
A synthetic approach to chrysophaentin F†
Jean-Baptiste
Vendeville
ab,
Rebecca F.
Matters
a,
Anqi
Chen
b,
Mark E.
Light
a,
Graham J.
Tizzard
a,
Christina L. L.
Chai
 *bc and
David C.
Harrowven
*a
*bc and
David C.
Harrowven
*a
aChemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: dch2@soton.ac.uk
bInstitute of Chemical and Engineering Sciences, Agency for Science, Technology and Research (A*STAR), 8 Biomedical Grove, Neuros, #07-01, 138665, Singapore
cDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, 117543, Singapore. E-mail: phacllc@nus.edu.sg
First published on 1st April 2019
Abstract
The chrysophaentins are a newly discovered natural product family displaying promising anti-infective activity. Herein we describe an approach to chrysophaentin F that uses an array of metal catalysed coupling reactions (Cu, Ni, Pd, W, Mo) to form key bonds.
The chrysophaentins were discovered by Bewley et al. in the methanoic extract of Chrysophaeum taylori alga during a screening study to identify promising new anti-infectives.1 Eight closely related bioactive macrocycles were identified in the extract, named chrysophaentins A–H (1–5, 8–10), which were subsequently joined by the linear chrysophaentins E2 (6) and E3 (7) (Fig. 1).2 From a structural perspective they define a new class of marine natural products, the bis-diarylbutenes. Their core resembles that of the bis-bibenzyl family of natural products,3,4 but with two additional carbon centres and unsaturation in each of the chains linking the diaryl ether units.
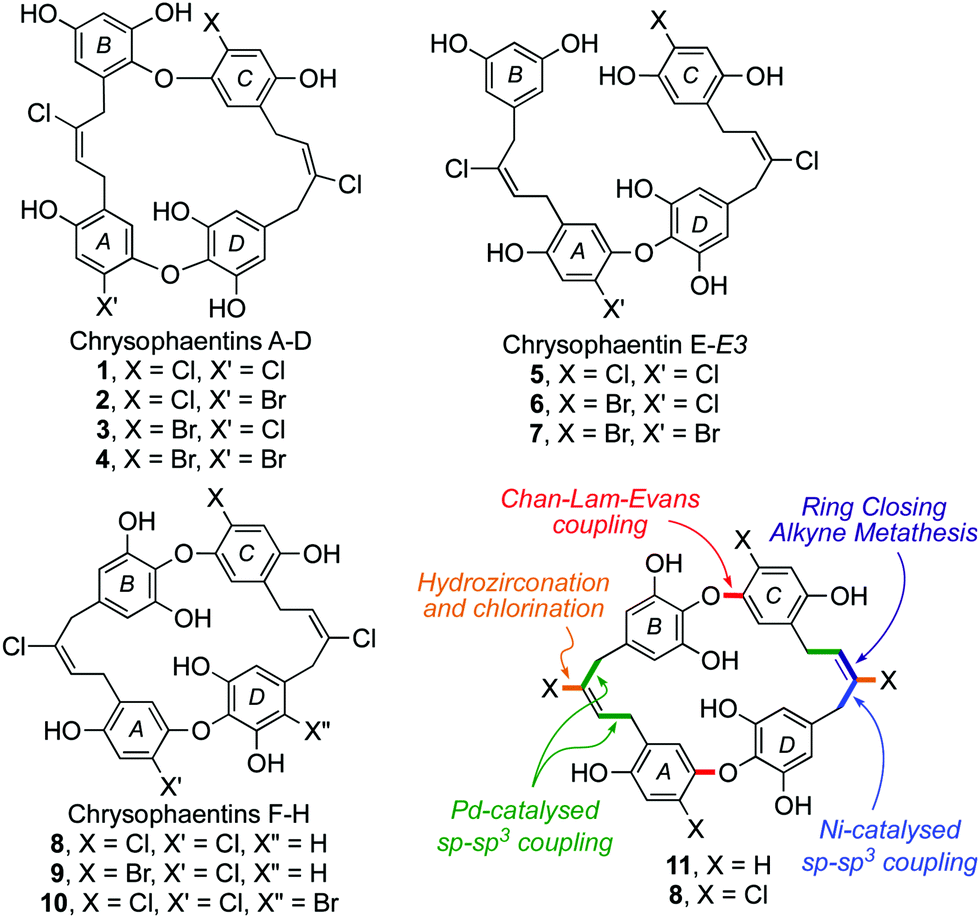 | ||
| Fig. 1 The chrysophaentins and our approach to their total synthesis. | ||
In assays against Staphylococcus aureus (SA), methicillin-resistant SA (MRSA) and multidrug-resistant SA (MDR-SA), chrysophaentins A (1), F (8) and H (10) were found to have useful potency, with minimum inhibitory concentrations (MIC50) in the range of 0.8–9.5 μg mL−1.1 By contrast, the acyclic chrysophaentin E 5, and those with a bromide on arene A or arene C (2–4 and 9), showed greatly reduced activity. Chrysophaentin A (1) was also found to inhibit the bacterial cell division protein FtsZ, a popular target in antimicrobial drug discovery programmes.5
From a synthetic perspective the chrysophaentins have proven to be elusive targets. To date, all of the approaches described have sought to make one of the symmetrical chrysophaentins using a cyclodimerization strategy, yet none has succeeded in gaining access to the macrocyclic core.2,4–6 Herein we describe our work on the development of a general synthetic approach to the natural product family, exemplified by syntheses of the dehalogentated core 11 and, tentatively, of chrysophaentin F (8) in impure form. Our approach, summarized in Fig. 1, has both divergent and convergent phases and uses an array of metal catalyzed reactions (Cu, Ni, Pd, W and Mo) to construct key bonds.
To test the validity of our approach, we first targeted the chrysophaentin F–H core 11 lacking all halogen substituents. Our synthesis began with the preparation of benzo-1,3-dioxane 15 and phenol 16 using standard protocols (Scheme 1).7–9 Their union to diaryl ether 18 was then achieved using a Chan–Lam–Evans coupling procedure.10 After examining a range of catalysts, solvents and reaction conditions, we found that it was best to employ Cu(OTf)2 with 4 Å molecular sieves in ethanol under an oxygen atmosphere. In that way diaryl ether 18 could be formed reliably in 76% yield. Sequential hydrolysis of the acetal to phenol 17; protection as benzyl ether 19 and conversion of the benzylic alcohol to the corresponding chloride,11 gave the pivotal diaryl ether 20 in high overall yield.
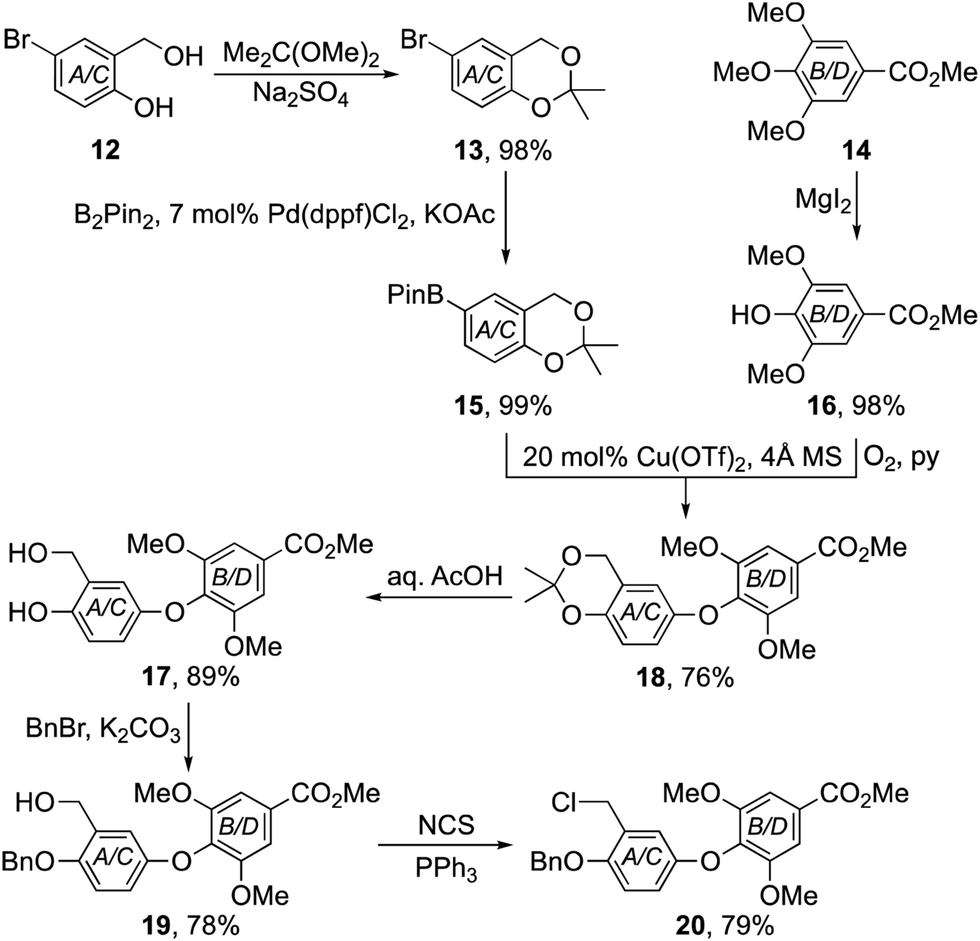 | ||
| Scheme 1 Synthesis of the pivotal diaryl ether 20. | ||
At this juncture our synthesis entered a divergent phase where diaryl ether 20 was advanced to both the B–O–C and A–O–D subunits, 26 and 28 respectively (Scheme 2). Thus, using a Pd-catalysed procedure developed by Buchwald et al.12 diaryl ether 20 was coupled with TMS-acetylene and 1-hexyne respectively, to afford alkynes 21 and 22 in excellent yield. The esters in each of these products were then reduced with LiAlH4 to facilitate their conversion to halides 25 and 26.11 Alas, attempts to effect the coupling of benzyl bromide 25 and 1-hexyne using the aforementioned Buchwald procedure also induced alkyne to allene isomerisation.12 However, by switching to a nickel catalyzed coupling reaction developed by Gau et al.,13 it was successfully coupled with hexynylAlEt2 to give diyne 28 in good yield after deprotection of the silyl acetylene 27 with catalytic silver triflate.14 Pleasingly, the Buchwald procedure proved effective for the coupling of diaryl ether fragments 26 and 28 providing the macrocyclic precursor 29 in 50% yield.12
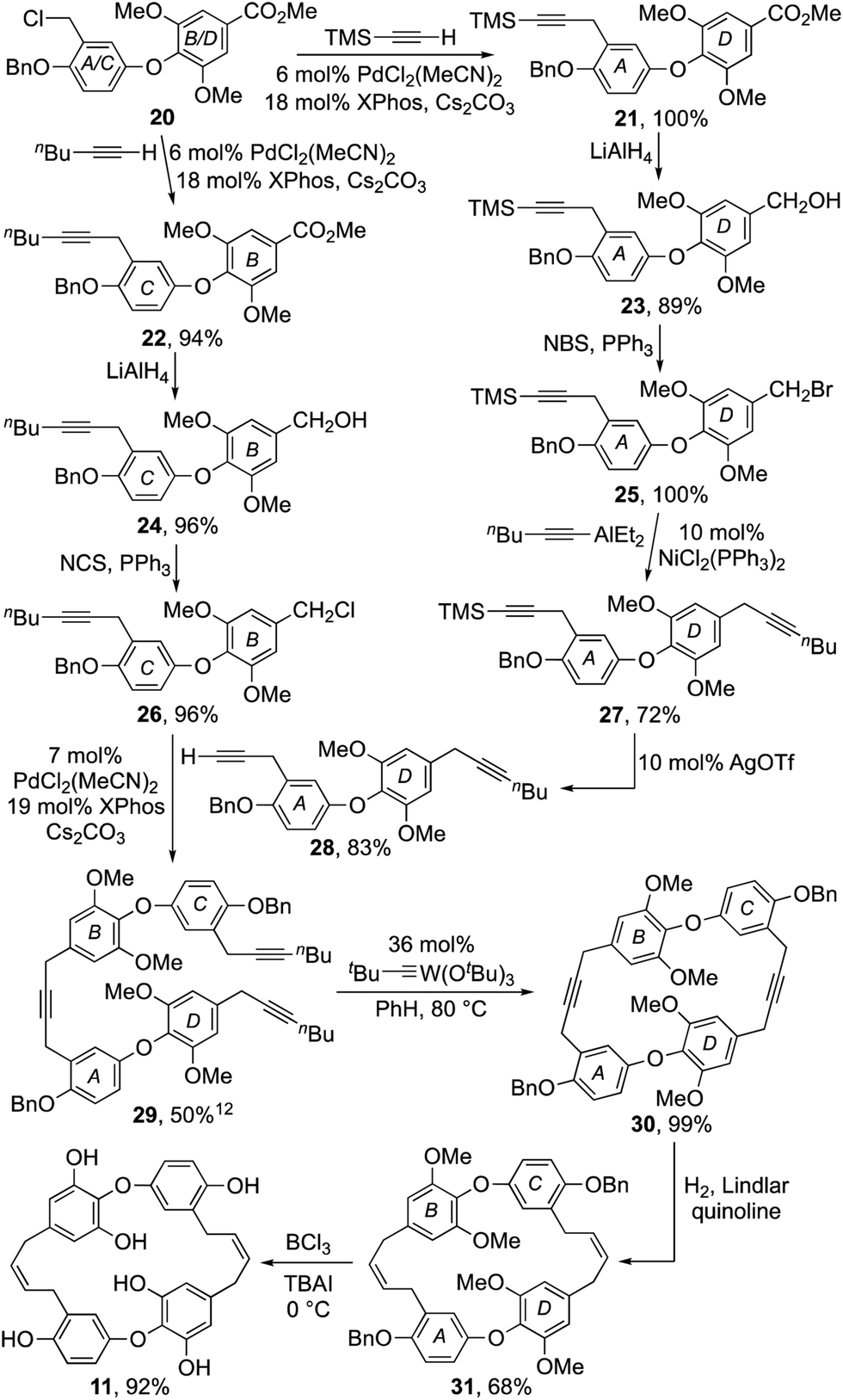 | ||
| Scheme 2 Synthesis of dehalogenated chrysophaentin 11. | ||
The stage was now set to enact our endgame strategy, which sought to use alkyne metathesis for the critical macrocyclization reaction.15,16 Pleasingly, this proved remarkably efficient as, after some optimization, triyne 29 was transformed into macrocyclic diyne 30 in near quantitative yield using Schrock's alkylidyne catalyst in toluene at 80 °C for 12 h under high dilution.16 Success was confirmed by X-ray crystallographic analysis (Fig. 2). Selective hydrogenation of 30 using Lindlar's catalyst in the presence of quinoline next provided bis-cis-alkene 31, leaving us the task of unmasking the six phenol residues. Although the double bonds in 31 proved sensitive to an array of standard deprotection protocols, a combination of BCl3 and tetrabutylammonium iodide in DCM gave our target 11 in 92% yield (Scheme 2).17
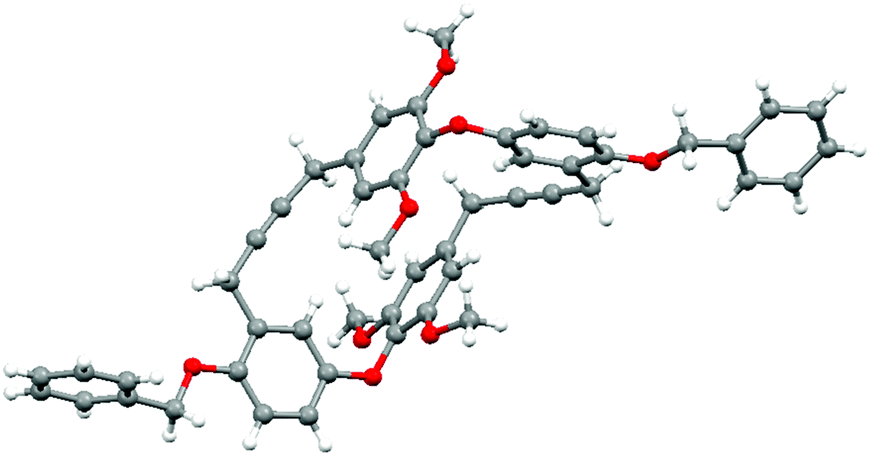 | ||
| Fig. 2 X-ray crystal structure of macrocyclic diyne 30. | ||
The total synthesis of chrysophaentin F 8 became our next target with the preparation of the keystone diaryl ether 38 becoming the immediate goal (Scheme 3). To that end, benzoic acid 32 was reduced with LiAlH4 to diol 33, which in turn was protected as its benzyl ether 34. Arene bromination to 35 then facilitated a Miyaura borylation to 37 enabling its coupling to phenol 16 using a Chan–Lam–Evans procedure. Finally, conversion of the resulting benzyl alcohol 36 to the corresponding chloride 38 delivered the required diaryl ether.
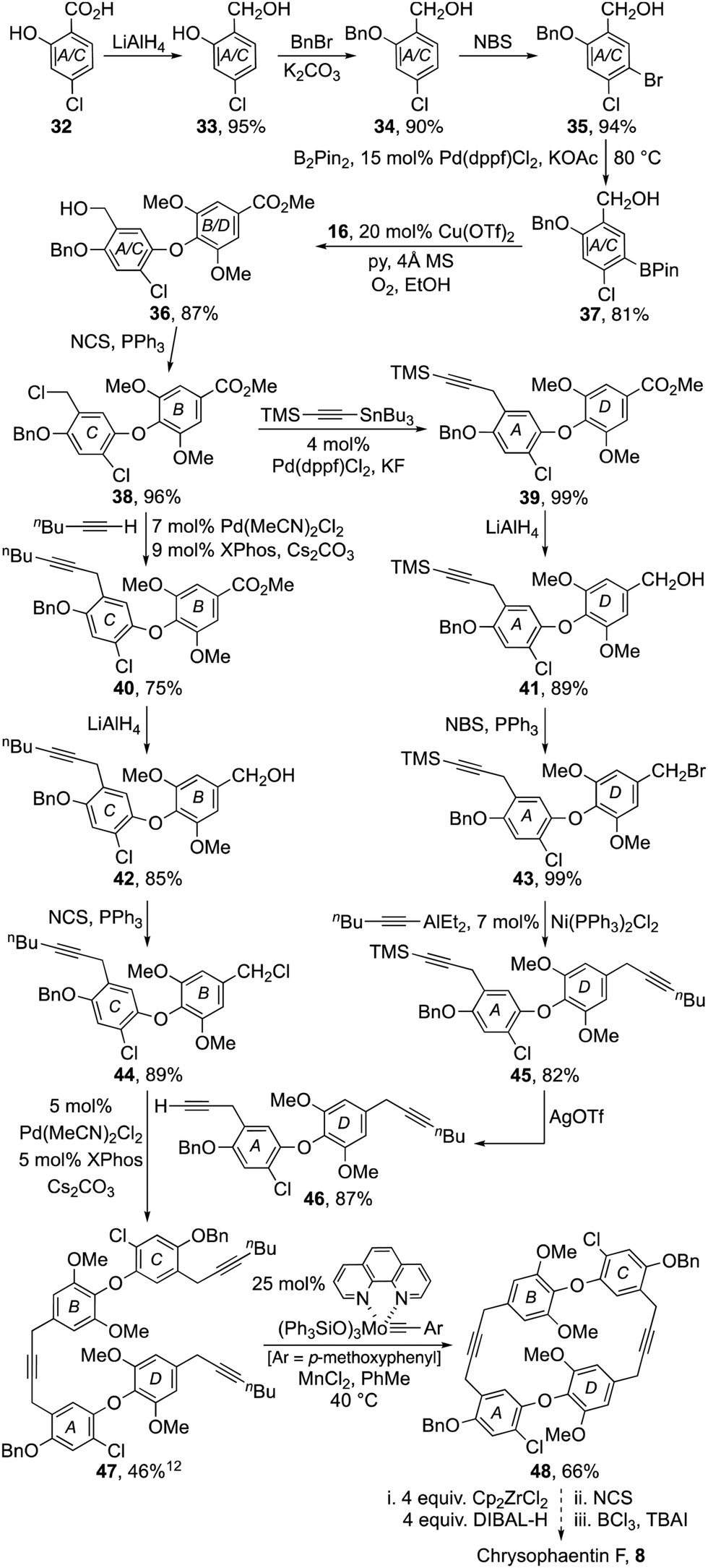 | ||
| Scheme 3 A tentative synthesis of chrysophaentin F. | ||
The divergent strategy used to prepare dehalochrysophaentin 11 was now applied to the synthesis of chrysophaentin F 8. While most of the steps were easy to replicate, it proved advantageous to use a Stille coupling to advance chloride 38 to alkyne 39 as it proceeded reliably in near quantitative yield.18 All other steps in the sequence mirrored those demonstrated previously, readily providing the B–O–C and A–O–D diaryl ether subunits 44 and 46 (Scheme 3). These were coupled using the Buchwald procedure to give triyne 47, setting the stage for our end-game strategy.
Pleasingly, after some optimization, triyne 47 was transformed into macrocyclic diyne 48 in modest yield using Fürstner's molybdenum·phenanthroline pre-catalyst in toluene at 40 °C for 12 h.15 Hydrozirconation of 48 with Schwartz reagent (generated in situ by reduction of Cp2ZrCl2) followed by chlorination with NCS gave a complex mixture of products,19,20 that was partially separated by column chromatography (see ESI† p. S124 and S125). The main fractions were then combined and treated with BCl3 and tetrabutylammonium iodide to unmask the phenolic residues. Purification of the product mixture by column chromatography gave a major fraction exhibiting spectral characteristics consistent with the formation of chrysophaentin F 8 in an impure state (see ESI† for LRMS, 1H and 13C NMR data).1 Alas, attempts to purify the natural product further by preparative TLC and HPLC proved unrewarding with material losses putting paid to further endeavours.
In summary, we have developed a synthetic approach to chrysophaentin F 8 with the flexibility to allow all members of this family to be targeted. The approach demonstrates the value of (i) Chan–Lam–Evans coupling reactions for the preparation of diaryl ethers with high steric demand;10 (ii) Buchwald's, Gau's and Stille's procedures for effecting sp–sp3 coupling reactions between alkynes and benzyl halides;12,13 and (iii) Fürtsner's alkylidyne pre-catalyst for macrocyclisation through ring-closing alkyne metathesis.15,16
We thank Prof. A. Fürstner for his kind donation of the molybdenumphenanthroline pre-catalyst. The University of Southampton VC Scholarship Scheme, the A*STAR ARAP Scheme, the ERDF (LabFact: InterReg V project 121) and the EPSRC [EP/K039466/1, EP/P013341/1 and EP/L003325/1] are thanked for their financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Plaza, J. Keffer, G. Bifulco, J. Lloyd and C. Bewley, J. Am. Chem. Soc., 2010, 132, 9069 CrossRef CAS PubMed; J. R. Davison and C. Bewley, J. Nat. Prod., 2019, 82, 148 CrossRef PubMed.
- J. L. Keffer, J. T. Hammill, J. R. Lloyd, A. Plaza, P. Wipf and C. A. Bewley, Mar. Drugs, 2012, 10, 1103 CrossRef PubMed.
- D. C. Harrowven and S. L. Kostiuk, Nat. Prod. Rep., 2012, 29, 223 RSC . For our work on macrocyclic bis-bibenzyl total synthesis see: F. A. Almalki and D. C. Harrowven, Eur. J. Org. Chem., 2016, 5738 CrossRef CAS; S. L. Kostiuk, T. Woodcock, L. F. Dudin, P. D. Howes and D. C. Harrowven, Chem. – Eur. J., 2011, 17, 10906 CrossRef PubMed; D. C. Harrowven, T. Woodcock and P. D. Howes, Angew. Chem., Int. Ed., 2005, 44, 3899 CrossRef PubMed.
- H. P. Erikson, Cell, 1995, 80, 367 CrossRef CAS; M. H. Foss, Y.-J. Eun and D. B. Weibel, Biochemistry, 2011, 50, 7719 CrossRef PubMed.
- A. Brockway, C. Grove, M. Mahoney and J. Shaw, Tetrahedron Lett., 2015, 56, 3396 CrossRef CAS PubMed.
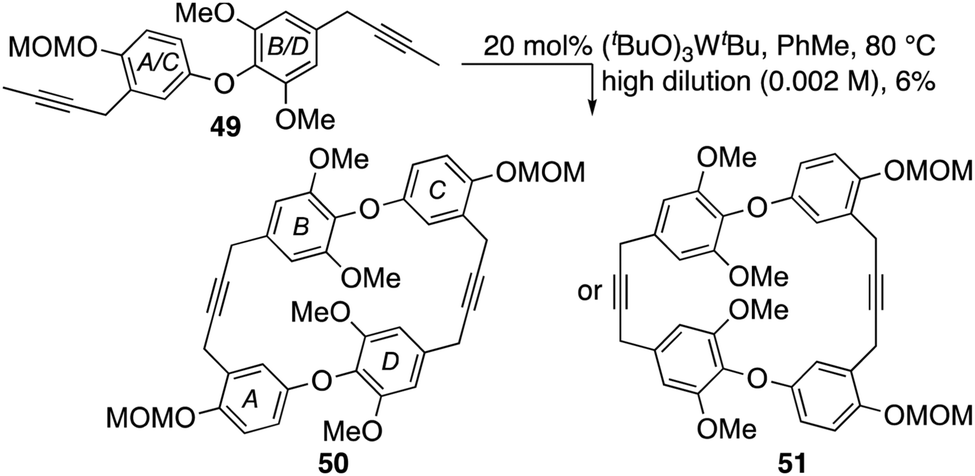
- In common with others, our first approach to chrysophaentin F was based on a cyclodimerization strategy, e.g.49 → 50. Indeed, after much optimization this gave a cyclodimer in 6% yield that could not be assigned unambiguously to either dimer 50 or its regioisomer 51 using the spectral data attained.
. - N. Gisch, J. Ballzarini and C. Meier, J. Med. Chem., 2007, 50, 1658 CrossRef CAS PubMed.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS.
- K. Bao, A. Fan, Y. Dai, L. Zhang, W. Zhang, M. Cheng and X. Yao, Org. Biomol. Chem., 2009, 7, 5084 RSC.
- D. Chan, K. Monaco, R. Wang and M. Winters, Tetrahedron Lett., 1998, 39, 2933 CrossRef CAS; D. Evans, J. Katz and T. West, Tetrahedron Lett., 1998, 39, 2937 CrossRef; P. Lam, C. Clark, S. Saubern, J. Adams, M. Winters, D. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941 CrossRef . See also S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef PubMed.
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801 CrossRef.
- C. H. Larsen, K. W. Anderson, R. E. Tundel and S. L. Buchwald, Synlett, 2006, 2941 CAS . The optimised procedure reported used the alkyne in slight excess (1.3 equiv.) and was adopted for advancing benzyl chloride 20 to alkynes 21 and 22. However, it was not deemed appropriate to use alkynes 28 and 46 in excess in their respective couplings with benzyl chlorides 26 and 44, as each was prepared by multi-step sequences. When using equimolar quantities of each component, some benzyl chloride was usually recovered (∼25%) but no alkyne. The known isomerisation of the alkyne product to an allene was also observed as a side reaction in many cases.
- D. Biradar and H. Gau, Chem. Commun., 2011, 47, 10467 RSC.
- A. Orsini, A. Vitérisi, A. Bodlenner, J.-M. Weibel and P. Pale, Tetrahedron Lett., 2005, 46, 2259 CrossRef CAS.
- J. Heppekausen, R. Stade, R. Goddard and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 11045 CrossRef CAS PubMed; A. Fürstner, Chem. Commun., 2011, 47, 6505 RSC; A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC.
- M. L. Listemann and R. R. Schrock, Organometallics, 1985, 4, 74 CrossRef CAS.
- P. R. Brooks, M. C. Wirtz, M. G. Vetelino, D. M. Rescek, G. F. Woodworth, B. P. Morgan and J. W. Coe, J. Org. Chem., 1999, 64, 9719 CrossRef CAS.
- R. R. Singidi and T. V. RajanBabu, Org. Lett., 2008, 10, 3351 CrossRef CAS PubMed; D. C. Harrowven, D. P. Curran, S. L. Kostiuk, I. L. Wallis-Guy, S. Whiting, K. J. Stenning, B. Tang, E. Packard and L. Nanson, Chem. Commun., 2010, 46, 6335 RSC; D. C. Harrowven and I. L. Guy, Chem. Commun., 2004, 1968 RSC.
- Z. Huang and E. Negishi, Org. Lett., 2006, 8, 3675 CrossRef CAS PubMed.
- D. W. Hart and J. Schwartz, J. Am. Chem. Soc., 1974, 96, 8115 CrossRef CAS; D. W. Hart, T. F. Blackburn and J. Schwartz, J. Am. Chem. Soc., 1975, 97, 679 CrossRef; J. Schwartz and J. A. Labinger, Angew. Chem., Int. Ed. Engl., 1976, 15, 333 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental accounts with spectral details and copies of NMR spectra. CCDC 1899398 and 1898679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc01666j |
| This journal is © The Royal Society of Chemistry 2019 |