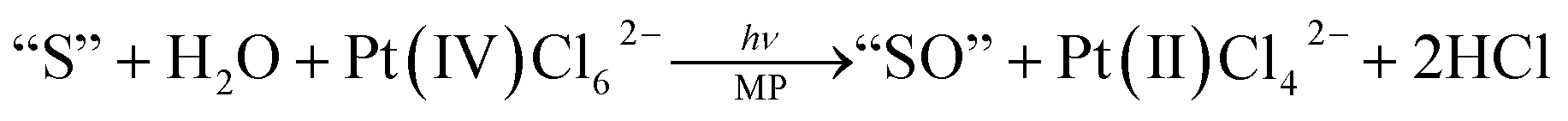Two-electron oxidation of water to form hydrogen peroxide catalysed by silicon-porphyrins†
Received
2nd March 2018
, Accepted 23rd March 2018
First published on 27th March 2018
Abstract
The second most earth abundant atom incorporated meso-tetrapyridylporphyrinatesilicon(IV) (SiTPyP) as an electrochemical catalyst was newly synthesized. The two axially ligated water molecules on SiTPyP and the four meso-substituted pyridyl nitrogens suffer eight-step protonations and deprotonations. Our detailed pKa study indicated that the two axially ligated water molecules were fully deprotonated to exist as O−–Si–O− under the condition of pH > 2. Under basic conditions, SiTPyP(O−)2 exerted the two-electron oxidation of water to form hydrogen peroxide as the primary product in aqueous acetonitrile, which was initiated through a one-electron oxidation process on the electrode with faradaic yield of 84–92%.
Introduction
The findings reported by Honda–Fujishima on light induced water splitting into hydrogen and oxygen catalysed by TiO2 in 1972,1 the chemical oxidation of water reported by Meyer2 and photochemical reduction of CO2 reported by Lehn3 have triggered extensive studies towards artificial photosynthesis.4,5 Among the various approaches for artificial photosynthesis such as (1) biological modification,6 (2) semiconductor photocatalysis,7–13 (3) molecular catalysis14–17 and (4) system integration,18–21 there are still crucial subjects to be challenged in the molecular catalyst approach in spite of the recent advances. One of the subjects is the “photon-flux-density problem” which causes the bottle neck in water oxidation via step-wise four-photon processes to evolve oxygen.22 Ordinarily, only one electron can be transferred from/to an excited molecule and the four-electron oxidation indicates the step-wise four-photon excitation of the molecular catalyst/sensitizer. Under the rarefied intensity of sunlight, the molecular catalyst should wait for the next photon's arrival to step up to its higher nth oxidation state (n+) such as (0) → (1+) → (2+) → (3+) → (4+). The time interval of each photon's arrival can be estimated to be ∼0.2 s depending on the cross-section of light absorption by the molecules.22 The stepping up processes to the four-electron oxidized state (4+) thus require ∼1 s, which is an extremely long period of time from the viewpoint of the time scale of molecules. The molecular catalyst in the higher oxidation state with high reactivity will suffer transformation/decomposition with much faster time constants before the next photon's arrival. This is termed as the “photon-flux-density problem”.22 To overcome or bypass this problem, we have been focusing our efforts towards developing a one-photon induced two-electron oxidation process with water as both the electron and oxygen atom donor.23–31 In 1986, our group reported the photochemical epoxidation of alkenes with water catalysed by metalloporphyrins upon visible-light irradiation as the first example of this type of reaction as shown in eqn (1):23| |  | (1) |
where “S”, “SO” and MP denote the substrate, oxygenated substrate and metalloporphyrins, respectively. We have also demonstrated that the photochemical two-electron oxygenation proceeds through the one-photon excitation of MP in the reaction cycle by laser flash photolysis to detect key intermediates.28 Recently, we have further succeeded in using the water molecule itself as “S” (substrate)32,33 and found the efficient formation of hydrogen peroxide as the two-electron oxidized product in the electrolysis of water catalysed by earth-abundant aluminium-porphyrins, which was initiated by one-electron oxidation of the molecular catalyst.33 The reaction mechanism was also revealed using a laser flash photolysis study through the direct detection of the key intermediates.34 This strongly prompted us to further explore whether or not the second most earth-abundant element, viz., silicon-based porphyrins could be used in the two-electron oxidation of water.35 Tuning the oxidation potential of MP has been recognized to be one of the crucial factors for the system to be designed.35 The introduction of an electron-withdrawing substituent into the meso-position of the porphyrin ring is expected to positively shift the oxidation potential of MP.35 Silicon-porphyrins having a pyridyl group on the ring are expected to have an oxidation potential more positive than 1 V vs. SHE as compared to those observed for various Si-porphyrins.35
We report herein that meso-tetrapyridyl-porphyrinatesilicon(IV) (SiTPyP) with appropriate oxidation potential exhibits good catalytic reactivity towards the two-electron oxidation of water to form hydrogen peroxide through one-electron oxidation as the initial step.
Results and discussion
Synthesis of the new silicon-porphyrin, SiTPyP
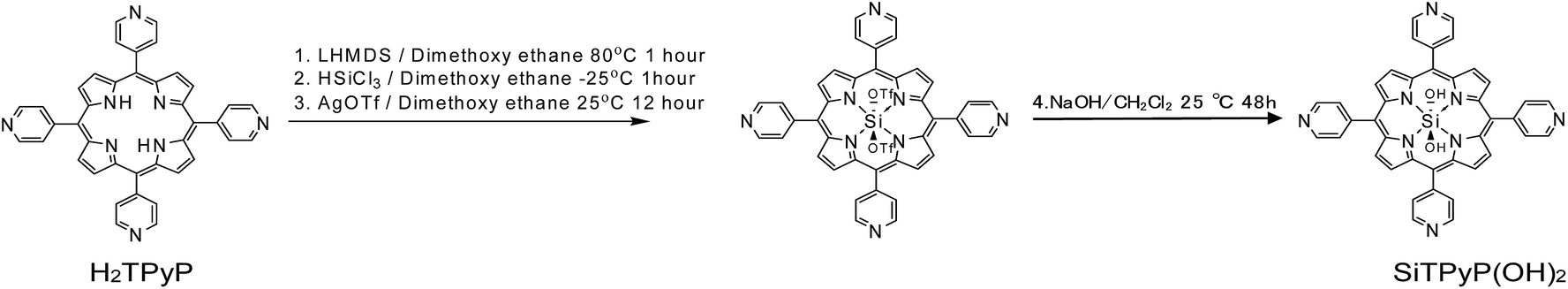
trans-Dihydroxo [5,10,15,20-tetra(4-pyridyl)porphyrinato]silicon(IV):Si(IV)TPyP(OH)2 as a new compound was synthesized using a method similar to that reported by Kane et al.36 The free base of 5,10,15,20-tetra(4-pyridyl)porphyrin (H2TPyP) was treated with lithium bis(trimethylsilyl)amide (LHMDS), trichlorosilane (HSiCl3), and silver(I) trifluromethanesulfonate (AgOTF) and further with NaOH in a one pot procedure to form SiTPyP(OH)2. The detailed procedures are described in the Experimental section below.
Protolytic behaviour of the axial ligands in tetra(4-pyridyl) porphyrinatosilicon(IV) (Si(IV)TPyP(OH)2)
The isolated SiTPyP was characterized to have two hydroxy groups as the axial ligands (see Experimental). Since SiTPyP is almost insoluble in water, the electrolysis of water is preferably carried out in aqueous acetonitrile, in which sufficient solubility of SiTPyP is obtained. To understand the nature of the axial ligands of SiTPyP in aqueous acetonitrile, first, the ligation behaviour for SiTPyP was carefully examined spectroscopically in dry acetonitrile, where the λmax = 415.8 nm corresponds to the Soret band. Upon the addition of water the λmax gradually shifts to 416.2 nm with an isosbestic point at λ = 417.5 nm (ESI 1†). Analysing the change in the absorbance upon the addition of water using a Benesi–Hidebrand type plot indicates that two water molecules ligate in one-step with an equilibrium constant K = 1.4 M−2. This indicates that the concentration of water higher than 15% was required for the almost quantitative coordination (>99%) of the two water molecules on SiTPyP (ESI 1†).
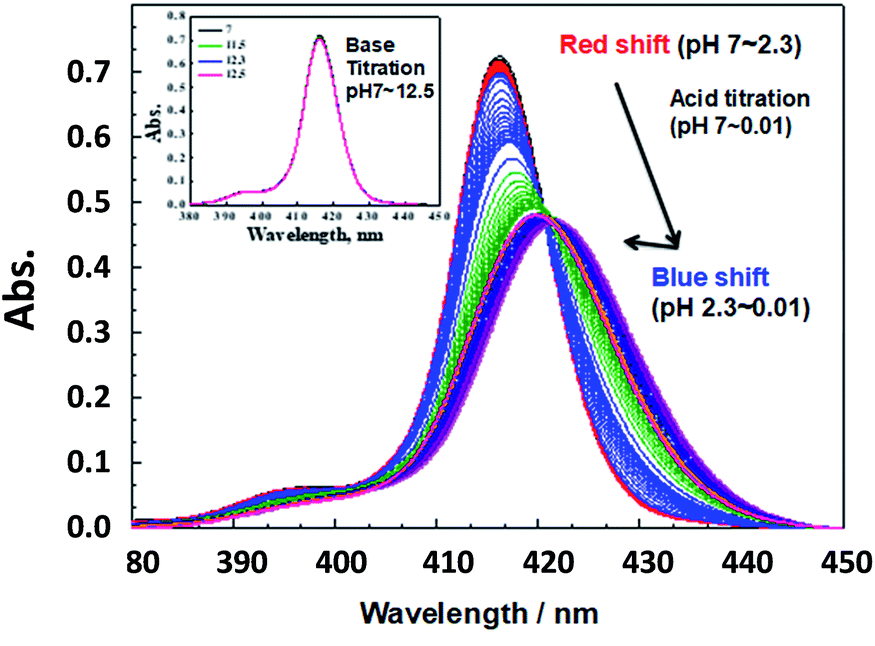
To assure SiTPyP has two water molecules as the axial ligands, the experiments hereafter were all carried out in CH3CN/H2O (8/2 v/v or 7/3 v/v). Since the axial ligand water molecules on metalloporphyrins often exhibit protolytic reactions of protonation/deprotonation,33,37 an acid/base titration was carried out for the water coordinated SiTPyP in CH3CN/H2O (8/2 v/v) as a typical condition. Hereafter, the pH value denotes the value of the aqueous solution added to the mixture. The absorption spectra upon acid titration from neutral down to strongly acidic conditions (pH = 0.01) exhibit a drastic change as shown in Fig. 1. An acid titration from pH 7 to 2.3 induced a gradual red-shift in the λmax of the Soret band from 416.2 nm to 421.7 nm with a decrease in the OD and broadening of the spectral width, indicating the decrease in electron density of the porphyrin ring.35 A further decrease of pH from 2.3 to 0.01 caused an inverted blue-shift with a slight increase in the OD, indicating the protonation on the axial ligands (Fig. 1).33,37
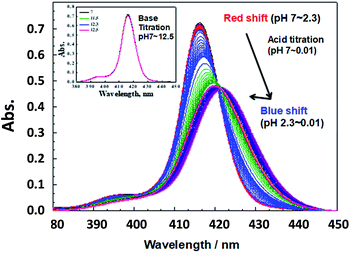 |
| | Fig. 1 Absorption spectral change of SiTPyP (1.5 × 10−6 M) upon acid titration using H2SO4 (pH 7–0.01) in CH3CN/H2O (8/2 v/v). Inset: absorption spectra of SiTPyP (1.5 × 10−6 M) upon the addition of KOH (pH 7–12.5). | |
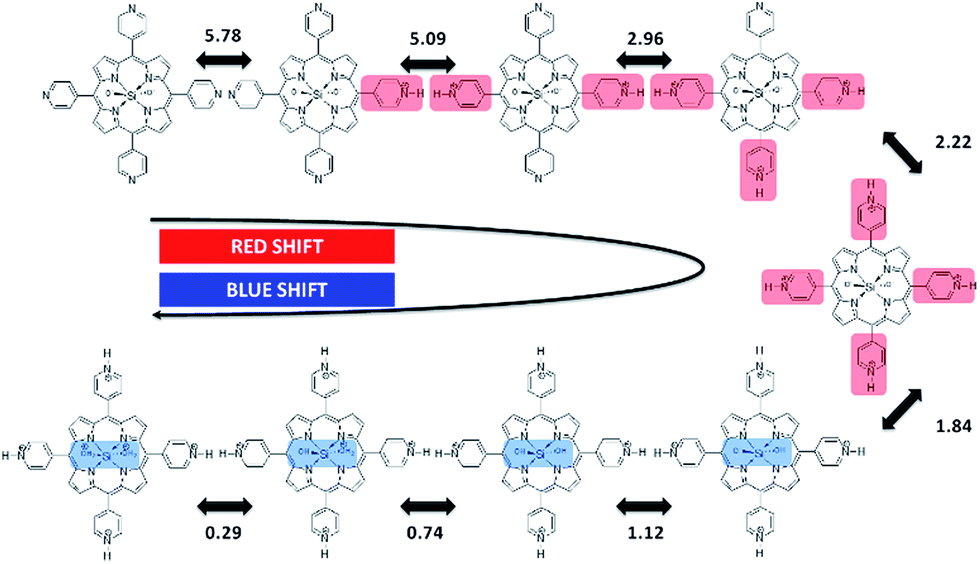
Careful observation of the gradual red shifts revealed a clear four-step change with a different isosbestic point in each step (ESI 2-1†). In the subsequent blue shifts, another four-step change was also observed, indicating the eight-step change during the acid titration experiment (ESI 2-2†). Moreover, in the basic titration (pH > 7) no change was observed, indicating that SiTPyP has an eight-step change in total only under acidic conditions. For each change in the absorption spectra, eight clear inflection points were observed in the plot between the absorbance at a fixed wavelength vs. pH value (ESI 3†). The inflection points were determined by extrapolation of the two linear lines for the narrow region of pH. Though the definition of the pKa value in aqueous acetonitrile is not straightforward due to the activity of the protons being dependent on the solvent composition, the relative pKa was assigned herein as the inflecting pH value of the added aqueous solution. Each inflecting pH value in the red shift region (pH 7–2.3) was thus assigned as the pKa of the four-step protonation to the peripheral pyridyl nitrogen (pKa = 5.78, 5.09, 2.96 and 2.18), which decreases the electron density of the porphyrin ring. In the blue shift region (pH 2.3–0.01) the pKa's of the four-step protonation (pKa = 1.84, 1.12, 0.74 and 0.29) were also determined by the inflecting pH values, which were assigned as the stepwise protonation processes of the two axial ligands. The protolytic behaviour of SiTPyP is thus depicted in Scheme 2. It should be noted that the axial OH ligands on SiTPyP are fully deprotonated even in the acidic region (pH > 2), which is contrary to the case of Al-porphyrins, which allow full deprotonation only under basic conditions.33,37
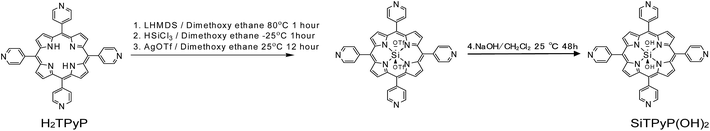 |
| | Scheme 1 Synthesis of SiTPyP(OH)2. | |
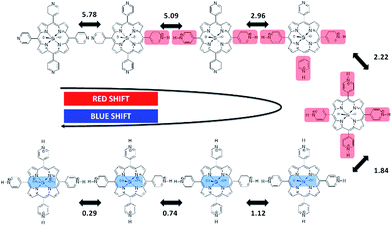 |
| | Scheme 2 Protolytic equilibria of SiTPyP and their corresponding pKa values. | |
Electrochemical behaviour of SiTPyP
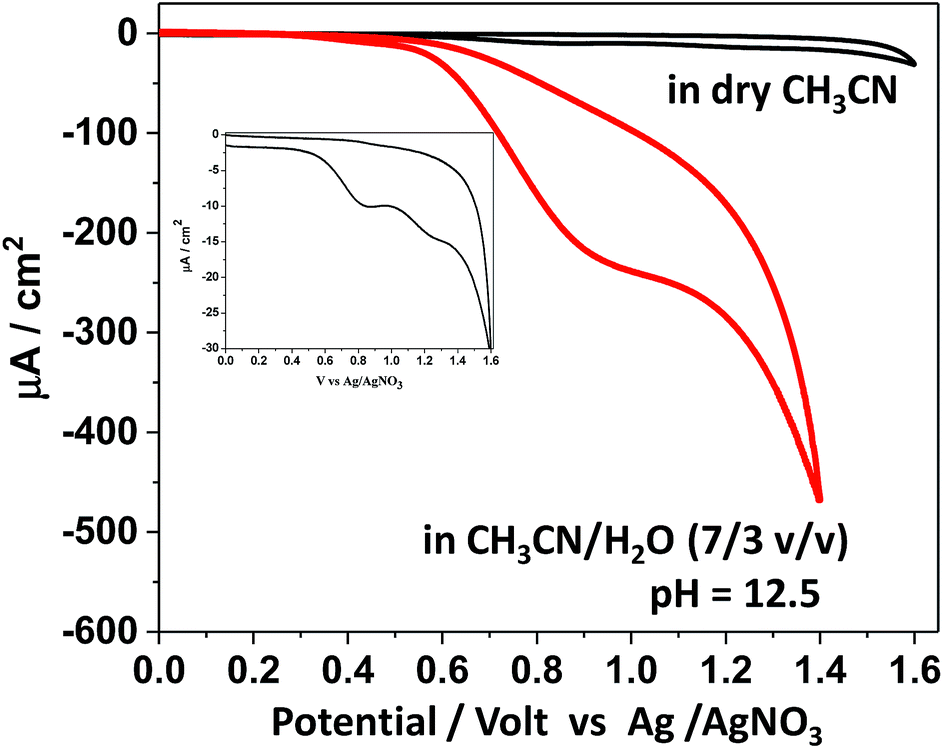
Based on the protolytic behaviour revealed above, the electrochemical behaviour of SiTPyP was further studied. To examine the possibility of water oxidation, the experiment was focused on the electrochemical oxidation behaviour of SiTPyP(O−)2 (1) in basic aqueous acetonitrile to compare with the case of meso-tetra(4-methylpyridiniumyl)porphyrinatealuminum (AlTMPYP(O−)2), which was limited to the basic conditions.33 As a reference, the cyclic voltammetry (CV) of SiTPyP in dry acetonitrile was also conducted using boron-doped diamond (BDD) as the working electrode.38 Ordinarily metalloporphyrins exhibit a two-step reversible oxidation wave in CV. In the case of AlTMPyP, a quasi-reversible two-step oxidation wave was observed in dry acetonitrile.33 However, SiTPyP in dry acetonitrile did not show this reversible wave, but displayed a rather irreversible-like wave with a peak potential of 0.87 V vs. Ag/AgNO3 (Fig. 2 inset). The addition of water up to 30% v/v induced drastic change in the CV curves to show a distinct catalytic oxidation wave with a peak potential at 0.92 V vs. Ag/AgNO3 with a significant increase in current (Fig. 2). In the case of meso-tetramesitylporphyrinatesilicon (SiTMP) with minor water oxidation ability, the quasi-reversible waves were observed in dry acetonitrile, which we reported recently.35 These results imply that SiTPyP will surely have high reactivity for water oxidation. Even in dry acetonitrile contaminated with a small amount of water, SiTPyP may exhibit an irreversible-like oxidation wave in its CV curve. According to the modified Randles–Sevcik equation,39 the observed current should be independent of the scan rate when the electrochemical processes undergo sufficient amount of catalytic cycles at the electrode. The oxidation wave of SiTPyP in CH3CN/H2O (7/3 v/v) was thus further examined to observe the effect of the scan rate on the applied potential to investigate whether or not it has a truly catalytic process on the electrode. A saturation phenomena of the oxidation current against the scan rate of the applied potential was observed to confirm that the oxidation wave of SiTPyP in aqueous acetonitrile was actually a catalytic wave (ESI 4†).
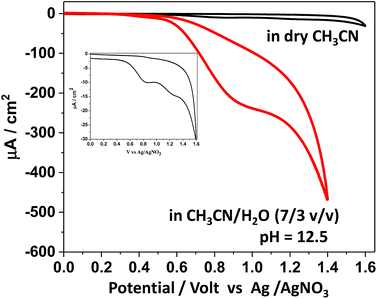 |
| | Fig. 2 Cyclic voltammogram of SiTPyP in dry CH3CN and in CH3CN/H2O (7/3 v/v) at pH = 12.5; magnified CV curve in dry CH3CN is shown in the inset. Conditions: [SiTPyP] = 0.15 mM, (C4H9)NPF6 (0.1 M) as electrolyte, BDD as the working electrode, Ag/AgNO3 as the reference electrode, Pt as the counter electrode, scan rate of 100 mV s−1. | |
Electrolysis of water catalysed by SiTPyP(O−)2
Since the catalytic oxidation wave was confirmed in the CV measurements, the bulk electrolysis of water catalysed by SiTPyP(O−)2 (1) was carried out under basic conditions at pH = 12.5. As we have already reported the two-electron water oxidation catalysed by Aluminum-porphyrin under basic conditions,33 it should be meaningful to compare the water oxidation processes among the Si- and Al-porphyrins.
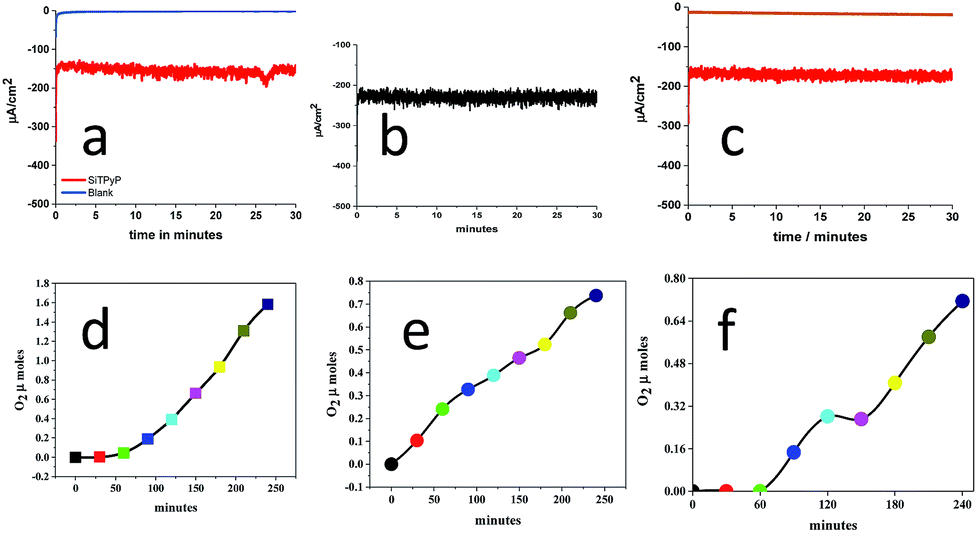
The applied potential of the anode, BDD, was set at 0.92 V vs. Ag/AgNO3 in the bulk electrolysis of SiTPyP(O−)2 in CH3CN/H2O (7/3 v/v) with a three-electrode system using BDD as the anode, Ag/AgNO3 as the reference electrode and Pt wire as the counter electrode in two chambers each connected with a salt bridge. As shown in Fig. 3, moderate and constant anodic currents at around 150–220 μA cm−2 were observed on the BDD electrode depending upon the conditions with/without additives, such as Na2CO3 and (C4H9)4N+(HPO4)−, which are a bit smaller than ∼350 μA cm−2 at 0.93 V vs. Ag/AgCl in the case of AlTMPyP(O−)2 in water33 (Table 1).
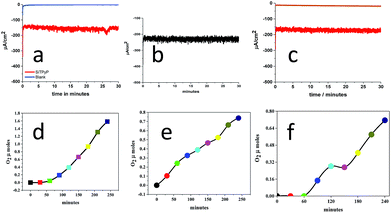 |
| | Fig. 3 Controlled potential electrolysis of water catalysed by SiTPyP(O−)2 (0.15 mM) in CH3CN/H2O (7/3 v/v) at an applied potential of 0.92 V vs. Ag/AgNO3 with [(Bu)4NPF6] = 0.1 M. (a), (b) and (c) show the anodic current in Run 1, 2 and 3, respectively (Table 1). (d), (e) and (f) show the amount of oxygen evolution in Run 1, 2 and 3, respectively. | |
Table 1 Product distribution in the controlled potential electrolysis of SiTPyP(O−)2 in CH3CN/H2O (7/3 v/v) at 0.92 V vs. Ag/AgNO3
| Run |
pH |
Additive/μmol |
Anodic current at 30 min/μA cm−2 |
O2/μmol (FYa/%) |
Peroxides (H2O2)/μmol (FYa/%) |
FYa/% |
|
Faradaic yield.
Tetrabutylammoniumphosphate.
|
| 1 |
12.5 |
None |
150 |
1.58 (53.6) |
1.82 (30.9) |
84.5 |
| 2 |
12.5 |
Na2CO3, 1 mM |
220 |
0.74 (32.3) |
2.76 (60.3) |
92.6 |
| 3 |
12.5 |
Bu4NHPO4b, 10 mM |
170 |
0.71 (30.2) |
2.19 (55.8) |
92.0 |
Three types of electrolysis products were detected. The gaseous product was analysed to be oxygen by GC-MS. Free hydrogen peroxide was also colorimetrically detected and quantitatively analysed using the TiTPyP method (ESI 5†).40 Titration by KMnO4 also indicated that a large amount of peroxide was formed in the reaction system, strongly suggesting that hydrogen peroxide was stored as the hydroperoxide of Si-porphyrin in the form of SiTPyP(O−)(OOH) as similarly observed in the case of AlTMPyP (ESI 6†).37 Thus, the total amount of hydrogen peroxide produced was taken to be the total concentration of peroxides determined by the titration of KMnO4 as indicated by eqn (2).
| | | [Peroxides titrated by KMnO4] = [H2O2]free + [SiTPyP(O−)(OOH)] | (2) |
The product distribution and the faradaic yield are summarized in Table 1. The faradaic yield (FY) of water oxidation was calculated by the amount of oxidation products (4e− for oxygen and 2e− for total peroxides in the unit of electrons per mole) against the integrated number of electrons per mole estimated from the anodic current and exhibit pretty good FY (84–92%), which are slightly lower than those obtained using AlTMPyP (almost quantitative FY).
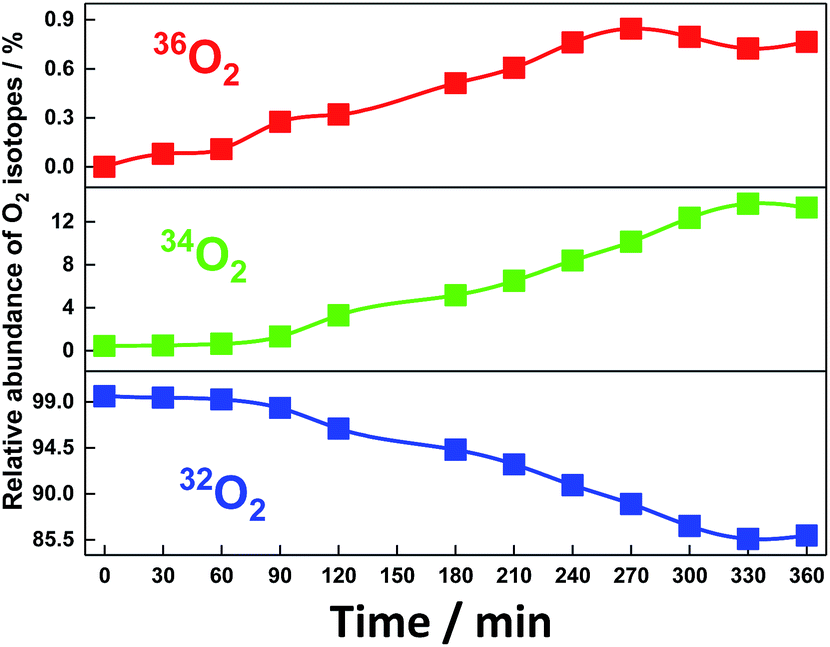
After electrolysis for 240 min, the shape of the absorption spectrum remained almost unchanged with a small decrease in the absorbance to 93% as a typical case for Run 2 (ESI 7†). When the surface of the anode BDD was thoroughly washed with aqueous acetonitrile after the electrolysis and used again in the electrolysis in fresh aqueous acetonitrile without SiTPyP under the same conditions, no anodic current was observed, clearly indicating that any active materials for water oxidation were not deposited on the anode during the electrolysis and the SiTPyP(O−)2 in solution does truly cause the water oxidation. Since oxygen and hydrogen peroxide were detected as the major oxidation products in the electrolysis, an 18O isotope experiment using H218O was carried out to check whether the water acts as the electron donor to form molecular oxygen and hydrogen peroxide. As shown in Fig. 4, the evident incorporation of 18O into molecular oxygen during the electrolysis indicates that water itself serves as both the electron and oxygen atom donor to evolve molecular oxygen. The isotopic analysis of gaseous oxygen in the electrolysis cell was carried out using GC-MS, revealing essentially quantitative isotopic incorporation into the evolved oxygen gas during prolonged electrolysis for 360 min. The observed isotopic distributions, 32O2 (85.9%), 34O2 (13.3%) and 36O2 (0.7%), correspond well to the calculated ratios, 32O2 (84%), 34O2 (15.4%) and 36O2 (0.67%), for 3.1 cycles of the catalytic reaction as estimated from the number of electrons against the number of catalyst molecules in the system (Table 1, ESI 8†).
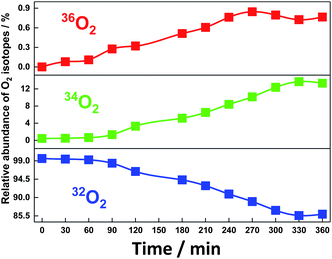 |
| | Fig. 4 Incorporation of isotope 18O into molecular oxygen evolved during the electrolysis of water catalysed by SiTPyP(O−)2 (0.2 mM) in CH3CN/H2O (7/3 v/v) at pH = 12.5. Conditions: BDD as the working electrode, Ag/AgNO3 as the reference electrode, Pt as the counter electrode, Bu4NPF6 (0.1 M) as the electrolyte and the applied potential controlled at 0.92 V vs. Ag/AgNO3. | |
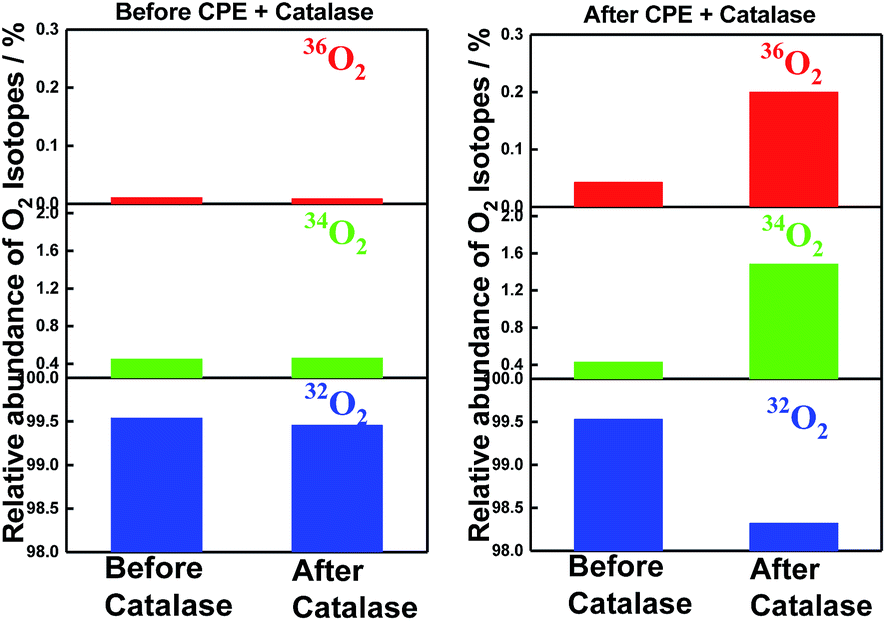
The incorporation of 18O into the evolved molecular oxygen provides definite evidence that water was oxidized during the electrolysis. Moreover, very convincingly, the addition of enzyme catalase, which converts hydrogen peroxide into molecular oxygen,41 verified the incorporation of 18O into the converted molecular oxygen as shown in Fig. 5. The reaction mixtures before and after electrolysis were once degassed by repeated freeze–thaw procedures to remove molecular oxygen and then treated with catalase under phosphate buffered conditions (pH = 6.86). The relative intensities of the GC-MS peaks at 32 (16O16O), 34 (16O18O) and 36 (18O18O) were compared before and after the treatment with the enzyme, catalase. Since the contamination of air (32O2) was not completely avoided during the injection procedure of enzyme catalase via a micro-syringe, the contamination of air (32O2) decreased the relative percentage of 36O2 (0.2%) from the calculated ratio (0.67%) for the 3.1 cycles of TON in the catalase treated sample. The data in Fig. 5 clearly indicates that hydrogen peroxide formed in the reaction mixture also comes from the water molecules during the electrolysis catalysed by SiTPyP(O−)2 (ESI 9†).
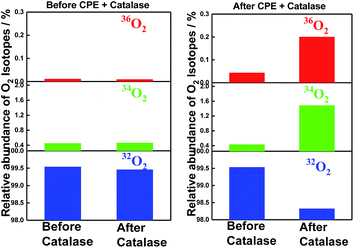 |
| | Fig. 5 Incorporation of isotope 18O into molecular oxygen evolved upon treatment with the enzyme, catalase, before (left) and after (right) the controlled potential electrolysis (CPE). The reaction mixtures before (left) and after (right) the electrolysis were once degassed by repeated freeze–thaw procedures and treated with catalase under phosphate buffered conditions (pH = 6.86). The relative intensities of the GC-MS peaks at 32 (16O16O), 34 (16O18O), and 36 (18O18O) were compared between before and after the treatment with the enzyme, catalase. Conditions: [SiTPyP(O−)2] = 0.2 mM in CH3CN/H2O (7/3 v/v) at pH = 12.5, BDD as the working electrode, Ag/AgNO3 as the reference electrode, Pt as the counter electrode, Bu4NPF6 (0.1 M) as the electrolyte and the applied potential controlled at 0.92 V vs. Ag/AgNO3. | |
Effect of additives on the selectivity of the oxidation products
In the water oxidation catalysed by AlTMPyP(O−)2, we have already found that Na2CO3 suppressed oxygen evolution to increase the selectivity toward hydrogen peroxide formation.33 Since Na2CO3 is known to stabilize hydrogen peroxide by forming the peroxycarbonate/ion complex,42,43 the further oxidation of free hydrogen peroxide on the anode into molecular oxygen was considered to be prevented. It was also observed in a laser flash photolysis experiment that the hydroperoxy complex with AlTMPyP (AlTMPyP(OOH)(O−)) liberated hydrogen peroxide to revert to the starting AlTMPyP through a dynamic reaction with Na2CO3.34 These results led to the conclusion that the primary product of water oxidation catalysed by AlTMPyP(O−)2 was hydrogen peroxide (two-electron oxidized product) and molecular oxygen (four-electron oxidized one) was produced by the further oxidation of hydrogen peroxide on the electrode.33 As listed in Table 1, the explanation is also well picturized for SiTPyP(O−)2. The addition of Na2CO3 (Run 2 in Table 1) increased the amount of peroxides/H2O2, but decreased the amount of oxygen evolved when compared to the electrolysis without Na2CO3 (Run 1 in Table 1). Interestingly, another ionic molecule, tetrabutylammonium-phosphate (Bu4NHPO4) also induced the effect similar to that of Na2CO3 (Run 3 in Table 1), implying that Bu4NHPO4 will also stabilize hydrogen peroxide in solution to prevent the further oxidation of free hydrogen peroxide.
Reaction mechanism of water oxidation catalysed by SiTPyP(O−)2
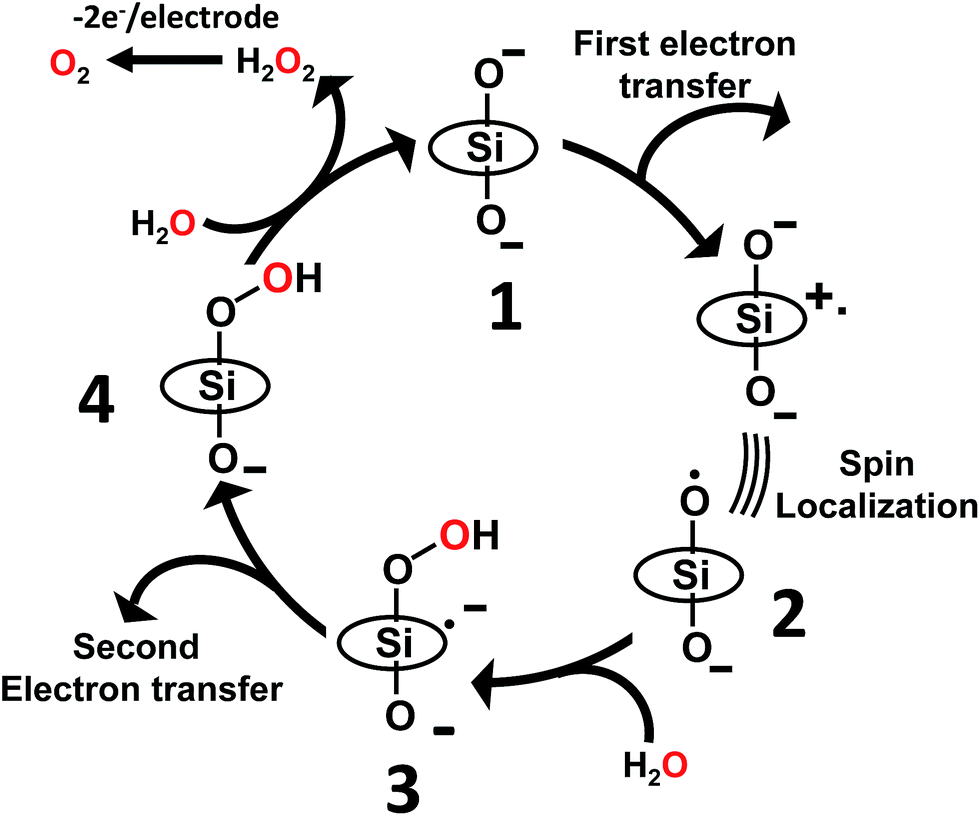
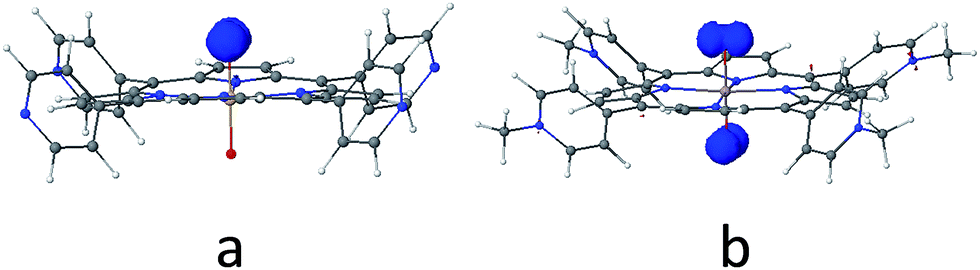
As discussed above, the primary product of water oxidation catalysed by SiTPyP(O−)2 (1) is considered to be the two-electron oxidized product, viz., hydrogen peroxide. The process is initiated through a one-electron oxidation of SiTPyP(O−)2 (1) at the electrode potential of the first oxidation wave (0.92 V vs. Ag/AgNO3). An oxygen molecule is produced by the further oxidation of free hydrogen peroxide, which is liberated from the hydroperoxy complex, SiTPyP(OOH)(O−) through substitution with an OH− ion. The reaction mechanism is depicted in Scheme 3, which is similar to that proposed for AlTMPyP(O−)2.33 DFT calculations (Gaussian 09 B3LYP/6-31G*) on the one-electron oxidized state of SiTPyP(O−) (1), [SiTPyP(O−)2]+˙, indicate that one of the two axial ligand oxygen atoms has a predominant spin density as shown in Fig. 6a. The axial oxygen atom has “oxyl radical” character, expressed as SiTPyP(O−)(O˙) (2). This will rationalize the reaction of 2 with OH− ions to form the adduct radical anion, [SiTPyP(OOH)(O−)]−˙ (3), of which its formation is also supported by the DFT calculations.
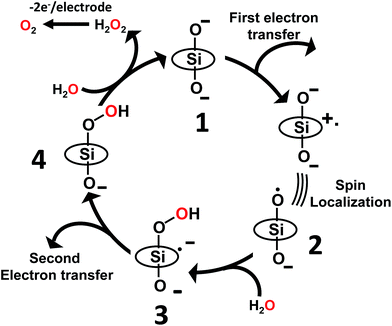 |
| | Scheme 3 Proposed reaction mechanism for electrochemical water oxidation catalysed by SiTPyP(O−)2. TPyP denotes the porphyrin ring. | |
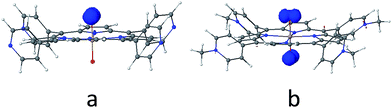 |
| | Fig. 6 Electron spin population of the (a) one-electron-oxidized form of [SiTPyP(O−)2], where the electron spin is mostly localized on the one of the two axial ligand oxygen atoms with oxyl-radical character and (b) one-electron-oxidized form of [AlTMPyP(O−)2], where the electron spin is situated on both sides of axial ligand oxygen atoms. The spin populations were calculated by Gaussian 09 (Rev. D01, UB3LYP/6-31G(d), SCRF (PCM, solvent = water)). | |
The excess electrons in the SOMO of 3 are mostly delocalized on the porphyrin ring similar to the electrons in the radical anion of porphyrins and should be easily oxidized on the electrode at the applied potential (0.92 V vs. Ag/AgNO3) to form the hydroperoxy complex, SiTPyP(O−)(OOH) (4). Free hydrogen peroxide is liberated from 4 through a substitution reaction with OH−. Molecular oxygen is evolved on further oxidation of the free hydrogen peroxide on the BDD electrode. The hydroperoxy complex formation between 1 and H2O2 was independently studied and the equilibrium constant K = 1.2 × 102 M−1 at pH = 12.5 in CH3CN/H2O (7/3 v/v), which is smaller than that obtained for AlTMPyP (K = 2.4 × 103 M−1) (ESI 10†). The smaller K value indicates that SiTPyP(O−)(OOH) more easily liberates free hydrogen peroxide than AlTMPyP. Regrettably, the turnover frequency of the catalytic cycle in Scheme 3 using the modified Randles–Sevcik equation was not able to be deduced this time because the non-catalytic wave was not observed in the CV measurement as discussed above (Fig. 2 inset). The catalytic reactivity of SiTPyP is expected to have a superior point in the K value though the overall current density in the electrolysis was smaller than that obtained using AlTMPyP (Table 1).33 Another interesting point may be the delicate difference in the electronic structure of the one-electron oxidized species as shown in Fig. 6. The spin density of the one-electron oxidized form of SiTPyP is mostly localized on one axial ligand oxygen atom and the attack of OH− ion is limited on one-side, while two axial ligands oxygen atoms share the electron spin in the case of AlTMPyP to allow the attack of OH− on both sides. The reaction mechanism shows the three subsequent processes: (1) attack of the OH− ion against 2 to form the radical anion, 3, (2) the second electron transfer from 3 to the electrode to form hydroperoxy complex, 4, and (3) the liberation of H2O2 from 4. The catalytic reactivity, that is, the speed of the reaction cycles, should depend on which process is the rate determining step. SiTPyP may have some limitation in step (1), but is superior in the step (3). A more detailed study on the reaction mechanism is now in progress.
Experimental
Materials
The chemical 5,10,15,20-tetrakis(tetra(4-pyridyl))porphyrin (H2TPyP) was purchased from TCI chemicals. DCM (purchased from TCI chemicals) was stored over molecular sieves (4A) overnight over K2CO3, dried over CaH2 and vacuum transferred immediately prior to use. Lithium bis(trimethylsilyl)amide (LHMDS) solid was purchased from Aldrich and stored under dry conditions. Trichlorosilane (HSiCl3) was purchased from TCI chemicals. AgOTf (silver trifluoromethanesulfonate) was purchased from TCI chemicals. Acetonitrile (HPLC grade) was used as received from Nakalai Tesque. Distilled water was passed through an ion exchange column (G-10, ORGANO Co.). The electrical conductivity of the water was below 0.1 μS cm−1.
Synthesis of SiTPyP
The synthesis of SiTPyP was carried out as a multi-step one-pot synthesis until step (3) as shown in Scheme 1. H2TPyP (160 mg, 0.25 mmol) was added to the reaction pot, kept under vacuum for 30 min and then purged with N2. Further, 25 mL of DCM was vacuum transferred to the reaction vessel and the resultant mixture stirred at room temperature for 1 h. To this mixture, LHMDS (85 mg, 0.5 mmol) was added and stirred at room temperature for 1 hour. The completion of step (1) was confirmed by a red shift in the UV spectrum (414 nm to 432 nm) of the reaction mixture. The reaction mixture was then cooled to −30 °C and then TEA (0.1 mL) and HSiCl3 (25 μL, 0.25 mmol) were added carefully. The reaction mixture was then slowly warmed to room temperature and stirred at room temperature for 1 h. The completion of step (2) was confirmed by a blue shift in the UV spectrum (432 nm to 427 nm) of the reaction mixture. Then, a slight excess of AgOTf was added to the reaction mixture and stirred overnight at room temperature. The completion of step (3) was confirmed by a blue shift in the UV spectrum (427 nm to 418 nm) of the reaction mixture. The reaction mixture was then filtered through 0.1 μm PTFE and Celite to remove the solid inorganic impurities. The crude crystals was further stirred for 48 hours in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DCM/aqueous solution of NaOH (1 M). The organic layer was separated and purified by silica gel column chromatography using DCM as eluent.
:1 mixture of DCM/aqueous solution of NaOH (1 M). The organic layer was separated and purified by silica gel column chromatography using DCM as eluent.
Elemental analysis.
Observed C 67.77%, H 4.58%, N 14.92%, calculated for [Si(IV)TPyP(OH)2](2CH3OH), C 67.91%, H 4.61%, N 15.08% 1H-NMR (CD3OD): δ = 9.00 (d, J = 5 Hz, 8H), 8.97 (s, 8H), 8.15 (d, J = 5 Hz, 8H) 13C-NMR (CD3OD): δ = 115.15 (s)a, 129.18 (s)b, 131.88 (s)c, 142.84 (s)d, 147.90 (s)e, 148.92 (s)f, 149.43 (s)g, −1.45 (s)h. 29Si-NMR (CD3OD): δ = −200.37 (s) IR in cm−1 (KBr method): 794 cm−1 C-hoop (aromatic), 1011 cm−1 Si–OH stretching, 1406 cm−1 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching 1593 cm−1 CN stretching, 3000 cm−1 C–H stretching, 3400 cm−1 O–H stretching UV-vis in ethanol: ε = 4.54 × 105 M−1 dm−3 (λmax = 419 nm), 1.80 × 104 M−1 dm3 (λ = 550 nm) ESI-MS (positive mode in methanol): m/z = 661 (SiTPyP(OH)).
C stretching 1593 cm−1 CN stretching, 3000 cm−1 C–H stretching, 3400 cm−1 O–H stretching UV-vis in ethanol: ε = 4.54 × 105 M−1 dm−3 (λmax = 419 nm), 1.80 × 104 M−1 dm3 (λ = 550 nm) ESI-MS (positive mode in methanol): m/z = 661 (SiTPyP(OH)).
Methods
The oxidation peak potentials of SiTPyP were measured by cyclic voltammetry on an electrochemical analyzer (Model 611DST, BAS) with boron-doped diamond/glassy carbon used as the working electrode, Ag/AgNO3 as the reference electrode, and Pt wire as the counter electrode in acetonitrile or aqueous acetonitrile containing (C4H9)4N+ PF6− (0.1 M) as the supporting electrolyte. The UV-visible spectra were measured on a Shimadzu UV-2550 spectrophotometer. The experimental set-up for the controlled potential electrolysis in a single cell and separated cell is described in ESI 11.† Oxygen was quantitatively analyzed using gas chromatography techniques (Shimadzu GC-TCD (GC-2014)) equipped with molecular sieves 13× (GL Scientific) and a thermal conductivity GC TCD detector using nitrogen as the carrier gas (conditions: temperature of the detector: 80 °C, pressure: 55.6 kPa, flow rate of nitrogen: 30 mL min−1 and injected volume: 0.5 mL). The amount of gas was estimated using a pre-calibrated standard curve plotted using standard gas samples prepared through manometer adjustments. The isotope labelled experiments were carried out in water containing 10% isotope labelled water (97% 18O-labelled water (H2O18); Cambridge Isotopes). The incorporation of isotope labelled oxygen molecules was analyzed using GC-MS (GC-2010-Plus) with RESTEK (Rt-Msieve5A) as the molecular sieves (conditions: temperature of the detector: 200 °C, He pressure: 40 kPa, flow rate: 50 mL min−1 and ionization temperature: 200 °C). The conditions were calibrated using standard gas samples (injected volume: 0.2 mL). Spectroscopic hydrogen peroxide analysis was carried out according to a literature method.40 The detailed procedure is described in ESI 5.† The procedure used in the isotope labeled experiment for hydrogen peroxide detection using the catalase enzyme is described in ESI 9.†
Conclusions
The second most earth abundant atom incorporated meso-tetrapyridylporphyrinatesilicon(IV) (SiTPyP) as an electrochemical catalyst was newly synthesized and its catalytic behaviour for water oxidation to form hydrogen peroxide as the primary two-electron oxidized product was examined. The formation of hydrogen peroxide in aqueous acetonitrile was initiated through a one-electron oxidation process on the electrode with a faradaic yield of 84–92%. Though the turnover frequency, which characterizes the catalytic reactivity, could not be determined, different aspects from those of AlTMPyP were observed in the electronic structure of the one-electron oxidized form and the formation constant of the hydroperoxy complex with the porphyrins. The former may affect the reactivity of the one-electron oxidized species of SiTPyP against OH− ion through the different electron spin densities on the two axial oxygen atoms. The latter may also induce different reactivity of liberating hydrogen peroxide from hydroperoxy complex formed during the reaction cycle.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was partly supported by JSPS KAKENHI Grant Number 17H06439 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)” and H. T. thanks the Tokyo Ohka Foundation for their support.
Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS.
-
POWERING THE WORLD WITH SUNLIGHT, A White Paper Describing the Discussions and Outcomes of the 1st Annual Chemical Sciences and Society Symposium (CS3) Kloster Seeon, Germany, July 23–25, 2009, https://www3.csj.jp/news/cs3-whitepaper.pdf Search PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS PubMed.
-
(a) H. Sakurai, H. Masukawa, M. Kitashima and K. Inoue, J. Photochem. Photobiol., C, 2013, 17, 1 CrossRef CAS;
(b) H. Masukawa, H. Sakurai, R. P. Hausinger and K. Inoue, Appl. Microbiol. Biotechnol., 2017, 101, 2177 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2016, 440, 295 CrossRef PubMed.
- X. Wu, F. Li, B. Zhang and L. Sun, J. Photochem. Photobiol., C, 2015, 25, 71 CrossRef CAS.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465 CrossRef CAS PubMed.
- R. Kuriki, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645 CrossRef CAS PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593 CrossRef CAS PubMed.
-
(a) S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240 CrossRef CAS PubMed;
(b) T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998 RSC.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 121 Search PubMed.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173 CAS.
- H. Inoue, M. Sumitani, A. Sekita and M. Hida, J. Chem. Soc., Chem. Commun., 1987, 1681 RSC.
- S. Takagi, M. Suzuki, T. Shiragami and H. Inoue, J. Am. Chem. Soc., 1997, 119, 8712 CrossRef CAS.
- S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734 CrossRef CAS PubMed.
- H. Inoue, S. Funyu, Y. Shimada and S. Takagi, Pure Appl. Chem., 2005, 77, 1019 CrossRef CAS.
- S. Funyu, M. Kinai, D. Masui, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2010, 9, 931–936 CAS.
- T. Shimada, A. Kumagai, S. Funyu, S. Takagi, D. Masui, Y. Nabetani, H. Tachibana, D. A. Tryk and H. Inoue, Faraday Discuss., 2012, 155, 145 RSC.
- K. Kurimoto, T. Yamazaki, Y. Suzuri, Y. Nabetani, S. Onuki, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2014, 13, 154 CAS.
- S. Mathew, F. Kuttassery, Y. Gomi, D. Yamamoto, R. Kiyooka, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2015, 313, 137 CrossRef CAS.
- S. N. Remello, T. Hirano, F. Kuttassery, Y. Nabetani, D. Yamamoto, S. Onuki, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2015, 313, 176 CrossRef CAS.
- T. Shiragami, H. Nakamura, J. Matsumoto, M. Yasuda, Y. Suzuri, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2015, 313, 131 CrossRef CAS.
- F. Kuttassery, S. Mathew, S. Sagawa, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemSusChem, 2017, 10, 1909 CrossRef CAS PubMed.
- S. Mathew, F. Kuttassery, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemPhotoChem, 2018, 2, 240 CrossRef CAS.
- S. N. Remello, F. Kuttassery, T. Hirano, Y. Nabetani, D. Yamamoto, S. Onuki, H. Tachibana and H. Inoue, Dalton Trans., 2015, 44, 20011 RSC.
- K. M. Kane, C. R. Lorenz, D. M. Heilman and F. R. Lemke, Inorg. Chem., 1998, 37, 669 CrossRef CAS.
- F. Kuttassery, S. Mathew, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, Electrochemistry, 2014, 82, 475 CrossRef CAS.
-
Diamond electrochemistry, ed. A. Fujishima, Y. Einaga, T. N. Rao and D. A. Tryk, Elsevier, 2005 Search PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Method: Fundamentals and Applications, Wiley, 2nd edn, 2001 Search PubMed.
- K. Takamura and C. Matsubara, Bull. Chem. Soc. Jpn., 2003, 76, 1873 CrossRef CAS.
-
A. C. Maehly, Methods Biochem. Anal., John Wiley & Sons, Inc., 2006, pp. 357–424 Search PubMed.
- A. McKillop and W. R. Sanderson, Tetrahedron, 1995, 51, 6145 CrossRef CAS.
- E. J. Ruiz, Y. Meas, R. Ortega and J. L. Jurado, ECS Trans., 2007, 3, 29 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Coordination of water to SiTPyP, eight-step change upon acid titration to SiTPyP, catalytic anodic current observed in CV, experimental procedures to analyse free H2O2 and peroxides, isotope experiment using catalase and experimental set-up for the controlled potential electrolysis. See DOI: 10.1039/c8se00102b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Siby
Mathew
c,
Siby
Mathew