
Highly active dinuclear cobalt complexes for solvent-free cycloaddition of CO2 to epoxides at ambient pressure†
Zafar A. K.
Khattak
 ab,
Hussein A.
Younus
*ac,
Nazir
Ahmad
ad,
Habib
Ullah
ab,
Suleman
Suleman
ab,
Md. Shahadat
Hossain
abe,
Mirella
Elkadi
g and
Francis
Verpoort
*abfg
ab,
Hussein A.
Younus
*ac,
Nazir
Ahmad
ad,
Habib
Ullah
ab,
Suleman
Suleman
ab,
Md. Shahadat
Hossain
abe,
Mirella
Elkadi
g and
Francis
Verpoort
*abfg
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: francis.verpoort@ghent.ac.kr
bSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China
cDepartment of Chemistry, Faculty of Science, Fayoum University, Fayoum 63514, Egypt. E-mail: hay00@fayoum.edu.eg
dDepartment of Chemistry, G. C. University Lahore, 54000, Pakistan
eDepartment of Chemistry, Faculty of Science, Cumilla University, Cumilla 3506, Bangladesh
fNational Research Tomsk Polytechnic University, Lenin Avenue 30, 634050 Tomsk, Russian Federation
gCollege of Arts and Sciences, Khalifa University of Science and Technology, PO Box 127788, Abu Dhabi, United Arab Emirates
First published on 19th June 2019
Abstract
Dinuclear Co-based catalysts are used for the coupling reaction of epoxides and CO2 in the presence of a cocatalyst. The easily recyclable catalysts efficiently complete the coupling of CO2 with various epoxides into industrially important cyclic carbonates at low catalyst loading and displayed high catalytic activity under relatively low CO2 pressure and solvent-free conditions. The maximum TON (168![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600) and TOF (3333 h−1) obtained in this work are the highest among the reported Co-complexes.
600) and TOF (3333 h−1) obtained in this work are the highest among the reported Co-complexes.
One of the world's primary energy uses is based on fossil fuels. This energy use is the dominant contributor to CO2 that exists in our atmosphere and other greenhouse gas emissions. Since the pre-industrial era, the level of CO2 has been continually increasing, being a component of greenhouse gas, primarily responsible for global warming.1 One of the active goals in chemistry is to harness a part of this CO2 stream to produce useful chemical products.2 The coupling of epoxides with CO2 into cyclic carbonates is one of the most promising as well as an eco-friendly method used for the chemical fixation of CO2.3 This reaction is one of the few known commercially important reactions that utilize CO2 as a chemical feedstock and has the potential to provide a sustainable basis for the future chemical industry. Even though the coupling reaction of epoxides and CO2 is highly exothermic, due to the kinetic and thermodynamic stability of carbon dioxide this coupling does not occur spontaneously because of the high activation energy requirement.4 To lower the energy of activation of this coupling reaction, various types of metal free5 and metal-based catalysts have been developed including alkali metal salts,6 main-group7 and transition metal complexes,8 metal–organic frameworks (MOFs)9 and metal oxides10etc.
In catalyst designing, the choice of an appropriate ligand in addition to the metal centre is very important and critical for fine-tuning the catalytic activities of metal complexes. For these coupling reactions, the most frequently investigated complexes are that of cobalt,11 iron,12 chromium,13 and zinc14 metals. Most of the catalytic work based on cobalt complexes is related to porphyrin,15 salen,16 bis(phenoxyiminato),17 and amine-bis(phenolato)18 ligands. To the best of our knowledge, the highest TON obtained using cobalt complexes is 2930 by Jing and co-workers,15 using a high pressure of 20 bar at 120 °C, while the highest value of TOF is 662 h−1 obtained by Ghosh and co-workers19 using a high pressure of 20.68 bar at 130 °C.
Literature studies show that in CO2 cycloaddition to epoxides, hydrogen bond donor groups on the catalyst facilitate the activation of the epoxides and enhance the progress of the reaction.20 Thus, diols were found to be more efficient to promote the reaction than monohydroxyl alcohols. Furthermore, diols in which the two hydroxyl groups exist on two adjacent carbons (vicinal carbons) are more active than other diols.
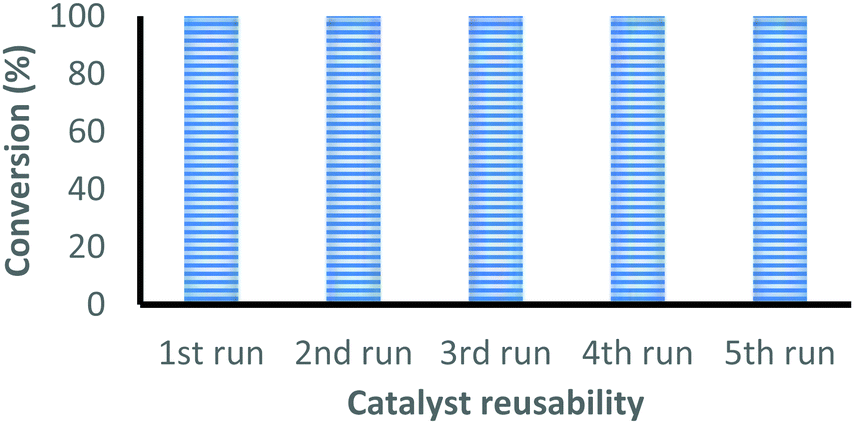 | ||
| Fig. 1 Recyclability of the complex 2 for the coupling reaction of CO2 with epichlorohydrin under optimal reaction conditions. | ||
Also, it is reported that multinuclear complexes are more efficient towards CO2 fixation than mononuclear complexes.7b,21 Due to the facile synthesis and wide-ranging flexibility in the choice and design of Schiff base ligands, polyhydroxy Schiff base ligands were selected aiming at their dinuclear cobalt complexes. These rich hydrogen-bond donor complexes exhibited exceptional activity towards the solvent-free cycloaddition of CO2 to epoxides at ambient pressure with and without a cocatalyst. To the best of our knowledge, this is the most active catalytic system based on cobalt complexes with the highest TON and TOF being reported for CO2 coupling with epoxides.
The Schiff base ligands were firstly synthesized via a condensation reaction of various aldehydes with 2-amino-2-hydroxymethyl-propane-1,3-diol, and then their corresponding cobalt complexes were synthesized from the reaction of these ligands with CoCl2·6H2O in the presence of triethylamine (TEA) as shown in Scheme 1. The obtained products were fully characterized using NMR (1H- and 13C-NMR), FT-IR, ESI-MS, and elemental analysis. The mass spectra of the complexes are in agreement with the formulae [Co(H2L1)]2, [Co(H2L2)]2, and [Co(H2L3)]2. The mass spectrum of the complex 1 shows a base peak at a mass-to-charge ratio (m/z) of 597.9928, corresponding to the dinuclear formula of [Co2(H2L1)2H]− (calculated m/z 597.9969). While, complexes 2 and 3 show base peaks at a mass-to-charge ratio (m/z) of 658.0127 and 698.0222 corresponding to formulae of [Co2(H2L2)2H]− (calculated m/z 658.0180) and [Co2(H2L3)2H]− (calculated m/z 698.0282), respectively (Fig. S10–S12, ESI†).
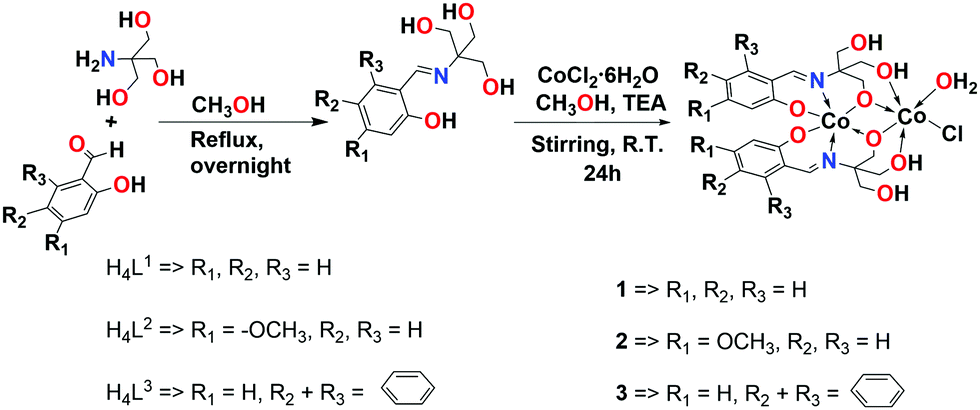 | ||
| Scheme 1 Synthesis of Schiff base ligands (H4L1–H4L3) and their corresponding dinuclear cobalt complexes (1–3). | ||
In the FT-IR spectra, the coordination of the Schiff base ligand to the cobalt metal centre via the N-atom is expected to decrease the electron density in the azomethine linkage and hence lower the absorption frequency of ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) (C
(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibrations.22 Strong and sharp absorption bands appearing in the range of 1635–1638 cm−1 in the free ligands are shifted to lower wavenumbers (1626–1633 cm−1) after complexation suggesting the participation of the azomethine group of the ligands in binding to the cobalt metal centre23 (Fig. S7–S9, ESI†). The powder XRD of complex 2 is in close agreement with the simulated powder XRD pattern of the reported complex (see Fig. S7 in ESI†).
N) stretching vibrations.22 Strong and sharp absorption bands appearing in the range of 1635–1638 cm−1 in the free ligands are shifted to lower wavenumbers (1626–1633 cm−1) after complexation suggesting the participation of the azomethine group of the ligands in binding to the cobalt metal centre23 (Fig. S7–S9, ESI†). The powder XRD of complex 2 is in close agreement with the simulated powder XRD pattern of the reported complex (see Fig. S7 in ESI†).
In order to evaluate the catalytic potential of the synthesized dinuclear cobalt complexes (1–3) for the coupling reactions, epichlorohydrin (substrate) was treated with carbon dioxide starting with 1 bar pressure of CO2 and 120 °C under solvent-free conditions using 1 mol% (0.05 mmol) of each complex as a catalyst (Table 1, entries 1–3). After 5 hours of reaction time, good conversions were observed for all the catalysts, though catalyst 2 has a relatively high conversion as compared to the other two catalysts, which may be due to the electron donation properties of methoxy groups.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Cat.:cocat.:substrate |
P (bar) | Temp. (°C) | Time (h) | Conversion (%) | TON | TOF (h−1) |
| TBAB was used as a cocatalyst unless mentioned otherwise.a DMAP as a cocatalyst. TON and TOF values are calculated per mole of complex. | ||||||||
| 1 | 1 | 1:0:100 |
1 | 120 | 5 | 80 | 80 | 16 |
| 2 | 2 | 1:0:100 |
1 | 120 | 5 | 90 | 90 | 18 |
| 3 | 3 | 1:0:100 |
1 | 120 | 5 | 55 | 55 | 11 |
| 4 | 2 | 1:0:100 |
1 | 100 | 24 | 74 | 74 | 3 |
| 5 | 2 | 1:0:100 |
1 | 75 | 24 | 11 | 11 | 0.5 |
| 6 | 2 | 1:0:100 |
5 | 75 | 12 | — | — | — |
| 7 | 2 | 1:0:100 |
5 | 120 | 5 | 80 | 800 | 16 |
| 8a | 2 | 1:1:1000 |
1 | 120 | 4 | 77 | 770 | 192 |
| 9 | 2 | 1:1:1000 |
1 | 120 | 4 | 81 | 810 | 202 |
| 10 | 2 | 1:1:1000 |
1 | 120 | 6 | 100 | 1000 | 167 |
| 11 | 2 | 1:1:1000 |
1 | 120 | 3 | 58 | 580 | 193 |
| 12 | 2 | 1:1:1000 |
2 | 120 | 3 | 64 | 640 | 213 |
| 13 | 2 | 1:1:1000 |
5 | 120 | 3 | 73 | 730 | 243 |
| 14 | 2 | 1:1:1000 |
10 | 120 | 3 | 81 | 810 | 270 |
| 15 | 2 | 1:1:10000 |
1 | 120 | 4 | 46.5 | 4650 | 1162 |
| 16 | 2 | 1:1:50000 |
1 | 120 | 12 | 37.1 | 18550 |
1546 |
| 17 | 2 | 1:1:100000 |
1 | 120 | 24 | 62.2 | 62200 |
2592 |
| 18 | 2 | 1:1:200000 |
1 | 120 | 3 | 5 | 10000 |
3333 |
| 19 | 2 | 1:1:200000 |
1 | 120 | 6 | 10 | 20000 |
3333 |
| 20 | 2 | 1:1:200000 |
1 | 120 | 12 | 19 | 38000 |
3167 |
| 21 | 2 | 1:1:200000 |
1 | 120 | 24 | 35 | 70000 |
2917 |
| 22 | 2 | 1:1:200000 |
1 | 120 | 48 | 66 | 132000 |
2750 |
| 23 | 2 | 1:1:200000 |
1 | 120 | 72 | 84.3 | 168600 |
2342 |

Generally, cocatalysts significantly improve the activity of several catalytic systems towards cyclic carbonate formation from epoxides.24 Therefore, we used the most commonly used cocatalysts, 4-dimethylaminopyridine (DMAP) and tetrabutylammonium bromide (TBAB), along with complex 2 to test the catalytic activities in the presence of these cocatalysts (Table 1, entries 8 and 9). The catalytic activity of complex 2 was nearly the same with both cocatalysts, though slightly better with TBAB. We then optimized the other reaction conditions, like temperature (°C) and pressure of CO2 (bar), relative to the molar ratio of catalyst:cocatalyst:substrate, and time (h) for the coupling reaction. For the optimization purpose, epichlorohydrin was used as a substrate. The temperature has a profound effect on the catalytic activity. With the negligible conversion of epichlorohydrin at 75 °C, the conversion increases from 11% (in 24 hours) to 90% (in 5 hours) with increasing the temperature from 75 to 120 °C (Table 1, entries 3–5). However, the catalytic activity is less sensitive to CO2 pressure increase in comparison to temperature. Thus, increasing the gas pressure from 1 to 10 bar, while keeping other parameters fixed, the conversion of epoxides into cyclic products increases from 58 to 81% in 3 h (Table 1, entries 11–14). Also see Table S1 (ESI†) for the ligands, complexes cocatalysts' conversions.
Based on these observations, 120 °C and 1 bar pressure of CO2 were adopted as the optimum reaction conditions and TBAB as a cocatalyst for further studies. At catalyst and TBAB loadings of 0.1 mol% each, we were able to achieve the complete conversion of an epichlorohydrin substrate at 120 °C and 1 bar pressure of CO2 after 6 h (Table 1, entry 10). We then investigated the use of reduced amounts of complex 2 (Table 1, entries 15–23) and observed the TON value of 168600 (Table 1, entry 23) and the TOF of up to 3333 h−1 (Table 1, entry 18), which are the highest values of TON and TOF reported for cobalt-based catalysts (Table S2, ESI†).
Due to the industrial importance of various cyclic carbonates, different epoxide substrates were used for this coupling reaction under the optimized catalytic conditions. Most importantly, the catalyst is effective in the conversion of terminal as well as challenging internal epoxides. The results are summarized in Table 2 (see Fig. S14–S24, ESI† for NMR spectra). It is observed that epichlorohydrin, epibromohydrin, allyl glycidyl ether, butyl glycidyl ether, and glycidyl isopropyl ether are fully converted into the corresponding cyclic carbonates in 6–8 hours (Table 2, entries 1–5). Also, good conversions were obtained in the case of styrene oxide, propylene oxide, and 1,2-epoxyhexane (Table 2, entries 6–8). We also tested the catalyst for internal epoxides. Relatively low conversion was observed at 1 bar. However, at high pressure (10 bar), excellent conversion was observed for all three internal epoxides used (Table 2, entries 9–11).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conversion (%) | TON | TOF (h−1) |
| Conditions: 0.1 mol% (0.01 mmol) of complex 2, 0.1 mol% (0.01 mmol) of TBAB. Reaction time is 8 hours unless stated otherwise. TON and TOF values are calculated per mole of complex.a Reaction time 6 h.b Reaction time 24 h and 0.5 mol% of TBAB.c 10 bar pressure of CO2. | ||||
| 1a | Epichlorohydrin | 100 | 1000 | 167 |
| 2 | Allyl glycidyl ether | 100 | 1000 | 125 |
| 3 | Butyl glycidyl ether | 100 | 1000 | 125 |
| 4 | Glycidyl iso-propyl ether | 100 | 1000 | 125 |
| 5 | Epibromohydrin | 100 | 1000 | 167 |
| 6 | Styrene oxide | 94 | 940 | 118 |
| 7 | Propylene oxide | 81 | 810 | 116 |
| 8 | 1,2-Epoxyhexane | 77 | 770 | 96 |
| 9b | 3,6-Dioxabicyclo [3,1.0] hexane | 56 (100)c | 560 (1000)c | 23 (42)c |
| 10b | Cyclopentene oxide | 45 (100)c | 450 (1000)c | 19 (42)c |
| 11b | Cyclohexene oxide | 20 (84)c | 200 (840) | 8 (35)c |
The general mechanism was proposed based on the literature.25 The key feature of the mechanism is the activation of both reaction components, the epoxy ring and carbon dioxide, by the dinuclear complex, and cocatalyst, TBAB. The role of TBAB in the mechanism might be the in situ generation of tributylamine, which helps in the activation of CO2.25,26 The coordination of the epoxide to the metal centre (Lewis acid) is followed by ring opening by bromide from the cocatalyst.26 This intermediate formed is supposed to react with a carbamate salt formed by the decomposition of TBAB to tributylamine and further reacted with CO2 to afford a complex in which both the epoxide and the CO2 are activated.26 The displacement of the tributylammonium group finally affords a metal-coordinated carbonate, which undergoes subsequent ring-closure to form the cyclic carbonate and regenerates the catalyst.26 Therefore, the complex and TBAB take part together to open the epoxy ring and then the subsequent addition of CO2 to give the corresponding cyclic carbonates in ring opening and re-cyclization processes.
The catalyst is easily recovered from the reaction mixture by centrifugation, washed with acetone and dried under vacuum at 50 °C for 4 hours before starting the next cycle. The conversion after each cycle was determined by 1H-NMR and the catalyst was characterized by FT-IR. This process was successfully repeated five times without any loss in the catalytic activity, see Fig. 1. The FT-IR spectrum of the recovered catalyst after five cycles closely matches with the fresh catalyst, which indicates that the catalyst retains its identity even after five reaction cycles (Fig. S21, ESI†).
In order to establish the applicability and effectiveness of the catalytic system used in this study, the performance of these catalysts was compared with several examples of molecular catalysts that are based on different transition metals. However, a direct comparison is complex due to different investigated conditions.27 It is difficult to draw a clear comparison with the reported catalytic systems, as in most cases the epoxides, cocatalysts and the experimental conditions such as temperature and pressure are not the same. But to give a rough idea of the efficacy of the catalytic system used in this study with the reported complexes, especially with those of Co-complexes, the results are summarized in Table S2 in the ESI.† Though very high TON and TOF values are reported in the case of Al-, Mg-, Fe- and Zn-complexes (Table S2, entries 12–19, ESI†), the TON and TOF values obtained in this study are significantly higher than the highest TON and TOF values reported for cobalt complexes, and also it competes well with other state of the art catalysts (Table S2, entries 1–11, ESI†) with the advantage of working under ambient pressure conditions.
In summary, dinuclear cobalt complexes have been successfully synthesized and used as active catalysts for the synthesis of cyclic carbonates from the coupling reaction of epoxides and carbon dioxide in the presence of TBAB as a cocatalyst. These catalysts efficiently performed the insertion of carbon dioxide into various terminal as well as challenging internal epoxides at low catalyst loading (0.01 mol%) at an ambient gas pressure of CO2, though high pressure is favourable for internal epoxides, under solvent-free conditions. Notably, the highest values of TON (168600) and TOF (3333 h−1) are achieved at a low pressure of CO2 (1 bar) as compared to the other reported cobalt complexes. The reuse of the recycled catalyst for at least five cycles giving the same activity revealed the remarkable stability of these compounds.
The authors gratefully acknowledge “State Key Lab of Advanced Technology for Materials Synthesis and Processing” for financial support. F. V. acknowledges the support from Tomsk Polytechnic University Competitiveness Enhancement Program grant (VIU-2019). Z. A. K. K. acknowledges funding (2014GXZ330) from the Chinese Scholarship Council (CSC) for PhD studies.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. F. M. Orr, Energy Environ. Sci., 2009, 2, 449 RSC.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281 RSC.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497 RSC.
- L. Wang, X. Jin, P. Li, J. Zhang, H. He and S. Zhang, Ind. Eng. Chem. Res., 2014, 53, 8426 CrossRef CAS.
- (a) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375 RSC; (b) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966 RSC.
- (a) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228 CrossRef CAS PubMed; (b) Z. A. K. Khattak, H. A. Younus, N. Ahmad, B. Yu, H. Ullah, S. Suleman, A. H. Chughtai, B. Moosavi, C. Somboon and F. Verpoort, J. CO2 Util., 2018, 28, 313 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D’Elia, ACS Catal., 2018, 8, 419 CrossRef CAS.
- (a) B. Mousavi, S. Chaemchuen, B. Moosavi, Z. Luo, N. Gholampour and F. Verpoort, New J. Chem., 2016, 40, 5170 RSC; (b) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462 RSC.
- (a) C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469 CrossRef CAS; (b) A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118 RSC; (c) M. Taherimehr, J. P. C. C. Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034 CrossRef CAS PubMed; (d) D. Alhashmialameer, J. Collins, K. Hattenhauer and F. M. Kerton, Catal. Sci. Technol., 2016, 6, 5364 RSC; (e) F. Della Monica, S. V. C. Vummaleti, A. Buonerba, A. D. Nisi, M. Monari, S. Milione, A. Grassi, L. Cavallo and C. Capacchione, Adv. Synth. Catal., 2016, 358, 3231 CrossRef.
- (a) D. Adhikari, S. T. Nguyen and M.-H. Baik, Chem. Commun., 2014, 50, 2676 RSC; (b) Y. Xie, R.-X. Yang, N.-Y. Huang, H.-J. Luo and W.-Q. Deng, J. Energy Chem., 2014, 23, 22 CrossRef CAS; (c) J. A. Castro-Osma, M. North and X. Wu, Chem. – Eur. J., 2016, 22, 2100 CrossRef CAS PubMed; (d) J. A. Castro-Osma, K. J. Lamb and M. North, ACS Catal., 2016, 6, 5012 CrossRef CAS.
- (a) Y.-M. Shen, W.-L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559 CrossRef CAS PubMed; (b) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC; (c) H. Vignesh Babu and K. Muralidharan, Dalton Trans., 2013, 42, 1238 RSC; (d) C. Martin, C. J. Whiteoak, E. Martin, M. Martinez Belmonte, E. C. Escudero-Adan and A. W. Kleij, Catal. Sci. Technol., 2014, 4, 1615 RSC; (e) M. A. Fuchs, C. Altesleben, S. C. Staudt, O. Walter, T. A. Zevaco and E. Dinjus, Catal. Sci. Technol., 2014, 4, 1658 RSC; (f) Y. Ren, J. Chen, C. Qi and H. Jiang, ChemCatChem, 2015, 7, 1535 CrossRef CAS; (g) E. Mercadé, E. Zangrando, C. Claver and C. Godard, ChemCatChem, 2016, 8, 234 CrossRef.
- X. Jiang, F. Gou, F. Chen and H. Jing, Green Chem., 2016, 18, 3567 RSC.
- (a) X.-B. Lu, X.-J. Feng and R. He, Appl. Catal., A, 2002, 234, 25 CrossRef CAS; (b) A. Berkessel and M. Brandenburg, Org. Lett., 2006, 8, 4401 CrossRef CAS PubMed.
- A. Sibaouih, P. Ryan, K. V. Axenov, M. R. Sundberg, M. Leskelä and T. Repo, J. Mol. Catal. A: Chem., 2009, 312, 87 CrossRef CAS.
- M. Reiter, P. T. Altenbuchner, S. Kissling, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2015, 1766 CrossRef CAS.
- P. Ramidi, N. Gerasimchuk, Y. Gartia, C. M. Felton and A. Ghosh, Dalton Trans., 2013, 42, 13151 RSC.
- (a) J. Song, Z. Zhang, B. Han, S. Hu, W. Li and Y. Xie, Green Chem., 2008, 10, 1337 RSC; (b) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131 RSC.
- (a) S. He, F. Wang, W.-L. Tong, S.-M. Yiu and M. C. W. Chan, Chem. Commun., 2016, 52, 1017 RSC; (b) M. V. Escárcega-Bobadilla, M. Martínez Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2013, 19, 2641 CrossRef PubMed.
- A. Kilic, M. Ulusoy, E. Aytar and M. Durgun, J. Ind. Eng. Chem., 2015, 24, 98 CrossRef CAS.
- A. Kilic, M. Ulusoy, M. Durgun and E. Aytar, Inorg. Chim. Acta, 2014, 411, 17 CrossRef CAS.
- R. L. Paddock and S. T. Nguyen, Chem. Commun., 2004, 1622 RSC.
- W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828 CrossRef CAS PubMed.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS PubMed.
- J. Rintjema and A. W. Kleij, ChemSusChem, 2017, 10, 1274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytical data and NMR spectra. See DOI: 10.1039/c9cc02626f |
| This journal is © The Royal Society of Chemistry 2019 |