
Lipid bilayer formation on organic electronic materials†
Yi
Zhang
a,
Shofarul
Wustoni
a,
Achilleas
Savva
a,
Alexander
Giovannitti
 b,
Iain
McCulloch
bc and
Sahika
Inal
*a
b,
Iain
McCulloch
bc and
Sahika
Inal
*a
aBiological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: sahika.inal@kaust.edu.sa
bDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK
cPhysical Sciences and Engineering Division, KAUST Solar Center (KSC), KAUST, Thuwal 23955-6900, Kingdom of Saudi Arabia
First published on 23rd April 2018
Abstract
The lipid bilayer is the elemental structure of a cell membrane, forming a stable barrier between the interior and exterior of the cell while hosting membrane proteins that enable selective transport of biologically important compounds and cellular recognition. Monitoring the quality and function of lipid bilayers is thus essential and can be performed using electrically active substrates that allow for transduction of signals. Such a promising electronic transducer material is the conducting polymer poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS) which has provided a plethora of novel bio transducing architectures. The challenge is, however, in assembling a bilayer on the conducting polymer surface, which is defect-free and has high mobility. Herein, we investigate the fusion of zwitterionic vesicles not only on a variety of PEDOT:PSS films, but also on an electron transporting, negatively charged organic semiconductor, in order to understand the surface properties that trigger vesicle fusion. The PEDOT:PSS films are prepared from dispersions containing different concentrations of ethylene glycol included as a formulation additive, which gives a handle to modulate the surface physicochemical properties without a compromise on the chemical composition. A strong correlation between the polarity of the surface, the fusion of the vesicles and the mobility of the resulting bilayer aids in extracting the design principles for the development of future conducting polymers that will enable the formation of lipid bilayers.
Introduction
Biomembranes act as an essential barrier that separates tissues or cells from their surrounding environment. For instance, a number of toxins secreted by pathogenic bacteria attack the biomembranes by disrupting the integrity of their structure, causing various diseases.1 The biomembrane integrated with its proteins is also essential in maintaining cellular communication and function by enabling selective transportation of certain compounds and ions in and out of the cell. Investigating the integrity and the function of a biomembrane is therefore not only crucial to understand biological phenomena but also to develop diagnostic tools for pharmaceutical screening and in designing therapeutics against pathogen attack. As the natural biomembrane is highly complex, its major structural component, i.e. the phospholipid bilayer, has provided a framework to access information regarding cellular machinery including critical membrane interactions among the constituents and the properties of membrane proteins.2–6 In particular, planar lipid bilayers supported by substrates provide a stable and robust platform. These supported lipid bilayers (SLBs) are self-assembled upon the adsorption and fusion of lipid vesicles on hydrophilic and smooth surfaces such as glass,7 mica,8 and silica.9 SLBs assembled on such surfaces are compatible with a variety of surface sensitive characterization techniques including total internal reflection fluorescence microscopy (TIRF),10 atomic force microscopy (AFM),11 quartz crystal microbalance with dissipation monitoring (QCM-D),12 and surface plasmon resonance (SPR).13Electrical methods can allow for high-throughput and label-free sensing of the integrity of the bilayer and the functionality of biological membrane receptors or channels using chip-based arrays.14 They rely on the intrinsically high electrical resistance of the bilayer and as such can monitor changes in membrane conductance associated with the transport of ions through ion channels or in membrane surface potential associated with membrane function and protein binding events.15 SLBs have been formed on conducting or semiconducting substrates such as single-walled carbon nanotubes (SWNTs),16 gold,17 indium tin oxide (ITO)18 or polypyrrole.19 In a recent example, Zhou et al. recorded currents from individual ion channels using transistors based on SWNTs functionalized with polymeric cushions, relying on the ion channel current charging directly the quantum capacitance of a single NT in a network of purified semiconducting NTs.16 Despite the high potential of electrical methods for monitoring lipid bilayers, a challenge remains in generating high resistance SLBs on electronic surfaces which can efficiently transduce signals across the membrane while allowing for integration with functional transmembrane proteins.
A major drawback of traditional metal or metal oxide materials is in fact related to the limited space between the lipids and the substrate surface, which restricts the insertion of large membrane proteins or leads to their denaturation due to the direct contact with the rigid surface and therefore limiting the biomimetic properties of the SLB.15 A promising material to interface the SLB is the conducting polymer, poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS). PEDOT:PSS is not only an efficient ion-to-electron transducer,20 but can also act as a hydrated polymer cushion providing a fluid environment for the bilayer. In fact, due to its compatibility with biological systems and good electrochemical stability in aqueous electrolytes, PEDOT:PSS has been the material of choice for a variety of electronic devices applied at the interface in biological systems. Examples include PEDOT:PSS based microelectrodes and organic electrochemical transistors (OECTs) for recording electrophysiological signals,21–23 monitoring cell coverage,24 as well as barrier tissue formation.25 The OECT is an electrolyte-gated transistor with record high transconductance, implying amplification of small biological signals into electronic current.26 Owens and co-workers recently showed, for the first time, the formation of a SLB on a PEDOT:PSS film and an organic electrochemical transistor (OECT) using PEDOT:PSS in its channel via vesicle fusion.27 These bilayers were characterized using QCM-D, fluorescence recovery after photobleaching (FRAP) and impedance spectroscopy. Due to the inherent amplification of the OECT, the integrity of the bilayer and the effects of a pore-forming toxin (α-hemolysin) on the bilayer integrity could be monitored.27 However, the resistance of the SLB was lower than the state-of-the-art, indicating that the vesicles did not completely transform into a bilayer. Instead, a mixed population of vesicles and bilayers existed on the conducting polymer surface.27 In order to achieve high sensitivity and obtain high throughput electrical measurements, a high resistivity and a defect-free structure of the bilayer are prerequisites.
With recent advances in the microfabrication and patterning technologies of conducting polymers,28 assembling transmembrane protein incorporated lipid bilayers on PEDOT:PSS surfaces shows great promise for the development of highly sensitive bio transducers such as OECTs. However, little is known about what triggers vesicles to form SLBs on these surfaces. Surface structures and the physicochemical properties of the substrate play an important role in vesicle fusion, and the structure and properties of the SLB.29–32 The interactions of the vesicles with the substrate are susceptible to the properties of the substrate such as surface charge, roughness and composition, as well as to the properties of the vesicles (e.g. composition, charge and size) and the buffer environment (pH, ionic strength and composition). As the field of organic bioelectronics expands beyond PEDOT:PSS and lipid bilayers are extremely interesting for the development of diagnostic tools for cells as well as designing therapeutics, it is crucial to understand what type of organic semiconductor surfaces are required to promote vesicle fusion. In this work, we investigate the fusion of zwitterionic vesicles on a variety of conjugated polymer films. We investigate PEDOT:PSS films, which have the same chemical composition but different surface physicochemical characteristics. We obtain these films via spin-cast dispersions of PEDOT:PSS containing different amounts of a solution additive, i.e., ethylene glycol (EG). Varying the EG content in the dispersion leads to different surface properties. We monitor vesicle fusion and subsequent lipid bilayer formation on these films using FRAP and QCM-D and find that the mobility of the bilayers depends strongly on the polarity of the surface, regulated by the EG content. Moreover, intact vesicles on an electropolymerized PEDOT:PSS film and an n-type organic semiconductor film reveal the importance of surface roughness and charge density for vesicle fusion, respectively. As such, we identify the surface properties that promote vesicle fusion and shed light on the vesicle fusion process on conjugated polymers as well as the factors controlling the resulting structure of the SLBs. These results will aid in extracting design rules for the development of future materials which will enable defect-free SLBs.
Experimental
Materials
PEDOT:PSS (Clevios PH 1000) was purchased from Heraeus Clevios GmbH. The n-type semiconducting polymer p(OH-gNDI-gT2) is based on the electron deficient 2,6-dibromonaphthalene-1,4,5,8-tetracarboxylic diimide (NDI). The polymer was synthesized starting from polymer p(gNDI-gT2)33 and the synthesis is described in the ESI.† The polymer can be dissolved in organic acids such as trifluoroacetic acid (TFA) and the 1H NMR spectrum is given in Fig. S1 (ESI†). 4-Dodecylbenzenesulfonic acid (DBSA), (3-glycidyloxypropyl)trimethoxysilane (GOPS), ethylene glycol (EG), chloroform, phosphate buffered saline (PBS) and trifluoroacetic acid were purchased from Sigma-Aldrich. The lipids, 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) and 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (DPhPE), were purchased from Avanti Polar Lipids (Alabaster, USA). N-(4,4-Difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (BODIPY-DHPE) was purchased from Thermo Fisher Scientific. The hydrodynamic diameter of the vesicles was measured to be ca. 118 nm using dynamic light scattering measurements.Film preparation
Microscope glass slides and cover glasses (24 × 32 mm; No. 1.5; VWR) were cleaned with Piranha solution (75% H2SO4 and 25% H2O2, v/v) followed by O2 plasma cleaning at 150 W for 2 min. PEDOT:PSS dispersions were prepared by mixing EG (between 0 and 20% by volume of dispersion as noted in the text/figures), DBSA (0.002% v/v), GOPS (1% w/w) and Clevios PH 1000 before ultra-sonication (30 min) and filtration through 1 μm glass fiber filters. PEDOT:PSS films were spin-cast on the glass substrates at 3000 rpm for 45 seconds unless otherwise stated. After spin-coating, the films were baked at 140 °C for 1 hour.The semiconducting polymer, p(OH-gNDI-gT2), was dissolved in trifluoroacetic acid at 2.5 mg mL−1 and drop-cast on a parylene C-coated glass substrate. Parylene C was used as a hydrophobic coating to promote the stability of the polymer film in aqueous media.
For the preparation of the electropolymerized PEDOT:PSS, we used a three-electrode system of a PEDOT:PSS-coated glass substrate as the working electrode, a platinum mesh counter electrode, and an Ag/AgCl reference electrode, all connected to a potentiostat (Metrohm Autolab). The monomer, EDOT, (10 mM) and the dopant PSS (0.8 wt%) were mixed in DI water in a glass cell. Electropolymerization of PEDOT:PSS was carried out using the potentiostatic mode, where the sample was biased at 1 V vs. Ag/AgCl for 300 s at room temperature. The polymer deposited electrode was then washed with DI water, followed by spraying with N2.
Before incubating the polymer films with lipid vesicles, all samples were immersed in DI water for 24 hours and dried with N2 spray, followed by O2 plasma activation at 25 W for 2 min. The PDMS wells were then assembled on top of the polymer-coated glass substrates and used to define the area for lipid bilayers.
Vesicle preparation
DPhPC and DPhPE were mixed in chloroform (molar ratio 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) with 0.5 mol% BODIPY-DHPE. Bulk chloroform was evaporated under a stream of N2 and any remaining solvent was removed under vacuum for at least 3 hours. PBS was then added to the dried lipid film which was re-suspended in a sonication bath for 30 minutes. The resulting multi-lamellar vesicles were extruded through a polycarbonate membrane (Whatman Nucleopore) with a pore size of 50 nm using a mini-extruder (Avanti Polar Lipids).
:3) with 0.5 mol% BODIPY-DHPE. Bulk chloroform was evaporated under a stream of N2 and any remaining solvent was removed under vacuum for at least 3 hours. PBS was then added to the dried lipid film which was re-suspended in a sonication bath for 30 minutes. The resulting multi-lamellar vesicles were extruded through a polycarbonate membrane (Whatman Nucleopore) with a pore size of 50 nm using a mini-extruder (Avanti Polar Lipids).
Fluorescence recovery after photobleaching (FRAP)
FRAP experiments were performed using a Zeiss LSM 800 or LSM 880 inverted microscope (Zeiss Germany) to determine the mobility of the SLB. With the lipid mixture, 0.5 mol% BODIPY was used as a fluorescent label. The formed bilayers were gently scratched using a needle to remove a small portion of the bilayer material. The scratches facilitated focusing at the plane of the bilayer for microscope imaging. Following this step, the bilayer was rinsed with PBS for at least 1 min to wash away the removed lipid material. A laser beam was used to bleach a circular spot in the bilayer membrane. The recovery of the fluorescence intensity of the bleached spot was recorded to determine the mobility of the lipid bilayer.34,35 After background subtraction and normalization, the recovery data was fitted using a Bessel function followed by a Soumpasis method.36 The diffusion coefficient (D) was calculated using the following equation:where w is the radius of the photobleached spot and t1/2 is the time required to achieve half of the maximum recovery intensity.
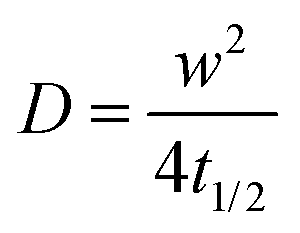
An electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D)
Combined QCM-D/electrochemical impedance spectroscopy measurements for PEDOT:PSS (dispersions with varying EG content)-coated SiO2 sensors were performed on a QSense Analyzer system (Biolin Scientific AB, Sweden) equipped with an electrochemistry module. The measurements were carried out at 24 °C with a flow rate of 100 μL min−1 controlled by a peristaltic pump. After stabilization of the baseline in PBS, the vesicles in PBS (0.5 mg mL−1) were injected into the sensors. The adsorption of the vesicles was monitored through changes in the resonance frequency (Δf) and energy dissipation (ΔD) as a function of time using several overtones. All QCM-D data presented in this work were recorded at the 7th overtone. During these measurements, we also simultaneously measured the impedance of the sensors placed in an electrochemistry module which was connected to a potentiostat (Autolab PGSTAT128N). The sensor was used as the working electrode while a platinum foil present in the module acted as the counter electrode and an Ag/AgCl electrode was used as the reference electrode. The impedance spectra of the films were acquired during different stages of bilayer formation under an AC potential of 10 mV and a DC voltage that corresponds to the open-circuit potential (Voc) before sending vesicles. The impedance spectra of these samples were also recorded at −0.05 V vs. Voc after the formation of lipid membranes. We performed a second set of measurements, this time for only PEDOT:PSS, without adding vesicles. The films were stabilized in PBS and then impedance spectra were recorded first at V = Voc and then V = −0.05 V vs. Voc. In order to exclude slight differences between the two PEDOT:PSS films studied for each EG content, we normalized the spectra at Voc with that recorded for PEDOT:PSS (no lipids, at Voc) and a calibration curve was obtained for each sample. This calibration curve was then used to derive the corresponding impedance spectra of the films at −0.05 V vs. Voc.Contact angle measurements
The wettability of the polymer films was determined using a goniometer (KRUSS, DSA100, Germany). The mean value was obtained by measuring water contact angles at three different locations of the substrate. Each measurement took place 15 s after a 5 μL deionized water droplet was deposited on the substrate.Surface zeta potential
The surface zeta potentials of the spin-cast PEDOT:PSS films, the electropolymerized PEDOT:PSS film and the p(OH-gNDI-gT2) film were determined using a Zetasizer Nano ZS (Malvern Instruments Ltd, UK) equipped with a surface zeta potential cell (ZEN1020). A small piece of the polymer-coated glass substrate was attached to the sample holder and placed in the measurement cell. Each measurement was performed at 25 °C in 10 mM KCl (pH 7.4) containing Coffee-mate® (Nestle S.A.) as tracer particles. The electrophoretic mobility of the tracer particles was measured at four different distances from the surface of the sample. The values were used to determine the surface zeta potential by the Zetasizer software. Three measurements were carried out for each sample.Atomic force microscopy (AFM)
The surface topography of the polymer samples was studied using an Agilent 5500 SPM system (Agilent Technologies, USA). All measurements were carried out in tapping mode in air and PBS.X-ray photoelectron spectroscopy (XPS)
XPS experiments were performed using a Kratos Axis Ultra DLD instrument equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) operating at a power of 150 W and under UHV conditions in the range of ∼10−9 mbar. All spectra were recorded in hybrid mode using electrostatic and magnetic lenses and an aperture slot of 300 μm × 700 μm. Survey and high-resolution spectra were acquired at fixed analyzer pass energies of 160 eV and 20 eV, respectively. The samples were mounted in floating mode in order to avoid differential charging. The spectra were acquired using charge neutralization and referenced by using neutral carbon (C 1s) at 284.8 eV. The background subtraction was carried out by the Tougaard method and convolution of all the component peaks was fitted by a mixed Gaussian and Lorentzian method.Results and discussion
Monitoring the formation of supported lipid membranes
It is well known that lipid bilayers spontaneously assemble on siliceous substrates via vesicle fusion. Though not completely understood, vesicle fusion on a solid substrate is thought to follow a two-phase process (Fig. 1a).5,29 The vesicles first adsorb onto the substrate. When a critical coverage of the adsorbed vesicles is reached, they start to deform, rupture and fuse, forming bilayer patches on the substrate. The edges of the SLBs spread and further induce adjacent vesicles to rupture forming a continuous bilayer.9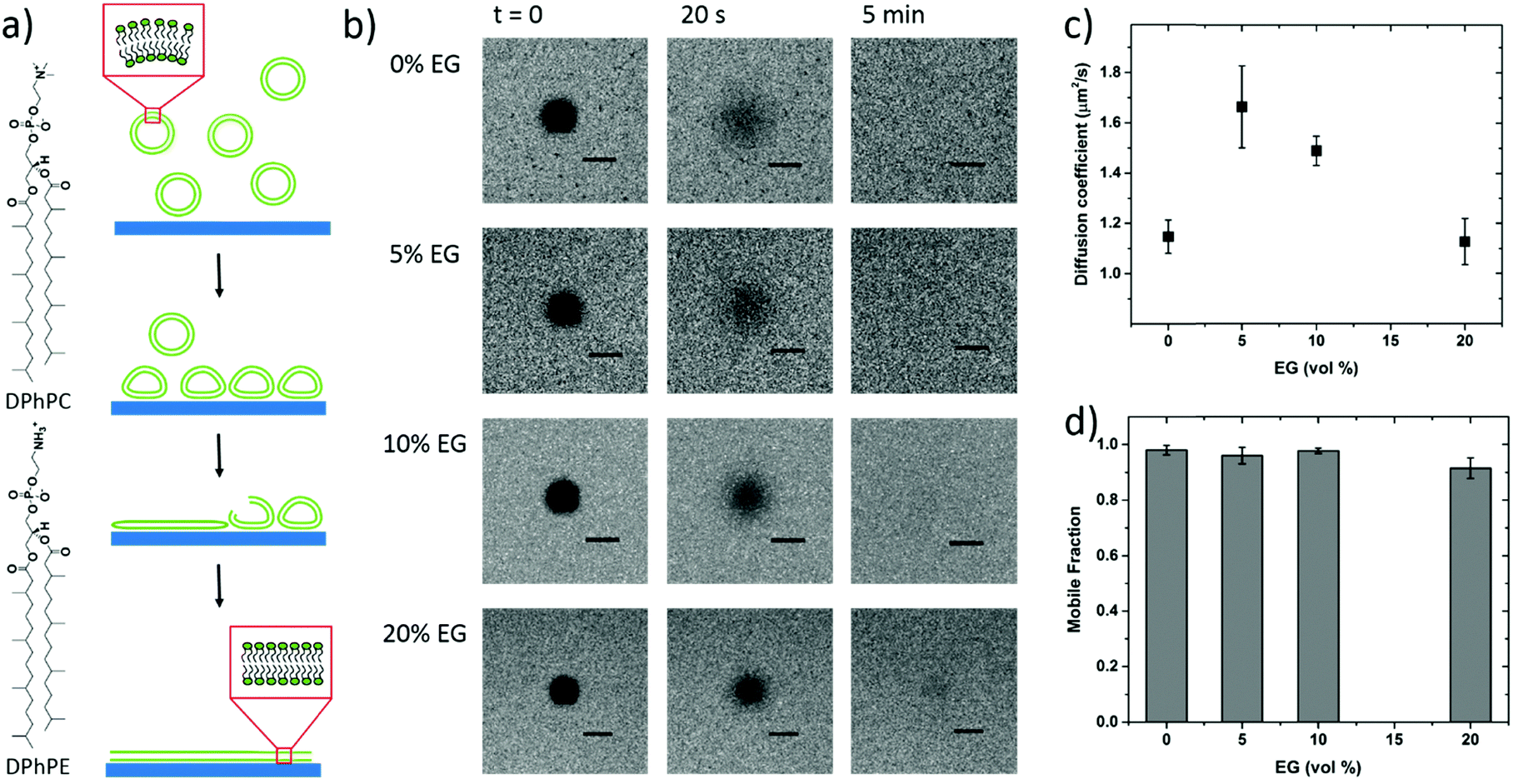 | ||
| Fig. 1 (a) Schematics of vesicle fusion, and (b) FRAP images of the lipid membranes DPhPC/DPhPE (7:3) assembled on the PEDOT:PSS films cast from dispersions containing various EG concentrations (0 vol%, 5 vol%, 10 vol% and 20 vol%). These images are treated to extract diffusion coefficients (c), and mobile fractions (d) of the membrane lipids; scale bar = 20 μm. | ||
To mimic the natural biomembrane, a key property is the two-dimensional mobility of the lipid molecules within the bilayer. The fluidity of the lipid membrane is a critical factor for interactions between the membrane constituents, membrane transport and enzyme activities.37,38 For instance, the integral membrane enzymes such as Ca2+-ATPase, Na, K-ATPase and cytochrome oxidase are less active when the lipid membrane becomes less fluid, consequently affecting the reaction rates.37 We, therefore, firstly characterize the SLBs using FRAP which allows for estimating the homogeneity and lateral mobility of the layer formed upon vesicle fusion. Fig. 1b shows the typical fluorescence images of the lipid membranes assembled from zwitterionic vesicles, DPhPC and DPhPE, on the PEDOT:PSS films over time after photobleaching. DPhPC and DPhPE belong to a class of phospholipids known as phytanyl lipids which are derived from archaebacterial membranes.39 Due to their high mechanical and chemical stability and low ion permeability,40 they are commonly used to mimic the cell membranes for ion-channel characterization41,42 and to investigate molecule/peptide–lipid interactions.43,44 For the bilayers formed with a particular composition of DPhPC mixed with DPhPE, high electrical resistances varying between 2 and 10 GΩ were reported.45 High resistance is necessary for studying small ionic fluctuations across the bilayer as in the case of functional ion channels. The recovery of the fluorescence indicated the formation of the lipid bilayer on all of these surfaces. Next, we investigate the properties of the lipid bilayer and its correlation to the EG content. The diffusion coefficient (D) and mobile fraction (MF) for each sample were determined based on the imaging data and are plotted in Fig. 1c and d, respectively. The diffusion coefficient gives an estimation of how fast the lipid molecules diffuse within the membrane. The mobile fraction is indicative of whether the lipid molecules are bound to the surface and hence, are unable to diffuse.
The lipid membrane formed on the PEDOT:PSS film cast from the dispersion containing 5 vol% EG exhibited the highest diffusion coefficient (D = 1.67 μm2 s−1) among all samples. The lowest coefficient was observed for the film with 20 vol% EG (D = 1.13 μm2 s−1), similar to the diffusion properties of the bilayer on the film that does not contain EG. We found that at least 91% of the labelled lipid molecules were laterally mobile in the membranes formed on all the PEDOT:PSS films, regardless of the EG content, suggesting continuity of the bilayers. The mobility of the SLB formed on glass was measured for comparison (Fig. S2, ESI†). The mobile fraction of the SLB on glass was 97% and the diffusion coefficient was determined to be 1.5 ± 0.1 μm2 s−1, comparable to the values reported for the PEG-cushioned DPhPC bilayer.46 We note that the addition of DPhPE lipids to the composition has been shown to reduce the fluidity further.47 The diffusion coefficient of the bilayer on glass is similar to that measured for the lipid membrane formed on PEDOT:PSS with 5 vol% and 10 vol% EG, validating our analysis.
To further confirm vesicle fusion on the bilayers and monitor its dynamics, we used the QCM-D technique. QCM-D is a surface sensitive technique that allows for real-time monitoring of the interactions between the adsorbed vesicles and the film and for quantifying mass changes in the film associated with vesicle fusion.30 For instance, a decrease in the oscillation frequency of the quartz crystal coated with the polymer film accompanied by an increase in the dissipation of its energy (dampening of the crystal) indicates that the film up-takes mass. Fig. 2 shows the QCM-D signals of the PEDOT:PSS films upon introduction of the vesicles – once they are completely swollen in PBS. For all the films, we observe a decrease in frequency upon addition of the lipid vesicles (Δfaverage = −245 Hz), the value of which is highest for the 5 vol% sample (Table S1, ESI†). The dissipation change, on the other hand, is lowest for this sample. The high dissipation changes of the PEDOT:PSS films (ΔDaverage = 85 × 10−6) suggest that the adsorbed vesicles enhance the viscoelasticity of the surfaces. Such high Δf and ΔD values were reported for SLBs formed on polyelectrolyte coated surfaces, attributed to the extensive hydration of waterborne regions.48
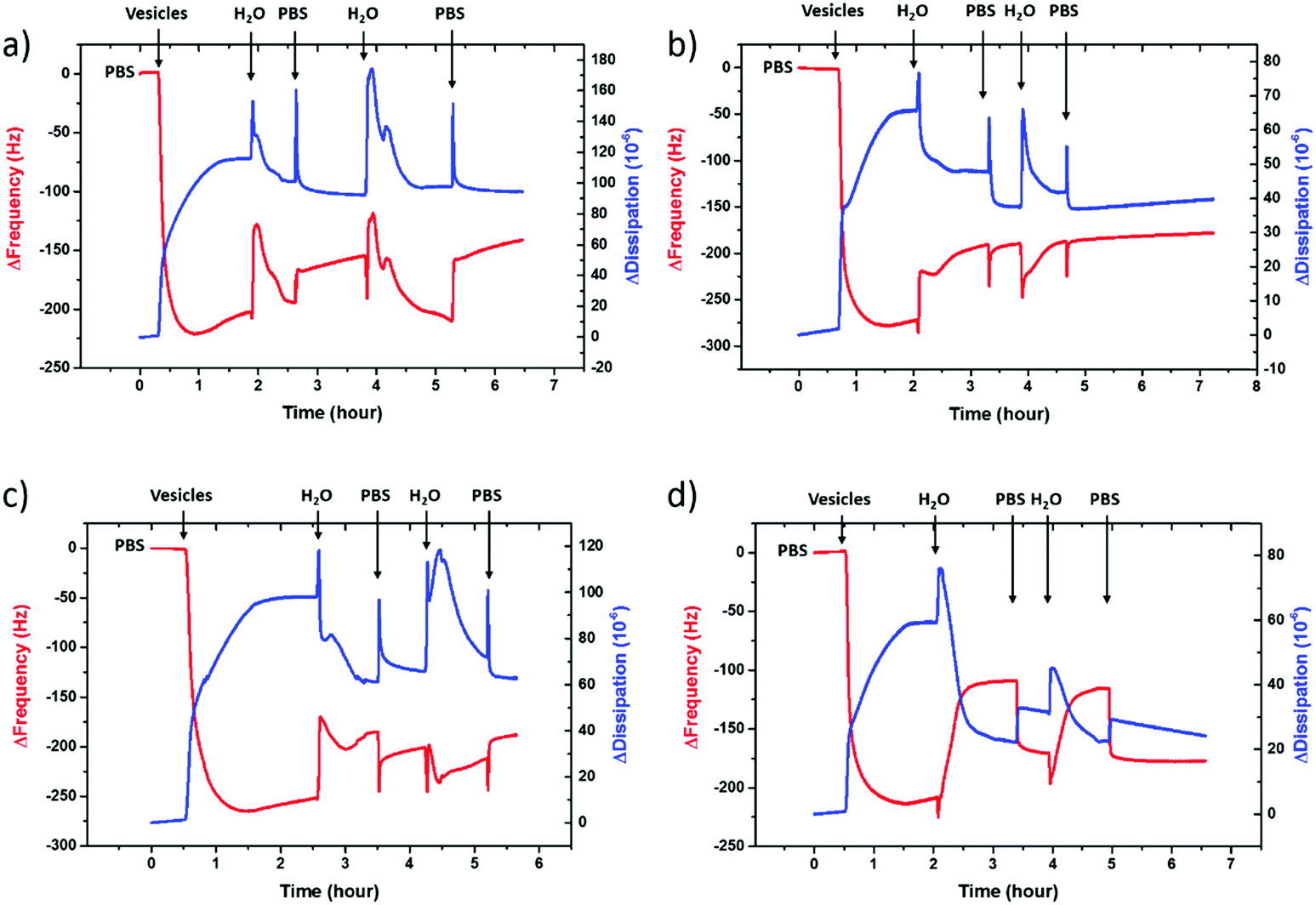 | ||
| Fig. 2 QCM-D monitoring of zwitterionic SLB formation on the PEDOT:PSS surfaces. The vesicles are injected after the baseline buffer (PBS) stabilization followed by two sets of water shocks each ending with buffer injection. The net effect of water is a rapid increase in the frequency and a decrease in the dissipation. Data presented here are recorded at the 7th overtone. From a to d, the EG content increases from 0 to 20 vol%. | ||
It was shown that incubating the intact vesicles on the PEDOT:PSS films in DI water creates an osmotic pressure which then triggers vesicle fusion.27 We therefore applied two consecutive water shocks to rupture the vesicles adsorbed on the surface. For all the films, we observe that these two waves of DI water addition lead to a rapid increase in the frequency (loss of mass), as well as a decrease of dissipation (reduced viscoelasticity) (Fig. 2). For the 0 and 5 vol% EG samples, while most of the fusion happens during the first water injection, for the 10 and 20 vol% EG samples, the 2nd water shock seems to be more effective. Table S1 (ESI†) summarizes the variations in the QCM-D signatures in terms of the initial adsorption of the vesicles and the vesicle rupture with subsequent bilayer formation after the 1st and final water shocks. These variations indicate vesicle rupture on the PEDOT:PSS surfaces, as evidenced by FRAP measurements. The QCM-D signals of all the films, higher than those reported for inorganic surfaces,27 and the complex nature of the vesicle–surface interactions suggest that the bilayers that are formed are not defect-free, in fact comprising a population of intact vesicles and bilayer patches, in agreement with a previous study with PEDOT:PSS.27 The low final ΔD value of the 5 vol% EG film, however, shows that the bilayer formed therein is reflecting a relatively more rigid and less water containing layer when compared to other films under investigation.
To be able to compare the sealing properties of the SLBs, we coupled an electrochemical impedance spectrometer to the QCM-D sensor. The impedance properties of the films are not significantly affected when going from the pristine PEDOT:PSS film to that after bilayer formation, except that for the formulation with 5 vol% EG, we observe an increase in the magnitude of impedance with the bilayer (Fig. S3, ESI†). We attribute this to the large area of the studied films (area of the EQCM-D sensor is 0.79 cm2), i.e., parts of the film that may not be covered with vesicles or bilayers and therefore solution ions are not impeded by a continuous lipid bilayer. Nonetheless, in order to understand the impact of this particular EG concentration (5 vol%) on vesicle fusion and the peculiar trend of lipid bilayer fluidity with EG, we sought to investigate the physicochemical characteristics of these film surfaces.
Surface hydrophilicity and zeta potential
Surface hydrophilicity is a prerequisite for the rupture of vesicles and the subsequent formation of bilayers.49 Fully fluid lipid membranes are noted to form on only highly hydrophilic surfaces,8,29,50 albeit a fluid bilayer could as well be assembled on a plasma-modified PDMS surface with a contact angle as high as 30°.49 Note that all our films were immersed in DI water overnight, followed by O2 plasma activation before depositing the vesicles on the films. In fact, we do not observe fluorescence recovery when lipids are deposited on the as-prepared films. We soak the films in DI water over night to remove any low molecular mass components from the film, inherent to the commercially available dispersion of PEDOT:PSS as these might interfere with the accuracy of the electrical recordings that require long term exposure of the films to water. Additionally, PEDOT:PSS uptakes a considerable amount of water,51 which softens the film, envisaged to make the substrate surface more hydrophilic and therefore more suitable for integration with lipids. We determine the wettability of our polymer films by water contact angle measurements. The films after water treatment have a higher contact angle (Table S2, ESI†). This is in agreement with previous reports demonstrating a water rinse52 or water immersion53 to remove the excess PSS moieties from the surface of the PEDOT:PSS films.The increased hydrophobicity, which is not favored for vesicle fusion, is counteracted by O2 plasma treatment, which improves the wettability of polymer surfaces by introducing polar functional groups containing oxygen.54–56 Ohayon et al. demonstrated that the water contact angle of a PEDOT:PSS film (identically prepared to the ones studied here) reduced from 45° to less than 5° after O2 plasma activation as a result of the introduction of hydroxyl (–OH) groups at the surface.28 After this “fusion treatment”, i.e., water immersion and plasma activation, proven to be essential to drive vesicle fusion on PEDOT:PSS, all of our films become superhydrophilic (contact angle = 0°) (Table S2, ESI†). However, a hydrophilic surface by itself is insufficient to induce bilayer formation through vesicle fusion,29,57 and does not guarantee a high quality (e.g. high lateral mobility) of the bilayer formed as seen for the PEDOT:PSS films. Previous studies showed the adsorption of intact vesicles on hydrophilic TiO2 surfaces,31,58,59 which is ascribed to weak vesicle–surface interactions. On the other hand, the SLB formation is highly influenced by electrostatic interactions between the vesicles and the substrate.60–64 Cha and coworkers investigated the role of electrostatic interactions in the rupture of zwitterionic liposomes on charged surfaces, demonstrating the critical role of surface charge density in controlling the fusion.65
To evaluate the charge density on the film surfaces, zeta potentials of the samples were measured using a Malvern Zetasizer Nano ZS system. Fig. 3 shows the surface zeta potential values of the spin-cast PEDOT:PSS films with various EG concentrations before and after the fusion treatment. The as-prepared films had negative surface potentials varying between −74.5 mV and −46.4 mV, indicating that the anionic PSS-rich layers are at the outmost surface (Fig. 3, A). After the treatment, the surface zeta potential decreases for all samples (Fig. 3, B). Negatively charged surfaces such as mica, silica and glass are widely used for forming zwitterionic lipid bilayers via vesicle fusion,66 suggesting that all of these films are potentially suitable for bilayer formation from the zwitterionic vesicles.
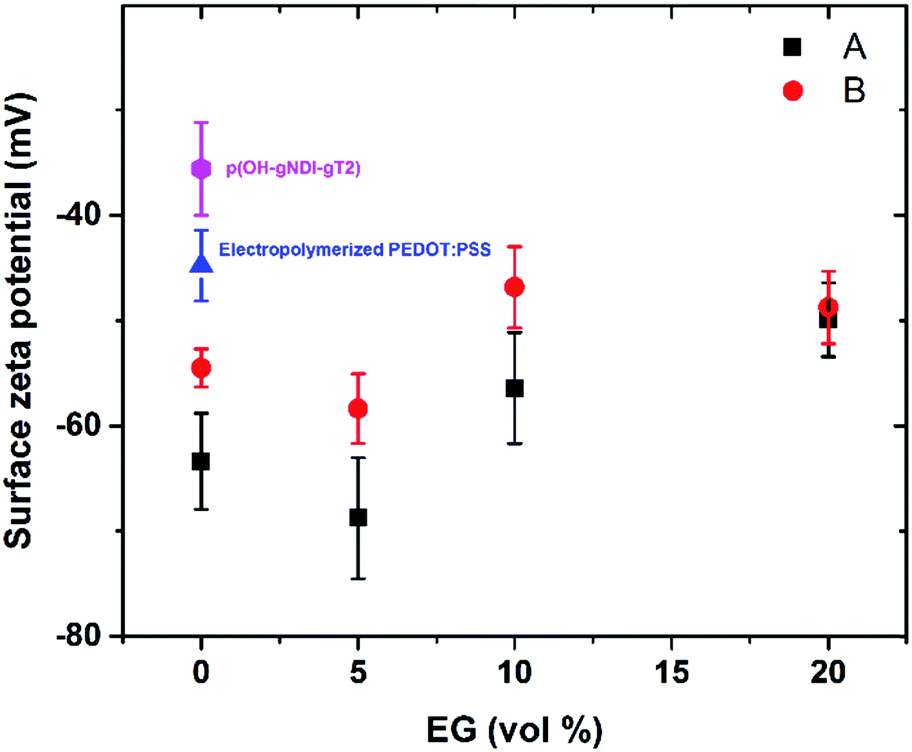 | ||
| Fig. 3 Surface zeta potential measurements of the PEDOT:PSS films with various EG concentrations before (A: black squares) and after (B: red dots) fusion treatment, i.e. immersion of the films in DI water and O2 plasma activation, before incubating the samples with vesicles. Also shown are the surface zeta potential measurements of electropolymerized PEDOT:PSS (blue triangle) and p(OH-gNDI-gT2) (pink hexagon) films exposed to the same treatment. | ||
The PEDOT:PSS film with 5 vol% EG has the highest surface zeta potential of −58.4 ± 3.3 mV, being the most negatively charged surface among all the samples. It is interesting to note however that although the zeta potentials do not vary broadly (from −61.7 mV to −43 mV), this most negatively charged surface is also the one that enables the most mobile lipid bilayer (Fig. 1c). However, we would like to understand the effect of surface charges on vesicle fusion itself. For this purpose, we fabricated an electropolymerized PEDOT:PSS film which is an alternative conducting polymer surface to test vesicle fusion without altering the chemical structure. The zeta potential of this electropolymerized film was −44.8 ± 3.4 mV after the water and plasma treatment (Fig. 3, blue triangle). Although this value is only slightly less than the film cast from the 10 vol% EG containing PEDOT:PSS dispersion, we observe no bilayer formation on the electropolymerized film (Fig. S4a, ESI†). It is most likely that the intact vesicles are adsorbed on the polymer surface. This result is intriguing as this negatively charged surface is also hydrophilic with a contact angle of ca. 10°. Neither the surface charge nor the wettability solely determines the vesicle fusion, therefore we analyze the topography of the films.
Surface roughness
We characterized the surface topography of the hydrated PEDOT:PSS films with tapping-mode AFM in PBS as the films have undergone fusion treatment. Large domains were observed for the PEDOT:PSS films cast from EG-containing dispersions (Fig. 4a). It is suggested that EG addition leads to the partial removal of PSS from the surface, accompanied by reorientation of the PEDOT chains from a coiled to linear structure and the formation of denser PEDOT aggregates.67–69 These drastic morphological changes enable better transport pathways for charges, and as such EG improves the electrical conductivity of the PEDOT:PSS films.68,70 The surface roughness (root mean square), Sq, was determined for each sample based on the AFM images with a scan area of 2 μm × 2 μm. PEDOT:PSS with 5 vol% EG shows the greatest roughness (Sq = 3.1 nm) while PEDOT:PSS without EG has the smoothest surface (Sq = 2.2 nm) (Fig. 4b, A). Despite these differences, the surfaces with such roughness values are deemed to be smooth.71 On the other hand, the Sq value for electropolymerized PEDOT:PSS in PBS is 7.2 nm (Fig. 4b, blue triangle, and see Fig. S4b (ESI†) for the AFM image of this film). Although roughness was considered to be strongly influential in the adsorption of vesicles72 and spreading of lipid membranes,61,62 several groups reported the formation of lipid bilayers on rough surfaces.19,73–75 In fact, for such rough surfaces, if the vesicle–substrate interactions are strong enough, roughness no longer plays a predominant role in vesicle fusion. The electropolymerized PEDOT:PSS film presents surface charges and the wettability in favor of vesicle fusion. However, the interactions between the vesicle and the film are inadequate (note that the zeta potential is at the lowest end of the tested range) to overcome the adverse influence of a rough surface.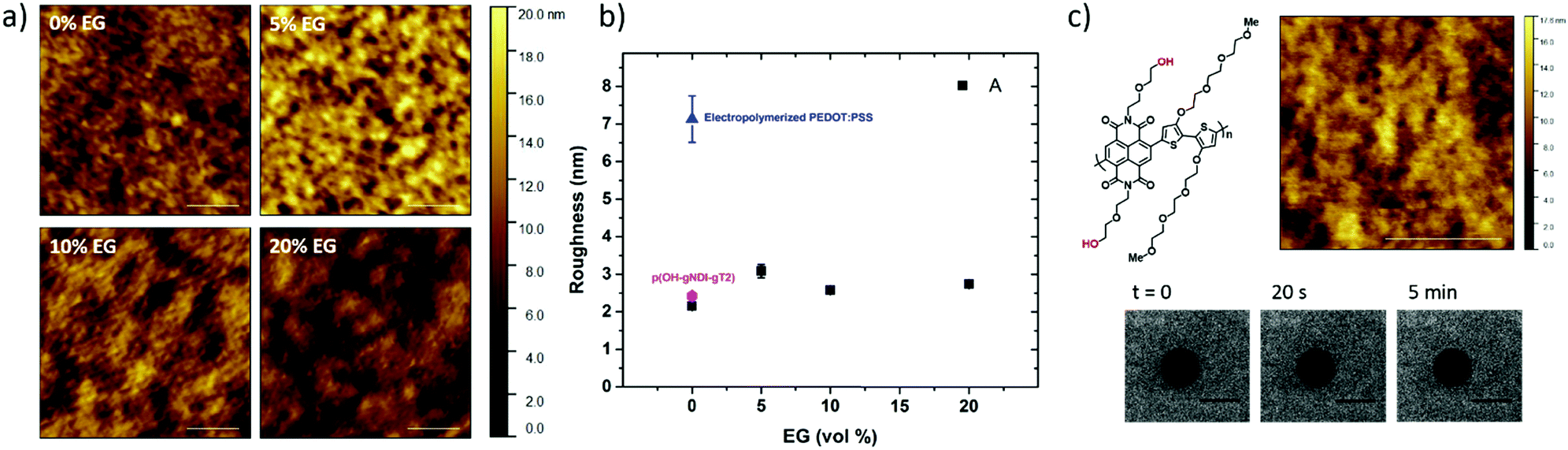 | ||
| Fig. 4 (a) AFM topography images of the PEDOT:PSS films in PBS at pH 7.4. The films were cast from dispersions with various EG concentrations and the images were taken after DI water immersion and O2 plasma activation. Scale bar = 500 nm; (b) surface roughnesses of the hydrated films with various EG contents (A: black squares) and those of the electropolymerized PEDOT:PSS (blue triangle) and p(OH-gNDI-gT2) (pink hexagon) after treatment and (c) AFM topography image of the p(OH-gNDI-gT2) film in PBS at pH 7.4 after water immersion and O2 plasma activation. The FRAP images show no recovery of fluorescence when lipids were deposited on such a polymer surface. The scale bars for the AFM and FRAP images are 400 nm and 20 μm, respectively. | ||
In order to distinguish the combined effects of roughness and surface charges, we also evaluated a semiconducting polymer containing hydroxyl groups, p(OH-gNDI-gT2), which is an analogue of the polymer p(gNDI-gT2), recently demonstrated as the first electron transporting polymer used in an accumulation mode OECT.33 The accumulation mode OECTs comprising this n-type polymer exhibit a high signal ON/OFF response (translating into high gain) and a low-power operation in aqueous electrolytes. Working in the opposite regime to PEDOT:PSS based devices (i.e. depletion mode, signal ON to OFF upon the application of a positive gate voltage), the n-type channel of this OECT becomes conducting only when cations from the solution enter the film, rendering the system a very promising platform to sense the bilayer functionality, e.g., opening of an ion channel for the transport of cations. For the synthesis of p(OH-gNDI-gT2), the ester group was cleaved to form free –OH groups (see the chemical structure in the ESI,† and Fig. S1 for NMR spectrum), resulting in an increase in surface hydrophilicity. The polymer p(OH-gNDI-gT2) exhibits similar roughness to the PEDOT:PSS films cast from dispersions (2.4 nm, see the AFM image in Fig. 4c) and has a contact angle of 21°–22° after water and plasma treatment. However, the lipids deposited on this polymer were immobile, deduced from the FRAP experiment (Fig. 4c). The corresponding surface zeta potential for the p(OH-gNDI-gT2) polymer film was measured to be −35.6 ± 4.4 mV (Fig. 3, pink hexagon), which is the lowest among all the samples investigated so far. We postulate that the vesicles are intact on the surface due to weak vesicle–substrate interactions incapable of overcoming the bending energy of the vesicle membrane. These findings are in agreement with the results of Cha et al. who reported the critical role of surface charge density on the rupture of adsorbed zwitterionic vesicles to form SLBs.65 On either positively or negatively charged self-assembled monolayer surfaces, there is a critical charge density below which zwitterionic vesicles readily fuse into SLBs. Above this value, (in our work, this would correspond to a zeta potential value higher than ca. −40 mV), the zwitterionic vesicles do not rupture no matter how smooth or hydrophilic the surface is.
XPS characterization
We used XPS to characterize the chemical composition of the PEDOT:PSS surfaces. We particularly assessed the S 2p spectra as shown in Fig. 5. In the spectra, the high binding energy region (BE = ca. 169 eV) is attributed to the sulfur atoms in the PSS, whereas the lower binding energy area (BE = ca. 164 eV) is associated with the sulfur atoms in the thiophene ring of the PEDOT chains.76 The S 2p core level of the PEDOT and PSS chains consists of a spin-split component of S 2p1/2 and S 2p3/2. Doublet peaks observed at around 168 eV and 169 eV originate from the sulfonate groups (ionic species, S 2p3/2) and from sulfonic acids of PSS (neutral species, S 2p1/2), respectively.77 We first investigate the surface composition of the PEDOT:PSS films as they are exposed to the fusion treatment, i.e., water immersion and plasma treatment. After the fusion treatment of the film cast from the dispersion with 5 vol% EG, we observe a significant decrease in the measured area of PSS to PEDOT (Fig. S5a, ESI† and Fig. 5b, respectively). Moreover, the ionic sulfur atoms of PSS become weaker compared to the neutral sulfur atoms due to the treatment. This is due to the removal of excess PSS in the treated films, supporting the results obtained from the surface zeta potential and contact angle measurements above.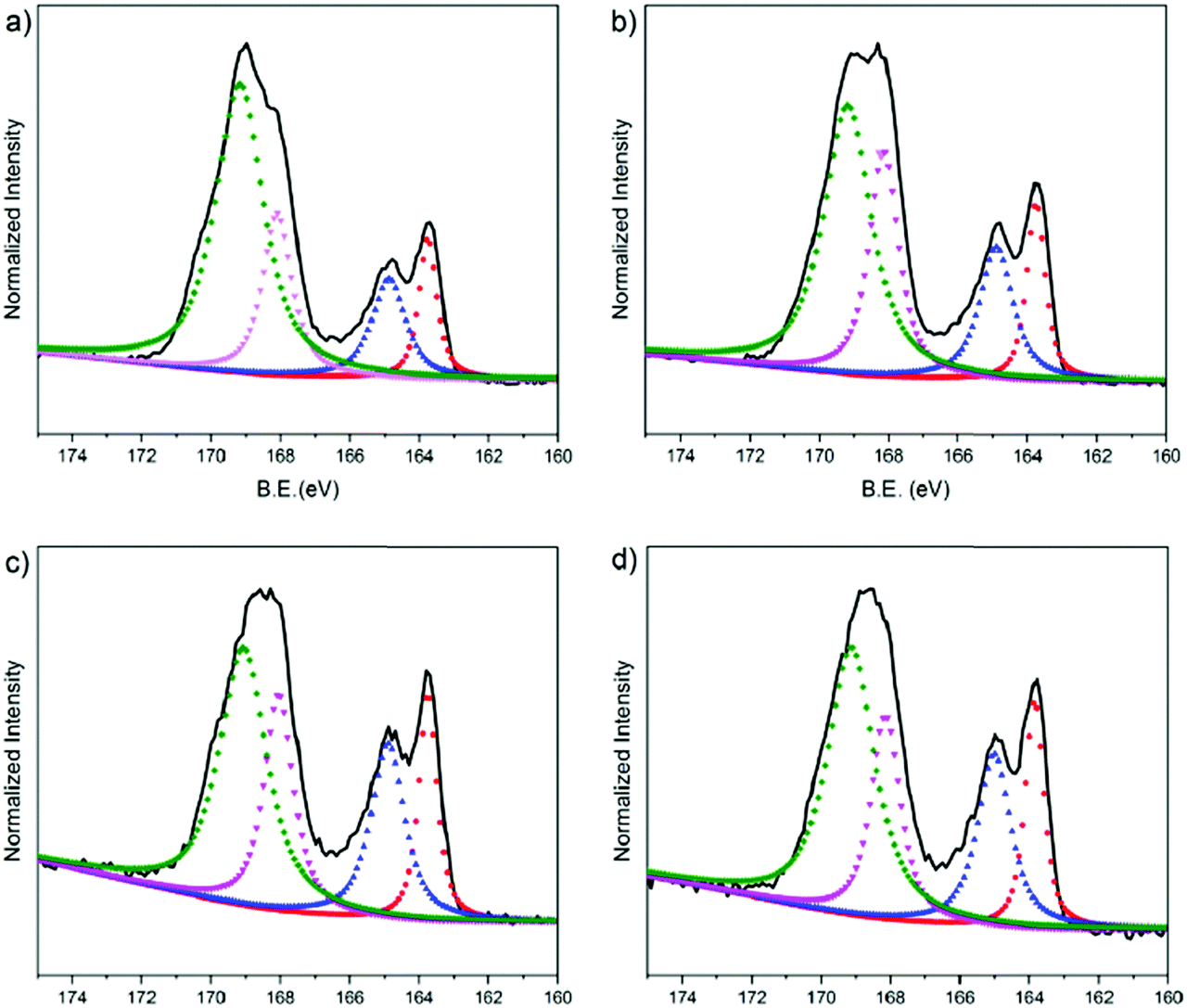 | ||
| Fig. 5 XPS S 2p spectra of the PEDOT:PSS films prepared from various compositions of EG (a: 0 vol%, b: 5 vol%, c: 10 vol% and d: 20 vol%). The spectra were normalized to the maximum intensity and binding energies (B.E., x axis) were referenced to the C 1s core level at 284.8 eV. The S 2p spectra (black lines) were deconvoluted into four distinct peaks (dashed curves), representing two types of sulfur atoms, one is on the thiophene group of PEDOT (ca. 164 eV) and the other is on the sulfonate group of PSS (ca. 169 eV). The red and blue dashed curves correspond to S 2p3/2 and S 2p1/2 PEDOT, whereas the pink and green dashed curves correspond to S 2p3/2 and S 2p1/2 PSS. | ||
Next, we investigate the effect of EG on the surface composition of the films. The relative area of the deconvoluted components of these spectra are summarized in Table S3 (ESI†). The film with 5 vol% EG has the highest fraction (area) of S 2p3/2 (24.3%), meaning that there are more ionic species of PSS in the surface of this film. The S 2p3/2 area decreases for the 10 vol% EG film (22.1%), followed by 20 vol% and 0 vol% EG films (20.8% and 19.5%, respectively). This trend is identical to the relationship of the EG content with the diffusion coefficient of the bilayers: PSS contributes to the fluidity of the lipid bilayer, while 5 vol% in the dispersion renders the film optimal. The largest fraction of S 2p3/2 PSS from 5 vol% EG, which corresponds to the highest negative charge on the surface, is also in complete agreement with our zeta potential measurements. On the other hand, the XPS spectrum of the electropolymerized PEDOT:PSS film shows clear differences (Fig. S5b, ESI†). PEDOT is dominating the surface composition of this film. For instance, for the 5% EG sample, the PEDOT to PSS ratio is 0.397, and this ratio increases up to 1.322 for the electropolymerized sample. The higher crystallinity of PEDOT compared to PSS and the relatively more rigid surface due to the lack of PSS makes the electropolymerized film surfaces much rougher compared to the spin cast samples. We suggest that such a surface accumulated by PEDOT is incompatible for triggering the fusion of vesicles and does not support the assembly of the lipid bilayer.
Conclusions
In this work, we aim to understand the basic surface properties that control vesicle rupture on conjugated polymers and investigate the formation of SLBs via fusion of zwitterionic vesicles on a variety of conjugated polymer films including an n-type semiconductor and the PEDOT:PSS prepared either via electrospinning or cast from different dispersion formulations. Among the two major types of conjugated polymers studied (PEDOT:PSS and p(OH-gNDI-gT2)), we could, first of all, form bilayers only on the PEDOT:PSS films cast from the commercially available dispersion. The surface properties of these films were modified using different concentrations of a co-solvent additive, EG, in the dispersion. The resulting films showed interesting correlations between their surface charge density (modulated by the EG content) and the diffusion coefficient and the viscoelasticity of the bilayers assembled on top. In particular, the lipid membrane formed on the PEDOT:PSS cast from the dispersion containing 5 vol% EG exhibits the highest bilayer diffusion coefficient and sealing behaviour. The underlying characteristic of this film is its high negative charge density due to a high contribution of the ionic PSS. Moreover, although the electropolymerized PEDOT:PSS film has the same chemical composition as the spin cast films, the uppermost surface comprises mostly PEDOT-rich regions, which leads to increased roughness. The vesicles do not rupture on such rough surfaces although the overall hydrophilicity and surface charge are comparable to the spin-cast PEDOT:PSS samples. In the case of the hydrophilic and smooth surface of the semiconductor p(OH-gNDI-gT2), the surface charge density was inadequate to promote vesicle fusion. Overall, we found that the surface hydrophilicity, charge, roughness and chemical composition play crucial roles in determining the efficiency of vesicle fusion. Although the bilayers formed contain defects, tuning the EG content in the PEDOT:PSS dispersions influences the properties of the SLB. A hydrophilic, smooth and highly negatively charged conjugated polymer surface is promising for the fusion of zwitterionic vesicles and the formation of fluid lipid bilayers. This material design guideline to tune the communication between the vesicles and supporting surfaces will lead to effective SLB formation on organic electronic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Nimer Wehbe at the Surface Analysis Laboratory for conducting XPS measurements.Notes and references
- J. E. Alouf, Folia Microbiol., 2003, 48, 5 CrossRef CAS.
- K. S. Phillips and Q. Cheng, Anal. Chem., 2005, 77, 327–334 CrossRef CAS PubMed.
- V. Kiessling, M. K. Domanska, D. Murray, C. Wan, L. K. Tamm and T. P. Begley, Wiley Encyclopedia of Chemical Biology, John Wiley & Sons, Inc., 2007, DOI:10.1002/9780470048672.wecb663.
- Y.-H. M. Chan and S. G. Boxer, Curr. Opin. Chem. Biol., 2007, 11, 581–587 CrossRef CAS PubMed.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS.
- C.-Y. Hsia, M. J. Richards and S. Daniel, Anal. Methods, 2015, 7, 7076–7094 RSC.
- K. L. Weirich, J. N. Israelachvili and D. K. Fygenson, Biophys. J., 2010, 98, 85–92 CrossRef CAS PubMed.
- R. P. Richter and A. R. Brisson, Biophys. J., 2005, 88, 3422–3433 CrossRef CAS PubMed.
- T. H. Anderson, Y. Min, K. L. Weirich, H. Zeng, D. Fygenson and J. N. Israelachvili, Langmuir, 2009, 25, 6997–7005 CrossRef CAS PubMed.
- J. M. Moran-Mirabal, J. B. Edel, G. D. Meyer, D. Throckmorton, A. K. Singh and H. G. Craighead, Biophys. J., 2005, 89, 296–305 CrossRef CAS PubMed.
- H. Schönherr, J. M. Johnson, P. Lenz, C. W. Frank and S. G. Boxer, Langmuir, 2004, 20, 11600–11606 CrossRef PubMed.
- B. Seantier and B. Kasemo, Langmuir, 2009, 25, 5767–5772 CrossRef CAS PubMed.
- A. B. Dahlin, N. J. Wittenberg, F. Höök and S.-H. Oh, Nanophotonics, 2013, 2, 83–101 CrossRef CAS PubMed.
- S. Terrettaz, M. Mayer and H. Vogel, Langmuir, 2003, 19, 5567–5569 CrossRef CAS.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656–663 CrossRef CAS PubMed.
- W. Zhou, Y. Y. Wang, T.-S. Lim, T. Pham, D. Jain and P. J. Burke, Sci. Rep., 2015, 5, 9208 CrossRef CAS PubMed.
- R. Naumann, S. M. Schiller, F. Giess, B. Grohe, K. B. Hartman, I. Kärcher, I. Köper, J. Lübben, K. Vasilev and W. Knoll, Langmuir, 2003, 19, 5435–5443 CrossRef CAS.
- S. Gritsch, P. Nollert, F. Jähnig and E. Sackmann, Langmuir, 1998, 14, 3118–3125 CrossRef CAS.
- Y. Shao, Y. Jin, J. Wang, L. Wang, F. Zhao and S. Dong, Biosens. Bioelectron., 2005, 20, 1373–1379 CrossRef CAS PubMed.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Gurfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Sanaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575 CrossRef PubMed.
- W. Lee, D. Kim, J. Rivnay, N. Matsuhisa, T. Lonjaret, T. Yokota, H. Yawo, M. Sekino, G. G. Malliaras and T. Someya, Adv. Mater., 2016, 28, 9722–9728 CrossRef CAS PubMed.
- X. T. Cui and D. D. Zhou, IEEE Trans. Neural. Syst. Rehabil. Eng., 2007, 15, 502–508 CrossRef PubMed.
- P. Lin, F. Yan, J. Yu, H. L. W. Chan and M. Yang, Adv. Mater., 2010, 22, 3655–3660 CrossRef CAS PubMed.
- L. H. Jimison, S. A. Tria, D. Khodagholy, M. Gurfinkel, E. Lanzarini, A. Hama, G. G. Malliaras and R. M. Owens, Adv. Mater., 2012, 24, 5919–5923 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- Y. Zhang, S. Inal, C.-Y. Hsia, M. Ferro, M. Ferro, S. Daniel and R. M. Owens, Adv. Funct. Mater., 2016, 26, 7304–7313 CrossRef CAS.
- D. Ohayon, C. Pitsalidis, A.-M. Pappa, A. Hama, Y. Zhang, L. Gallais and R. M. Owens, Adv. Mater. Interfaces, 2017, 4, 1700191 CrossRef.
- C. A. Keller and B. Kasemo, Biophys. J., 1998, 75, 1397–1402 CrossRef CAS PubMed.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Hook, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- E. Reimhult, F. Höök and B. Kasemo, Langmuir, 2003, 19, 1681–1691 CrossRef CAS.
- R. Tero, Materials, 2012, 5, 2658–2680 CrossRef CAS.
- A. Giovannitti, C. B. Nielsen, D.-T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Nat. Commun., 2016, 7, 13066 CrossRef CAS PubMed.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef CAS PubMed.
- D. E. Koppel, D. Axelrod, J. Schlessinger, E. L. Elson and W. W. Webb, Biophys. J., 1976, 16, 1315–1329 CrossRef CAS PubMed.
- D. M. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef CAS PubMed.
- T. Heimburg and D. Marsh, in Biological Membranes: A Molecular Perspective from Computation and Experiment, ed. K. M. Merz and B. Roux, Birkhäuser Boston, Boston, MA, 1996, pp. 405–462 DOI:10.1007/978-1-4684-8580-6_13.
- E. J. M. Helmreich, Biophys. Chem., 2002, 100, 519–534 CrossRef.
- J. L. C. M. van de Vossenberg, A. J. M. Driessen and W. N. Konings, Extremophiles, 1998, 2, 163–170 CrossRef CAS PubMed.
- T. Baba, Y. Toshima, H. Minamikawa, M. Hato, K. Suzuki and N. Kamo, Biochim. Biophys. Acta, Biomembr., 1999, 1421, 91–102 CrossRef CAS.
- M. Raja and E. Vales, Biophys. Chem., 2009, 142, 46–54 CrossRef CAS PubMed.
- M. Andersson, H. M. Keizer, C. Zhu, D. Fine, A. Dodabalapur and R. S. Duran, Langmuir, 2007, 23, 2924–2927 CrossRef CAS PubMed.
- F.-Y. Chen, M.-T. Lee and H. W. Huang, Biophys. J., 2002, 82, 908–914 CrossRef CAS PubMed.
- A. V. Sokolov, V. S. Sokolov, T. B. Feldman and M. A. Ostrovsky, Biochemistry, 2008, 2, 404–411 Search PubMed.
- M. Andersson, J. Jackman, D. Wilson, P. Jarvoll, V. Alfredsson, G. Okeyo and R. Duran, Colloids Surf., B, 2011, 82, 550–561 CrossRef CAS PubMed.
- J. Lin, J. Szymanski, P. C. Searson and K. Hristova, Langmuir, 2010, 26, 3544–3548 CrossRef CAS PubMed.
- K. J. Seu, L. R. Cambrea, R. M. Everly and J. S. Hovis, Biophys. J., 2006, 91, 3727–3735 CrossRef CAS PubMed.
- M. Wlodek, M. Szuwarzynski and M. Kolasinska-Sojka, Langmuir, 2015, 31, 10484–10492 CrossRef CAS PubMed.
- P. Lenz, C. M. Ajo-Franklin and S. G. Boxer, Langmuir, 2004, 20, 11092–11099 CrossRef CAS PubMed.
- A. A. Brian and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6159–6163 CrossRef CAS.
- M. ElMahmoudy, S. Inal, A. Charrier, I. Uguz, G. G. Malliaras and S. Sanaur, Macromol. Mater. Eng., 2017, 302, 1600497 CrossRef.
- D. M. DeLongchamp, B. D. Vogt, C. M. Brooks, K. Kano, J. Obrzut, C. A. Richter, O. A. Kirillov and E. K. Lin, Langmuir, 2005, 21, 11480–11483 CrossRef CAS PubMed.
- C. Duc, A. Vlandas, G. G. Malliaras and V. Senez, Soft Matter, 2016, 12, 5146–5153 RSC.
- A. Mata, A. J. Fleischman and S. Roy, Biomed. Microdevices, 2005, 7, 281–293 CrossRef CAS PubMed.
- J. Chai, F. Lu, B. Li and D. Y. Kwok, Langmuir, 2004, 20, 10919–10927 CrossRef CAS PubMed.
- F. D. Egitto and L. J. Matienzo, IBM J. Res. Dev., 1994, 38, 423–439 CrossRef CAS.
- J. T. Groves, N. Ulman and S. G. Boxer, Science, 1997, 275, 651–653 CrossRef CAS PubMed.
- E. Reimhult, F. Höök and B. Kasemo, J. Chem. Phys., 2002, 117, 7401–7404 CrossRef CAS.
- I. Reviakine, F. F. Rossetti, A. N. Morozov and M. Textor, J. Chem. Phys., 2005, 122, 204711 CrossRef PubMed.
- P. Nollert, H. Kiefer and F. Jähnig, Biophys. J., 1995, 69, 1447–1455 CrossRef CAS PubMed.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- J. Raedler, H. Strey and E. Sackmann, Langmuir, 1995, 11, 4539–4548 CrossRef CAS.
- C. Hennesthal and C. Steinem, J. Am. Chem. Soc., 2000, 122, 8085–8086 CrossRef CAS.
- Y.-H. Kim, M. M. Rahman, Z.-L. Zhang, N. Misawa, R. Tero and T. Urisu, Chem. Phys. Lett., 2006, 420, 569–573 CrossRef CAS.
- T. Cha, A. Guo and X. Y. Zhu, Biophys. J., 2006, 90, 1270–1274 CrossRef CAS PubMed.
- G. J. Hardy, R. Nayak and S. Zauscher, Curr. Opin. Colloid Interface Sci., 2013, 18, 448–458 CrossRef CAS PubMed.
- J. Ouyang, Q. Xu, C.-W. Chu, Y. Yang, G. Li and J. Shinar, Polymer, 2004, 45, 8443–8450 CrossRef CAS.
- Q. Wei, M. Mukaida, Y. Naitoh and T. Ishida, Adv. Mater., 2013, 25, 2831–2836 CrossRef CAS PubMed.
- H. Shi, C. Liu, Q. Jiang and J. Xu, Adv. Electron. Mater., 2015, 1, 1500017 CrossRef.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- N. P. Mellott, S. L. Brantley, J. P. Hamilton and C. G. Pantano, Surf. Interface Anal., 2001, 31, 362–368 CrossRef CAS.
- A. A. Duarte, S. L. Filipe, L. M. G. Abegão, P. J. Gomes, P. A. Ribeiro and M. Raposo, Microsc. Microanal., 2013, 19, 867–875 CrossRef CAS PubMed.
- S. Nirasay, A. Badia, G. Leclair, J. Claverie and I. Marcotte, Materials, 2012, 5, 2621 CrossRef CAS.
- K. C. Weng, J. J. R. Stålgren, D. J. Duval, S. H. Risbud and C. W. Frank, Langmuir, 2004, 20, 7232–7239 CrossRef CAS PubMed.
- K. Mulligan, Z. J. Jakubek and L. J. Johnston, Langmuir, 2011, 27, 14352–14359 CrossRef CAS PubMed.
- K. Z. Xing, M. Fahlman, X. W. Chen, O. Inganäs and W. R. Salaneck, Synth. Met., 1997, 89, 161–165 CrossRef CAS.
- J. Y. Kim, J. H. Jung, D. E. Lee and J. Joo, Synth. Met., 2002, 126, 311–316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of p(OH-gNDI-gT2), NMR spectrum of p(OH-gNDI-gT2), FRAP images of a bilayer formed on glass, QCM-D signals of bilayers formed on various PEDOT:PSS surfaces, impedance spectra of PEDOT:PSS before and after vesicle fusion, water contact angle of a PEDOT:PSS film before and after fusion treatment, additional FRAP and AFM images of the electropolymerized PEDOT:PSS, and a summary of the XPS results. See DOI: 10.1039/c8tc00370j |
| This journal is © The Royal Society of Chemistry 2018 |