
Fe(II)-Catalyzed alkenylation of benzylic C–H bonds with diazo compounds†
Jiang-Ling
Shi
a,
Qinyu
Luo
a,
Weizhi
Yu
a,
Bo
Wang
a,
Zhang-Jie
Shi
 b and
Jianbo
Wang
*a
b and
Jianbo
Wang
*a
aBeijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wangjb@pku.edu.cn
bDepartment of Chemistry, Fudan University, Shanghai 200433, China
First published on 6th March 2019
Abstract
We report herein an alkenylation of benzylic C(sp3)–H bonds with diazo compounds via carbon cation intermediates with DDQ as the oxidant in the presence of a catalytic amount of Fe(II). Diphenylmethane, toluene, benzyl methyl sulfide and their derivatives could be applied as substrates to afford the tetra-substituted olefin products, which may serve as useful building blocks in organic synthesis.
Transition-metal-catalyzed oxidative transformation of C(sp3)–H bonds has emerged as one of the important tools in synthetic organic chemistry.1–4 In particular, Fe(II)-catalyzed oxidative coupling of benzylic C(sp3)–H bonds has attracted considerable attention.5,6 The general pathway of this type of transformation is shown in Scheme 1a. In the presence of an oxidant and Fe(II) catalyst, a benzylic cation is generated through hydrogen abstraction followed by single electron oxidation of the generated benzylic radical species. The benzylic cation is then trapped by a nucleophile to deliver the product. With the Fe(II)/2,3-dichloro-5,6-dicyanoquinone (DDQ) reaction system, various nucleophiles have been explored in the benzylic C(sp3)–H bond functionalization.5–11 For example, Shi et al. reported an oxidative coupling of C(sp3)–H bonds with arenes and vinyl acetates with Fe(II) catalyst and DDQ as the oxidant.8,9 Jiao et al. explored azides as the nucleophiles in the amidation reaction of diphenylmethane through benzylic C(sp3)–H bond cleavage and Schmidt rearrangement.10 In 2015, Song et al. used 1,3-dicarbonyl compounds as the nucleophiles in a similar reaction system with arylmethanes.11 While significant progress has been made in this arena, the scope of this type of transformation is still limited. Thus, further exploration of other nucleophiles is highly desirable.
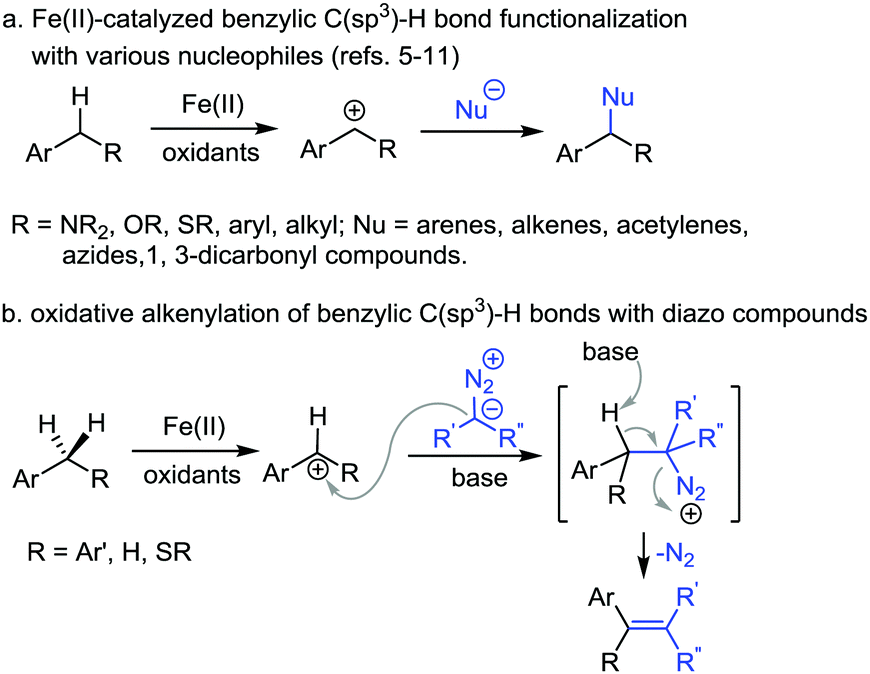 | ||
| Scheme 1 Fe(II)-Catalyzed oxidative coupling of benzylic C(sp3)–H bonds. | ||
On the other hand, diazo compounds have found wide applications as carbene precursors in transition-metal-catalyzed reactions.12 The diazo compounds have also been used as nucleophiles in various transformations.13 In particular, the reaction of diazo compounds with carbon cations is well-known in the literature.14 We thus conceived that for the above-mentioned Fe(II)-catalyzed oxidative cleavage of benzylic C(sp3)–H bonds, it might be possible to apply diazo compounds as the nucleophiles to trap the benzylic carbon cation. The reaction is expected to generate diazonium intermediates followed by dinitrogen extrusion to afford olefin products (Scheme 1b). We report herein the study along this line, namely the FeCl2-catalyzed alkenylation of benzylic C(sp3)–H bonds with diazoesters in the presence of DDQ as the oxidant.
To begin with, we have evaluated the compatibility of the diazo compounds under the reaction conditions with FeCl2 catalyst and DDQ oxidant (Scheme 2). Diphenylmethane 1a was chosen as the substrate to investigate the benzylic C(sp3)–H bond transformation with diazo compounds. We first tested the reaction between 1a (0.4 mmol) and phenyldiazoacetate 2a (0.2 mmol) with 10 mol% FeCl2 and DDQ (0.5 mmol) in 1,2-dichloroethane (DCE, 1.0 mL) at 90 °C for 24 h. Gratifyingly, the expected methyl 2,3,3-triphenylacrylate product was obtained, albeit in a low yield. However, with methyl 2-diazopropanoate 2b as the nucleophile, the reaction failed to afford the expected olefin product. Similarly, the reaction with (diazomethylene)dibenzene 2c also gave no olefin product. The low yields or no product formation with the diazo substrates 2a–c were attributed to the side reactions of the diazo substrates under the reaction conditions, mainly the dimerization and oxidation of the diazo compounds. We then turned our attention to dimethyl 2-diazomalonate 2d, which was known to be the diazo compound with relatively high stability. As expected, the dimethyl 2-diazomalonate 2d was compatible with the reaction conditions and afforded the olefin product in 88% yield. Furthermore, control experiments showed that no C–H bond insertion product could be detected in the absence of DDQ. However, in the absence of FeCl2 catalyst, the olefin product could be obtained in 30% yield.
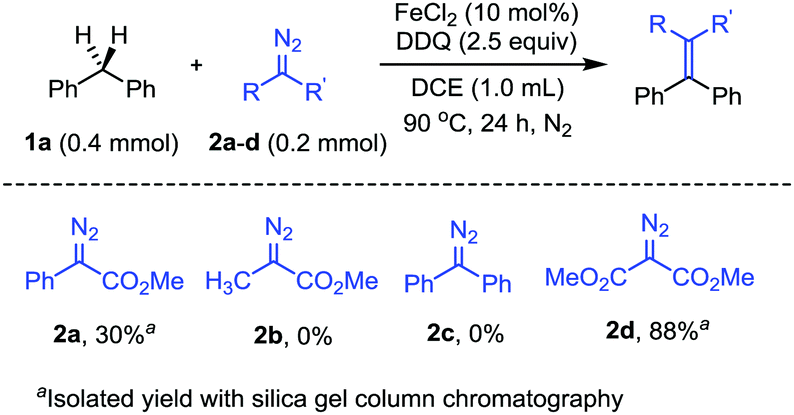 | ||
| Scheme 2 Evaluation of the compatibility of diazo compounds. | ||
With dimethyl 2-diazomalonate 2d as the nucleophile, we then investigated the scope of a series of substituted diphenylmethanes 1a–p (Scheme 3). The substrates with electron-withdrawing and electron-donating groups at the ortho position of the phenyl ring both afforded the corresponding products 3a–d in good yields. The results indicated that a steric effect did not affect the reaction efficiency. The substrates with electron-rich or weakly electron deficient substituents at the meta position of the phenyl ring could also smoothly participate in the reaction (3e, 3h). In addition, the substituted groups with various electronic properties on the para phenyl ring were also tolerated (3g–k). The reaction with other diphenylmethane derivatives, such as symmetrical and unsymmetrical disubstituted derivatives, also afforded the corresponding products 3l–p in moderate to good yields under the standard conditions.
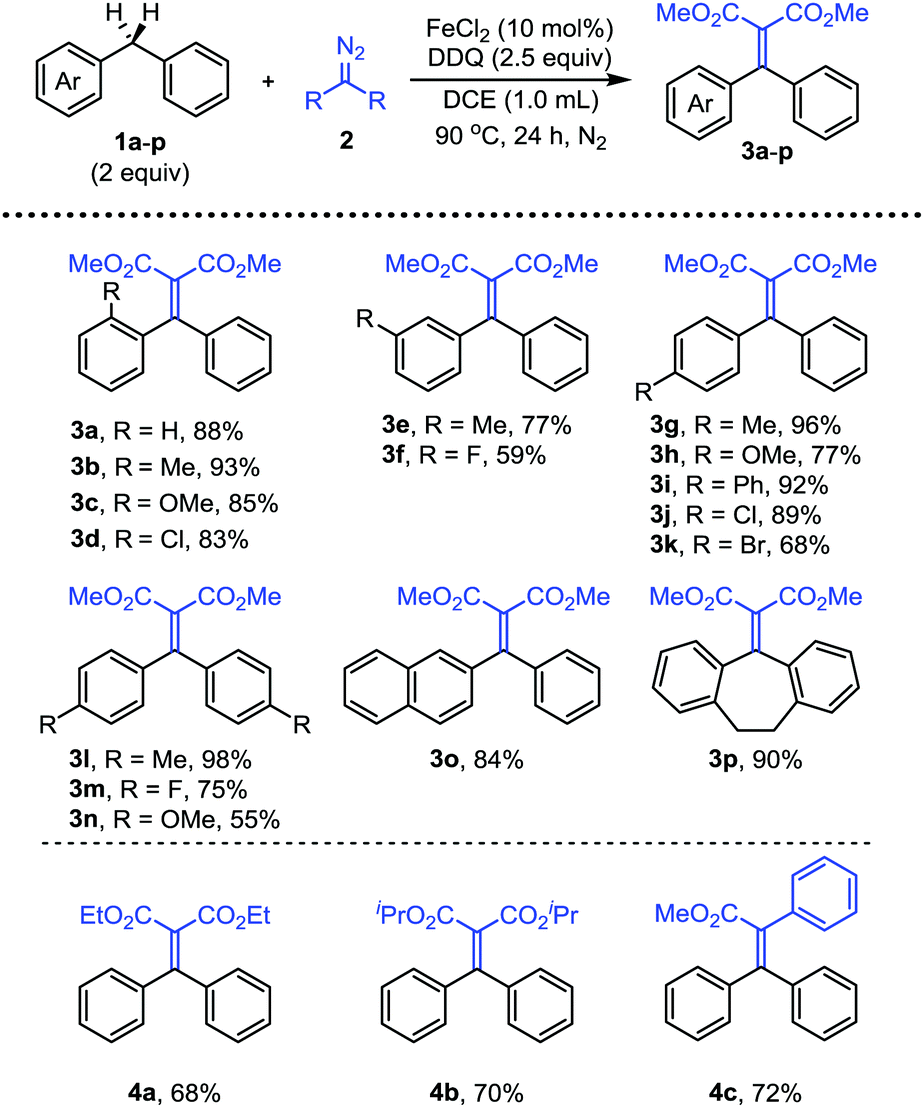 | ||
| Scheme 3 Evaluation of substrate scope of diphenylmethanes. | ||
Next, we have examined the substrate scope of the diazo compounds (Scheme 3). The reactions with diethyl 2-diazomalonate 2e and diisopropyl 2-diazomalonate 2f afforded the products in similar yields (4a, 4b). Notably, for the reaction with methyl phenyldiazoacetate 2a, when the diazo substrate was added using a syringe pump over a period of 30 min, the corresponding product 4c could be obtained in 72% yield. Slow addition of the diazo substrate is necessary in this case for avoiding the oxidation of the diazo substrate. Other aryldiazoacetates were also subjected to the reaction under the same conditions, however, the reactions only offered complicated mixtures.
Furthermore, we have explored the reactivity of other benzylic C(sp3)–H bonds under the same conditions. We have found that the reaction with benzyl methyl sulfide 5a could also afford the alkenylation product 6a in 70% yield. We then evaluated the scope of a series of benzyl sulfide derivatives 5a–h (Scheme 4). The substrates with different substituents on sulfur, such as ethyl, phenyl, p-tolyl and p-FC6H4−, could afford the corresponding alkenylation products in moderate yields (6b–d, 6h). The substituents on the benzyl aromatic ring also tolerated the reaction conditions (6e–g). Benzyl methyl sulfoxide failed to afford the alkenylation products, presumably due to its oxidation to sulfone. It is worth mentioning that alkenyl sulfides are valuable building blocks widely used as enolate surrogates, Michael acceptors, or intermediates in organic synthesis.15
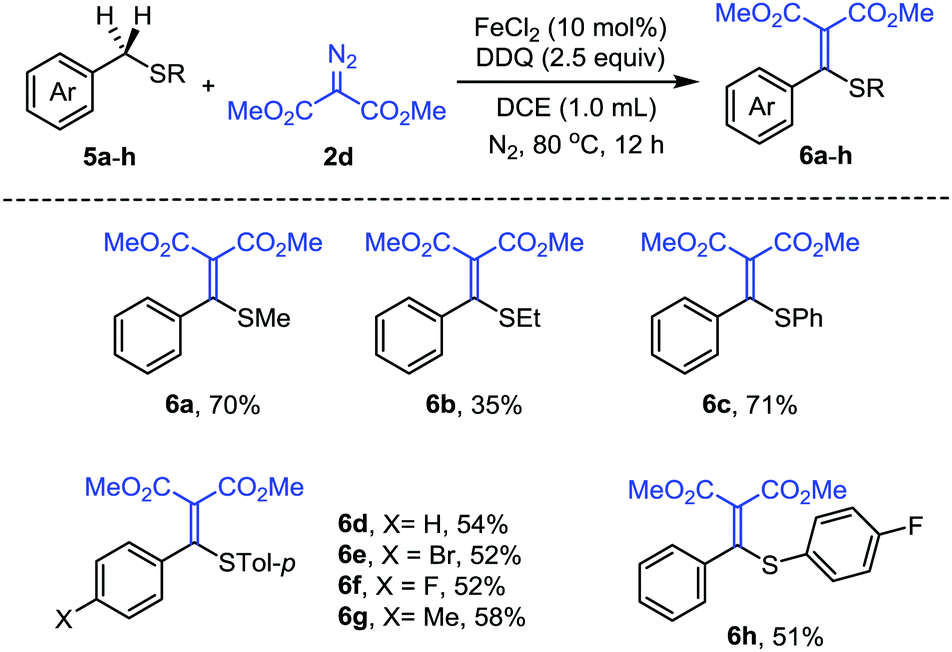 | ||
| Scheme 4 Reaction with benzyl sulfide derivatives. | ||
Interestingly, when toluene 7a was submitted to the reaction under slightly modified conditions (20 mol% FeCl2 and 1.0 mmol DDQ in 1.0 mL DCE at 90 °C for 24 h), the benzylic cation intermediate first underwent Friedel–Crafts reaction with the substrate to give diphenylmethane, which was further followed by alkenylation with diazo substrate 1d to deliver the products 8a and 8b in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The toluene substrates bearing substituents 7b–e were also examined. The reactions with these substrates all proceeded similarly, affording products 9a,b, 10a,b and 11a,b, respectively. The regioselectivity was found relevant to the position and steric bulk of the substituents (Scheme 5).
:1 ratio. The toluene substrates bearing substituents 7b–e were also examined. The reactions with these substrates all proceeded similarly, affording products 9a,b, 10a,b and 11a,b, respectively. The regioselectivity was found relevant to the position and steric bulk of the substituents (Scheme 5).
 | ||
| Scheme 5 Reaction with toluene and its derivatives. | ||
Based on the previous reports, we have proposed a plausible reaction mechanism, as shown in Scheme 6.5–11 First, radical A is generated through hydrogen abstraction from the substrate 1a by FeCl2 and DDQ. In this process, hydrogen is reduced to a proton through single electron transfer to Fe(II)/DDQ, generating Fe(III) intermediate B. Subsequently, single electron transfer (SET) from A to B leads to the formation of carbon cation C, which reacts with nucleophilic diazo substrate 2d to form D. From D deprotonation and elimination of N2 occur to deliver the product 3a, with the regeneration of Fe(II) and 2,3-dichloro-5,6-dicyanohydroquinone E (Scheme 6).
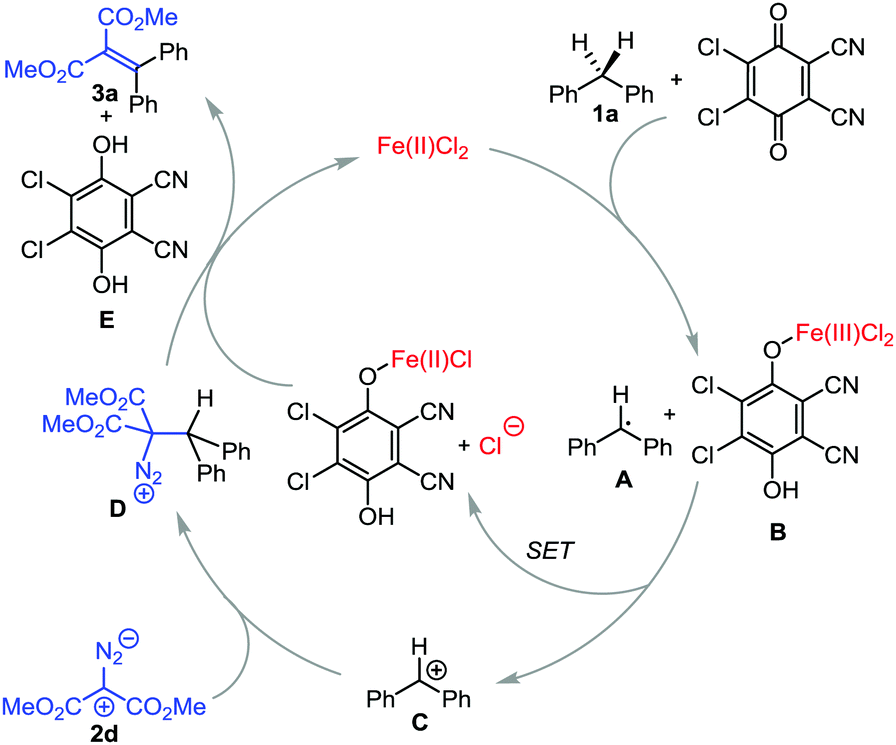 | ||
| Scheme 6 The proposed reaction mechanism. | ||
In summary, we have developed the first alkenylation of benzylic C(sp3)–H bonds with diazo compounds under the FeCl2/DDQ reaction system. The oxidative reaction conditions are compatible with a wide range of diazo substrates. The reaction provides an efficient and unique method for the synthesis of substituted olefins.
The project is supported by 973 Program (No. 2015CB856600) and NSFC (Grant 21871010).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (b) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- P. Xie, Y. J. Xie, B. Qian, H. Zhou, C. G. Xia and H. M. Huang, J. Am. Chem. Soc., 2012, 134, 9902–9905 CrossRef CAS PubMed.
- R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724–5727 CrossRef CAS PubMed.
- T. Stopka, L. Marzo, M. Zurro, S. Janich, E. U. Wurthwein, C. G. Daniliuc, J. Aleman and O. G. Mancheno, Angew. Chem., Int. Ed., 2015, 54, 5049–5053 CrossRef CAS PubMed.
- (a) Z. P. Li, L. Cao and C.-J. Li, Angew. Chem., Int. Ed., 2007, 46, 6505–6507 CrossRef CAS PubMed; (b) Y. H. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242–4243 CrossRef CAS PubMed.
- (a) Z. P. Li, R. Yu and H. J. Li, Angew. Chem., Int. Ed., 2008, 47, 7497–7500 CrossRef CAS PubMed; (b) S. G. Pan, J. H. Liu, H. R. Li, Z. Y. Wang, X. W. Guo and Z. P. Li, Org. Lett., 2010, 12, 1932–1935 CrossRef CAS PubMed; (c) F. Jia and Z. P. Li, Org. Chem. Front., 2014, 1, 194–214 RSC.
- (a) F. Kakiuchi, K. Tsuchiya, M. Matsumoto, E. Mizushima and N. Chatani, J. Am. Chem. Soc., 2004, 126, 12792–12793 CrossRef CAS PubMed; (b) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509–15512 CrossRef CAS; (c) J. Liu, H. Zhang, H. Yi, C. Liu and A. W. Lei, Sci. China: Chem., 2015, 58, 1323–1328 CrossRef CAS; (d) S. B. Guo, Y. X. Li, Y. Wang, X. Guo, X. Meng and B. H. Chen, Adv. Synth. Catal., 2015, 357, 950–954 CrossRef CAS.
- Y. Z. Li, B. J. Li, X. Y. Lu, S. Lin and Z. J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817–3820 CrossRef CAS.
- C. X. Song, G. X. Cai, T. R. Farrell, Z. P. Jiang, H. Li, L. B. Gan and Z. J. Shi, Chem. Commun., 2009, 6002–6004 RSC.
- C. Qin, W. Zhou, F. Chen, Y. Ou and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 12595 CrossRef CAS PubMed.
- K. Yang and Q. L. Song, Org. Lett., 2015, 17, 548–551 CrossRef CAS.
- For reviews on diazo compounds, see: (a) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS; (b) M. P. Doyle, M. A. McKervey and T. Ye, in Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed; (c) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577–6605 CrossRef CAS; (d) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS.
- (a) Y. Zhao and J. Wang, Synlett, 2005, 2886–2892 CAS; (b) Y. Zhang and J. Wang, Chem. Commun., 2009, 5350–5361 RSC.
- (a) T. Stopka, L. Marzo, M. Zurro, S. Janich, E.-U. Würthwein, C. G. Daniliuc, J. Alemán and O. G. Mancheño, Angew. Chem., Int. Ed., 2015, 54, 5049–5053 CrossRef CAS PubMed; (b) T. Bug, M. Hartnagel, C. Schlierf and H. Mayr, Chem. – Eur. J., 2003, 9, 4068–4076 CrossRef CAS PubMed; (c) H. Jangra, Q. Chen, E. Fuks, I. Zenz, P. Mayer, A. R. Ofial, H. Zipse and H. Mayr, J. Am. Chem. Soc., 2018, 140, 16758–16772 CrossRef CAS.
- (a) Y. Dong, M. Wang, J. Liu, W. Ma and Q. Liu, Chem. Commun., 2011, 47, 7380–7382 RSC; (b) N. Velasco, C. Virumbrales, R. Sanz, S. Suárez-Pantiga and M. A. Fernández-Rodríguez, Org. Lett., 2018, 20, 2848–2852 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data. See DOI: 10.1039/c9cc01060b |
| This journal is © The Royal Society of Chemistry 2019 |