Fig. 2 Illustration of the overlapping dynamics in an LFA system, by which its physical (transport dynamics), chemical (reaction kinetics), and optical (signal generation) properties are interconnected in a highly complex, time-dependent fashion. State-of-the-art improvements to LFA sensitivity must accommodate these interconnected system dynamics. | ||
In this review, we have categorized improvements in LFA analytical sensitivity from a high-level systems perspective using three organizing principles—reaction (section 2), transport (section 3), and signal (section 4). Furthermore, we have categorized the transformative improvements that connect at least two of these organizing principles (section 5) and noted how these interconnected improvements invite a systems approach to LFA development. We have constrained the scope of this review, by use of these organizing principles, to be within the operational bounds of the LFA. Therefore, we have not discussed improvements to sensitivity that arise from innovations in reagent or material manufacturing processes,4,5 or non-integrated sample preparation steps,6,7 for example.
All the same, the variety of approaches that fit within our framework is quite large. The magnitude of recent literature on this topic speaks not only to the importance of the LFA and its broad applicability, but also to the complexity of the conventional LFA, in that it can support such a wide range of modifications. Due to the breadth of that range, which can include differences in test format, analytical target, read-out method, instrumentation, etc., we chose to provide—where reported—the fold-change difference observed in relation to an unmodified, conventional LFA system.
In addition to our categorization of recent advances in LFA sensitivity enhancement, we have endeavoured to critically assess the limitations on approaches within each of our categories. We have found it often to be the case that innovations designed to improve sensitivity in LFAs have degraded other important performance characteristics (e.g. test time, user steps, test autonomy) and/or have not been tested in a fully implemented test format against a representative set of clinical or environmental samples. Although the focus of our review is on sensitivity enhancements, it is these general observations that back our advocacy for a system-level approach to LFA development and innovation.
2. Reaction
The reactions that occur within an LFA strip are most often in the same format as a “sandwich” ELISA. In this format, an immobilized capture reagent (typically an antibody) binds an analyte (typically a protein antigen with multiple epitopes) to which binds a labelled detection reagent (typically one or more different antibodies conjugated to a nanoparticle label). Ideally, either a significant amount of detection reagent or none is localized to the capture reagent at equilibrium, depending on analyte presence or absence, respectively.Perhaps the most obvious way to improve the performance of the LFA reaction with respect to these conditions is to introduce capture and detection reagents with high affinity and/or avidity for the analyte. Less obvious is what the best approach to obtaining or creating new, high-affinity reagents should be. Examples of demonstrated approaches include the adaptation of naturally occurring high-affinity reagents and the design of synthetic, de novo high-affinity reagents.
Natural
Adaptations of streptavidin and biotin as assay reagents in the LFA format take advantage of a binding pair that is one of the strongest found in nature, is highly resistant to biochemical interference, and is naturally multivalent in that wild-type streptavidin binds four biotins. BioProto has commercialized the generic RapidAssay Device (gRAD), in which analyte binds to a labelled detection reagent and a non-immobilized, biotinylated capture reagent upstream of the test line. This complex then binds to an immobilized, biotin-binding reagent at the test line. Yang et al. demonstrated a fast (10 minutes) gRAD for foot-and-mouth disease with ELISA-like sensitivity.8 Similarly, Expedeon Inc. (San Diego, CA, USA) has launched a Universal Lateral Flow Assay Kit, in which the non-immobilized capture reagent contains a “ULFA-Tag” and the LFA test line is made of immobilized anti-ULFA-Tag antibody.Synthetic
The creation of de novo binding reagents takes advantage of increasingly powerful computational models that can target specific epitopes, search over multiple interdependent reagent characteristics (e.g. size, stability, charge), and introduce multivalent binding. For example, Strauch et al. demonstrated the use of two computationally designed hemagglutinin-binding proteins, specifically designed to bind to different analyte epitopes, as detection and capture probes in an influenza LFA.9 However, these reagents were not reported as tested with clinical influenza samples, or against an existing influenza LFA for the purpose of demonstrating improved sensitivity.Limitations
The reaction category covers the high-affinity interactions between analyte and the capture and/or detection reagents, whether implemented in a sandwich or a competitive assay. In seeking to introduce the highest possible affinity reagents to conventional LFA systems, the two approaches categorized here ultimately suffer from a problem of scale. Currently, the number of natural high-affinity reagents commonly used in an LFA is limited to a single affinity pair (streptavidin–biotin) that can only be used in one place within an LFA reaction. Conversely, the number de novo proteins that are candidates for use as high-affinity reagents in LFAs is unknown, but likely astronomical. Unfortunately, while the tools for designing, producing, and screening de novo proteins are continuously improving, the development effort behind even a single de novo reagent is currently considerable. In addition, this extensive development process is primarily underwritten by therapeutic applications, which pose a set of design specifications that are not necessarily compatible with diagnostic applications. Nanobodies are examples of therapeutic agents with obvious diagnostic applications,10 and they are easier to produce, smaller, and more stable than antibodies, but they still require extensive optimization and screening similar to that for antibodies. Aptamers are examples of “programmable,” nucleic-acid-based binding reagents that have shown a range of sensitivities from mid-picomolar to high-micromolar, which argues a continued need for case-by-case optimization even though—or especially because—they introduce new reaction sets to the LFA format.11,12 Ultimately, the production process of any LFA reagent—antibody, de novo protein, or otherwise—must align with the most desirable characteristics of the LFA itself: integrated, low-cost, and rapid. Without this alignment the innovations in this category are unlikely to make an impact on the practice of LFA development.3. Transport
The transport of sample, buffers, and assay reagents in an LFA strip is provided by the capillary action that occurs within its constituent porous materials. Reaction and transport are inextricably linked, in that changes to transport characteristics such as volumes or flow rates necessarily change reaction kinetics. Many fluidic controls have been introduced to tune transport dynamics, and therefore to tune reaction kinetics, and have been demonstrated to improve sensitivity in paper-based systems. Examples include modified sample and conjugate pad geometries (8-fold sensitivity improvement in a human IgG LFA),13 modified flow rates using a sponge shunt (10-fold sensitivity improvement in a nucleic acid LFA),14 and modified flow rates using wax pillars (3-fold sensitivity improvement in a human IgG LFA).15 Many other paper-based fluidic controls were recently reviewed elsewhere.16However, one area in which physical, transport-based approaches to sensitivity improvement can be largely distinguished from biochemical, reaction-based approaches is sample preparation. Several on-board sample preparation methods that selectively transport analyte—through the physical separation and/or concentration of analyte from sample—to improve sensitivity have been demonstrated on LFAs. For example, the size-based separation of sample components by filtration is easily achieved in a system of porous materials, like an LFA. Commercial vendors, such as GE Healthcare Life Sciences (Pittsburgh, PA USA) and Pall Life Sciences (Ann Arbor, MI USA), offer plasma separation membranes can be integrated into the LFA strip format without affecting the standard LFA workflow.17,18
Other selective transport methods that have recently been applied to LFAs include separation and/or concentration of analyte from sample by electrophoresis, aqueous two-phase systems, and dialysis membranes.
Electrophoretic
Electrophoretic methods drive transport of charged particles with applied electric fields. Demonstrated electrophoretic sample separation methods in paper-based devices include the direct application of electric fields, ion concentration polarization (ICP), and isotachophoresis (ITP). Wu et al. applied a direct electric field to selectively enhance the transport of nucleic acids, and improve sensitivity 400-fold, in an LFA that detected PCR products from an H5N1 influenza PCR assay.19 Although the application of ICP to micro/nanofluidic platforms was recently reviewed,20 there are some examples of ICP in paper-based devices. Gong et al. demonstrated ICP in a saturated paper-based device, which improved sensitivity 5-fold for a FITC–BSA conjugate and 4-fold for bromocresol green dye compared to without ICP; while Liu et al. demonstrated ICP in a Parafilm- and paper-based device, which concentrated FITC.21 Lastly, Moghadam et al. demonstrated ITP and improved sensitivity 400-fold in a goat anti-human IgG LFA.22Phasic
Aqueous two-phase systems (ATPS) use two different water-soluble materials (e.g. polymers, salts, or micelles) at high concentration to form two immiscible aqueous phases, allowing concentration of a target protein in one phase.23 While ATPS were demonstrated to work as a pre-concentration step before the application of sample to a bacteriophage M1324 and a transferrin25 LFA, Pereira et al. integrated the APTS step into a “three-dimensional” design to improve sensitivity 10-fold in a Plasmodium lactate dehydrogenase (pLDH) LFA.26Dialytic
Dialysis is a common technique applied to concentrate nucleic acids and proteins, and to deplete a sample of small contaminants. Tang et al. demonstrated nucleic acid dialysis and protein dialysis in separate, one-step LFA devices. They integrated a dialysis membrane and PEG-based water absorption onto LFA strips, and concentrated (separately) HIV nucleic acid and myoglobin analytes, which improved sensitivity 10-fold and 4-fold in the respective LFAs.27Limitations
The transport category contains an important set of optimization parameters for an LFA. Transport dynamics in an LFA should be tuned during development to optimize the integrated binding reactions (specific and non-specific) and optical signal generation. There are several design elements that can be changed to tune LFA transport dynamics, including the type, geometry, and layout of porous materials, but also the constitution of various buffers and the physical and chemical characteristics of reagents. Selective transport, on the other hand, represents a set of sensitivity enhancement approaches in which initial sample preparation steps are prepended to the standard LFA workflow. Here, as in the more interconnected reaction/transport and reaction/transport/optical approaches discussed below, more sophisticated transport systems support more sophisticated workflows, but while the additional sophistication might lead to improved sensitivity, it is often at the cost of added test time, user steps, and/or instrumentation. Future innovations in this category should look to minimize these trade-offs.4. Signal
The signal coming from an LFA test strip is most often a change in optical intensity generated by the analyte-associated aggregation (or disaggregation, in a competitive format) of detection reagents on the test line. In the simplest case, such as when a coloured label is part of the detection reagent, the signal can be interpreted as colorimetric signals, directly by eye of the operator under ambient conditions. The earliest and most commonly used coloured labels are colloidal gold (typically 20–40 nm)1 and coloured latex28 labels. These simple, spherical labels do not directly bind analyte and must be conjugated to an analyte-binding reagent to form the detection reagent. Despite the ubiquity of these basic labels, many other types of labels,29 some of which generate signal by alternate mechanisms, have improved sensitivity in LFAs.Colorimetric labels
For example, various shapes of gold labels have been explored. While alternative gold label shapes—including nanorods, nanowires, nanoshells, and nanocubes—are commercially available from vendors such as Nanopartz (Loveland, CO USA) and Sono Nanotech (Halifax, Nova Scotia Canada), reports of their use as labels in LFAs are limited. However, a study comparing thermal contrast and optical contrast detection methods indicated that gold nanorods and nanoshells may improve sensitivity of LFAs compared to standard colloidal gold nanospheres.30 Likewise, gold nanorod labels were investigated during development of a competitive LFA for breast cancer testing. The developers characterized monodispersed gold nanorods with various aspect ratios, and tested methods of bio-functionalization (to create detection reagents) and immobilization (to create capture reagents).31 Separately, Yang et al. reported that a proof-of-concept IgG LFA with gold nanocage labels yielded 2.5-fold improved sensitivity over the same system with colloidal gold labels.32Multimeric gold labels can also improve assay sensitivity. For example, Shen and co-authors used dendrimers to assemble gold label multimers, which improved their assay sensitivity by 20-fold.33 Hu et al. used complementary oligonucleotides to create gold label multimers.34 Their analysis found that the colour intensity of the multimers was stronger than either monomer, which led to 2.5-fold improved sensitivity in their HIV nucleic acid LFA. Micron-sized labels, created from long (3–7 μm) silica nanorods decorated with smaller (10 nm) gold nanoparticles, improved sensitivity 50-fold in a rabbit IgG LFA.35 Similarly, a large multi-material label, composed of many small (16 nm) gold labels immobilized to individual chitosan-coated Fe2O3 nanoparticles (chosen to enhance reagent preparation through magnetic separation), improved sensitivity 3-fold for a competitive aflatoxin B2 assay in a qualitative LFA.36
A variety of other colorimetric labels have improved sensitivity compared to gold or latex labels. Dye-filled liposomes can improve sensitivity compared to gold and latex labels, due to the large amount of dye molecules they can contain, but they can be difficult to stabilize.5 NanoAct labels (Asahi Kasei, Miyazaki, Japan) are cellulose nanobeads that were reported to yield 10-fold higher sensitivity than gold or latex labels in an influenza LFA.37 The authors attribute the improved sensitivity to the larger surface area of the NanoAct labels (330 nm), and more intense dye colours available for impregnation into the cellulosic material. Carbon nanoparticle labels were reported to improve sensitivity 100-fold over gold label in a dengue antigen NS1 LFA.38 They provide high visual contrast to the analytical membrane39 and are inexpensive to produce, but they are only available from a limited number of vendors.5 Comparatively, carbon nanotube labels form high surface area test lines, which were reported to improve sensitivity 5-fold in an LFA with DNA oligo detection40 and 10-fold in an hCG LFA.41
Non-colorimetric labels
Yet other, non-colorimetric labels can generate signals other than a change in visible-by-eye optical intensity, such as fluorescent dyes, luminescent molecules, quantum dots, upconverting phosphors, and magnetic and SERS particles; these have been recently reviewed for improvement of sensitivity in LFAs.42 Advantages of these labels over colorimetric labels include enhanced signal range, and added objectivity and potential quantification provided by associated reader instrumentation; disadvantages include detection reagent complexity, and the cost and complexity of the reader.Commercial readers are available for detection of colorimetric and non-colorimetric labels. They are sold as stand-alone units, for example from Qiagen Lake Constance (Stockach, Germany), LRE (Munich, Germany), Hamamatsu (Tokyo, Japan), and Axxin (Fairfield, Australia);28 or integrated into the LFA itself, for example the BD Veritor43 and Clearblue tests.44 In addition, to reduce reader-associated test costs, particularly for low-resource settings, recent research has been conducted on the use of smartphones as LFA readers.45–47
Near-infrared (NIR) fluorescent dyes were reported to allow the sensitivity of IL-6 and C-reactive protein LFAs to match that of traditional ELISAs. Additionally, as these dyes absorb and emit fluorescence at wavelengths outside of typical biological matrices, these results were obtained for analytes spiked into 10% plasma matrices.48 Chen and Wu used a biotin-functionalized antibody, biotin-functionalized polylysine scaffolds loaded with multiple fluorescent dyes, and streptavidin linkages to create a detection reagent with tens to hundreds of fluorescent dye labels.49 These multivalent labels increased the assay sensitivity 100-fold compared to gold labels in a Cry1Ab toxin LFA. Silver nanoparticles were used to quench carbon dots in a zearalenone LFA that was ten times more sensitive than the LFA with silver nanoparticle labels alone.50
Up-converting phosphor labels were reported to yield greater than 10-fold sensitivity in a Schistosoma LFA compared to a traditional ELISA,51 and equivalent sensitivity to a liquid chromatography tandem mass spectrometry test in morphine and methamphetamine LFAs.52 Luminescent labels were reported to return a 10-fold improvement in a lysozyme LFA using a time-resolved reader;53 luminescent quantum dots have contributed two orders of magnitude improved sensitivity over colloidal gold in an LFA for aflatoxin in maize extract.54
Non-optical labels
Non-optical signals from LFAs become measurable with readers added as instrumentation. Thermal contrast imaging readers optically stimulate metallic (such as gold) nanoparticle labels and measure the generated heat in areas of metallic label aggregation, such as the test line of an LFA. Measuring thermal contrast increased sensitivity 32-fold in an FDA-approved cryptococcal antigen LFA30 and 8-fold over visual or colorimetric readers for influenza, malaria, or Clostridium difficile.55 Advantages for thermal contrast readers include compatibility with already available commercial tests. Additionally, thermal contrast imaging readers with low-cost components have been designed for application in low-resource settings,55 reducing cost for implementation. Our FlowDx group at Intellectual Ventures Laboratory has developed a field-rugged, stand-alone thermal reader with an on-board advanced algorithm for data analysis.56 Gold labels provide opportunities for detection of other signal modalities, including photoacoustic57 and X-ray58 imaging. The former technique was reported to yield a 100-fold improvement over colorimetric detection in a cryptococcal antigen LFA, while the latter technique has not been applied to the LFA format to our knowledge.Other non-optical signals become measurable with readers and alternative labels. For example, the use of surface-enhanced Raman scattering (SERS) improved the sensitivity of a staphylococcal enterotoxin B LFA by four orders of magnitude compared to a commercial point-of-care test for the same analyte.59 Similarly, gold nanoparticle labels coated with 4-aminothiophenol enabled SERS detection in a neomycin and quinolones antibiotics LFA, with sensitivity improved three orders of magnitude over visual detection.60 Another group reported SERS detection of gold nanostars in a multiplexed LFA that distinguished between non-structural protein 1 from Zika and dengue viruses, with 15-fold and 7-fold improved sensitivity for the respective targets.13,61
Magnetic labels can be detected with a magnetic reader, as demonstrated by Shi et al.62 with a Listeria monocytogenes LFA. These authors used superparamagnetic nanoparticle labels and a magnetic reader (Magna Biosciences, San Diego, CA) to generate a 10-fold improvement in assay sensitivity. A similar degree of sensitivity improvement was reported for a 3-amino-2-oxazolidinone (AOZ) LFA that used magnetic labels.63
Catalytic labels are most often used for the amplification of colorimetric signals (which we have categorized in the reaction/signal section below), but Lin et al. showed that platinum label-mediated catalysis of hydrogen peroxide can produce a measurable change in pressure inside gas-tight containers. They reported comparable sensitivity, having tested 45 clinical blood samples for myoglobin, for their pressure-based LFA and a chemiluminescent microparticle immunoassay (CMIA), which is the clinical gold standard.64
Limitations
The signal category covers signals that can be generated from a conventional LFA reaction, which is fundamentally limited to a proportional measure of the amount of analyte in a sample. As such, the sensitivity of a conventional LFA, even after enhancement by alternative labels and reader instrumentation, will never approach the sensitivity of a nucleic acid amplification test, for example, in which the signal is amplified exponentially. The possibility of enhancing the sensitivity of an LFA by several orders of magnitude is what has led to extensive research on integrating signal amplification reactions into the LFA format. We discuss these signal amplification approaches in the reaction/signal category below, although they have their own drawbacks due to their typically substantial deviations from the conventional LFA workflow. Those drawbacks keep the door open for innovations in label and reader technology that do not require deviations from the conventional LFA workflow, as they may be generically applied to current LFA technology. A high degree of compatibility with current LFA development and manufacturing practices are advantages for new labels and readers that may outweigh fundamental limitations on sensitivity improvements until more transformative, system-level innovations are commercially proven.5. Transformative innovations occur in areas of overlap for reaction, transport, and signal
5.1 Reaction/transport
The integration of assay reactions and reagent transport enables LFAs to serve as an automated, single-step platform for running what are multi-step assays in other formats, such as plate-based ELISAs. The sensitivity of LFAs can be improved by carefully balancing reaction kinetics and transport dynamics. For example, screening the assay reactions on analytical membranes with various fluid flow rates is typically an early optimization step in a standard LFA development process. However, more transformative innovations go beyond the standard optimization efforts to explore non-standard arrangements of reaction and transport.A non-standard order is sequential delivery, in which analyte is transported to the test line before (instead of with) the detection reagent. Sequential delivery requires a non-standard arrangement of materials to automate, because the detection reagent cannot simply be rehydrated by the added sample. However, it can be more sensitive than the premix delivery, as demonstrated by Liang et al. in a paper-based malaria assay.65 This demonstration included an analysis of the intensity across the test line (in the direction of flow) for premix compared to sequential delivery. Premix delivery led to a uniform distribution of labels on the test line. Sequential delivery led to a non-uniform distribution of labels on the test line, with more labels located upstream than the downstream. The authors make the argument, based on these distributions, that the kinetics of the reaction between capture reagents and the analyte/detection reagent complexes are slower than the kinetics of the reactions between the capture reagents and the analytes, and the captured analytes and the detection reagents.
These and other researchers in the Yager laboratory have demonstrated more non-standard reaction and transport dynamics in LFAs by moving from the strip format to a two-dimensional paper network (2DPN) format.66 For example, they modelled transport in 2DPNs67 and with sequential delivery,68 and produced 2DPNs with the sequential delivery of signal amplification reagents for 4-fold sensitivity improvement of a Plasmodium falciparum histidine-rich protein 2 (PfHRP2) LFA.69
Similarly, ChemBio Diagnostic Systems Inc. (Medford, NY) introduced their Dual Path Platform Technology, which enabled sequential and orthogonal delivery of sample and detection reagents to the test line in a T-shaped LFA format. They reported an 8-fold sensitivity improvement in a tuberculosis LFA compared to a standard strip format.70 Cho et al. reported an orthogonal delivery scheme to implement an enzymatic amplification step that yielded 30-fold improvement over a conventional Hepatitis B surface antigen LFA.71 However, this scheme was not automated and required an additional user step, which is a common limitation of signal amplification schemes, as discussed below.
Alternate formats exist for other test types as well, such as serological LFAs. For example, Sotnikov et al. performed a comparative theoretical and experimental study for two alternate LFA formats applied to the serodiagnosis of human pulmonary tuberculosis.72 Detection of low-affinity antibodies in human sera samples was 6-fold more sensitive for the alternate format predicted by their theoretical model.
Oh et al. refactored a competitive cortisol LFA such that capture reagents replaced the test and control lines to form “deletion” and “detection” zones, respectively. The deletion zone comprised cortisol–BSA conjugates, which captured detection reagents (anti-cortisol antibodies with gold labels) not bound to cortisol analyte. The detection zone comprised anti-mouse IgG antibodies, which captured detection reagents that flowed through the deletion zone due to cortisol analyte binding to the anti-cortisol antibodies. By tuning the concentration of cortisol–BSA conjugates in the deletion zone, and applying a ratiometric imaging method, the authors reported a 30-fold improvement over a conventional cortisol LFA.75
5.2 Transport/signal
The integration of reagent transport and signal generation in a standard LFA provides an automated mechanism for accumulating signal at a test line. However, the sensitivity of LFAs can be degraded by undesirable noise arising from the incomplete transport (i.e. non-specific binding or adsorption) of detection reagents. Detection reagents immobilized at and around the test line, but not by analyte, represent increased signal noise, which decreases the range of the actual signal. The reduction of sensitivity by assay interferents in human clinical sample matrices, such as blood and urine, due to the non-specific immobilization of detection reagents, has been reviewed extensively.76–79More recently, active blocking reagents were developed to further reduce noise, and thus improve sensitivity in LFAs. Commercial examples of active blocking reagents include MAB33 IgG1/IgG1 Poly from Roche Custom Biotech (Indianapolis, IN), TRU Block from Meridian Life Science, Inc (Memphis, TN), HAMA Blocking Reagent from Fitzgerald Industries International (Acton, MA), and the recently launched Morffi from BBI Solutions (Cardiff, UK).
Todd et al.80 tested HeteroBlock (Omega Biologicals, Bozeman, MT), heterophilic blocking reagent (HBR, Scantibodies Laboratory, Santee, CA), and immunoglobulin-inhibiting reagent (IIR, BioIVT, Baltimore, MD) for depletion of serum rheumatoid factor, using multiplex immunoassay platforms that detect cytokine and chemokine analytes. These tests showed a significant improvement in signal when the active blocking reagents were used. Generally, active blocking reagents have formulations recommended for LFAs or rapid tests, but the appropriate blocking reagent and blocking conditions will depend on the clinical sample type, sample processing methods, target analyte, and other LFA characteristics.
5.3 Reaction/signal
The integration of assay reactions and signal generation in a standard LFA is achieved in a detection reagent comprised of analyte-binding antibodies conjugated to a colorimetric label. However, a common strategy for improving sensitivity in LFAs is signal amplification, whereby alternative detection reagents support additional reactions that substantively enhance signal. Various approaches to implementing signal amplification in LFAs include the addition of enzymatic, chemical, aggregative, or proximity-dependent reactions.Strategies for improving reaction and signal performance separately can be applied to improving integrated reaction/signal performance as well. For example, improving the enzyme amplification reaction by forming the detection reagent from a biotin–antibody to streptavidin–poly-HRP reaction at the test line allowed a PfHRP2 LFA to show equivalent sensitivity to a commercial ELISA due to the fast, strong, and multivalent biotin–streptavidin interaction.84 The use of multivalent poly-HRP also contributed to the signal amplification. Likewise, the use of a viral nanoparticle as a multivalent detection reagent scaffold, to which multiple HRPs and multiple analyte-specific antibodies were conjugated, improved sensitivity 100-fold in a norovirus GI.1 (Norwalk) LFA40 and nearly 1000-fold in a bacteriophage MS2 LFA.85
While HRP is a popular choice for enzymatic signal amplification reactions, alkaline phosphatase (ALP) was used in an LFA for potato virus X to improve sensitivity 27-fold over a conventional LFA. The substrate nitro blue tetrazolium (5-bromo-4-chloro-3-indolyl phosphate) was added directly to the test strip over the test and control lines, where it converted to the insoluble, dark-violet precipitate diformazan.86 Gao et al. used platinum (coated less than ten atomic layers thick around gold nanoparticle labels) as a peroxidase-like catalyst to enhance sensitivity by 2 orders of magnitude in a human prostate-specific antigen LFA.87 Similarly, Loynachan et al. used porous platinum core−shell nanocatalyst labels and CN/DAB (4-chloro-1-naphthol/3,3′-diaminobenzidine, tetrahydrochloride) substrate in the presence of hydrogen peroxide to obtain a 20-fold improvement over commercial HIV p24 LFAs when run with human sera in the dipstick format.88
While many of these recent signal amplification approaches have used smaller substrate volumes for minimal disruption of the LFA format and workflow84,87,89 we are unaware of embodiments with dry reagent storage reported in the literature. However, Huang et al. constructed a device that provided long-term wet storage of reagents and automated delivery of those reagents to an influenza A/B LFA after sample introduction. Nonetheless, the additional complexity of signal amplification reactions is a barrier to the widespread application of these methods to LFA development.
All these examples, which clearly demonstrated sensitivity improvements from the use of silver stain on LFAs, nonetheless required freshly made silver solutions to be pipetted onto an LFA at specific times in specific volumes. Fridley et al. dried a commercial gold enhancement (salt-based) solution onto a malaria PfHRP2 LFA, and demonstrated a 3-fold improvement in sensitivity, but required a device that modified transport to achieve this modest result.95 As with the enzymatic signal amplification approach, reaction/signal modifications can trigger the need to deploy complementary transport modifications as well. We will return to this topic below, where we discuss interconnection of reaction, transport, and signal in LFA systems.
A reported alternative chemical approach is polymerization-based signal amplification (PBA), where the detection reagent contains a photoinitiator that triggers hydrogel polymerization in the presence of light and acrylate monomers; a pH indicator converts the presence of the hydrogel into a colorimetric signal. This signal amplification reaction was introduced as part of a paper-based PfHRP2 assay with a reported LOD of 0.56 μg mL−1.96 The primary advantage of the PfHRP2 paper-based PBA assay was speed, with a time-to-result of 2–3 minutes, as PfHRP2 LFAs with enzymatic amplification have better sensitivities but with a typical time-to-result of 20–30 minutes.92
For example, a two-gold-label, multi-level aggregation system resulted in a 100-fold sensitivity improvement in a troponin LFA over the standard, one-gold-label system.97 In the two-gold-label system, a smaller (10 nm) gold label was functionalized with anti-analyte antibodies and blocked with BSA, then dried downstream of a larger (40 nm) gold label, which was functionalized with anti-BSA antibodies. Upon application to the LFA of a sample containing analyte, the smaller gold labels were pulled down to the test line first, by the analyte. The larger gold labels were then pulled down to the test line second, by the BSA on the smaller gold labels. Two variations on this method were described by Wiriyachaiporn et al., in that the larger gold labels were pulled down with a biotin–anti-biotin antibody interaction and two analytes were detected on the same test line.98 Capture of a second analyte increased the overall density of labels at the test line. The authors reported an 8-fold improvement in sensitivity over a conventional, single-analyte LFA. Taranova et al. used three labels (functionalized with specific antibody, biotin, and streptavidin, respectively) in a similar format to increase the sensitivity 30-fold in a procalcitonin LFA.99 Razo et al. used two different label types, magnetic nanoparticles conjugated to biotinylated antibodies and streptavidin-coated gold nanoparticles, and a magnetic concentration step to obtain 32-fold sensitivity improvement in an LFA for potato virus X over a conventional LFA without the magnetic concentration or the gold-label-mediated aggregation.100
A classic form of proximity-dependent reaction is fluorescence (or Förster) resonance energy transfer (FRET), in which energy from a fluorescently excited donor molecule is transferred non-radiatively to an acceptor molecule in close vicinity.101 This energy transfer significantly reduces the fluorescence signal from the donor molecule, and can increase the fluorescence signal from the acceptor molecule unless it is a fluorescence quencher. Either signal can be monitored with a fluorescence reader.
Recently, Wang et al. reported the use of gold nanoparticle labels to quench fluorescein-labelled capture antibodies in a carcinoembryonic antigen (CEA) LFA.102 The sensitivity increased 100-fold from the standard, non-FRET LFA with gold labels only. However, as with any non-colorimetric signal, instrumentation was required to measure the fluorescent signal. Additionally, signal calibrations showed that while the accuracy of the LFA was quite good for real patient samples, the coefficients of variation were high (in some cases greater than 20%).
More complicated reaction mechanisms such as proximity ligation and proximity extension have been recently reviewed.103 In these assays, oligonucleotides are conjugated to both capture and detection reagents, such that hybridization between oligos or to a third oligo can occur when capture and detection reagents are both bound to analyte. Proximity-dependent hybridization is followed by ligation or extension, then nucleic acid amplification. This reaction mechanism is sufficiently complicated to challenge the typical LFA workflow, but a variant of the oligonucleotide ligation assay (OLA) was reported with sensitivity equivalent to a plate-based OLA, but also with 3-fold reduction in assay time, in an HIV LFA for the detection of viral point mutations.104
5.4 Reaction/transport/signal
The integration of chemical, physical, and optical processes within an LFA produces the major advantages of the technology, which revolve around simple, rapid, and robust operation. However, as we have explored here, this integrated functionality results in a highly complex set of trade-offs that affect overall system performance. As previously mentioned, the approaches we discuss in this review have improved sensitivity relative to baseline performance, but in the process may have increased the number or complexity of user steps, increased overall test time or cost, decreased test robustness, or introduced expensive instrumentation.To put it another way, we quickly see the distinctions blur between the reaction/transport/signal categories for the more impactful innovations, but also that any modification within one or two of these categories will trigger complex trade-offs across all categories. Even in conventional LFA development, navigating trade-offs in system performance typically requires a long process of manual optimization over many experimental parameters, including those related to reagents and reaction conditions, materials, and test signals.105,106
For example, in conventional LFA development, reagent optimization requires (at least) screening for high-affinity antibodies, selecting colorimetric labels, assessing the efficacy of attachment chemistry, cross-testing the capture and detection reagents, and evaluating the impact of blocking, spotting, drying, and/or rehydrating reagents in porous materials. Reaction condition optimization can include evaluating with sample matrices, and various buffers (for storage, reaction, wash/rinse, blocking, and/or running), surfactants, and other additives; testing the impact of various temperature, salt, and pH conditions; and testing porous materials for specific and non-specific binding. Material optimization typically involves choosing porous material types, width, thicknesses, lengths and overlaps; adding a backing strip and optionally a cover tape; and adjusting the mechanical design of a cassette. During these and potentially other development steps, validation (as far as possible) of optimal conditions at each step should be performed with statistically relevant measurements of signal-to-noise ratios over a relevant range of analyte concentrations, in a realistic sample matrix when possible, which is a huge amount of work.
One of the earliest LFA modelling approaches predicted the non-linearity of signal response at high target analyte concentration in an LFA sandwich format, and defined reactions and equations to allow for computational—rather than empirical—optimization.109 Recent advances in LFA modelling include work by Ragavendar et al. to optimize placement of the test line on the LFA strip and sample volume,110 and by Sotnikov et al. to optimize serological LFAs in which the target is an antibody, rather than an antigen.111 For the latter, the presence of excess target antibody in the blood sample adds an additional layer of complexity to the LFA assay; the authors recommend high concentration of reagents and serum dilutions in the range of 10–100-fold to improve sensitivity.
Zhan et al. recently modelled and experimentally validated the effect of gold nanoparticle sizes on detection of an LFA signal by a thermal contrast reader.112 The model and empirical data showed that larger particles increased antibody load due to their higher surface area, and gave stronger thermal responses. The use of 100 nm gold labels in their LFA led to a 256-fold increase in assay sensitivity compared to 30 nm gold labels.
Recently, we have developed and validated a mechanistic model in the FlowDx group at Intellectual Ventures Laboratory to broadly improve LFA sensitivity.108 The model accepts input parameters that include kinetic binding constants for reactions, porosity of transport materials, and viscosity of fluids, which allows for in silico optimization of signal generation and/or reagent use. Furthermore, the model can identify certain types of non-specific binding, which allows for at least partial in silico optimization of noise reduction. To date, we have validated the model for a malaria LFA under development.
Conclusions
We have used a categorical framework to identify state-of-the-art innovations in LFA sensitivity enhancement, group them according to shared strategies, and critically assess their limitations from a system-level perspective. We have also argued that a system-level perspective is necessary to overcome limitations blocking many of these innovations from transforming the current practice of LFA development. In anticipation of this outcome, we look forward to a new generation of LFAs that have been systematically improved to remain specific, stable, rapid, inexpensive, and user-friendly; and to acquire sensitivities that rival expensive, instrumented, laboratory-based assays.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our thanks to our colleagues in the FlowDx group at Intellectual Ventures Laboratory, as well as to our partners at Diagnostic Consulting Network, Carlsbad, CA, USA. Funding provided by The Global Good Fund I, LCC (http://www.globalgood.com).Notes and references
- M. Sajid, A.-N. N. N. Kawde and M. Daud, J. Saudi Chem. Soc., 2015, 19, 689–705 CrossRef.
- A. Merkoc, C. Parolo, A. De Escosura-mun, A. de la Escosura-Muñiz, A. Merkoçi, A. Merkoc, C. Parolo and A. de la Escosura-Muñiz, Biosens. Bioelectron., 2013, 40, 412–416 CrossRef PubMed.
- S. Qian and H. H. Bau, Anal. Biochem., 2004, 326, 211–224 CrossRef CAS PubMed.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210–2251 RSC.
- T. C. Tisone and B. O'Farrell, in Lateral Flow Immunoassay, ed. R. C. Wong and H. Y. Tse, Humana Press, New York, 2009, pp. 131–156 Search PubMed.
- H. Qi, Z. Zhong, H. X. Zhou, C. Y. Deng, H. Zhu, J. F. Li, X. L. Wang and F. R. Li, Int. J. Nanomed., 2011, 6, 3033–3039 CAS.
- P. L. A. M. Corstjens, R. K. Nyakundi, C. J. De Dood, T. M. Kariuki, E. A. Ochola, D. M. S. Karanja, P. N. M. Mwinzi and G. J. Van Dam, Parasites Vectors, 2015, 8, 241 CrossRef PubMed.
- M. Yang, M. Goolia, W. Xu, H. Bittner and A. Clavijo, Virol. J., 2013, 10, 125 CrossRef PubMed.
- E. M. Strauch, S. M. Bernard, D. La, A. J. Bohn, P. S. Lee, C. E. Anderson, T. Nieusma, K. K. Lee, A. B. Ward, P. Yager, D. H. Fuller, I. A. Wilson and D. Baker, Nat. Biotechnol., 2017, 35, 667–671 CrossRef CAS PubMed.
- S. Y. Doerflinger, J. Tabatabai, P. Schnitzler and C. Farah, mSphere, 2016, 1, 1–6 CrossRef PubMed.
- T. Schüling, A. Eilers, T. Scheper and J. Walter, AIMS Bioeng., 2018, 5, 78–102 Search PubMed.
- M. Jauset-Rubio, M. S. El-Shahawi, A. S. Bashammakh, A. O. Alyoubi and C. K. O'Sullivan, Aptamers Anal. Appl., 2018, pp. 273–299 Search PubMed.
- C. Parolo, M. Medina-Sánchez, A. De La Escosura-Muñiz and A. Merkoçi, Lab Chip, 2013, 13, 386–390 RSC.
- R. Tang, H. Yang, Y. Gong, Z. Liu, X. J. Li, T. Wen, Z. G. Qu, S. Zhang, Q. Mei and F. Xu, Sci. Rep., 2017, 7, 1360 CrossRef PubMed.
- L. Rivas, M. Medina-Sánchez, A. De La Escosura-Muñiz and A. Merkoçi, Lab Chip, 2014, 14, 4406–4414 RSC.
- X. Jiang and Z. H. Fan, Annu. Rev. Anal. Chem., 2016, 9, 203–222 CrossRef PubMed.
- T. Songjaroen, W. Dungchai, O. Chailapakul, C. S. Henry and W. Laiwattanapaisal, Lab Chip, 2012, 12, 3392–3398 RSC.
- B. D. Grant, C. A. Smith, P. E. Castle, M. E. Scheurer and R. R. Richards-Kortum, Vaccine, 2016, 34, 5656–5663 CrossRef PubMed.
- J. C. Wu, C. H. Chen, J. W. Fu and H. C. Yang, Sensors, 2014, 14, 4399–4415 CrossRef PubMed.
- L.-M. Fu, H.-H. Hou, P.-H. Chiu and R.-J. Yang, Electrophoresis, 2018, 39, 289–310 CrossRef CAS PubMed.
- N. Liu, D.-T. Phan and W. S. Lew, IEEE Trans. Biomed. Circuits Syst., 2017, 11, 1392–1399 Search PubMed.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2015, 87, 1009–1017 CrossRef CAS PubMed.
- S. Raja, V. R. R. Murty, V. Thivaharan, V. Rajasekar and V. Ramesh, Sci. Technol., 2012, 1, 7–16 Search PubMed.
- F. Mashayekhi, R. Y. T. Chiu, A. M. Le, F. C. Chao, B. M. Wu and D. T. Kamei, Anal. Bioanal. Chem., 2010, 398, 2955–2961 CrossRef CAS PubMed.
- R. Y. T. Chiu, A. V. Thach, C. M. Wu, B. M. Wu and D. T. Kamei, PLoS One, 2015, 10, e0142654 CrossRef PubMed.
- D. Y. Y. Pereira, R. Y. T. T. Y. T. Chiu, S. C. L. L. C. L. Zhang, B. M. M. Wu and D. T. T. Kamei, Anal. Chim. Acta, 2015, 882, 83–89 CrossRef CAS PubMed.
- R. Tang, H. Yang, J. R. Choi, Y. Gong, J. Hu, S. Feng, B. Pingguan-Murphy, Q. Mei and F. Xu, Talanta, 2016, 152, 269–276 CrossRef CAS PubMed.
- B. O'Farrell, in The Immunoassay Handbook, 2013, pp. 89–107 Search PubMed.
- J. Kim, M. A. A. Mohamed, K. Zagorovsky and W. C. W. Chan, Biomaterials, 2017, 146, 97–114 CrossRef CAS PubMed.
- Z. Qin, W. C. W. Chan, D. R. Boulware, T. Akkin, E. K. Butler and J. C. Bischof, Angew. Chem., Int. Ed., 2012, 51, 4358–4361 CrossRef CAS PubMed.
- M. Venkataramasubramani and L. Tang, in IFMBE Proceedings, 2009, vol. 24, pp. 199–202 Search PubMed.
- Y. Yang, M. Ozsoz and G. Liu, Microchim. Acta, 2017, 184, 2023–2029 CrossRef CAS PubMed.
- G. Shen, H. Xu, A. S. S. Gurung, Y. Yang and G. Liu, Anal. Sci., 2013, 29, 799–804 CrossRef CAS PubMed.
- J. Hu, L. Wang, F. Li, Y. L. Han, M. Lin, T. J. Lu and F. Xu, Lab Chip, 2013, 13, 4352–4357 RSC.
- H. Xu, J. Chen, J. Birrenkott, J. X. Zhao, S. Takalkar, K. Baryeh and G. Liu, Anal. Chem., 2014, 86, 7351–7359 CrossRef CAS PubMed.
- D. Tang, J. C. C. Sauceda, Z. Lin, S. Ott, E. Basova, I. Goryacheva, S. Biselli, J. Lin, R. Niessner and D. Knopp, Biosens. Bioelectron., 2009, 25, 514–518 CrossRef CAS PubMed.
- A. Sakurai, K. Takayama, N. Nomura, N. Yamamoto, Y. Sakoda, Y. Kobayashi, H. Kida and F. Shibasaki, J. Virol. Methods, 2014, 209, 62–68 CrossRef CAS PubMed.
- E. M. Linares, L. T. Kubota, J. Michaelis and S. Thalhammer, J. Immunol. Methods, 2012, 375, 264–270 CrossRef CAS PubMed.
- D. Quesada-González and A. Merkoçi, Biosens. Bioelectron., 2015, 73, 47–63 CrossRef PubMed.
- W. Qiu, H. Xu, S. Takalkar, A. S. Gurung, B. Liu, Y. Zheng, Z. Guo, M. Baloda, K. Baryeh and G. Liu, Biosens. Bioelectron., 2015, 64, 367–372 CrossRef CAS PubMed.
- W. Sun, X. Hu, J. Liu, Y. Zhang, J. Lu and L. Zeng, Biosci., Biotechnol., Biochem., 2017, 81, 1874–1882 CrossRef CAS PubMed.
- E. B. Bahadır and M. K. Sezgintürk, TrAC, Trends Anal. Chem., 2016, 82, 286–306 CrossRef.
- S. W. Ryu, J. H. Lee, J. Kim, M. A. Jang, J. H. Nam, M. S. Byoun and C. S. Lim, Br. J. Biomed. Sci., 2016, 73, 115–120 CrossRef PubMed.
- L. J. Kricka, eJIFCC, 2016, 27, 253–258 Search PubMed.
- D. J. You, T. S. Park and J. Y. Yoon, Biosens. Bioelectron., 2013, 40, 180–185 CrossRef CAS PubMed.
- E. Eltzov, S. Guttel, A. Low Yuen Kei, P. D. Sinawang, R. E. Ionescu and R. S. Marks, Electroanalysis, 2015, 27, 2116–2130 CrossRef CAS.
- J. T. T. Connelly, J. P. P. Rolland and G. M. M. Whitesides, Anal. Chem., 2015, 87, 7595–7601 CrossRef CAS PubMed.
- C. Swanson and A. D'Andrea, Clin. Chem., 2013, 59, 641–648 CrossRef CAS PubMed.
- C. Chen and J. Wu, Sensors, 2012, 12, 11684–11696 CrossRef CAS PubMed.
- S. Li, J. Wang, W. Sheng, W. Wen, Y. Gu and S. Wang, Microchim. Acta, 2018, 185, 388 CrossRef PubMed.
- P. L. A. M. Corstjens, L. Van Lieshout, M. Zuiderwijk, D. Kornelis, H. J. Tanke, A. M. Deelder and G. J. Van Dam, J. Clin. Microbiol., 2008, 46, 171–176 CrossRef CAS PubMed.
- Q. Hu, Q. Wei, P. Zhang, S. Li, L. Xue, R. Yang, C. Wang and L. Zhou, Analyst, 2018, 143, 4646–4654 RSC.
- A. S. Paterson, B. Raja, G. Garvey, A. Kolhatkar, A. E. V. Hagstrom, K. Kourentzi, T. R. Lee, R. C. Wilson, T. R. Lee and R. C. Willson, Anal. Chem., 2014, 86, 9481–9488 CrossRef CAS PubMed.
- M. Ren, H. H. Xu, X. Huang, M. Kuang, Y. Xiong, H. H. Xu, Y. Xu, H. Chen and A. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 14215–14222 CrossRef CAS PubMed.
- Y. Wang, Z. Qin, D. R. R. Boulware, B. S. S. Pritt, L. M. M. Sloan, I. J. Gonzalez, D. Bell, R. R. R. Rees-Channer, P. Chiodini, W. C. W. W. C. W. Chan, J. C. C. Bischof, I. J. González, D. Bell, R. R. R. Rees-Channer, P. Chiodini, W. C. W. W. C. W. Chan and J. C. C. Bischof, Anal. Chem., 2016, 88, 11774–11782 CrossRef CAS PubMed.
- D. Gasperino, M. P. Horning, K. P. F. Nichols, P. Rutschman, B. K. Wilson and O. E. Yildirim, US Pat., 9625385, 2017, p. 36 Search PubMed.
- Y. Zhao, Y. Huang, X. Zhao, J. F. McClelland and M. Lu, Nanoscale, 2016, 8, 19204–19210 RSC.
- S. Ahn, S. Jung, S. Lee, S. Ahn, S. Y. Jung and S. J. Lee, Molecules, 2013, 18, 5858–5890 CrossRef PubMed.
- J. Hwang, S. Lee and J. Choo, Nanoscale, 2016, 8, 11418–11425 RSC.
- Q. Shi, J. Huang, Y. Sun, R. Deng, M. Teng, Q. Li, Y. Yang, X. Hu, Z. Zhang and G. Zhang, Microchim. Acta, 2018, 185, 84 CrossRef PubMed.
- M. Sánchez-Purrà, M. Carré-Camps, H. De Puig, I. Bosch, L. Gehrke and K. Hamad-Schifferli, ACS Infect. Dis., 2017, 3, 767–776 CrossRef PubMed.
- L. Shi, F. Wu, Y. Wen, F. Zhao, J. Xiang and L. Ma, Anal. Bioanal. Chem., 2015, 407, 529–535 CrossRef CAS PubMed.
- L. Yan, L. Dou, T. Bu, Q. Huang, R. Wang, Q. Yang, L. Huang, J. Wang and D. Zhang, Food Chem., 2018, 261, 131–138 CrossRef CAS PubMed.
- B. Lin, Z. Guan, Y. Song, E. Song, Z. Lu, D. Liu, Y. An, Z. Zhu, L. Zhou and C. Yang, Lab Chip, 2018, 18, 965–970 RSC.
- T. Liang, R. Robinson, J. Houghtaling, G. Fridley, S. A. Ramsey and E. Fu, Anal. Chem., 2016, 88, 2311–2320 CrossRef CAS PubMed.
- E. Fu, B. Lutz, P. Kauffman and P. Yager, Lab Chip, 2010, 10, 918–920 RSC.
- E. L. Fu, S. A. Ramsey, P. C. Kauffman, B. Lutz and P. Yager, Microfluid. Nanofluid., 2011, 10, 29–35 CrossRef CAS PubMed.
- S. Dharmaraja, L. Lafleur, S. Byrnes, P. Kauffman, J. Buser, B. Toley, E. Fu, P. Yager and B. Lutz, in SPIE 8615, Microfluidics, BioMEMS, and Medical Microsystems XI, ed. H. Becker and B. L. Gray, International Society for Optics and Photonics, San Francisco, California, United States, 2013, vol. 8615, p. 86150X Search PubMed.
- E. Fu, T. Liang, P. Spicar-Mihalic, J. Houghtaling, S. Ramachandran and P. Yager, Anal. Chem., 2012, 84, 4574–4579 CrossRef CAS PubMed.
- J. Esfandiari, US Pat., 7189522, 2007, pp. 1–18 Search PubMed.
- J.-H. Cho, E.-H. Paek, I.-H. Cho and S.-H. Paek, Anal. Chem., 2005, 77, 4091–4097 CrossRef CAS PubMed.
- D. V. Sotnikov, A. V. Zherdev and B. B. Dzantiev, Sensors, 2018, 18(1), 36 Search PubMed.
- N. G. Welch, J. A. Scoble, B. W. Muir and P. J. Pigram, Biointerphases, 2017, 12, 02D301 CrossRef PubMed.
- E. A. Miller, S. Baniya, D. Osorio, Y. J. Al Maalouf and H. D. Sikes, Biosens. Bioelectron., 2018, 102, 456–463 CrossRef CAS PubMed.
- H. K. Oh, J. W. Kim, J. M. Kim and M. G. Kim, Analyst, 2018, 143, 3883–3889 RSC.
- J. Tate and G. Ward, Clin. Biochem. Rev., 2004, 25, 105–120 Search PubMed.
- L. J. Kricka, Clin. Chem., 1999, 45, 942–956 CAS.
- J. F. Emerson and K. K. Y. Lai, Lab. Med., 2013, 44, 69–73 CrossRef.
- H. de Puig, I. Bosch, L. Gehrke and K. Hamad-Schifferli, Trends Biotechnol., 2017, 35, 1169–1180 CrossRef CAS PubMed.
- D. J. Todd, N. Knowlton, M. Amato, M. B. Frank, P. H. Schur, E. S. Izmailova, R. Roubenoff, N. A. Shadick, M. E. Weinblatt, M. Centola and D. M. Lee, Arthritis Rheum., 2011, 63, 894–903 CrossRef CAS PubMed.
- Y. Fuchiwaki, K. Goya and M. Tanaka, Anal. Sci., 2018, 34, 57–63 CrossRef CAS PubMed.
- C. Zhang, Y. Zhang and S. Wang, J. Agric. Food Chem., 2006, 54, 2502–2507 CrossRef CAS PubMed.
- X. Mao, Y. Ma, A. Zhang, L. Zhang, L. Zeng and G. Liu, Anal. Chem., 2009, 81, 1660–1668 CrossRef CAS PubMed.
- B. D. Grant, C. A. Smith, K. Karvonen and R. Richards-Kortum, Anal. Chem., 2016, 88, 2553–2557 CrossRef CAS PubMed.
- J. Kim, M. Adhikari, S. Dhamane, A. E. V. Hagstrom, K. Kourentzi, U. Strych, R. C. Willson and J. C. Conrad, ACS Appl. Mater. Interfaces, 2015, 7, 2891–2898 CrossRef CAS PubMed.
- V. G. Panferov, I. V. Safenkova, Y. A. Varitsev, A. V. Zherdev and B. B. Dzantiev, Microchim. Acta, 2018, 185, 25 CrossRef PubMed.
- Z. Gao, H. Ye, D. Tang, J. Tao, S. Habibi, A. Minerick, D. Tang and X. Xia, Nano Lett., 2017, 17, 5572–5579 CrossRef CAS PubMed.
- C. N. Loynachan, M. R. Thomas, E. R. Gray, D. A. Richards, J. Kim, B. S. Miller, J. C. Brookes, S. Agarwal, V. Chudasama, R. A. McKendry and M. M. Stevens, ACS Nano, 2018, 12, 279–288 CrossRef CAS PubMed.
- A. E. V. Hagström, G. Garvey, A. S. Paterson, S. Dhamane, M. Adhikari, M. K. Estes, U. Strych, K. Kourentzi, R. L. Atmar and R. C. Willson, PLoS One, 2015, 10, e0126571 CrossRef PubMed.
- D.-J. Chiao, R.-H. Shyu, C.-S. Hu, H.-Y. Chiang and S.-S. Tang, J. Chromatogr., B, 2004, 809, 37–41 CrossRef CAS PubMed.
- M. O. Rodríguez, L. B. Covián, A. C. García and M. C. Blanco-lópez, Talanta, 2016, 148, 272–278 CrossRef PubMed.
- S. Lathwal, H. D. D. Sikes, S. Lathway, H. D. D. Sikes, S. Lathwal and H. D. D. Sikes, Lab Chip, 2016, 16, 1374–1382 RSC.
- V. G. Panferov, I. V. Safenkova, Y. A. Varitsev, N. V. Drenova, K. P. Kornev, A. V. Zherdev and B. B. Dzantiev, Talanta, 2016, 152, 521–530 CrossRef CAS PubMed.
- L. Anfossi, F. Di Nardo, C. Giovannoli, C. Passini and C. Baggiani, Anal. Bioanal. Chem., 2013, 405, 9859–9867 CrossRef CAS PubMed.
- G. E. Fridley, H. Le and P. Yager, Anal. Chem., 2014, 86, 6447–6453 CrossRef CAS PubMed.
- A. K. Badu-Tawiah, S. Lathwal, K. Kaastrup, M. Al-Sayah, D. C. Christodouleas, B. S. Smith, G. M. Whitesides and H. D. Sikes, Lab Chip, 2015, 15, 655–659 RSC.
- D. H. Choi, S. K. L. Lee, Y. K. Oh, B. W. Bae, S. D. Lee, S. Kim, Y.-B. Shin and M.-G. Kim, Biosens. Bioelectron., 2010, 25, 1999–2002 CrossRef CAS PubMed.
- N. Wiriyachaiporn, W. Maneeprakorn, C. Apiwat and T. Dharakul, Microchim. Acta, 2015, 182, 85–93 CrossRef CAS.
- N. A. Taranova, A. E. Urusov, E. G. Sadykhov, A. V. Zherdev and B. B. Dzantiev, Microchim. Acta, 2017, 184, 4189–4195 CrossRef CAS.
- S. C. Razo, V. G. Panferov, I. V. Safenkova, Y. A. Varitsev, A. V. Zherdev and B. B. Dzantiev, Anal. Chim. Acta, 2018, 1007, 50–60 CrossRef CAS PubMed.
- G. Bunt and F. S. Wouters, Biophys. Rev., 2017, 9, 119–129 CrossRef PubMed.
- J. Wang, F. Cao, S. He, Y. Xia, X. Liu, W. Jiang, Y. Yu, H. Zhang and W. Chen, Talanta, 2018, 176, 444–449 CrossRef CAS PubMed.
- C. Greenwood, D. Ruff, S. Kirvell, G. Johnson, H. S. Dhillon and S. A. Bustin, Biomol. Detect. Quantif., 2015, 4, 10–16 CrossRef CAS PubMed.
- N. Panpradist, I. A. Beck, M. H. Chung, J. N. Kiarie, L. M. Frenkel and B. R. Lutz, PLoS One, 2016, 11, e0145962 CrossRef PubMed.
- H. V. Hsieh, J. L. Dantzler, B. H. Weigl, H. V. Hsieh, J. L. Dantzler and B. H. Weigl, Diagnostics, 2017, 7, 29 CrossRef CAS PubMed.
- A. V. Zherdev and B. B. Dzantiev, Rapid Test - Adv. Des. Format Diagnostic Appl., 2017, DOI:10.5772/intechopen.76926.
- D. Gasperino, T. Huynh and B. Weigl, Proc. 21st Int. Conf. Miniaturized Syst. Chem. Life Sci., Savannah, GA, USA, 22–26 Oct. 2017, 2017, pp. 391–392 Search PubMed.
- D. Gasperino, T. Baughman, H. V. Hsieh, D. Bell and B. H. Weigl, Annu. Rev. Anal. Chem., 2018, 11, 219–244 CrossRef PubMed.
- S. Qian and H. H. Bau, Anal. Biochem., 2003, 322, 89–98 CrossRef CAS PubMed.
- M. S. Ragavendar and C. M. Anmol, in Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society, EMBS, 2012, vol. 20112, pp. 2408–2411 Search PubMed.
- D. V. Sotnikov, A. V. Zherdev and B. B. Dzantiev, Anal. Chem., 2017, 89, 4419–4427 CrossRef CAS PubMed.
- L. Zhan, S. Z. Z. Guo, F. Song, Y. Gong, F. Xu, D. R. R. Boulware, M. C. C. McAlpine, W. C. W. C. W. Chan and J. C. C. Bischof, Nano Lett., 2017, 17, 7207–7212 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |