
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed enantioselective direct vinylogous allylic alkylation of coumarins†‡
Rahul
Sarkar
 ,
Sankash
Mitra
and
Santanu
Mukherjee
*
,
Sankash
Mitra
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@iisc.ac.in; Fax: +91-80-2360-0529; Tel: +91-80-2293-2850
First published on 4th June 2018
Abstract
The first iridium-catalyzed enantioselective vinylogous allylic alkylation of coumarins is presented. Using easily accessible linear allylic carbonates as the allylic electrophile, this reaction installs unfunctionalized allyl groups at the γ-position of 4-methylcoumarins in an exclusively branched-selective manner generally in high yields with an excellent level of enantioselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er).
:1 er).
Introduction
Transition-metal-catalyzed asymmetric allylic substitution (AAS) reactions have emerged as an extremely powerful and versatile method for the synthesis of enantioenriched compounds from easily available starting materials through enantioselective construction of carbon–carbon and carbon–heteroatom bonds.1 In contrast to initially developed and more commonly used palladium catalysts, iridium-catalyzed AAS reactions enable the synthesis of branched products from unsymmetrical allylic electrophiles through preferential attack of nucleophiles at the more substituted terminus of the π-allyl–Ir intermediate (Scheme 1).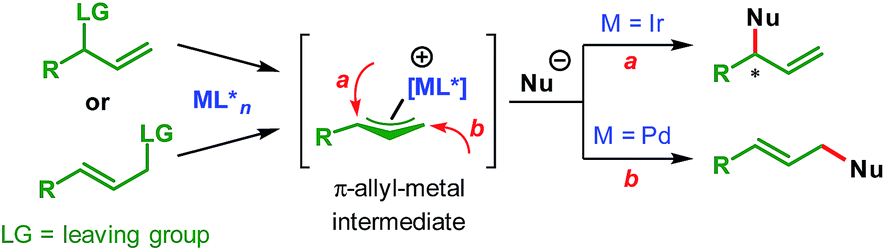 | ||
| Scheme 1 Transition-metal-catalyzed allylic substitution. | ||
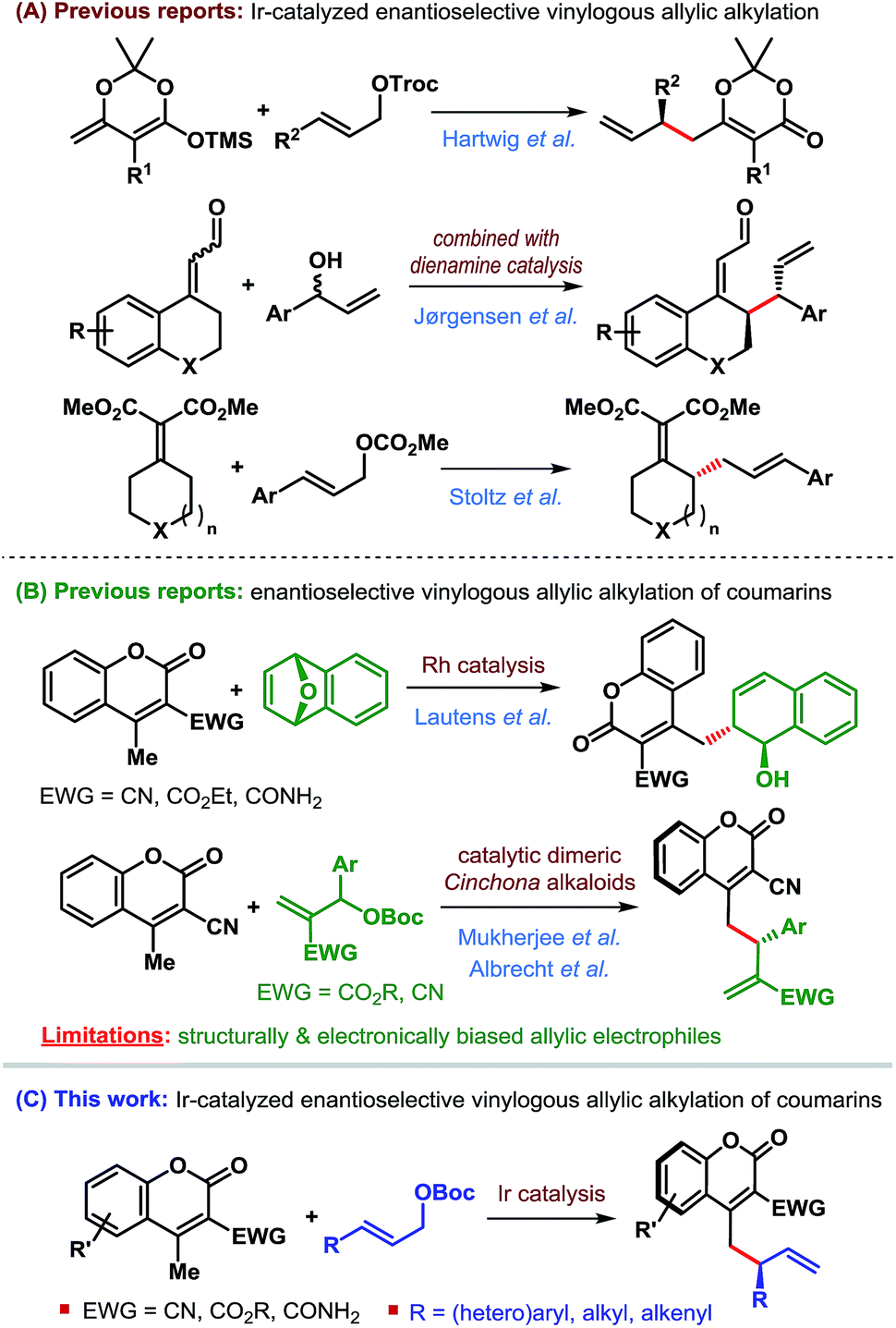
This characteristic of the Ir-catalyzed AAS overcomes the limitations associated with Pd-catalysis with respect to the scope of reaction partners and even allows for the use of achiral (non-prochiral) carbon and heteroatom nucleophiles in reactions with unsymmetrical allylic electrophiles.2 Consequently, a wide variety of nucleophiles have been applied3–5 to highly regioselective (branched-to-linear) and enantioselective allylic alkylation reactions ever since the introduction of Ir-catalysts in 1997.6 Despite these developments during the past two decades, the use of vinylogous nucleophiles in Ir-catalyzed asymmetric allylic alkylation (AAA) has received much less attention and only a handful of reports exist (Scheme 2A).7–9
 | ||
| Scheme 2 Enantioselective vinylogous allylic alkylation. | ||
In 2014, Hartwig et al. reported the first application of a vinylogous nucleophile, namely preformed silyl dienolates, in Ir-catalyzed AAA.7 The Jørgensen group combined the concept of dienamine activation with Ir-catalyzed AAA for regio-, diastereo- and enantioselective γ-allylic alkylation of α,β-unsaturated aldehydes.8 More recently, the Stoltz group developed a formal enantioselective γ-allylic alkylation of α,β-unsaturated malonates through a sequential Ir-catalyzed AAA/Cope rearrangement.9
With our own interest in vinylogous nucleophilic reactivity,10 we embarked on the development of direct catalytic enantioselective allylic alkylation of other vinylogous nucleophiles.
We became particularly interested in 4-methylcoumarins as the potential vinylogous nucleophile due to the wide abundance of coumarin derivatives in over 1000 natural products and bioactive targets as well as their utility in the dye industry.11 Although this class of nucleophiles received considerable attention since the introduction of 3-cyano-4-methylcoumarins by Xie et al. in 2010,12 enantioselective allylic alkylation of this potentially useful class of vinylogous nucleophiles remained underexplored until recently. Lautens' group developed the first enantioselective γ-allylic alkylation of 4-methylcoumarins in 2016 through a Rh-catalyzed desymmetrizing ring-opening reaction of oxabicycles (Scheme 2B).13 Very recently, our group14 and subsequently Albrecht et al.15 independently developed the first organocatalytic enantioselective γ-allylic alkylation of 3-cyano-4-methylcoumarins using Morita–Baylis–Hillman carbonates as the allylic electrophile (Scheme 2B). While an excellent level of enantioselectivity has been achieved, the success of these reactions is inherently dependent on the type of allylic electrophile – both structurally and electronically, thereby limiting their scope.
Since Ir-catalyzed allylic alkylation reactions are not constrained by such structural and electronic bias on the allylic electrophile, we believed that Ir-catalysis would provide a general strategy for the synthesis of coumarin derivatives that were previously challenging to access.
The purpose of this communication is to disclose the first Ir-catalyzed enantioselective vinylogous allylic alkylation of coumarins (Scheme 2C).
Results and discussion
We began our investigation with the optimization of the catalyst and reaction conditions16 for a model reaction between 3-cyano-4-methylcoumarin 1a and tert-butyl cinnamyl carbonate 2a in dichloroethane (DCE) at 50 °C (Table 1). A combination of [Ir(COD)Cl]2 and Feringa's phosphoramidite ligand L1,17 pioneered by Hartwig and co-workers,5i was initially tested. In the absence of any external base to activate 1a, the desired γ-allylated product 3aa was formed exclusively as a single regioisomer with a promising enantioselectivity, although in only 22% yield (entry 1). The tert-butoxide anion, generated in situ from the reaction between an Ir-complex and 2a, was anticipated to be the active base in this reaction.18 In line with the observation reported previously for Ir-catalyzed AAS reactions,3–5 the choice of external base was found to have a profound influence on both the reaction efficacy and enantioselectivity. Extensive exploration of various bases (entries 2–7), including inorganic and organic bases, revealed DABCO to be the optimum,19 affording the product in high yield and enantioselectivity (entry 7). A number of ligands for iridium were then tested under the influence of DABCO (entries 8–11). The use of ligand L2, a diastereomer of L1, failed to catalyze the reaction (entry 8). Ligand L3, an ortho-methoxy-derivative of L1, introduced by Alexakis et al.20 and generally known to act as a superior ligand compared to L1 in many AAS reactions, furnished 3aa with improved er but in poor yield (entry 9). No improvement in yield or enantioselectivity was observed with L4 or L5 (entries 10 and 11), and L1 remained the ligand of choice. A solvent screening (entries 12–14) at this point revealed dichloromethane as the optimum, providing the product in 86% isolated yield and with 98:2 er (entry 14).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Base | t [h] | Yieldb [%] | erc |
| a Reaction conditions: 3 mol% [Ir(COD)Cl]2, 6 mol% ligand, 0.24 mmol of 1a, 0.2 mmol of 2a and 0.2 mmol of base in 0.6 mL solvent. The catalyst was prepared via n-PrNH2 activation. b Yields were determined by 1H-NMR spectroscopy with mesitylene as the internal standard. Isolated yields are given in the parentheses. c The enantiomeric ratio (er) was determined by HPLC analysis on a chiral stationary phase; n.d. = not determined. DCE = 1,2-dichloroethane. | ||||||
| 1 | L1 | DCE | — | 48 | (22) | 92.5:7.5 |
| 2 | L1 | DCE | Cs2CO3 | 48 | <5 | n.d. |
| 3 | L1 | DCE | DBU | 48 | <5 | n.d. |
| 4 | L1 | DCE | i-Pr2NEt | 48 | 72 | 96:4 |
| 5 | L1 | DCE | Et3N | 48 | 74 | 97:3 |
| 6 | L1 | DCE | i-Pr2NH | 48 | 76 | 97:3 |
| 7 | L1 | DCE | DABCO | 48 | 84 | 97.5:2.5 |
| 8 | L2 | DCE | DABCO | 48 | <5 | n.d. |
| 9 | L3 | DCE | DABCO | 48 | 11 | 98:2 |
| 10 | L4 | DCE | DABCO | 48 | <5 | n.d. |
| 11 | L5 | DCE | DABCO | 48 | 28 | 92:8 |
| 12 | L1 | THF | DABCO | 48 | 11 | 96:4 |
| 13 | L1 | CHCl3 | DABCO | 48 | 67 | 97:3 |
| 14 | L1 | CH2Cl2 | DABCO | 36 | 88 (86) | 98:2 |

With the optimum ligand and reaction conditions (Table 1, entry 14) in hand, we chose to explore the scope and limitations of this direct vinylogous allylic alkylation protocol. We were pleased to note that the efficacy displayed by the Ir/L1 combination for the reaction between 1a and 2a under the optimum reaction conditions is indeed a general phenomenon and could be extended to other substrate combinations. As shown in Table 2, 3-cyano-4-methylcoumarin (1a) underwent facile allylic alkylation with an assortment of allylic carbonates (2a–u). Aryl-substituted allylic carbonates with diverse steric and electronic demands on the aryl ring (2a–i) were well tolerated, providing the products (3aa–ai) in high yields with excellent enantioselectivities (Table 2A, entries 1–9). When the substituent was present in either the meta- or para-position of the phenyl ring, products were formed with similar yields and enantioselectivities. In accordance with previously reported AAS reactions with L1,21 the ortho-substituent on the phenyl ring led to slow reaction, giving the product with moderate yield and er (entry 10). This adverse effect of ortho-substituent is clearly evident from the comparative outcome of the reactions with dichloro-substituted cinnamyl carbonates 2k and 2l (entries 11 and 12). We were also interested in incorporating pharmaceutically relevant heterocycles into our products, and found that allylic carbonates bearing dioxolane (2m), pyridine (2n), furan (2o) and thiophene (2p) provided the products in good to high yields with excellent er (entries 13–16).
| a Reaction conditions: 3 mol% [Ir(COD)Cl]2, 6 mol% L1, 0.24 mmol of 1a, 0.2 mmol of 2 and 0.2 mmol of DABCO in 0.6 mL CH2Cl2. The catalyst was prepared via n-PrNH2 activation. Yields correspond to the isolated product after chromatographic purification. er was determined by HPLC analysis on a chiral stationary phase. |
|---|
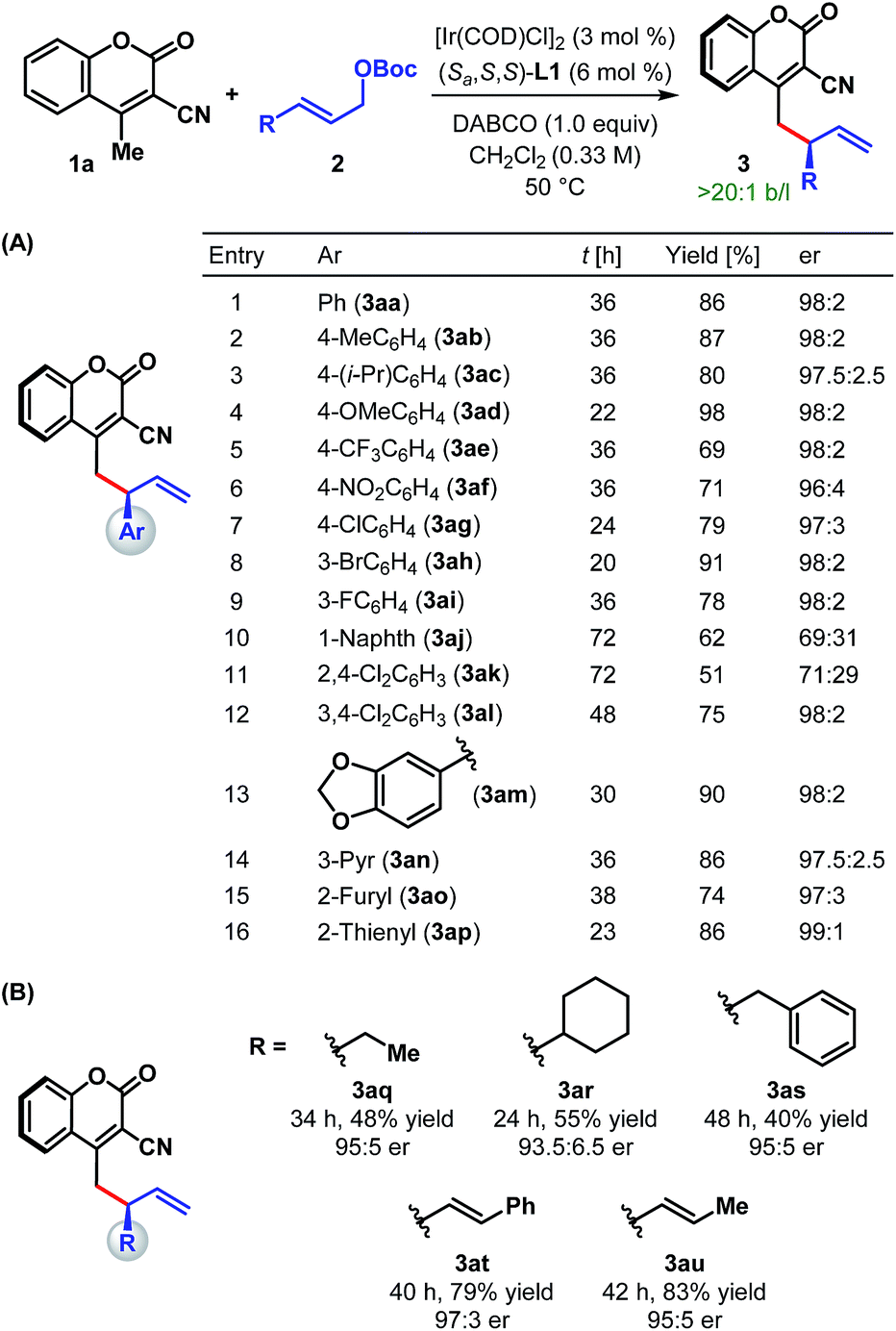
|
The scope of our protocol is not limited to (hetero)aromatic allylic carbonates. As illustrated by the examples in Table 2B, products derived from allylic carbonates containing a linear (2q) and a branched alkyl group (2r) as well as benzyl (2s) were obtained with good enantioselectivities, albeit in moderate yield. In addition, alkenyl-substituted allylic carbonates (2t and 2u) participated in this reaction and generated the single regioisomeric products (3at and 3au), out of three possible regioisomers, in good yield with good to high er.
The scope of the reaction with respect to other cyanocoumarin derivatives was examined next (Table 3). A number of substituted cyanocoumarins bearing either electron donating (e.g. Me and OMe) or electron withdrawing (e.g. F, Cl, and Br) groups were found to be equally suited under our standard reaction conditions, affording γ-allylated products (3ba–ga) with excellent yields and enantioselectivities. Disubstituted cyanocoumarin 1h also participated in this reaction with equal efficiency.
| a Reaction conditions: 3 mol% [Ir(COD)Cl]2, 6 mol% L1, 0.24 mmol of 1, 0.2 mmol of 2 and 0.2 mmol of DABCO in 0.6 mL CH2Cl2. The catalyst was prepared via n-PrNH2 activation. Yields correspond to the isolated product after chromatographic purification. er was determined by HPLC analysis on a chiral stationary phase. |
|---|
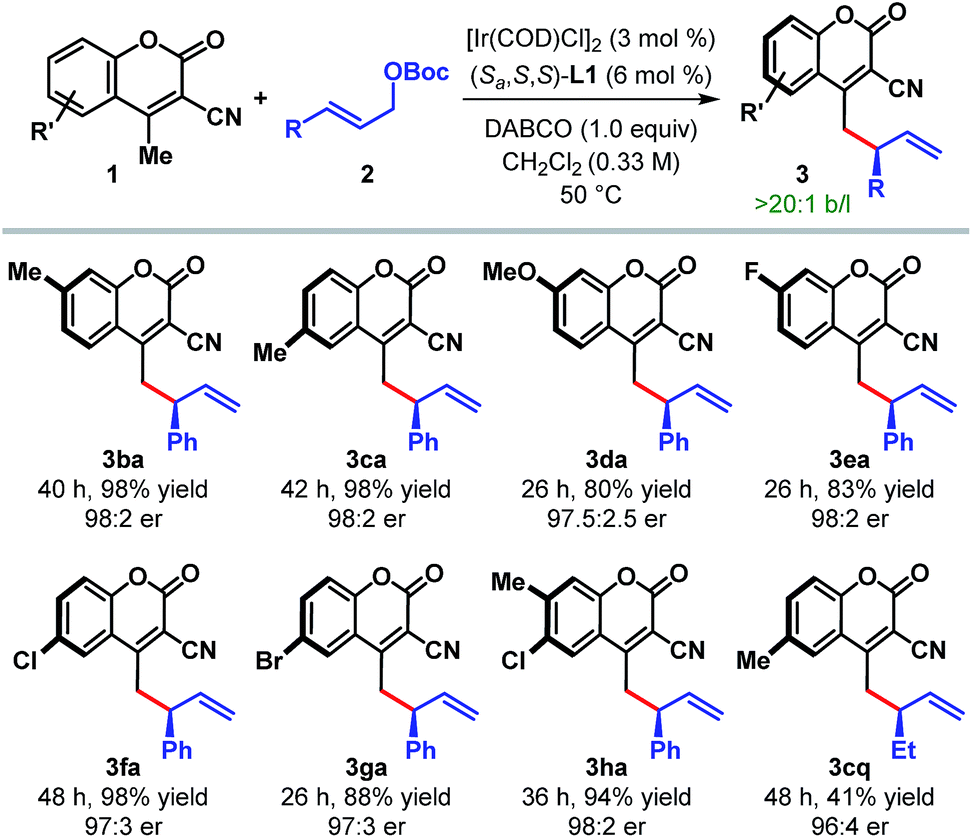
|
After successfully demonstrating the scope of the reaction with cyanocoumarins, we wondered whether the reactivity of the coumarin derivative could be retained by replacing the cyano group with other α-substituents. To our delight, when CN (in 1a) was replaced with an amide group (CONH2), the vinylogous allylic alkylation reaction indeed took place to furnish the corresponding γ-allylated product 3ia with a similar level of yield and enantioselectivity (Table 4, entry 2). Not only amide but also related esters (1j and 3k) could be employed as substrates and afforded the products (3ja and 3ka) in high yield and with excellent er (entries 3 and 4). However, α-carboxylatocoumarin (1l) failed to react in the desired fashion and instead resulted in 4-methylcoumarin 1m in 52% yield through decarboxylation (entry 5). Similarly, unsubstituted 4-methylcoumarin (1m) itself remained unreacted under our standard reaction conditions even after 48 h (entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | X | t [h] | 3 | Yieldb [%] | erc |
| a Reaction conditions: 3 mol% [Ir(COD)Cl]2, 6 mol% L1, 0.24 mmol of 1, 0.2 mmol of 2a and 0.2 mmol of DABCO in 0.6 mL CH2Cl2. The catalyst was prepared via n-PrNH2 activation. b Yields correspond to the isolated product after chromatographic purification. c er was determined by HPLC analysis on a chiral stationary phase. d 1m was isolated in 52% yield. | |||||
| 1 | CN (1a) | 36 | 3aa | 86 | 98:2 |
| 2 | CONH2 (1i) | 36 | 3ia | 81 | 98:2 |
| 3 | CO2Et (1j) | 48 | 3ja | 80 | 97.5:2.5 |
| 4 | CO2Bn (1k) | 48 | 3ka | 85 | 98:2 |
| 5 | CO2H (1l) | 42 | 3la | <5d | — |
| 6 | H (1m) | 48 | 3ma | <5 | — |
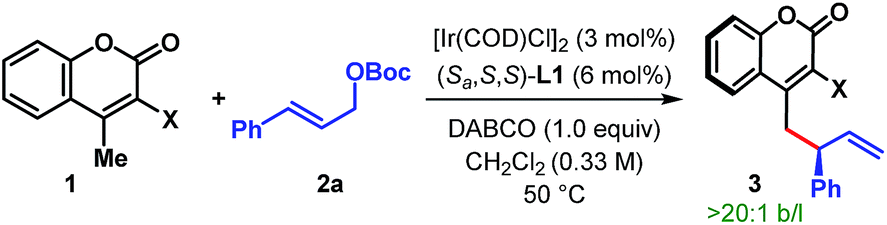
These studies clearly indicate that the presence of an electron withdrawing group at the α-position of 4-methylcoumarin is necessary to exert its vinylogous reactivity. This prerequisite is certainly a limitation of our protocol and prevents the direct vinylogous allylic alkylation of α-unsubstituted 4-methylcoumarin. Our attempts to access this motif (3ma) through a one-pot sequential Ir-catalyzed γ-allylic alkylation/deallylative decarboxylation of allyl esters (1n and 1o) proved futile and led only to the formation of 1m (Scheme 3A). Similarly, an attempted intramolecular decarboxylative migratory allyl transfer22 of 1n under Ir-catalysis also resulted in deallylative decarboxylation to generate 1m. Finally, a two-step sequence consisting of ester hydrolysis followed by decarboxylation delivered the desired α-unsubstituted γ-allylcoumarin 3ma in overall 60% yield from 3ja without any erosion of enantiopurity (Scheme 3B).
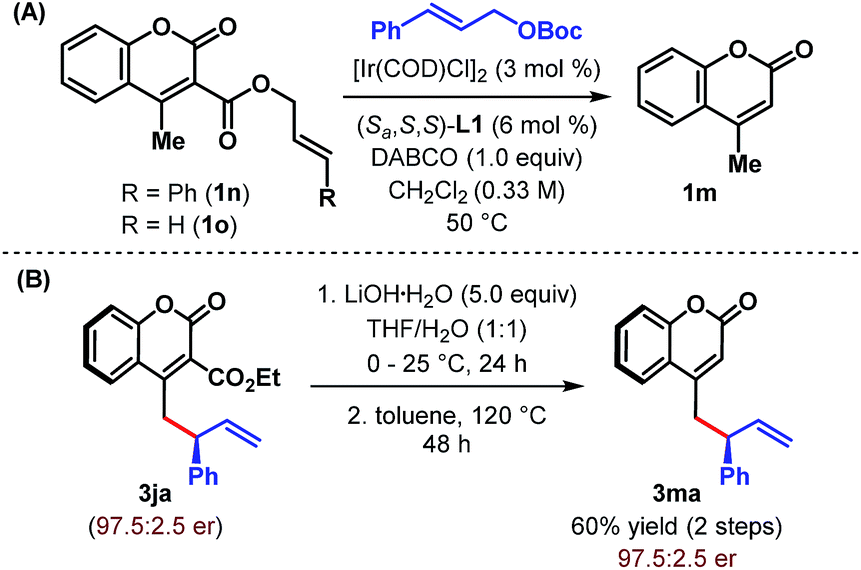 | ||
| Scheme 3 Attempted and successful synthesis of α-unsubstituted γ-allylcoumarin. | ||
The practicality of our enantioselective direct vinylogous allylic alkylation protocol is established by carrying out a gram-scale synthesis of 3aa, which gave the product in 88% yield but with somewhat diminished enantioselectivity (Scheme 4A). However, a single recrystallization restored the enantiopurity of 3aa to 98:2 er.
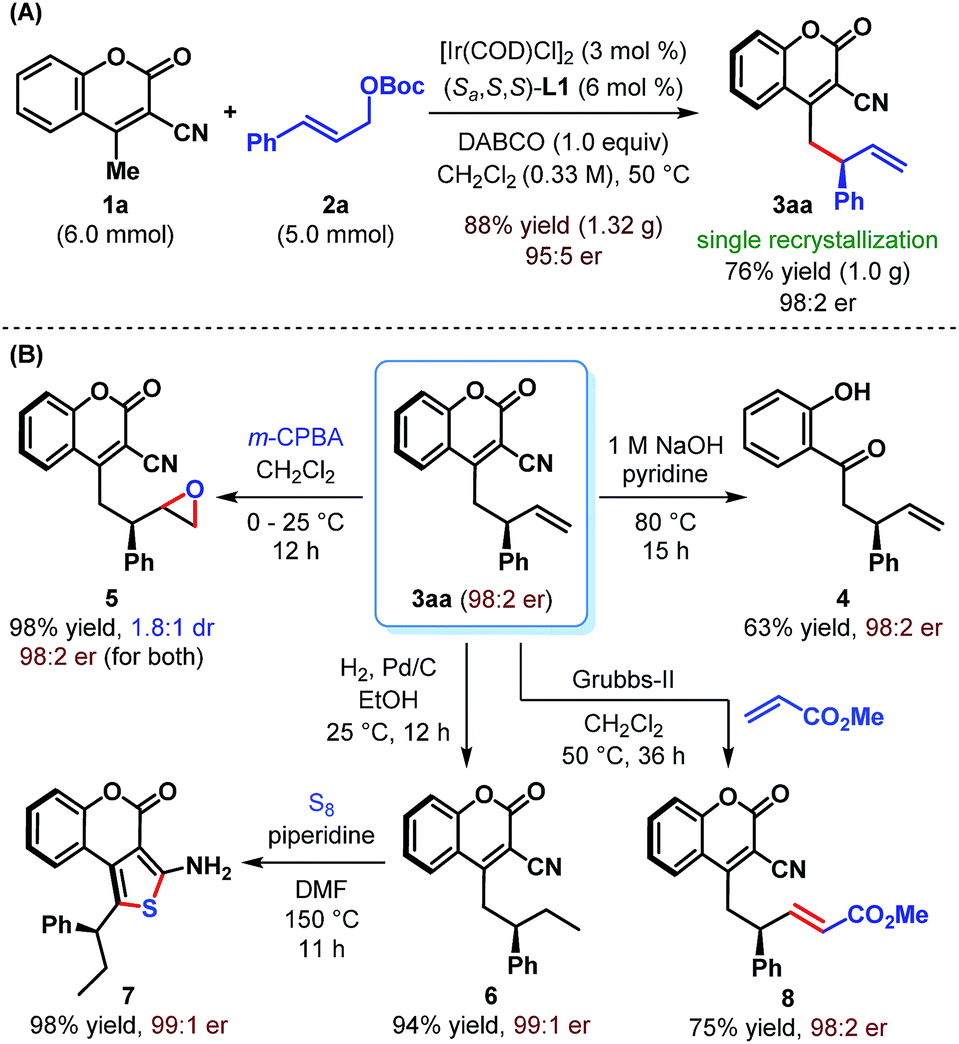 | ||
| Scheme 4 Gram-scale synthesis and synthetic elaboration of γ-allylcoumarin 3aa. | ||
We realized that the ability to transform the existing functionalities present in the products may further increase their synthetic potential. Accordingly, a base-mediated retro-Knoevenagel condensation/hydrolysis of 3aa was carried out to furnish α-allylated o-hydroxyacetophenone 4 in 63% yield (Scheme 4B). Treatment of 3aa with m-CPBA provided the corresponding epoxide 5 in excellent yield but with poor diastereoselectivity (1.8:1 dr). Selective hydrogenation of the terminal double bond of 3aa was possible under Pd/C and resulted in 6 in 94% yield. A base-catalyzed cyclization of 6 with sulfur furnished tricyclic aminothiophenocoumarin 7. This structural motif is known for its presence in antifungal agents.23 The absolute stereochemistry of 3aa, 6 and 7 has previously been confirmed by Waldmann et al.24 and in turn established the absolute configuration of our allylated products. Finally, an olefin cross-metathesis of 3aa with methyl acrylate in the presence of Grubbs' 2nd generation catalyst occurred smoothly to give terminally functionalized olefin 8 in 75% yield. In all these cases, the reactions proceeded with complete conservation of stereochemical integrity.
A tentative catalytic cycle based on the literature precedence25 is depicted in Scheme 5 and involves the intermediacy of the iridacycle intermediate A. Ligand dissociation from the coordinatively saturated species A is likely to generate species B having 16 valence electrons at Ir. Coordination of allylic carbonate (2) to B followed by oxidative addition-decarboxylation gives the π-allyl–Ir intermediate C. An enantiodetermining nucleophilic addition of dienolate D to C then produces complex E, product dissociation from which completes the catalytic cycle. The sense of stereoinduction was found to be the same as predicted by the Hartwig model.25b
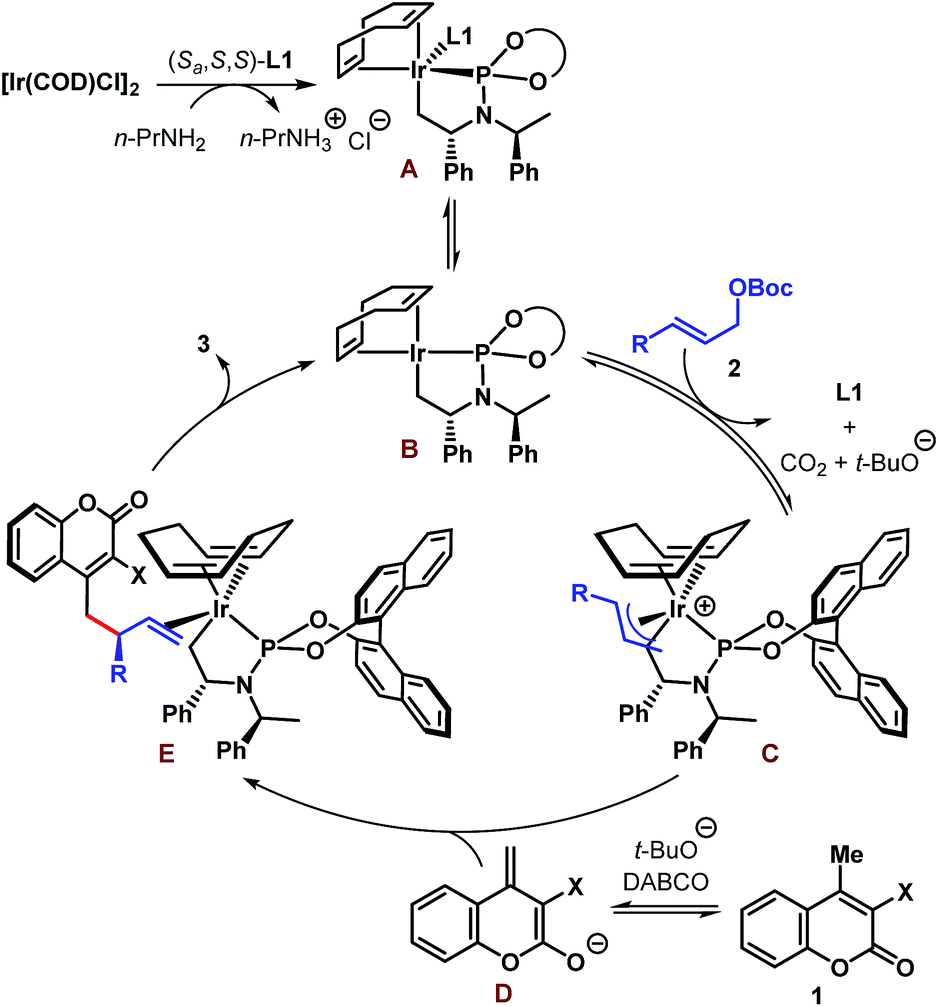 | ||
| Scheme 5 Tentative catalytic cycle for the Ir-catalyzed enantioselective allylic alkylation of coumarins. | ||
Conclusions
In conclusion, we have developed the first Ir-catalyzed enantioselective vinylogous allylic alkylation of coumarins. Our protocol does not require preactivation of 4-methylcoumarins and installs an unfunctionalized allyl group using easily accessible linear allylic carbonates as the allylic electrophile. The resulting Ir/phosphoramidite-catalyzed direct vinylogous allylic alkylation reaction produces γ-allylcoumarins in an exclusively branched-selective manner generally in high yields with an excellent level of enantioselectivity. An enantioselective synthesis of α-unsubstituted γ-allylcoumarin as well as synthetic elaboration of γ-allylcyanocoumarin to a diverse range of products have also been demonstrated. Future efforts in this direction from our laboratory would focus on the study of other classes of vinylogous nucleophiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the Science and Engineering Research Board (SERB), New Delhi [Grant No. EMR/2016/005045]. R. S. thanks the Council of Scientific and Industrial Research (CSIR), New Delhi for a doctoral fellowship.Notes and references
- For selected reviews, see: (a) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef PubMed; (b) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef PubMed; (c) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef PubMed; (d) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef PubMed; (e) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2007, 47, 258–297 CrossRef PubMed; (f) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef PubMed; (g) B. M. Trost, Chem. Pharm. Bull., 2002, 50, 1–14 CrossRef PubMed.
- For selected reviews, see: (a) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef PubMed; (b) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, ACS Catal., 2016, 6, 6207–6213 CrossRef PubMed; (c) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147–3163 RSC; (d) W.-B. Liu, J.-B. Xia and S.-L. You, Iridium-Catalyzed Asymmetric Allylic Substitutions, in Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, ed. U. Kazmaier, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 155–207 Search PubMed; (e) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef PubMed; (f) G. Helmchen, A. Dahnz, P. Dubon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675–691 RSC; (g) H. Miyabe and Y. Takemoto, Synlett, 2005, 1641–1655 Search PubMed.
- For selected recent examples using achiral (non-prochiral) carbon nucleophiles, see: (a) S. E. Shockley, J. C. Hethcox and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 11545–11548 CrossRef PubMed; (b) X. Jiang and J. F. Hartwig, Angew. Chem., Int. Ed., 2017, 56, 8887–8891 CrossRef PubMed; (c) X. J. Liu and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 4002–4005 CrossRef PubMed; (d) J. Liu, C.-G. Cao, H.-B. Sun, X. Zhang and D. Niu, J. Am. Chem. Soc., 2016, 138, 13103–13106 CrossRef PubMed; (e) S. Breitler and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 5296–5299 CrossRef; (f) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994–997 CrossRef PubMed.
- For selected recent examples using prochiral and chiral carbon nucleophiles, see: (a) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080–2084 CrossRef PubMed; (b) X. Jiang, P. Boehm and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1239–1242 CrossRef PubMed; (c) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef PubMed; (d) Y.-L. Su, Y.-H. Li, Y.-G. Chen and Z.-Y. Han, Chem. Commun., 2017, 53, 1985–1988 RSC; (e) R. He, P. Liu, X. Huo and W. Zhang, Org. Lett., 2017, 19, 5513–5516 Search PubMed; (f) X. Jiang, W. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 5819–5823 CrossRef PubMed; (g) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef PubMed.
- For selected examples using heteroatom nucleophiles, see: (a) Z.-P. Yang, R. Jiang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2018, 140, 3114–3119 CrossRef PubMed; (b) Z. P. Yang, C. Zheng, L. Huang, C. Qian and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 1530–1534 CrossRef PubMed; (c) F. Peng, H. Tian, P. Zhang, C. Liu, X. Wu, X. Yuan, H. Yang and H. Fu, Org. Lett., 2017, 19, 6376–6379 CrossRef PubMed; (d) W. B. Liu, X. Zhang, L. X. Dai and S. L. You, Angew. Chem., Int. Ed., 2012, 51, 5183–5187 CrossRef PubMed; (e) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 8652–8655 CrossRef PubMed; (f) M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470–3473 CrossRef PubMed; (g) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568–5571 CrossRef PubMed; (h) C. Shu and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4794–4797 CrossRef PubMed; (i) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164–15165 CrossRef PubMed.
- (a) R. Takeuchi and M. Kashio, Angew. Chem., Int. Ed., 1997, 36, 263–265 CrossRef; (b) J. P. Janssen and G. Helmchen, Tetrahedron Lett., 1997, 38, 8025–8026 CrossRef.
- M. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 12172–12176 CrossRef PubMed ; also see: M. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 11651–11655 CrossRef PubMed.
- L. Næsborg, K. S. Halskov, F. Tur, S. M. N. Mønsted and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 10193–10197 CrossRef PubMed.
- W.-B. Liu, N. Okamoto, E. J. Alexy, A. Y. Hong, K. Tran and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 5234–5237 CrossRef PubMed . Also see: S. Rieckhoff, J. Meisner, J. Kästner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2018, 57, 1404–1408 CrossRef PubMed.
- (a) A. K. Simlandy and S. Mukherjee, Org. Biomol. Chem., 2016, 14, 5659–5664 RSC; (b) M. S. Manna and S. Mukherjee, Chem. Sci., 2014, 5, 1627–1633 RSC; (c) V. Kumar and S. Mukherjee, Chem. Commun., 2013, 49, 11203–11205 RSC; (d) M. S. Manna and S. Mukherjee, Chem.–Eur. J., 2012, 18, 15277–15282 CrossRef PubMed; (e) A. Ray Choudhury and S. Mukherjee, Org. Biomol. Chem., 2012, 10, 7313–7320 RSC; (f) M. S. Manna, V. Kumar and S. Mukherjee, Chem. Commun., 2012, 48, 5193–5195 RSC.
- (a) K. A. Kumar, N. Renuka, G. Pavithra and G. V. Kumar, J. Chem. Pharm. Res., 2015, 7, 67–81 Search PubMed; (b) B. Y. Wang, X. Y. Liu, Y. L. Hu and Z. X. Su, Polym. Int., 2009, 58, 703–709 CrossRef; (c) S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3078 CrossRef PubMed; (d) A. Estevez-Braun and A. G. Gonzalez, Nat. Prod. Rep., 1997, 14, 465–475 RSC.
- X. Huang, Y.-H. Wen, F.-T. Zhou, C. Chen, D.-C. Xu and J.-W. Xie, Tetrahedron Lett., 2010, 51, 6637–6640 CrossRef.
- C. C. J. Loh, M. Schmid, B. Peters, X. Fang and M. Lautens, Angew. Chem., Int. Ed., 2016, 55, 4600–4604 CrossRef PubMed.
- S. Kayal and S. Mukherjee, Org. Lett., 2017, 19, 4944–4947 CrossRef PubMed.
- D. Kowalczyk and Ł. Albrecht, Adv. Synth. Catal., 2018, 360, 406–410 CrossRef.
- See the ESI‡ for details.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef PubMed.
- For an example of the use of an in situ generated alkoxide anion as a base in Ir-catalyzed AAS reaction, see: Z.-P. Yang, Q.-F. Wu, W. Shao and S.-L. You, J. Am. Chem. Soc., 2015, 137, 15899–15906 CrossRef PubMed.
- For selected examples of the use of DABCO as an external base, see: (a) J. C. Hethcox, S. Shockley and B. Stoltz, Angew. Chem., Int. Ed., 2018, DOI:10.1002/anie.201804820; (b) B. P. Bondzic, A. Farwick, J. Liebich and P. Eilbracht, Org. Biomol. Chem., 2008, 6, 3723–3731 RSC.
- K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426–2428 CrossRef PubMed.
- W.-B. Liu, C. Zheng, C.-X. Zhuo, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2012, 134, 4812–4821 CrossRef PubMed.
- For a Pd-catalyzed non-enantioselective intramolecular decarboxylative allylic alkylation of coumarins, see: R. Jana, J. J. Partridge and J. A. Tunge, Angew. Chem., Int. Ed., 2011, 50, 5157–5161 CrossRef PubMed ; for an Ir-catalyzed enantioselective intramolecular decarboxylative allylic alkylation, see: H. He, X.-J. Zheng, Y. Li, L.-X. Dai and S.-L. You, Org. Lett., 2007, 9, 4339–4341 CrossRef PubMed.
- P. S. Fogue, P. K. Lunga, E. S. Fondjo, J. D. D. Tamokou, B. Thaddée, J. Tsemeugne, A. T. Tchapi and J. R. Kuiate, Mycoses, 2012, 55, 310–317 CrossRef PubMed.
- H. Xu, L. Laraia, L. Schneider, K. Louven, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 11232–11236 CrossRef PubMed.
- (a) J. A. Raskatov, S. Spiess, C. Gnamm, K. Brödner, F. Rominger and G. Helmchen, Chem.–Eur. J., 2010, 16, 6601–6615 CrossRef PubMed; (b) S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228–7229 CrossRef PubMed; (c) D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 11680–11681 CrossRef PubMed . Also see: ref. 2e.
Footnotes |
| † Dedicated to Professor E. J. Corey on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. See DOI: 10.1039/c8sc02041h |
| This journal is © The Royal Society of Chemistry 2018 |