
A ruthenium-catalyzed free amine directed (5+1) annulation of anilines with olefins: diverse synthesis of phenanthridine derivatives†
Deepan
Chowdhury
,
Suman
Dana
,
Anup
Mandal
and
Mahiuddin
Baidya
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, Tamil Nadu, India. E-mail: mbaidya@iitm.ac.in
First published on 9th September 2019
Abstract
A ruthenium(II)-catalyzed cross-ring (5+1) annulation between 2-aminobiphenyls and activated olefins is disclosed for succinct synthesis of valuable phenanthridine scaffolds. The protocol avails a common organic functional group, free amine, as a directing group and represents a unique combination of C–H activation/annulation/C–C bond cleavage cascade that bodes well in the production of bioactive alkaloids including trisphaeridine and bicolorine.
Transition-metal-catalyzed annulation reactions exploiting ubiquitous and otherwise inactive C–H bonds represent an important synthetic strategy to fabricate polycyclic molecular frameworks.1,2 Over the years, chemists have compiled a ruthenium-catalyzed reaction compendium that consists of a series of (4+2),3a–e (3+2),3f–h (2+2+2),3i and (4+1)3j annulations, forging diverse carbocycles and heterocycles. Despite these achievements, to date, ruthenium-catalyzed (5+1) annulation has remained largely underdeveloped.4 In these annulation reactions, directing groups play fundamental roles in facilitating the C–H bond activation process and mitigate the problem of regioselectivity. Common organic functional groups like carboxylic acid, ester, amide, ketone, etc. are often employed as directing groups.5 However, the free amine group (NH2), one of the most valuable and widely abundant functionalities, has largely been ignored in ruthenium-catalyzed directed C–H bond activation reactions,6 probably owing to the challenges associated with its strong coordinating ability with metal catalysts along with the superior nucleophilic reactivity that result in pivotal issues of catalyst deactivation and unwarranted side reactions.6c,9b Thus, there is ample scope in the free amine directed ruthenium(II)-catalyzed regioselective C–H bond activation/annulation manifold and importantly, it could potentially lead to high-value N-heterocycles when the amine directing group becomes the critical component of the ring structure.
Phenanthridine and benzophenanthridine alkaloids signify an important class of organic molecules with promising biological activities.7,8 Some of the important natural products are presented in Fig. 1. The biological activities of such alkaloids range from anti-cancer to anti-fungal, and anti-bacterial, to name a few. Consequently, devising novel synthetic strategies towards such molecular frameworks is highly desirable.8 Arguably, a C–H bond activation based (5+1) cross-ring-annulation (CRA) reaction of biaryl-2-amines would be a succinct route to access these scaffolds (Scheme 1).
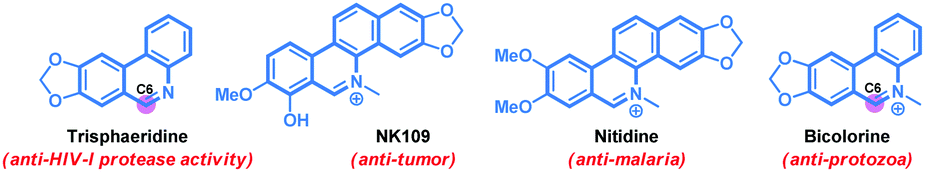 | ||
| Fig. 1 Biologically important phenanthridine alkaloids. | ||
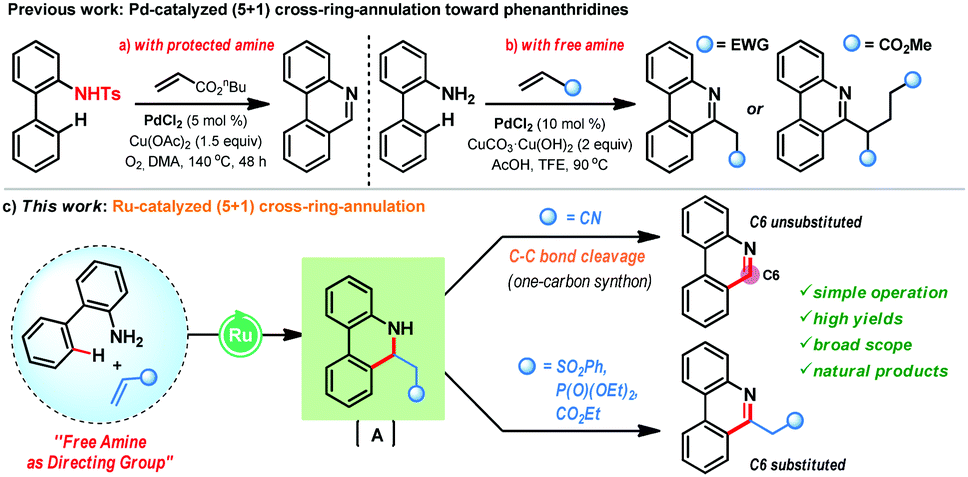 | ||
| Scheme 1 Ru(II)-catalyzed free amine directed cross-ring (5+1) annulation towards phenanthridine alkaloids. | ||
Furthermore, the majority of the naturally occurring phenanthridine alkaloids do not possess any substitution at the C6-position and hence, challenges lie in the strategic design of a suitable one-carbon synthon for the CRA reaction. In 2012, the Li group reported an intriguing Pd-catalyzed (5+1) CRA reaction of biaryl-2-amines with activated alkenes (butyl acrylate) that features the pivotal C–C bond cleavage to offer C6-unsubstituted phenanthridines in high yields (Scheme 1a).9a In this case, the use of N-protected biaryl-2-amines was necessary as N-unprotected biaryl-2-amines gave poor yields. In parallel, the Zhang group also reported (5+1) CRA reaction of biaryl-2-amines with alkenes under Pd-catalysis in trifluoroethanol (Scheme 1b).9b This reaction is effective with unprotected amines, however, they did not observe any C–C bond cleavage phenomenon and, in the case of acrylate coupling partner, a second Michael addition was proposed for the aromatization step en route to C6-substituted phenanthridines. Currently, such a CRA reaction manifold for the production of phenanthridines is unknown with Ru-catalysis and herein, we disclose the first example of free amine directed (5+1) CRA reaction of biaryl-2-amines with activated alkenes under Ru-catalysis (Scheme 1c). When acrylonitrile is used as a coupling partner, it acts as a C1-synthone and delivers C6-unsubstituted phenanthridines after the C–C bond cleavage. In contrast, other activated olefins, such as vinyl sulfone, vinyl phosphate, and acrylate, furnished C6-substituted phenanthridines in very high yields.
We commenced our investigations following the model reaction of 2-aminobiphenyl 1a with acrylonitrile 2a (Table 1). The choice of acrylonitrile as an olefin coupling partner is intriguing as initially formed dihydrophenanthridine intermediate A bearing a cyanomethyl (–CH2CN) functionality may experience a C–C bond cleavage phenomenon either through a radical pathway or a coordination assisted base promoted elimination mechanism to validate domino C–H activation based (5+1) annulation en route to the C6-unsubstituted phenanthridine scaffold (Scheme 1c). Accordingly, when we treated 1a and 2a in the presence of [Ru(p-cymene)Cl2]2 (5 mol%), Cu(OAc)2·H2O (2 equiv.), AgSbF6 (20 mol%), and CH3CO2H (1.2 equiv.) in THF solvent, we were delighted to find the desired 6-unsubstituted phenanthridine product 3a in 52% yield (Table 1, entry 1). Switching the reaction solvent to DCE, dioxane, and DME furnished inferior results (entries 2–4). Screening of the acid additives revealed mesitoic acid as the best choice, delivering the desired product 3a in 72% isolated yield (entry 5). Change of the reaction solvent from THF to higher boiling point 2-methyl tetrahydrofuran (2-Me-THF) gave only 37% yield of 3a (entry 6). Further tuning of the reaction conditions, such as use of 1-AdCO2H acid (entry 7), increasing or decreasing of reaction temperature (entry 8), utilization of AgBF4 additive and use of CuO oxidant (entry 9) had detrimental effects. Control experiments revealed that all the components were essential for the success of the reaction (entries 10 and 11). Yield also decreased in the presence of excess water in the reaction medium (entry 12). Other transition metals like Pd, Rh, and Ir based catalysts were ineffective under standard reaction conditions, highlighting the uniqueness of ruthenium in this protocol (entry 13).
|
|
||||
|---|---|---|---|---|
| Entry | Acid additive | Solvent | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.36 mmol), solvent (4.2 mL) for 48 h under an argon atmosphere. b Isolated yields. c AgBF4 (20 mol%) was used as an additive. d CuO (2 equiv.) was used as an oxidant. e Reaction without [Ru(p-cymene)Cl2]2 catalyst or Cu(OAc)2·H2O oxidant or AgSbF6 additive. f Reaction without MesCO2H (mesitoic acid) additive. g 2 equiv. of water was added. h With Pd(OAc)2. i With (Cp*RhCl2)2 catalyst. j With (Cp*IrCl2)2 catalyst. | ||||
| 1 | AcOH | THF | 80 | 52 |
| 2 | AcOH | DCE | 80 | 32 |
| 3 | AcOH | DME | 80 | 38 |
| 4 | AcOH | Dioxane | 80 | 46 |
| 5 | MesCO2H | THF | 80 | 72 |
| 6 | MesCO2H | 2-Me-THF | 80 | 37 |
| 7 | 1-AdCO2H | THF | 80 | 12 |
| 8 | MesCO2H | THF | 100/60 | 62/0 |
| 9 | MesCO2H | THF | 80 | 16c/11d |
| 10e | MesCO2H | THF | 80 | — |
| 11f | — | THF | 80 | <5 |
| 12g | MesCO2H | THF | 80 | 59 |
| 13 | MesCO2H | THF | 80 | 16h/0i/0j |
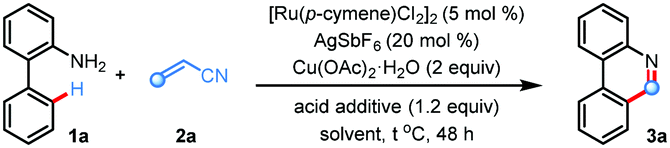
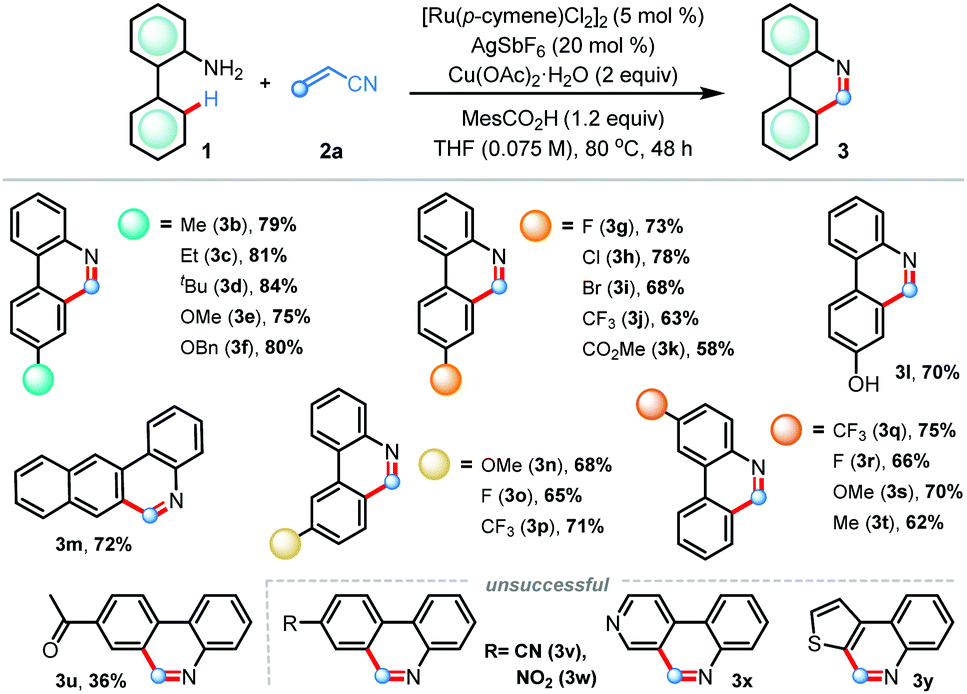
Having acquired the optimal conditions, we sought to explore the scope of the (5+1) annulation reaction varying the electronic and steric nature in the arene ring (Table 2). The presence of electron-releasing groups such as alkyl (3b–d) and alkoxy (3e–f) at the para-position gave desired products in uniformly high yields (75–84%). Substrates bearing electron-withdrawing groups, for example halogens (3g–i), trifluoromethyl (3j), and ester (3k) were smoothly reacted to produce C6-unsubstituted phenanthridines in good yields.
|
Pleasingly, coordinating free-hydroxyl groups did not hamper the reaction, furnishing compound 3l in 70% yield. When unsymmetrical meta-substitution was considered, annulations proceeded selectively at the sterically less hindered site to forge products 3n–p in good yields. The protocol also worked efficiently with the 2-naphthyl derivative, generating important fused polyaromatic heterocycle benzo[j]phenanthridine (3m) in 72% yield. The effect of substituents in the aniline ring was also examined; a host of electron-rich and electron-deficient anilines were effective for this reaction, delivering 3q–t in 62–75% isolated yields. Synthetically useful yield was also obtained with a sensitive ketone functionality (3u). Under the standard conditions, annulations did not take place with substrates having strongly electron withdrawing cyano (3v) and nitro (3w) groups as well as with anilines derived from heterocycles (3x–y).
After successful implementation of our hypothesis, we questioned whether other activated olefinic coupling partners would participate in this ruthenium(II)-catalyzed CRA reaction (Table 3).10 When phenyl vinyl sulfone 2b was reacted with 2-aminobiphenyl 1a under the conditions established with acrylonitrile 2a, the desired (5+1) annulation reaction did not take place effectively with the recovery of the starting materials, indicating that a revision of the reaction conditions was necessary. Delightfully, the same reaction proceeded smoothly when the reaction solvent was changed to dioxane; however, we did not observe the concomitant C–C bond cleavage in this case and 6-substituted phenanthridine derivative 4a was isolated in 75% yield. Other substituted 2-arylanilines also rendered products 4b–g in good to high yields (57–84%). Similarly, reactions with diethyl vinylphosphonate 2c were fruitful to offer alkyl phosphonate hinged phenanthridines 4h and 4i in 70% and 75% yields, respectively (Table 3). These findings reinforce the uniqueness of acrylonitrile in (5+1) cross-ring-annulation (CRA) for exclusive access of 6-unsubstituted phenanthridines.
| a Reaction conditions: 1 (0.3 mmol), 2b or 2c (0.36 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), Cu(OAc)2·H2O (2 equiv.), MesCO2H (1.2 equiv.), AgSbF6 (20 mol%), dioxane (4.2 mL) at 80 °C for 48 h under argon. |
|---|

|
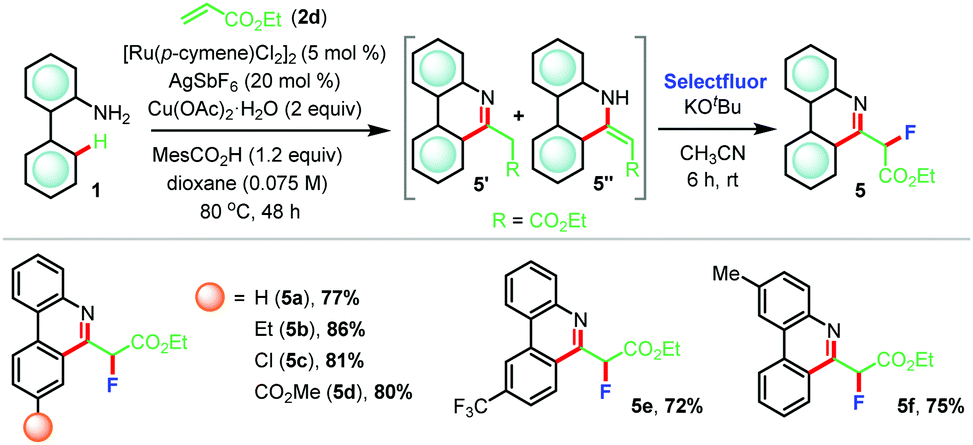
Furthermore, reaction of 2-aminobiphenyl 1a with ethyl acrylate 2d afforded a mixture of two products which were inseparable by column chromatography (Table 4). 1H-NMR analysis implied the presence of desired (5+1) annulated product 5a′ along with its tautomer 5a′′. At this juncture, we posited to use a suitable electrophile to functionalize the acidic C–H bond adjacent to the carboxylate group (R = CO2Et) that might compel the formation of a phenanthridine moiety. We focused on electrophilic fluorination since fluorinated analogues of phenanthridine might exert interesting pharmaceutical properties. Consequently, the crude reaction mixture thus obtained from the (5+1) annulation step was exposed to Selectfluor in the presence of KOtBu in anhydrous acetonitrile at room temperature and, to our satisfaction, the desired product 5a was formed in 77% yield (Table 4). Following the same sequence, fluorinated analogues 5b–f were prepared in very high yields (72–86%).
| a Reaction conditions: 1 (0.3 mmol), 2d (0.36 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), Cu(OAc)2·H2O (2 equiv.), MesCO2H (1.2 equiv.), AgSbF6 (20 mol%), dioxane (4.2 mL) at 80 °C for 48 h under argon. Then, KOtBu (1.2 equiv.) and Selectfluor (1.2 equiv.) were used in dry acetonitrile at room temperature for 6 h. |
|---|
|
The synthetic utility of this protocol was highlighted in the preparation of phenanthridine-based natural products. For example, trisphaeridine that displays excellent antiproliferative effects on both human and mouse cells was rapidly prepared from the reaction of 2-phenylaniline116 with acrylonitrile 2a under the standard conditions in 76% yield (Scheme 2). Subsequent methylation gave the natural product bicolorine 8 in 88% yield and synthesis of dihydrobicolorine and N-methyl crisanidine from bicolorine is a known process (Scheme 2).12a,b
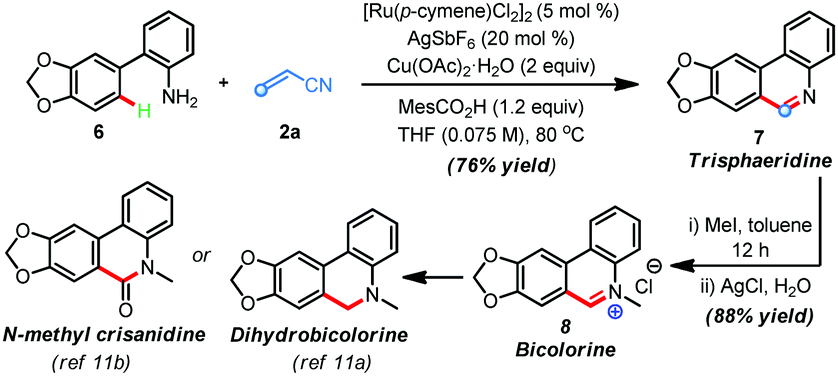 | ||
| Scheme 2 Synthesis of bioactive alkaloids trisphaeridine and bicolorine. | ||
To gain mechanistic insights, we performed a few control experiments. No significant deuterium incorporation was observed when bench-mark reaction of 1a and 2a was performed in the presence of excess D2O, approving an irreversible C–H metalation step (Scheme 3a). Kinetic isotope effect (KIE) studies through independent parallel (kH/kD = 2.80) and competitive (pH/pD = 2.45) experiments suggested that the C–H metalation could be the rate-determining step (Scheme 3b). Furthermore, the reaction was ineffective in the presence of TEMPO, but 3a was isolated in 52% yield in the presence of BHT, implying that TEMPO might hamper the Ru-catalysis and the involvement of a radical pathway is rather unlikely (Scheme 3c). While the exact reaction mechanism must await further investigations, we believe, in contrast to other alkenes, that the unique C–C bond cleavage in the case of acrylonitrile is facilitated through the coordination of the cationic Ru-catalyst followed by carboxylate assisted deprotonation as shown in Scheme 3d.9c,d,13
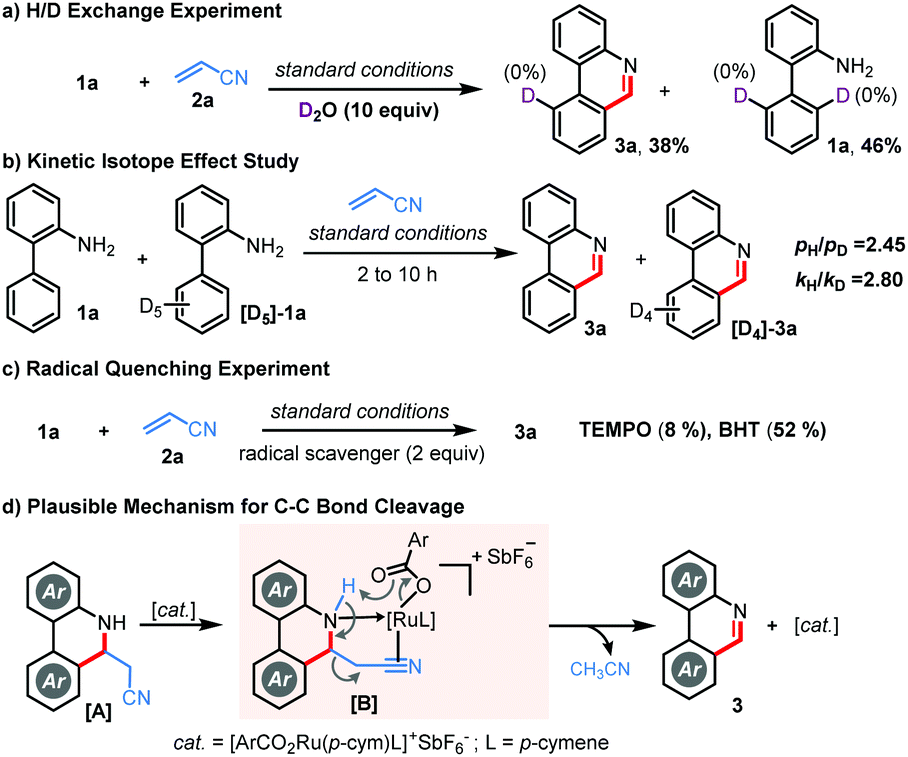 | ||
| Scheme 3 Control experiments. | ||
In conclusion, an efficient (5+1) cross-ring-annulation (CRA) reaction using readily available 2-aminobiphenyls and activated olefins under common functional group free amine assisted ruthenium(II) catalysis has been accomplished to prepare a library of high-value functionalized phenanthridines in very high yields. Identification of acrylonitrile as a one-carbon synthon was a critical parameter for achieving the concomitant C–C bond cleavage, furnishing 6-unsubstituted phenanthridines in a succinct manner. Also, the applications of this methodology in syntheses of bioactive alkaloids like trisphaeridine and bicolorine add to the fruitfulness of the protocol. Further applications of Ru(II)-catalyzed annulation are currently ongoing in our laboratory.
We gratefully acknowledge SERB-DST for financial support. M. B. also thanks IIT-Madras for the Institute Research Development Award (IRDA). We also thank the Department of Chemistry, IIT-Madras for instrumental facilities.
Conflicts of interest
There are no conflicts to declare.References
- For selected reviews: (a) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed; (b) D. M. D’Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095 RSC; (c) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (e) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (f) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Rev., 2015, 115, 5301 CrossRef CAS PubMed; (g) Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872 RSC; (h) M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000 CrossRef PubMed; (i) S. Prakash, R. Kuppusamy and C. H. Cheng, ChemCatChem, 2018, 10, 683 CrossRef CAS.
- (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (b) G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS; (c) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed; (d) S. Reddy Chidipudi, I. Khan and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 12115 CrossRef CAS PubMed; (e) C. Y. Wu, M. Hu, Y. Liu, R. J. Song, Y. Lei, B. X. Tang, R. J. Li and J. H. Li, Chem. Commun., 2012, 48, 3197 RSC; (f) C. Kornhaaß, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190 CrossRef PubMed; (g) M. Deponti, S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Org. Biomol. Chem., 2013, 11, 142 RSC; (h) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513 CrossRef CAS; (i) R. Mei, S. K. Zhang and L. Ackermann, Synlett, 2017, 1715 CAS.
- Selected examples for Ru(II)-catalyzed (4+2) annulation: (a) B. Li, H. Feng, S. Xu and B. Wang, Chem. – Eur. J., 2011, 17, 12573 CrossRef CAS PubMed; (b) L. Ackermann, L. Wang and A. V. Lygin, Chem. Sci., 2012, 3, 177 RSC; (c) J. D. Dooley, S. Reddy Chidipudi and H. W. Lam, J. Am. Chem. Soc., 2013, 135, 10829 CrossRef CAS PubMed; (d) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032 CrossRef CAS PubMed; (e) S. Nakanowatari and L. Ackermann, Chem. Eur. J., 2014, 20, 5409 CrossRef CAS PubMed ; For (3+2): ; (f) J. Zhang, A. Ugrinov and P. Zhao, Angew. Chem., Int. Ed., 2013, 52, 6681 CrossRef CAS PubMed; (g) Y. Q. Zhu and L. Dong, J. Org. Chem., 2015, 80, 9973 CrossRef CAS PubMed; (h) Y. Zhao, Z. He, S. Li, J. Tang, G. Gao, J. Lan and J. You, Chem. Commun., 2016, 52, 4613 RSC ; For (2+2+2): ; (i) H. Chen, L. Ouyang, J. Liu, W.-J. Shi, G. Chen and L. Zheng, J. Org. Chem., 2019 DOI:10.1021/acs.joc.9b00926 ; For (4+1): ; (j) X. Wu, B. Wang, S. Zhou, Y. Zhou and H. Liu, ACS Catal., 2017, 7, 2494 CrossRef CAS.
- For Ru-catalyzed [5+1] annulation: (a) P. Chen, J. Nan, Y. Hu, Q. Ma and Y. Ma, Org. Lett., 2019, 21, 4812 CrossRef CAS; (b) S. Reddy Chidipudi, M. D. Wieczysty, I. Khan and H. W. Lam, Org. Lett., 2013, 15, 570 CrossRef CAS PubMed ; Other examples of transition-metal-catalyzed [5+1] annulations: ; (c) D. J. Burns and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 9931 CrossRef CAS PubMed; (d) N. Casanova, A. Seoane, J. L. Mascareñas and M. Gulías, Angew. Chem., Int. Ed., 2015, 54, 2374 CrossRef CAS PubMed; (e) A. Cajaraville, J. Suárez, S. López, J. A. Varela and C. Saá, Chem. Commun., 2015, 51, 15157 RSC; (f) R. Kuppusamy, K. Muralirajan and C. H. Cheng, ACS Catal., 2016, 6, 3909 CrossRef CAS.
- (a) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (b) M. P. Drapeau and L. J. Gooßen, Chem. – Eur. J., 2016, 22, 18654 CrossRef PubMed; (c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- Selected examples on free-amine directed C–H bond functionalization; with Ru-catalysts: (a) C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2013, 15, 3990 CrossRef CAS PubMed; (b) P. Villuendas and E. P. Urriolabeitia, J. Org. Chem., 2013, 78, 5254–5263 CrossRef CAS; (c) C. Suzuki, K. Morimoto, K. Hirano, T. Satoh and M. Miura, Adv. Synth. Catal., 2014, 356, 1521 CrossRef CAS ; With Pd-catalysts: ; (d) Z. Zuo, J. Liu, J. Nan, L. Fan, W. Sun, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 15385 CrossRef CAS PubMed; (e) G. Jiang, S. Wang, J. Zhang, J. Yu, Z. Zhang and F. Ji, Adv. Synth. Catal., 2019, 361, 1798 CrossRef CAS ; With Rh-catalysts: ; (f) Q. Jiang, D. Duan-Mu, W. Zhong, H. Chen and H. Yan, Chem. – Eur. J., 2013, 19, 1903 CrossRef CAS PubMed; (g) P. Bai, X. F. Huang, G. D. Xu and Z. Z. Huang, Org. Lett., 2016, 18, 3058 CrossRef CAS PubMed . For Ir-catalysts: ; (h) K. Yan, Y. Lin, Y. Kong, B. Li and B. Wang, Adv. Synth. Catal., 2019, 361, 1570 CrossRef CAS.
- (a) M. Suffnes and G. A. Cordell, The Alkaloids, Academic, New York, 1985, vol. 25, p. 178 Search PubMed; (b) T. Ishikawa, Med. Res. Rev., 2001, 21, 61 CrossRef CAS; (c) T. Nakanishi, M. Suzuki, A. Saimoto and T. Kabasawa, J. Nat. Prod., 1999, 62, 864 CrossRef CAS PubMed; (d) D. Castillo, M. Sauvain, M. Rivaud and V. Jullian, Planta Med., 2014, 80, 902 CrossRef CAS PubMed; (e) I. Zupkó, B. Réthy, J. Hohmann, J. Molnár, I. Ocsovszki and G. Falkay, In Vivo, 2009, 23, 41 Search PubMed; (f) G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987 CrossRef CAS PubMed; (g) J. Bouquet, M. Rivaud, S. Chevalley, E. Deharo, V. Jullian and A. Valentin, Malar. J., 2012, 11, 1 CrossRef PubMed; (h) Y. Ding, D. Qu, K. M. Zhang, X. X. Cang, Z. N. Kou, W. Xiao and J. B. Zhu, J. Asian Nat. Prod. Res., 2017, 19, 53 CrossRef CAS PubMed.
- (a) L. M. Tumir, M. R. Stojković and I. Piantanida, Beilstein J. Org. Chem., 2014, 10, 2930 CrossRef PubMed; (b) V. Bisai, M. K. S. Shaheeda, A. Gupta and A. Bisai, Asian J. Org. Chem., 2019, 8, 946 CrossRef CAS.
- (a) Y. Y. Liu, R. J. Song, C. Y. Wu, L. Bin Gong, M. Hu, Z. Q. Wang, Y. X. Xie and J. H. Li, Adv. Synth. Catal., 2012, 354, 347 CrossRef CAS; (b) Z. Liang, L. Ju, Y. Xie, L. Huang and Y. Zhang, Chem. – Eur. J., 2012, 18, 15816 CrossRef CAS PubMed; (c) X. Bao, W. Yao, Q. Zhu and Y. Xu, Eur. J. Org. Chem., 2014, 7443 CrossRef CAS . For a C–C bond cleavage of phthalimide, see: ; (d) Y. C. Yuan, R. Kamaraj, C. Bruneau, T. Labasque, T. Roisnel and R. Gramage-Doria, Org. Lett., 2017, 19, 6404 CrossRef CAS PubMed.
- In the current scenario, annulation did not take place with unactivated olefins such as 1-octene and isopropenylbenzene.
- W. Guo, S. Li, L. Tang, M. Li, L. Wen and C. Chen, Org. Lett., 2015, 17, 1232 CrossRef CAS PubMed.
- (a) W. L. Chen, C. Y. Chen, Y. F. Chen and J. C. Hsieh, Org. Lett., 2015, 17, 1613 CrossRef CAS PubMed; (b) G. Wang, W. Hu, Z. Hu, Y. Zhang, W. Yao, L. Li, Z. Fu and W. Huang, Green Chem., 2018, 20, 3302 RSC.
- (a) M. Chenna Reddy and M. Jeganmohan, Chem. Commun., 2015, 51, 10738 RSC; (b) For a plausible reaction mechanism, see ESI† (Fig. S1).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05717j |
| This journal is © The Royal Society of Chemistry 2019 |