
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupercooling of functional alkyl-π molecular liquids†
Fengniu
Lu
 a,
Keumhee
Jang
a,
Izabela
Osica
b,
Keita
Hagiwara
c,
Michito
Yoshizawa
c,
Masashi
Ishii
d,
Yoshiaki
Chino
e,
Kazuchika
Ohta
e,
Kinga
Ludwichowska
b,
Krzysztof Jan
Kurzydłowski
b,
Shinsuke
Ishihara
a and
Takashi
Nakanishi
*a
a,
Keumhee
Jang
a,
Izabela
Osica
b,
Keita
Hagiwara
c,
Michito
Yoshizawa
c,
Masashi
Ishii
d,
Yoshiaki
Chino
e,
Kazuchika
Ohta
e,
Kinga
Ludwichowska
b,
Krzysztof Jan
Kurzydłowski
b,
Shinsuke
Ishihara
a and
Takashi
Nakanishi
*a
aInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan. E-mail: nakanishi.takashi@nims.go.jp
bMaterials Design Division, Faculty of Materials Science and Engineering, Warsaw University of Technology, Woloska 141, 02-507 Warsaw, Poland
cLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
dMaterials Data Platform Center, Research and Services Division of Materials Data and Integrated System (MaDIS), NIMS, 1-2-1 Sengen, Tsukuba 305-0047, Japan
eSmart Material Science and Technology, Interdisciplinary Graduate School of Science and Technology, Shinshu University, 1-15-1 Tokida, Ueda 386-8567, Japan
First published on 17th July 2018
Abstract
Metastable states of soft matters are extensively used in designing stimuli-responsive materials. However, the non-steady properties may obstruct consistent performance. Here we report an approach to eradicate the indistinguishable metastable supercooled state of functional molecular liquids (FMLs), which remains as a liquid for weeks or months before crystallizing, via rational molecular design. The phases (solid, kinetically stable liquid, and supercooled liquid) of a model FML, branched alkyl chain-substituted 9,10-diphenylanthracene (DPA), are found to be governed by subtle alterations of the molecular structure (alkyl-DPA ratio and bulkiness of the DPA unit). We thus outline molecular design principles to avoid supercooled FML formation. Moreover, we demonstrate a practical technique to rapidly discriminate supercooled FMLs (within 5 h) by accelerating their crystallization in differential scanning calorimetry heating via pre-annealing or relatively slow scanning.
Introduction
The skillful manipulation of metastable states in stimuli-responsive materials is extensively used for sensing, energy harvesting, biomedical engineering, etc.1 However, materials in metastable states would be destructive for applications requiring consistent performance under various conditions. Considering the metastable matter of supercooled liquids (SCLs) as an example, the kinetically trapped melts below their melting points often crystallize as energetically favoured solids.2 Such a phase transition in π-conjugated molecular systems always changes the optoelectronic properties.3 Therefore, SCLs obstruct long-term reliability when used as contributing components in optoelectronic devices. Despite decades of active study, the controllability of SCLs remains challenging because of their rich phenomenology and complicated phase transition behaviour.4Recently, room-temperature functional molecular liquids (FMLs),5 which are fluidic and optoelectronically active in nature, have been widely applied in luminescent inks,6 host–guest binding media,7 and flexible optoelectronic devices including organic light-emitting diodes,8 photovoltaics,9 and organic semiconductors.10 FMLs are generally prepared by wrapping a π-conjugated molecular unit with bulky and flexible side chains (alkyl,6,8,11 siloxane,10b,12 alkylsilyl,10a,13 or ethylene glycol7,9a,14), which suppress the intermolecular π–π interactions and reduce the melting point to below room temperature (20 ± 5 °C). To date, several strategies have been proposed for powerful liquefaction of solid π-molecules.15 However, some FMLs are SCLs with melting points above room temperature; even when obtained as fluids, they solidify over time.16 The latent crystallization of supercooled FMLs, which occurs over very long time periods because of the highly amorphous side chains, impedes the identification of SCLs. To guarantee reliable functioning of FMLs-based devices, techniques to disclose indistinguishable supercooled FMLs and molecular design strategies to produce kinetically stable FMLs are necessary.
In this study, we focus on the effects of side chain-content ratio and π-skeleton structure on the phase behaviours of FMLs, aiming to establish molecular design principles for FMLs with kinetic stability at room temperature. As a proof of concept, we report the liquefaction of a model π-conjugated molecule, namely, 9,10-diphenylanthracene (DPA) (melting point: 249–250 °C),17 with bulky and flexible branched alkyl chains. Guerbet alcohol-based18 branched alkyl chains of different lengths were attached to the 3,5-substituent positions of the phenyl units in DPA (Fig. 1a, DPAn, n = 2, 4, 6, 8, and 10, where n denotes the number of carbons in the branched hydrocarbon chain substituted at the β-position of the longer hydrocarbon chain). The compounds with n ≥ 6 were successfully liquefied (Fig. 1b). However, the kinetic stability of the resulting liquids depended strongly on the alkyl-DPA content ratio in the molecular system. Furthermore, by introducing bulky bromine atoms on the 2,6-positions of the anthracene unit, the kinetic stability was efficiently improved (Fig. 1a and b, DPA8Br).
 | ||
| Fig. 1 (a) Chemical structures of DPAn (n = 2, 4, 6, 8, and 10) and DPA8Br. (b) Photographs of freshly synthesized DPAn and DPA8Br in daylight at 20 °C, with a drop of fluid or powder of solid on a glass plate. (c) Photographs of DPA6, DPA8, and DPA10 as fresh (left) and after long-term of aging at ambient temperature (right) taken under irradiation of a UV lamp (365 nm). | ||
Results and discussion
Synthesis of DPA derivatives
DPAn compounds were synthesized via the Williamson ether reaction from 9,10-bis(3,5-dihydroxyphenyl)anthracene and alkyl bromides with yields over 49% (see details in ESI†). The 2,6-dibrominated anthracene DPA8Br was prepared via the bromination of 9,10-bis(3,5-dimethoxyphenyl)anthracene, followed by O-demethylation and O-alkylation with 2-octyldodecyl chains. All compounds were unambiguously characterized by 1H nuclear magnetic resonance (1H NMR), 13C NMR, and matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS). The 1H and 13C NMR spectra confirmed the absence of residual solvents in all fluidic samples.Crystallization of supercooled DPA8 and DPA10 promoted by thermal treatment
DPA8, previously reported as a room temperature liquid,5b unexpectedly solidified after aging at ambient conditions for over 6 months (Fig. 1c). DPA10 solidified within 4 weeks from its liquid form (Fig. 1c). To analyze the phase behaviours, we measured thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) for DPAn. TGA confirmed no degradation of any compound below 393 °C (Fig. S1 and Table S1, ESI†). In the DSC heating traces at 10 °C min−1, DPA2 and DPA4 exhibit endothermic melting peaks (Tm) at 71.0 °C and 60.5 °C, respectively, whereas DPA10 exhibits a broad glass-to-isotropic liquid transition (Tg) (<−50 °C), followed by an exothermic crystallization peak (Tc) at −3.9 °C and a Tm at 26.6 °C (Fig. 2a). According to the thermal behaviours of typical organic crystals summarized in Fig. S2 (ESI†), the fluidic sample of DPA10 was in a supercooled state below 26.6 °C (Fig. S2c, ESI†). For DPA6 and DPA8, no melting peaks, other than Tg (<−50 °C), are observed in their DSC heating traces (Fig. 2a). However, after annealing at −45 °C for 12 h, subsequent heating induces the crystallization of DPA8 at 10.2 °C, which thereafter melts at 21.6 °C (Fig. 2b(i)). Therefore, the fluidic sample of DPA8 at temperatures below 21.6 °C is also in its supercooled state, which crystallizes under cold conditions (Fig. 2b). This result contradicts the previous report that DPA8 at ambient temperature exists only in liquid form;5b thus, FMLs require careful attention to avoid the misuse of their SCL phase.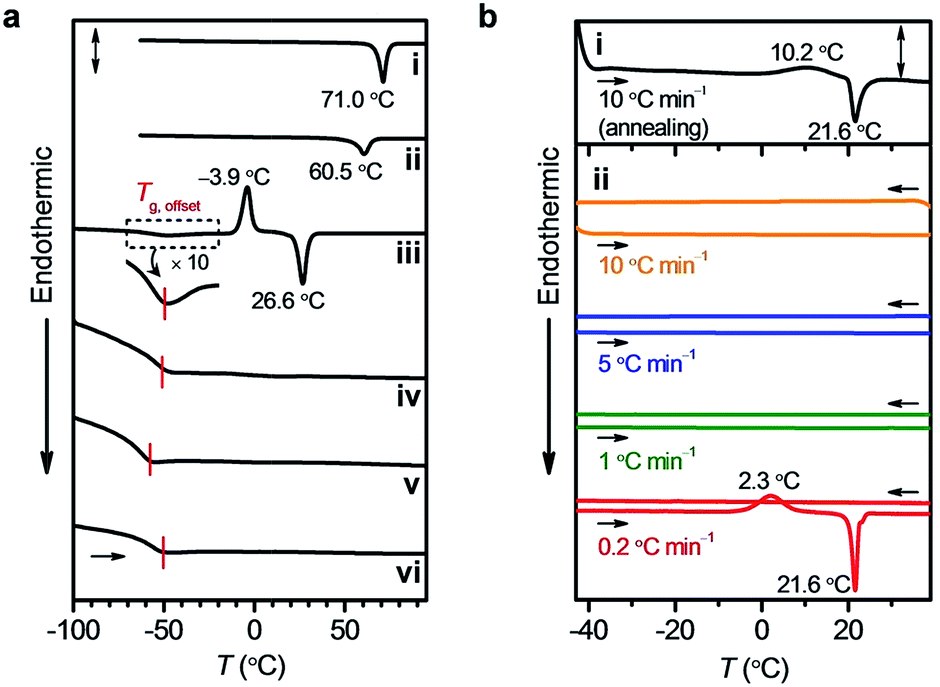 | ||
| Fig. 2 (a) DSC thermograms of (i) DPA2, (ii) DPA4, (iii) DPA10, (iv) DPA6, (v) DPA8, and (vi) DPA8Br during the first heating scan from −100 to 95 °C at 10 °C min−1 under N2 flow (Tg,offset, the offset temperature of the glass-to-isotropic liquid transition; scale bar (sb), 2.0 W g−1 for (i–iii) and 0.2 W g−1 for (iv–vi)). (b) DSC thermograms of DPA8 (i) during the heating scan from −45 to 40 °C at 10 °C min−1 after annealing at −45 °C for 12 h (sb, 0.1 W g−1) and (ii) during the heating/cooling cycles between −45 and 40 °C at 10 °C min−1 (orange; sb, 1.6 W g−1), 5 °C min−1 (blue; sb, 1.6 W g−1), 1 °C min−1 (green; sb, 0.4 W g−1), and 0.2 °C min−1 (red; sb, 0.1 W g−1) under N2 flow. | ||
The effect of annealing on the phase behaviour of DPA8 can be explained by the crystallization mechanism. The crystallization of an SCL occurs via two steps of nucleation and subsequent crystal growth.19 Nucleation can occur at any temperature below Tm, but the tendency increases as the temperature decreases between Tm and Tg. Therefore, annealing the DPA8 SCL at −45 °C, which is just above Tg, promotes nucleation. The generated nuclei grow spontaneously at an elevated temperature (Tc), at which the mobility is sufficient for molecular reorientation and self-organization. Since no Tc was observed in the second heating trace without annealing (Fig. S3, ESI†), we assumed that the time scale of the heating process at 10 °C min−1 was insufficient for DPA8 crystallization (Fig. S2e, ESI†). Our assumption was approved by the appearance of a crystallization peak at a long heating process with the scan rate reduced to 0.2 °C min−1 (Fig. 2b(ii)). In sharp contrast, the crystallization of DPA10 occurred even at 40 °C min−1 without annealing (Fig. S4, ESI†). Therefore, DPA10 exhibits a greater crystallization tendency than DPA8 does.
DPA6 showed no crystallization peak in the DSC traces even after annealing at −45 °C for 12 h or heating at 0.2 °C min−1 (Fig. S5, ESI†). In addition, no solidification was observed during storage at ambient temperature throughout our experimental period (exceeding 1 year) (Fig. 1c). Thus, we conclude that DPA6 is a kinetically stable liquid at ambient temperatures.20
Crystallization tendency governed by alkyl-π ratio and the bulkiness of the π-unit
Judging from the phase behaviours of DPA2 → DPA6, lengthening the alkyl chains reduces the melting point. According to the decreased entropy values from DPA4 (111.0 J mol−1 K−1) to DPA2 (131.8 J mol−1 K−1) at their respective melting temperatures (Table S1, ESI†), solid DPA4 has a larger amorphous content than solid DPA2. For DPA6, the amorphousness may be further enhanced by its bulkier and more random alkyl side chains, thus suppressing solidification.On the other hand, from DPA6 → DPA10, lengthening the alkyl chains enhances the crystallization tendency of the melts. To gain deeper insight, the distribution of DPA units in DPA6, DPA8, and DPA10 melts at 30 °C was investigated. All three melts were isotropic liquids, as confirmed by the absence of birefringence in polarized optical microscopic (POM) observations (Fig. S6, ESI†). Their liquid-like behaviours were suggested by the higher values for viscous loss moduli (G′′) than for storage elastic moduli (G′) throughout the measured angular frequency (ω) range (Fig. S7, ESI†). The subsequent small- and wide-angle X-ray scattering (SWAXS) measurements verified the lack of long-range molecular ordering, as no sharp crystalline peaks were detected (Fig. 3a). The average intermolecular DPA–DPA distances (taken from the top of the halo appearing in the small-angle region) in DPA6 (18.9 Å), DPA8 (21.0 Å), and DPA10 (22.0 Å) were much larger than that for active π–π interactions (3.4–3.5 Å), indicating negligible interactions among adjacent DPA units. We thus hypothesize that the increased crystallization tendency from DPA6 → DPA10 might originate from increased van der Waals interactions among alkyl chains of longer lengths.3b In addition, when the length of alkyl chains increases from DPA6 → DPA10, the low-q halo become narrower (Fig. 3a, full width at half maximum was calculated as 0.26 Å−1 for DPA6, 0.22 Å−1 for DPA8, and 0.19 Å−1 for DPA10), suggesting improved regularity of intermolecular distances. Such an improvement is probably caused by the more uniform molecular shape, e.g., more globular-like, with longer alkyl side chains, which may also contribute to the enhanced crystallization tendency.
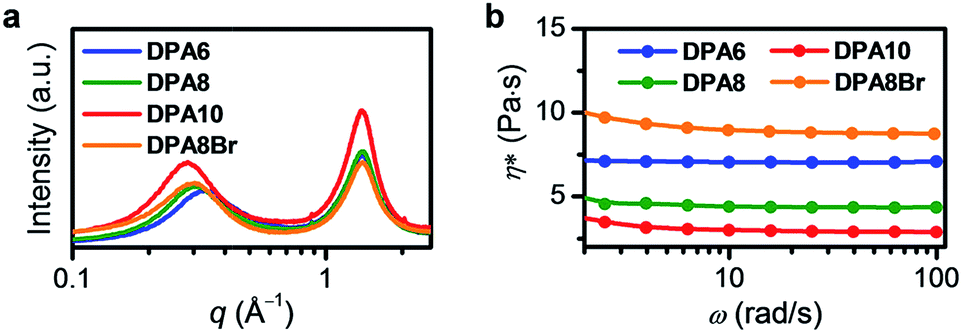 | ||
| Fig. 3 (a) SWAXS profiles and (b) complex viscosity (η*) of DPA6 (blue), DPA8 (green), DPA10 (red), and DPA8Br (orange) at 30 °C (ω, angular frequency). | ||
The viscosity of liquid is also influenced by the alkyl-DPA ratio. As confirmed by the order of complex viscosity (η*) of the melts at 30 °C (at angular frequency (ω) = 10 rad s−1) of DPA10 (3.0 Pa s) < DPA8 (4.4 Pa s) < DPA6 (7.1 Pa s) (Fig. 3b and Table S1, ESI†), higher alkyl chain contents promote greater molecular softening and thus increase fluidity. Consequently, DPA10 and DPA8 may experience faster molecular diffusion, which can facilitate crystallization. Therefore, it can be concluded that the kinetic stability of alkylated-π FMLs is governed by the alkyl-π ratio.
In addition, we introduced bromine atoms on the 2,6-positions of anthracene in DPA8, yielding the kinetically stable liquid of DPA8Br (Fig. 1a and b). No crystallization is observed either in the DSC measurements (Fig. 2a and S8, ESI†) or after long-term (>1 year) aging at ambient temperature. Possible reasons for the suppressed crystallization of DPA8Br include a decrease of π-skeleton symmetry from D2h to C2h and a higher η* (9.0 Pa s) than that of DPA8 (4.4 Pa s) (Fig. 3b and Table S1, ESI†). The increased viscosity may originate from the larger molecular size of DPA8Br. As supported by SWAXS analysis, the average core-to-core distance of DPA8Br (21.3 Å) is slightly larger than that of DPA8 (21.0 Å) (Fig. 3a).
Structural and photophysical changes of DPA8 and DPA10 upon SCL-to-crystal transition
To identify the molecular organization during crystallization, variable-temperature SWAXS and POM studies were performed for DPA10; the results agreed well with the DSC findings. As DPA10 was cooled from 30 to −30 °C, the broad halos in the SWAXS profile and the dark sample region in the POM image (Fig. 4a) were retained, indicating supercooling of the melt. Upon heating from −30 to −15 °C at 10 °C min−1, crystalline peaks were detected by SWAXS and birefringence was observed via POM in the SCL domain. The peaks in the SWAXS profiles sharpened and intensified, and the texture in the POM images gradually propagated throughout the sample area as the temperature was elevated from −15 to 15 °C. Further heating to 30 °C caused the collapse of all SWAXS sharp peaks and POM texture (see also Movie S1, ESI†) due to the melting of the crystalline phase. The profile at 15 °C exhibited multiple peaks (Fig. 4a). However, all peaks were relatively broad. Moreover, the broad halo at the wide-angle region (4.5 Å), which corresponds to the mean distance between molten alkyl chains (Fig. 3a), remained in the profile. Thus, the crystalline sample of DPA10 is partially amorphous, resembling a liquid crystalline phase. However, pressing and shear caused no changes in the POM texture (Fig. S10, ESI†). And the enthalpy values at Tc and Tm of the DSC curve are quite high (Table S1, ESI†). Therefore, DPA10 is not only partially amorphous but also partially crystalline.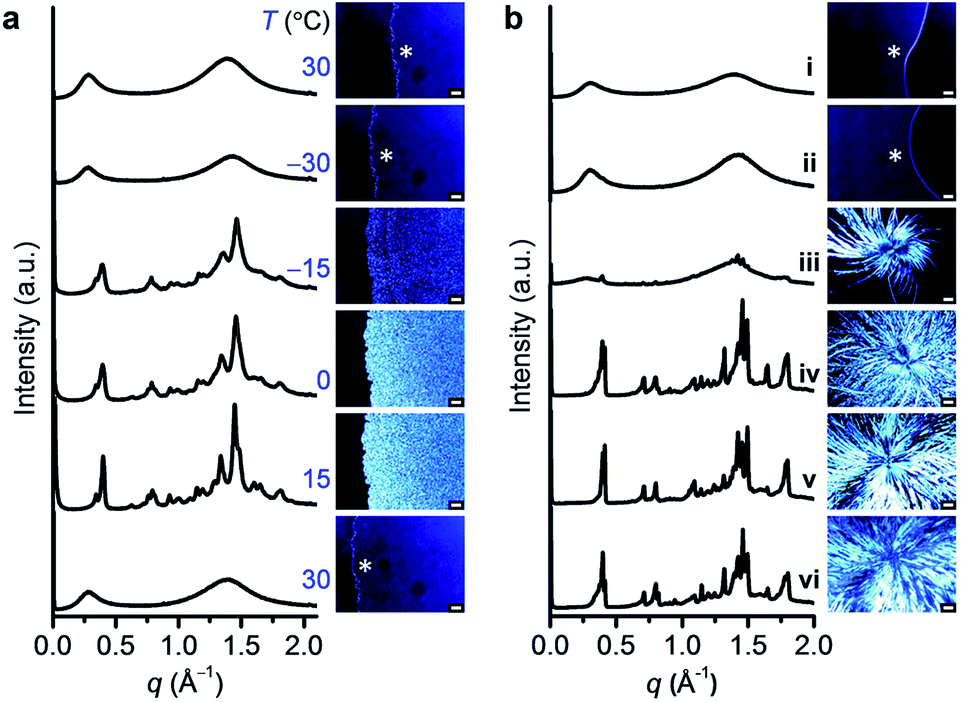 | ||
| Fig. 4 (a) Variable-temperature SWAXS profiles (left) and POM images (right; scale bar (sb), 50 μm) of DPA10 cooled from 30 to −30 °C, and then heated to −15, 0, 15, and 30 °C at 10 °C min−1 (top to down). (b) SWAXS profiles (left) and POM images (right; sb, 50 μm for (i–iii) and 100 μm for (iv–vi)) of DPA8: (i) freshly melted, (ii) annealed at −18 °C for 1 day, and subsequent aging at 15 °C for (iii) 1 day, (iv) 3 days, (v) 7 days, and (vi) 14 days. The sample area was denoted with asterisk (*) in the POM. | ||
The crystallization of DPA8 was also monitored via SWAXS and POM (Fig. 4b), by annealing the sample at −18 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 for 1 day and then holding it at 15 °C. Multiple peaks in the SWAXS profile and birefringence texture on the POM image appeared after aging at 15 °C for 1 day. After 7 days, crystallization ceased. POM images show a dendritic texture (Fig. 4b) with a larger domain size than the spherulite texture of DPA10 formed via a slow crystallization process by annealing the melted sample at −5 °C for 7 h (Fig. S9a, ESI†). As further reflected in the SWAXS profiles, crystalline DPA8 exhibited a larger number of sharp peaks and a less intense alkyl halo (Fig. 4b) than did DPA10 (Fig. S9b, ESI†). The molecular packing structures in the crystalline states of both DPA8 and DPA10 were difficult to determine because of their complex patterns, which showed larger numbers of peaks than those of any considerable liquid crystals, and both broader and fewer peaks than those of the presumed single-crystalline materials.
21 for 1 day and then holding it at 15 °C. Multiple peaks in the SWAXS profile and birefringence texture on the POM image appeared after aging at 15 °C for 1 day. After 7 days, crystallization ceased. POM images show a dendritic texture (Fig. 4b) with a larger domain size than the spherulite texture of DPA10 formed via a slow crystallization process by annealing the melted sample at −5 °C for 7 h (Fig. S9a, ESI†). As further reflected in the SWAXS profiles, crystalline DPA8 exhibited a larger number of sharp peaks and a less intense alkyl halo (Fig. 4b) than did DPA10 (Fig. S9b, ESI†). The molecular packing structures in the crystalline states of both DPA8 and DPA10 were difficult to determine because of their complex patterns, which showed larger numbers of peaks than those of any considerable liquid crystals, and both broader and fewer peaks than those of the presumed single-crystalline materials.
To analyze the influence of the SCL-to-crystal transition on the optical characteristics, the photophysical properties of DPAn were studied. The melts of DPA6, DPA8, and DPA10 showed optical features comparable to those of their dilute solutions (Fig. S11 and Table S2, ESI†), confirming the absence of significant intermolecular interactions among the solvent-free DPA chromophores. Fig. 5a shows good spectral consistence between liquid DPA6 and SCLs of DPA8 and DPA10, further indicating that the supercooled state is indistinguishable by spectroscopic features. The more vibronic structure of the fluorescence bands of DPA8 and DPA10 may originate from their lower viscosities. Upon crystallization, the emission bands of DPA8 and DPA10 become less vibronic (with the disappearance of the 411 nm shoulder peak) and slightly blue-shifted (Fig. 5a). The spectral shift is ascribed to increased molecular organization, rather than restricted molecular motion, in the solid crystalline state. As confirmed by the temperature-variable fluorescence spectra of DPA10, negligible spectral shift is observed when the sample is frozen into the amorphous glass state upon cooling from 10 to −110 °C (Fig. 5b, up). However, subsequent heating to above −50 °C (at which crystallization could occur) causes a hypsochromic shift of the emission band (Fig. 5b, below). According to these results, the crystallization of a luminescent supercooled FML leads to non-steady photophysical properties.
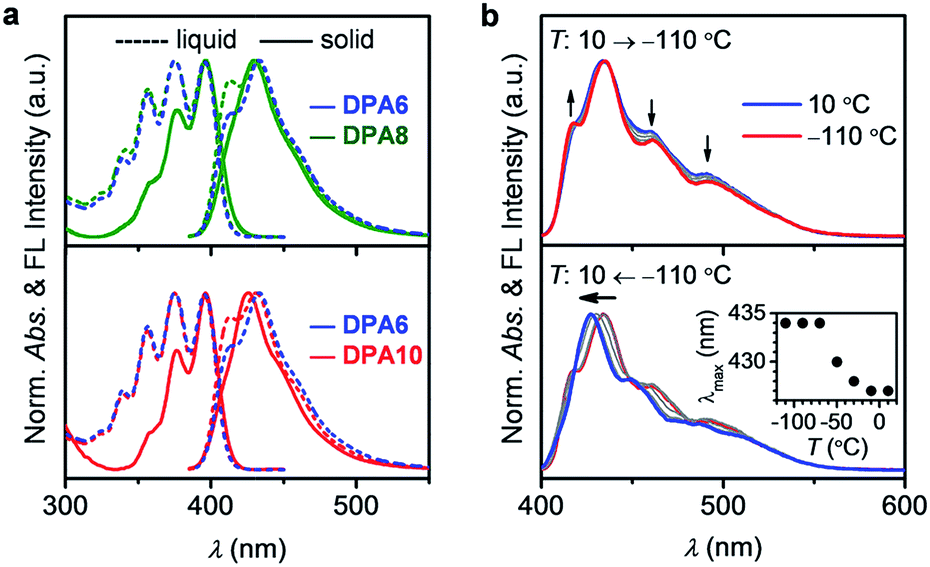 | ||
| Fig. 5 (a) Normalized UV-vis absorption (left) and fluorescence (right) spectra of DPA6 (blue), DPA8 (green) and DPA10 (red) as liquid (dashed line) and crystalline (solid line) at 20 °C. (b) Variable-temperature fluorescence spectra of DPA10 measured at 20 °C steps during cooling from 10 to −110 °C (up) and subsequent heating from −110 to 10 °C (below; inset, maximum emission wavelength at each temperature) at 10 °C min−1. | ||
Conclusions
We elucidate the issue of supercooling of FMLs for the first time and propose effective measures to distinguish and eradicate the undesired metastable matter. By liquefying a solid π-molecule, 9,10-diphenylanthracene, with branched alkyl chains and further modifying the anthracene core with bulky atoms, we clarify three fundamental factors to prohibit the formation of supercooled FMLs: (i) a delicate alkyl chain-to-π ratio, (ii) a bulkier π-unit, and (iii) a less symmetric π-skeleton. These findings are essential to avoid the misuse of SCLs with non-steady functions and facilitate long-term practical applications of FMLs. In addition, rapid discrimination of supercooled FMLs is enabled by appropriate thermal treatment that accelerates the crystallization process. Deeper studies of the effect of π-skeleton structure on the kinetic stability of FMLs and the organization of supercooled molecules during crystallization would aid the development of advanced molecular design methodologies for the production of more reliable functional soft materials. Research in this direction is in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (JSPS KAKENHI Grant Number JP25104011, JP15H03801, JP18H03922) from the MEXT, Japan. The authors thank the Soft Materials Line and the MANA TSS Group at NIMS for use of their facilities.Notes and references
- (a) H. Wang, H. K. Bisoyi, L. Wang, A. M. Urbas, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2018, 57, 1627–1631 CrossRef PubMed; (b) K. G. Gutierrez-Cuevas, L. Wang, Z. Zheng, H. K. Bisoyi, G. Li, L.-S. Tan, R. A. Vaia and Q. Li, Angew. Chem., Int. Ed., 2016, 55, 13090–13094 CrossRef PubMed; (c) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef PubMed.
- (a) P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef PubMed; (b) M. D. Ediger, C. A. Angell and S. R. Nagel, J. Phys. Chem., 1996, 100, 13200–13212 CrossRef.
- (a) S. Karasawa, R. Hagihara, Y. Abe, N. Harada, J.-i. Todo and N. Koga, Cryst. Growth Des., 2014, 14, 2468–2478 CrossRef; (b) K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94–102 CrossRef PubMed; (c) Y. Sagara, K. Kubo, T. Nakamura, N. Tamaowki and C. Weder, Chem. Mater., 2017, 29, 1273–1278 CrossRef; (d) K. Mase, Y. Sasaki, Y. Sagara, N. Tamaoki, C. Weder, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2018, 57, 2806–2810 CrossRef PubMed.
- (a) K. Paeng and L. J. Kaufman, Chem. Soc. Rev., 2014, 43, 977–989 RSC; (b) E. Brini, C. J. Fennell, M. Fernandez-Serra, B. Hribar-Lee, M. Lukšič and K. A. Dill, Chem. Rev., 2017, 117, 12385–12414 CrossRef PubMed.
- (a) T. Michinobu, T. Nakanishi, J. P. Hill, M. Funahashi and K. Ariga, J. Am. Chem. Soc., 2006, 128, 10384–10385 CrossRef PubMed; (b) P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056–19059 CrossRef PubMed; (c) M. J. Hollamby, M. Karny, P. H. H. Bomans, N. A. J. M. Sommerdjik, A. Saeki, S. Seki, H. Minamikawa, I. Grillo, B. R. Pauw, P. Brown, J. Eastoe, H. Möhwald and T. Nakanishi, Nat. Chem., 2014, 6, 690–696 CrossRef PubMed; (d) N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. C. Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 CrossRef PubMed.
- (a) S. S. Babu, J. Aimi, H. Ozawa, N. Shirahata, A. Saeki, S. Seki, A. Ajayaghosh, H. Möhwald and T. Nakanishi, Angew. Chem., Int. Ed., 2012, 51, 3391–3395 CrossRef PubMed; (b) S. S. Babu, M. J. Hollamby, J. Aimi, H. Ozawa, A. Saeki, S. Seki, K. Kobayashi, K. Hagiwara, M. Yoshizawa, H. Möhwald and T. Nakanishi, Nat. Commun., 2013, 4, 1969 CrossRef PubMed.
- (a) T. Ogoshi, T. Aoki, R. Shiga, R. Iizuka, S. Ueda, K. Demachi, D. Yamafuji, H. Kayama and T.-a. Yamagishi, J. Am. Chem. Soc., 2012, 134, 20322–20325 CrossRef PubMed; (b) J. Zhang, S.-H. Chai, Z.-A. Qiao, S. M. Mahurin, J. Chen, Y. Fang, S. Wan, K. Nelson, P. Zhang and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 932–936 CrossRef PubMed.
- (a) S. Hirata, K. Kubota, H. H. Jung, O. Hirata, K. Goushi, M. Yahiro and C. Adachi, Adv. Mater., 2011, 23, 889–893 CrossRef PubMed; (b) N. Kobayashi, T. Kasahara, T. Edura, J. Oshima, R. Ishimatsu, M. Tsuwaki, T. Imato, S. Shoji and J. Mizuno, Sci. Rep., 2015, 5, 14822 CrossRef PubMed.
- (a) H. J. Snaith, S. M. Zakeeruddin, Q. Wang, P. Péchy and M. Grätzel, Nano Lett., 2006, 6, 2000–2003 CrossRef PubMed; (b) T. J. Kramer, S. S. Babu, A. Saeki, S. Seki, J. Aimi and T. Nakanishi, J. Mater. Chem., 2012, 22, 22370–22373 RSC.
- (a) B. A. Kamino, T. P. Bender and R. A. Klenkler, J. Phys. Chem. Lett., 2012, 3, 1002–1006 CrossRef PubMed; (b) T. G. Plint, B. A. Kamino and T. P. Bender, J. Phys. Chem. C, 2015, 119, 1676–1682 CrossRef.
- (a) S. S. Babu and T. Nakanishi, Chem. Commun., 2013, 49, 9373–9382 RSC; (b) A. Ghosh and T. Nakanishi, Chem. Commun., 2017, 53, 10344–10357 RSC; (c) K. Masutani, M. Morikawa and N. Kimizuka, Chem. Commun., 2014, 50, 15803–15806 RSC; (d) M. J. Hollamby, A. E. Danks, Z. Schnepp, S. E. Rogers, S. R. Hart and T. Nakanishi, Chem. Commun., 2016, 52, 7344–7347 RSC; (e) F. Lu, T. Takaya, K. Iwata, I. Kawamura, A. Saeki, M. Ishii, K. Nagura and T. Nakanishi, Sci. Rep., 2017, 7, 3416 CrossRef PubMed; (f) Z. Agnieszka, T. Atsuro, S. Hiroya, S. Akinori, L. Marcin and N. Takashi, Chem.–Asian J., 2018, 13, 770–774 CrossRef PubMed; (g) B. Narayan, K. Nagura, T. Takaya, K. Iwata, A. Shinohara, H. Shinmori, H. Wang, Q. Li, X. Sun, H. Li, S. Ishihara and T. Nakanishi, Phys. Chem. Chem. Phys., 2018, 20, 2970–2975 RSC.
- (a) B. A. Kamino, J. B. Grande, M. A. Brook and T. P. Bender, Org. Lett., 2010, 13, 154–157 CrossRef PubMed; (b) E. M. Maya, A. W. Snow, J. S. Shirk, R. G. S. Pong, S. R. Flom and G. L. Roberts, J. Mater. Chem., 2003, 13, 1603–1613 RSC.
- M. Taki, S. Azeyanagi, K. Hayashi and S. Yamaguchi, J. Mater. Chem. C, 2017, 5, 2142–2148 RSC.
- C.-H. Shim, S. Hirata, J. Oshima, T. Edura, R. Hattori and C. Adachi, Appl. Phys. Lett., 2012, 101, 113302 CrossRef.
- (a) M. J. Hollamby and T. Nakanishi, J. Mater. Chem. C, 2013, 1, 6178–6183 RSC; (b) F. Lu and T. Nakanishi, Sci. Technol. Adv. Mater., 2015, 16, 014805 CrossRef PubMed.
- (a) T. Machida, R. Taniguchi, T. Oura, K. Sada and K. Kokado, Chem. Commun., 2017, 53, 2378–2381 RSC; (b) L. C. Kerkhof, K. M. Allan, K. M. McGrath, J. L. Spencer and J. M. Hodgkiss, Int. J. Nanotechnol., 2017, 14, 432 CrossRef.
- C. K. Ingold and P. G. Marshall, J. Chem. Soc., 1926, 129, 3080–3089 RSC.
- A. J. O'Lenick, J. Surfactants Deterg., 2001, 4, 311–315 CrossRef.
- F. Abraham, Homogeneous Nucleation Theory, Academic Press, New York, 1974 Search PubMed.
- For accuracy, “kinetically stable” was used in this manuscript, because it is difficult to prove that DPA6 is “thermodynamically stable”. Even under more strict conditions (annealing at −45 °C for 24 h or applying slow scan rate of 0.2 °C min−1 after annealing at −45 °C for 12 h), no crystallization peak was observed in the DSC heating trace.
- The lowest temperature we can regulate to stock the samples for SWAXS measurements and POM observation.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR, MALDI-TOF MS, additional figures and tables as indicated in the main text. See DOI: 10.1039/c8sc02723d |
| This journal is © The Royal Society of Chemistry 2018 |