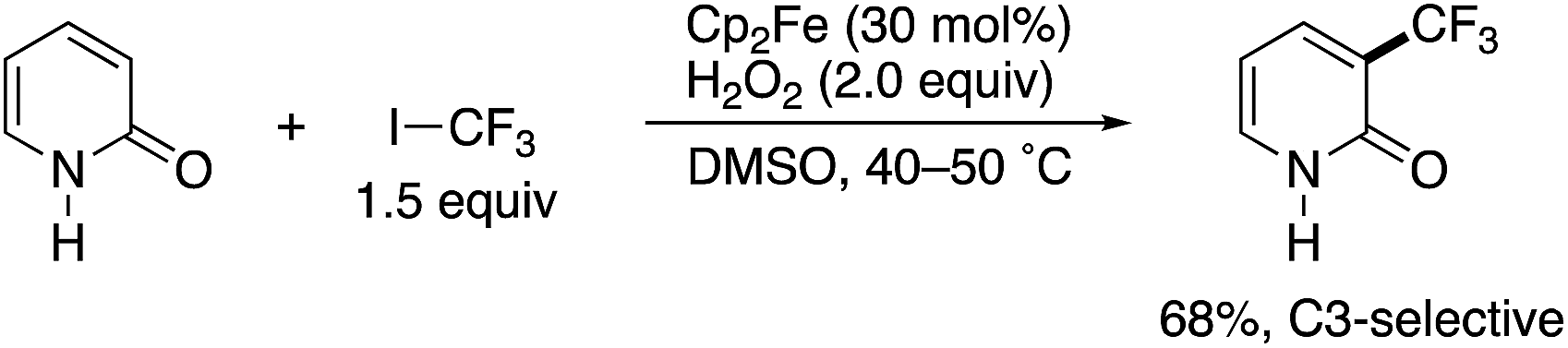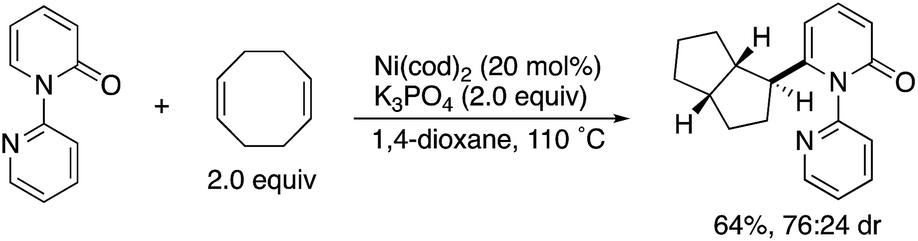DOI:
10.1039/C7SC04509C
(Minireview)
Chem. Sci., 2018,
9, 22-32
A lesson for site-selective C–H functionalization on 2-pyridones: radical, organometallic, directing group and steric controls
Received
18th October 2017
, Accepted 26th November 2017
First published on 27th November 2017
Abstract
A 2-pyridone ring is a frequently occurring subunit in natural products, biologically active compounds, and pharmaceutical targets. Thus, the selective synthesis of substituted 2-pyridone derivatives through decoration and/or formation of pyridone rings has been one of the important longstanding subjects in organic synthetic chemistry. This minireview focuses on recent advances in site-selective C–H functionalization on 2-pyridone. The reported procedures are categorized according to the site selectivity that is achieved, and the substrate scope, limitations, mechanism, and controlling factors are briefly summarized.
 Koji Hirano | Koji Hirano was born in Aichi, Japan, in 1980. He received his B.S. and Ph.D. degrees from Kyoto University in 2003 and 2008 under the supervision of Professor Koichiro Oshima. He served as a JSPS postdoctoral fellow, working with Professor Tamio Hayashi at Kyoto University from April to September in 2008. He joined the research group of Professor Miura at Osaka University as an assistant professor in October, 2008, and then was promoted to the rank of an associate professor in 2015. His current research interests are organic synthesis and organometallic chemistry, especially the development of new synthetic reactions catalyzed by transition metal catalysts. |
 Masahiro Miura | Masahiro Miura is a professor of the Graduate School of Engineering, Osaka University. He studied chemistry at Osaka University and received his Ph.D. degree in 1983 under the guidance of Prof. S. Kusabayashi and Prof. M. Nojima. After working in the chemical industry for one and a half years, he started his academic career as an assistant professor at Osaka University. He was promoted to associate professor in 1994 and to full professor in 2005. He also worked as a Humboldt fellow with Prof. K. Griesbaum at Karlsruhe University of Germany from 1990–1991. His current research interests include transition-metal catalysis and the synthesis of functional molecules, especially new π-conjugated aromatic substances. |
Introduction
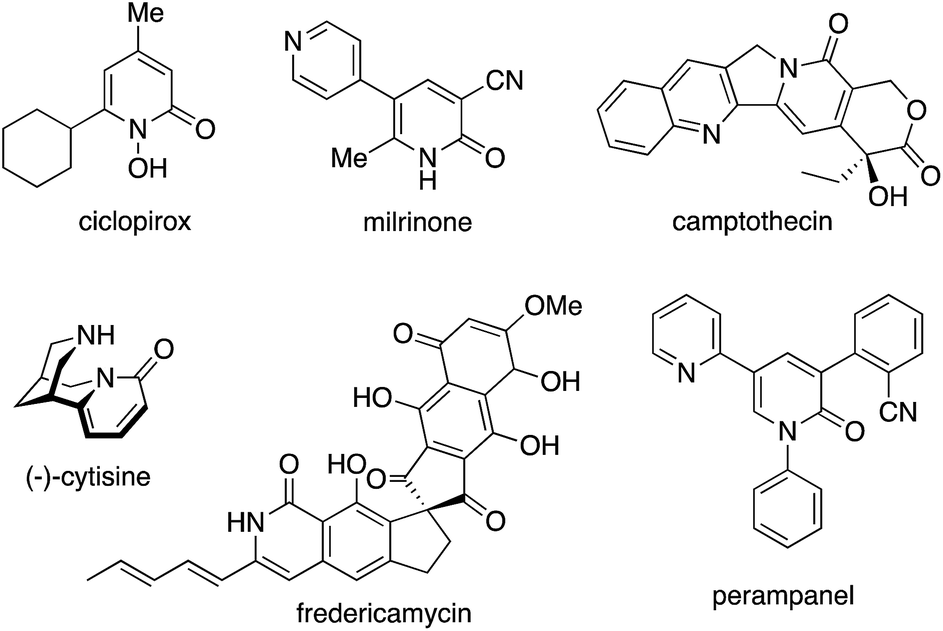
2-Pyridone, the tautomer of 2-hydroxypyridine, is one of the privileged heteroaromatic rings in natural products, bioactive molecules, and pharmaceutical agents. Such well known compounds include ciclopirox, milrinone, camptothecin, (−)-cytisine, fredericamycin, and perampanel (Fig. 1).1 Thus, the decoration and construction of pyridone rings for the preparation of multiply substituted 2-pyridone derivatives has been a longstanding research subject in organic chemistry. To date, synthetic chemists have developed numerous methodologies for the above purpose.
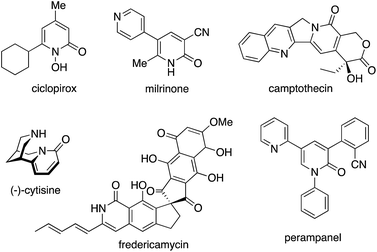 |
| | Fig. 1 2-Pyridone rings in natural products, bioactive molecules, and pharmaceutical agents. | |
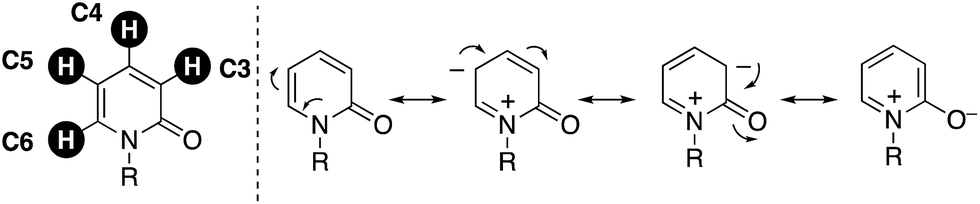
On the other hand, over the past two decades tremendously rapid and explosive progress has occurred in the research field of C–H activation because it can obviate the preactivation steps of starting substrates, such as halogenation and stoichiometric metalation, which are inevitable in traditional cross-coupling technology, to increase the efficiency and atom and step economy in chemical synthesis.2 Although the C–H functionalization strategy seems to be greatly beneficial for the preparation of substituted pyridones, there are four possible reactive C–H bonds on the pyridone ring (C3–C6), and thus control of the site selectivity is the most important and challenging issue (Fig. 2). Considering the resonance structures of 2-pyridone, there are relatively large electronic biases; the C3 and C5 positions are more electron-rich and thus reactive toward electrophiles. On the other hand, the reaction with nucleophiles is more favoured at the C4 and C6 positions, which are of a more electron-deficient nature. In this minireview, we focus on recent advances in the site-selective C–H functionalization of 2-pyridone. The reported procedures are categorized according to the reaction site, and the substrate scope, limitations, mechanism, and site-selectivity controlling factors are briefly summarized.
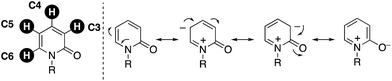 |
| | Fig. 2 Four possible reactive C–H bonds and electronic biases on the 2-pyridone ring. | |
C3-Selective functionalization
In 2010, Yamakawa and co-workers reported a radical trifluoromethylation reaction of some simple (hetero)arenes with CF3I under Fenton-type conditions using FeSO4 or Cp2Fe, H2O2, and DMSO. In the substrate scope investigation, the parent NH-pyridone (2-hydroxypyridine) was tested, and the corresponding 3-trifluoromethylated pyridone was successfully obtained with high site selectivity (Scheme 1).3 To the best of our knowledge, this is the first successful example of the C3-selective radical C–H functionalization of 2-pyridone. However, just a single substrate was employed, and the authors did not specifically mention the observed unique site selectivity in this report.
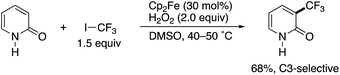 |
| | Scheme 1 The seminal example of C3-selective C–H functionalization of 2-pyridone reported by Yamakawa. | |
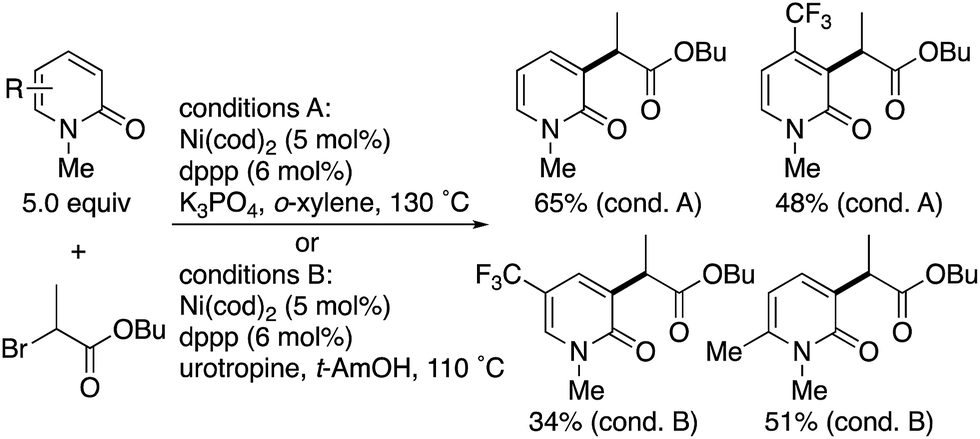
Radical-induced, more general C3-selectivity was observed in the nickel-catalysed direct alkylation with α-bromo carbonyl compounds developed by Hirano and Miura (Scheme 2).4 Regardless of the electronic and steric nature of the substituents on the pyridone, perfect C3-selectivity was uniformly observed. On the other hand, when the C3 position was blocked with the methyl group, no reaction occurred.
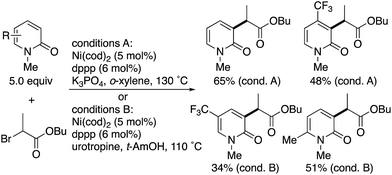 |
| | Scheme 2 Nickel-catalysed radical C3-selective alkylation of 2-pyridones with α-bromo carbonyl compounds. | |
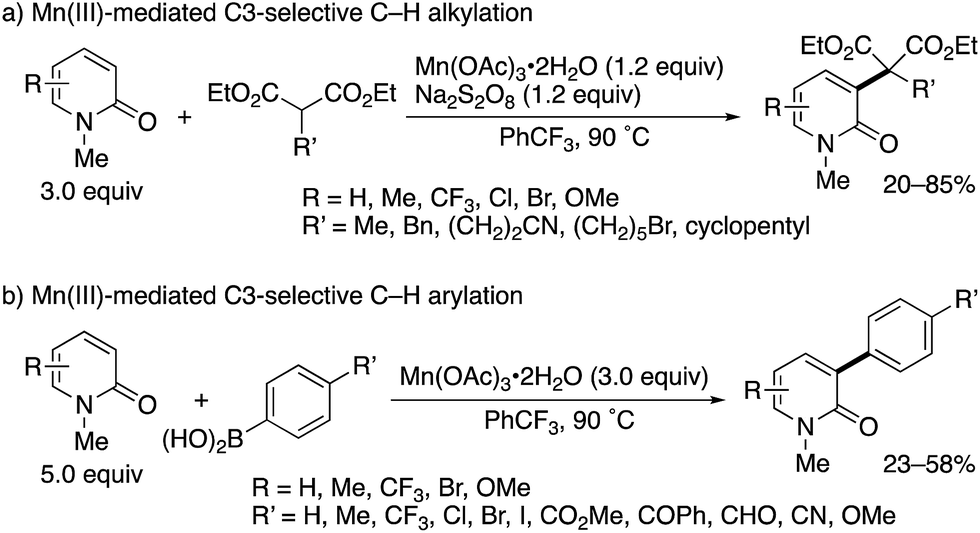
Subsequently, the same research group developed Mn(III)-based methodologies5 for direct alkylation and arylation with malonate esters and arylboronic acids, respectively (Scheme 3).6 Several 2-substituted diethyl malonates could be coupled with 2-pyridones to form the corresponding alkylated products in moderate to good yields (Scheme 3a). In the case of arylboronic acids, the reaction was compatible with a wide range of functional groups including halogen, ester, ketone, aldehyde, nitrile, and ether (Scheme 3b). Similar to Scheme 2, the exclusive C3-selectivity was observed in all cases.
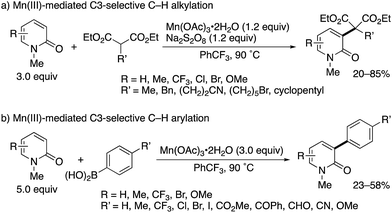 |
| | Scheme 3 Mn(III)-Mediated radical C3-selective alkylation and arylation of 2-pyridones with malonate esters and arylboronic acids, respectively. | |
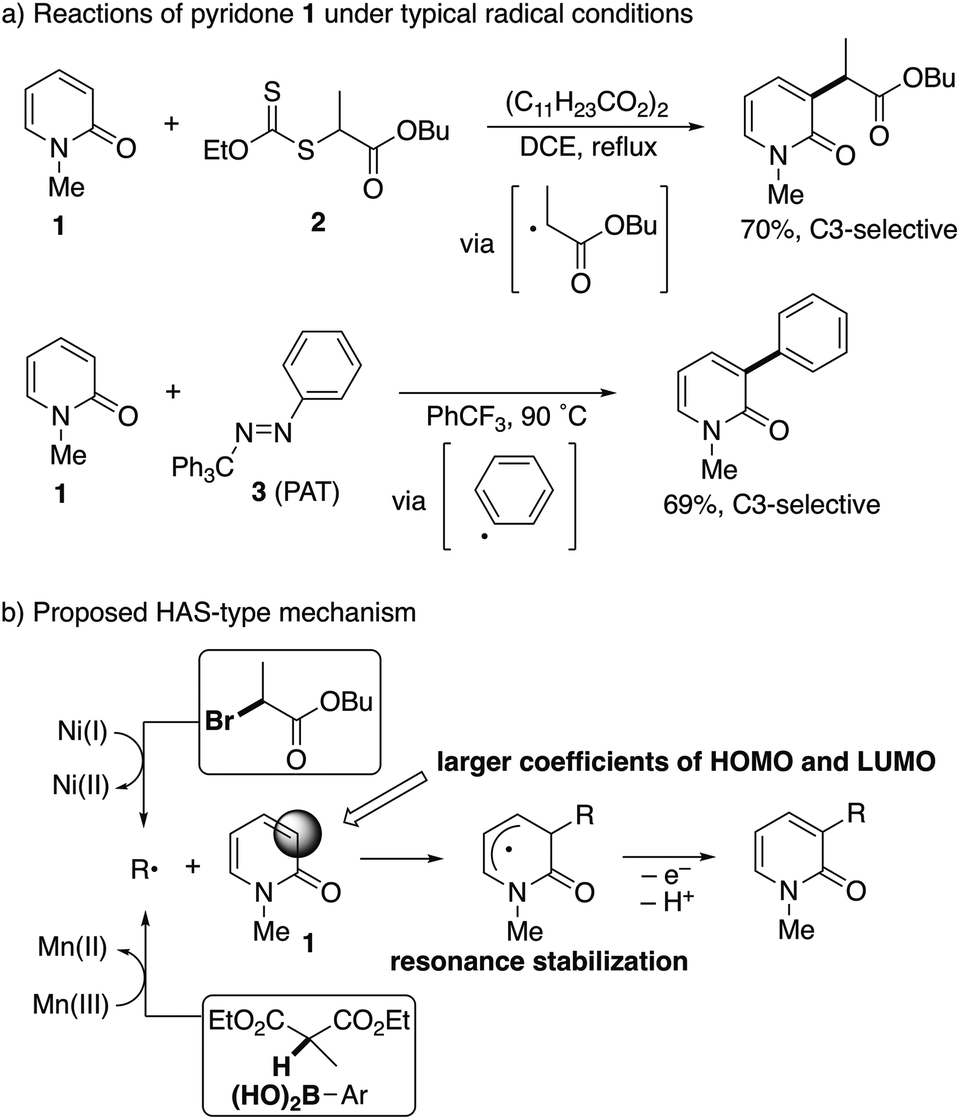
The radical intermediacy was supported by metal-free radical reactions of N-methyl-2-pyridone (1) with xanthate 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and phenylazo(triphenyl)methane8 (PAT, 3); both 2 and 3 readily reacted with 1 under typical radical conditions to deliver the corresponding C3-functionalized pyridones selectively (Scheme 4a). On the basis of these results, a homolytic aromatic substitution9 (HAS)-type mechanism is proposed (Scheme 4b). The C3-selectivity is determined in the radical addition step and controlled by resonance stabilization of the intermediate and a SOMO/HOMO or a SOMO/LUMO interaction between the radical species and pyridone; the larger coefficients of both the HOMO and LUMO at the C3 position of pyridone are also suggested by preliminary DFT calculations at the RB3LYP/6-31G(d) level of theory. In the case of the nickel catalyst, single electron transfer from the radical intermediate following the addition to Ni(II) regenerates the starting Ni(I) species.
7 and phenylazo(triphenyl)methane8 (PAT, 3); both 2 and 3 readily reacted with 1 under typical radical conditions to deliver the corresponding C3-functionalized pyridones selectively (Scheme 4a). On the basis of these results, a homolytic aromatic substitution9 (HAS)-type mechanism is proposed (Scheme 4b). The C3-selectivity is determined in the radical addition step and controlled by resonance stabilization of the intermediate and a SOMO/HOMO or a SOMO/LUMO interaction between the radical species and pyridone; the larger coefficients of both the HOMO and LUMO at the C3 position of pyridone are also suggested by preliminary DFT calculations at the RB3LYP/6-31G(d) level of theory. In the case of the nickel catalyst, single electron transfer from the radical intermediate following the addition to Ni(II) regenerates the starting Ni(I) species.
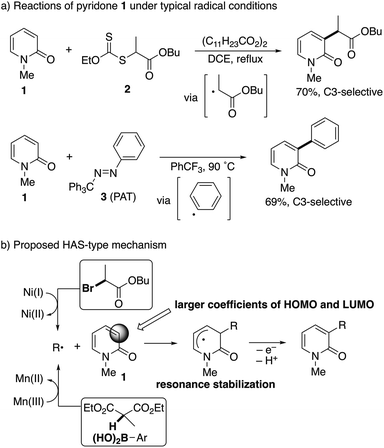 |
| | Scheme 4 Evidence of radical intermediacy and a proposed mechanism for radical-mediated C3-selective functionalization. | |
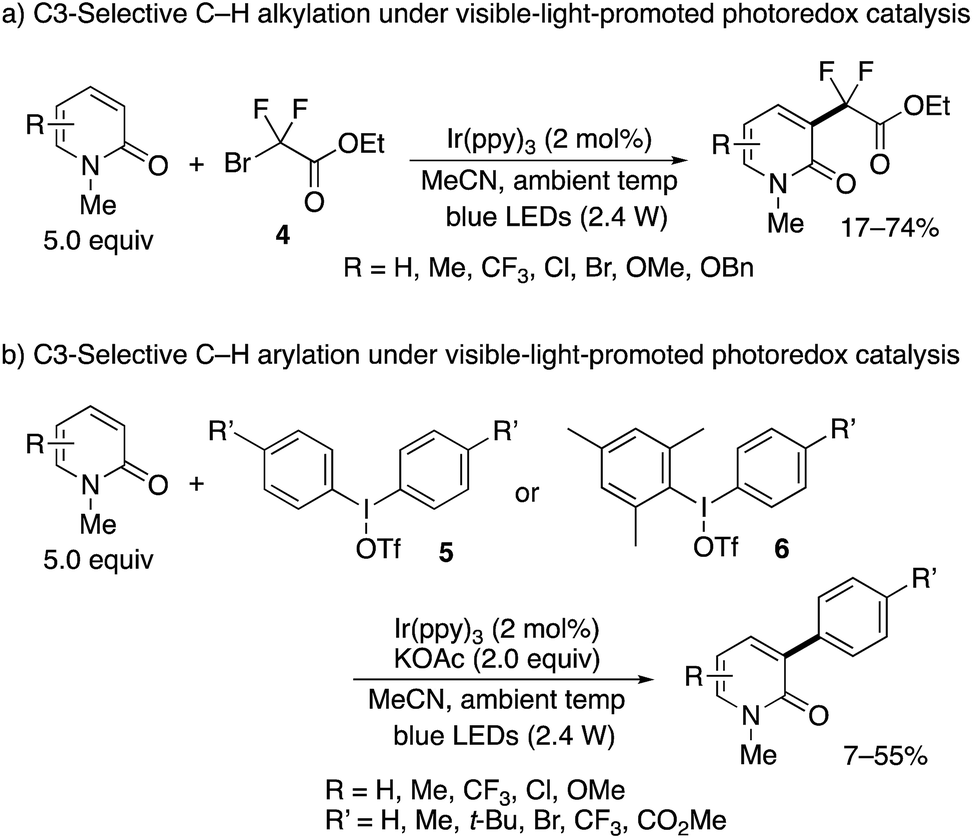
The nickel- and manganese-mediated processes showed excellent C3-selectivity but still suffered from the elevated reaction temperature (>90 °C) and use of excess Mn(III) salt. Hirano and Miura partially addressed these problems by using visible-light-promoted photoredox catalysis.10 In the presence of Ir(ppy)3 photosensitizer, similar alkyl and aryl radical species were generated from ethyl bromodifluoroacetate (4) and diaryliodonium triflates 5, respectively, under blue LED irradiation even at ambient temperature to deliver the substituted 2-pyridones (Scheme 5).11 Again, exclusive C3-selectivity was achieved irrespective of the substitution pattern on the pyridone ring. The unsymmetrical Mes–I(III)–Ar reagent 6 could also be employed; the bulky Mes substituent did not transfer at all, and the desired Ar group was selectively incorporated.
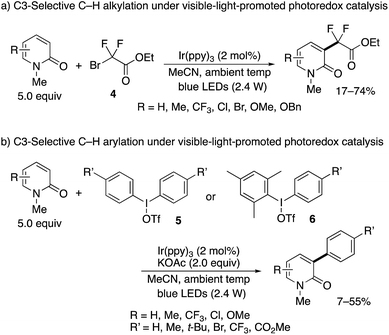 |
| | Scheme 5 C3-Selective alkylation and arylation of 2-pyridones under visible-light-promoted photoredox catalysis. | |
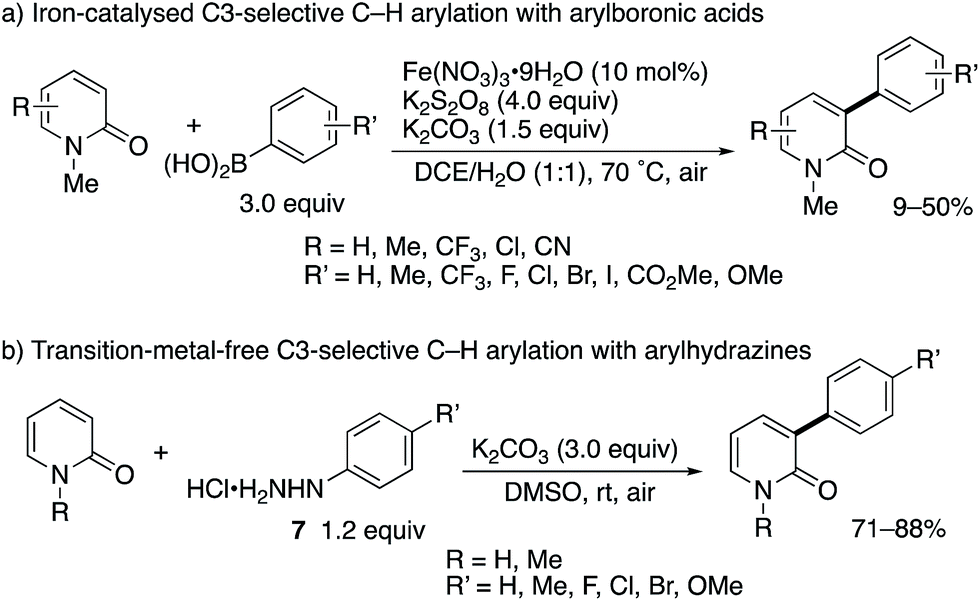
After the stimulating work by Hirano and Miura, some research groups reported greatly improved methodologies for radical C3-arylation (Scheme 6).12 Maiti and co-workers developed an iron-based catalytic system in conjunction with the K2S2O8 terminal oxidant (Scheme 6a). This is a catalytic variant of the Mn(III)-mediated protocol shown in Scheme 3b. Additionally, the 2-pyridone could be used as a limiting agent under the iron catalysis, which is beneficial because of the possibility of late-stage functionalization of 2-pyridones. More recently, Yadav reported the transition-metal-free, K2CO3/O2-promoted direct C3-arylation of 2-pyridone with arylhydrazine HCl salts 7 (Scheme 6b).13 The reaction proceeded smoothly under very mild conditions (at room temperature in air) to furnish a diverse set of C3-arylated pyridones in good to high yields even with the limiting pyridone substrates. Particularly notable is the comparable yield of NH-pyridones.
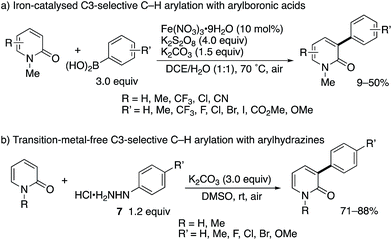 |
| | Scheme 6 Iron-catalysed and metal-free radical C3-selective arylation of 2-pyridones with arylboronic acids and arylhydrazines, respectively. | |
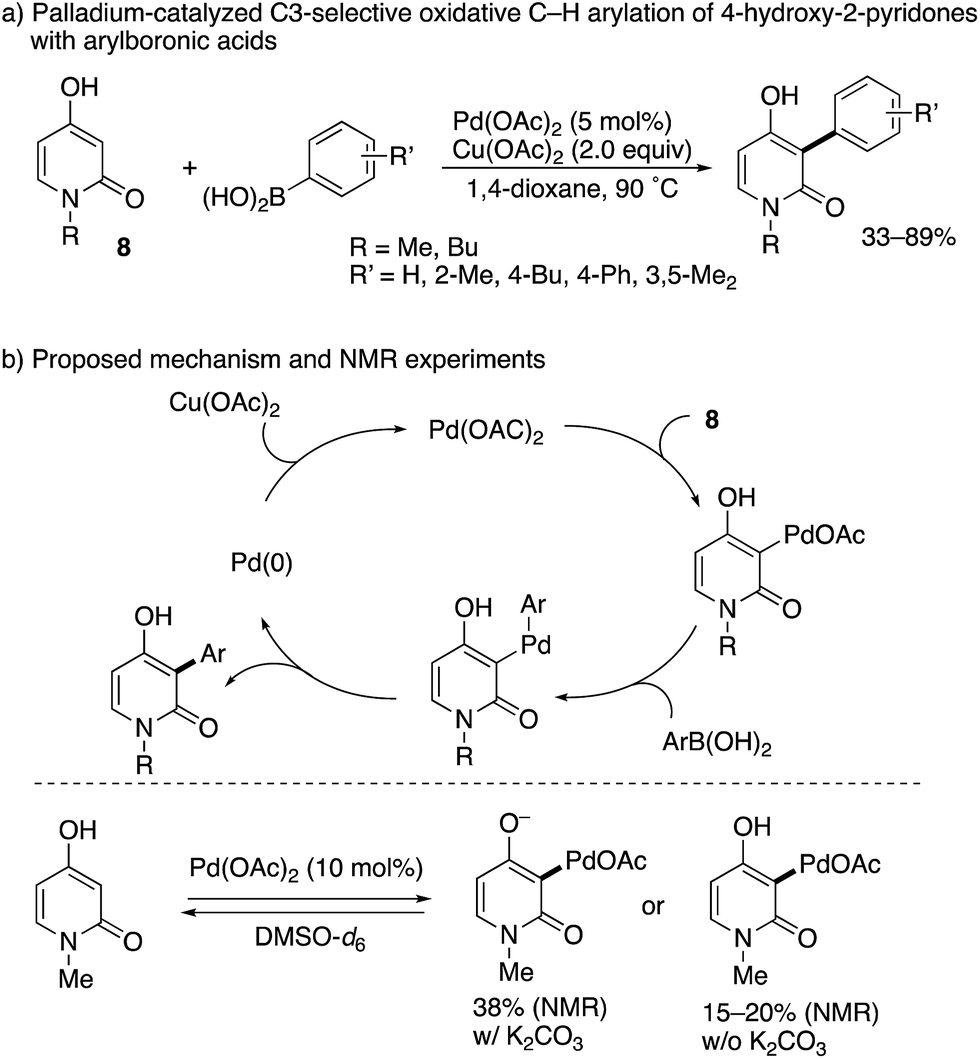
While limited in scope, a completely different approach to the C3-arylation of pyridone was developed by Zografos and co-workers (Scheme 7).14 They focused on the unique enol-like character of 4-hydroxy-2-pyridone 8, and developed a palladium-catalysed oxidative arylation with arylboronic acids. Their proposed catalytic cycle includes: (1) C–H palladation at the C3 position, (2) transmetalation with arylboronic acid, (3) reductive elimination and (4) reoxidation with Cu(OAc)2. The key elementary step is the site-selectivity-determining, electrophilic C–H palladation at the C3 position, which was supported by NMR experiments without the arylboronic acid and Cu(OAc)2 in DMSO-d6: in the presence or absence of K2CO3, C3-palladated pyridone was detected below 70 °C within 20 min.
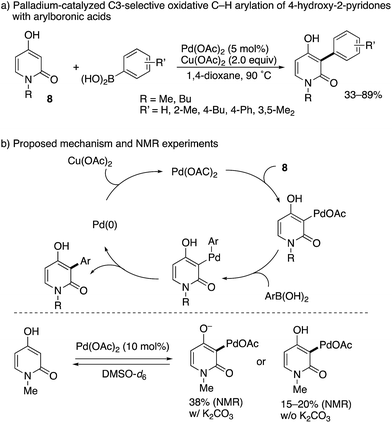 |
| | Scheme 7 Pd(II)-Catalysed oxidative C3-selective arylation of 4-hydroxy-2-pyridones with arylboronic acids. | |
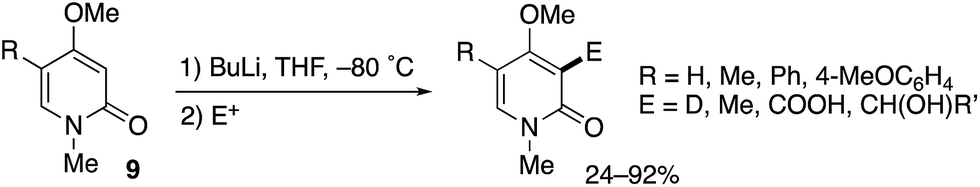
A related MeO-directed ortho-metalation was reported in 1992 (Scheme 8).15 The C–H lithiation of 4-methoxy-2-pyridones 9 at −80 °C was followed by trapping with electrophiles to furnish various C3-functionalized pyridones in good yields.
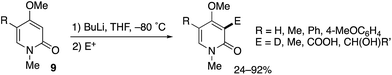 |
| | Scheme 8 MeO-Directed ortho-lithiation for C3-selective functionalization of 4-methoxy-2-pyridones. | |
C5-Selective functionalization
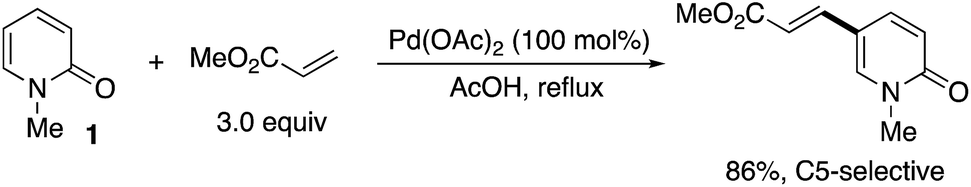
The first successful C5-selective functionalization was reported by Itahara in 1984 (Scheme 9).16 Although a stoichiometric amount of Pd(OAc)2 was essential, the Fujiwara–Moritani-type direct alkenylation17 with acrylates occurred selectively at the C5 position.
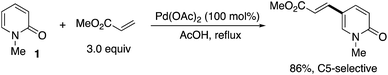 |
| | Scheme 9 The first successful example of palladium-mediated C5-selective alkenylation by Itahara. | |
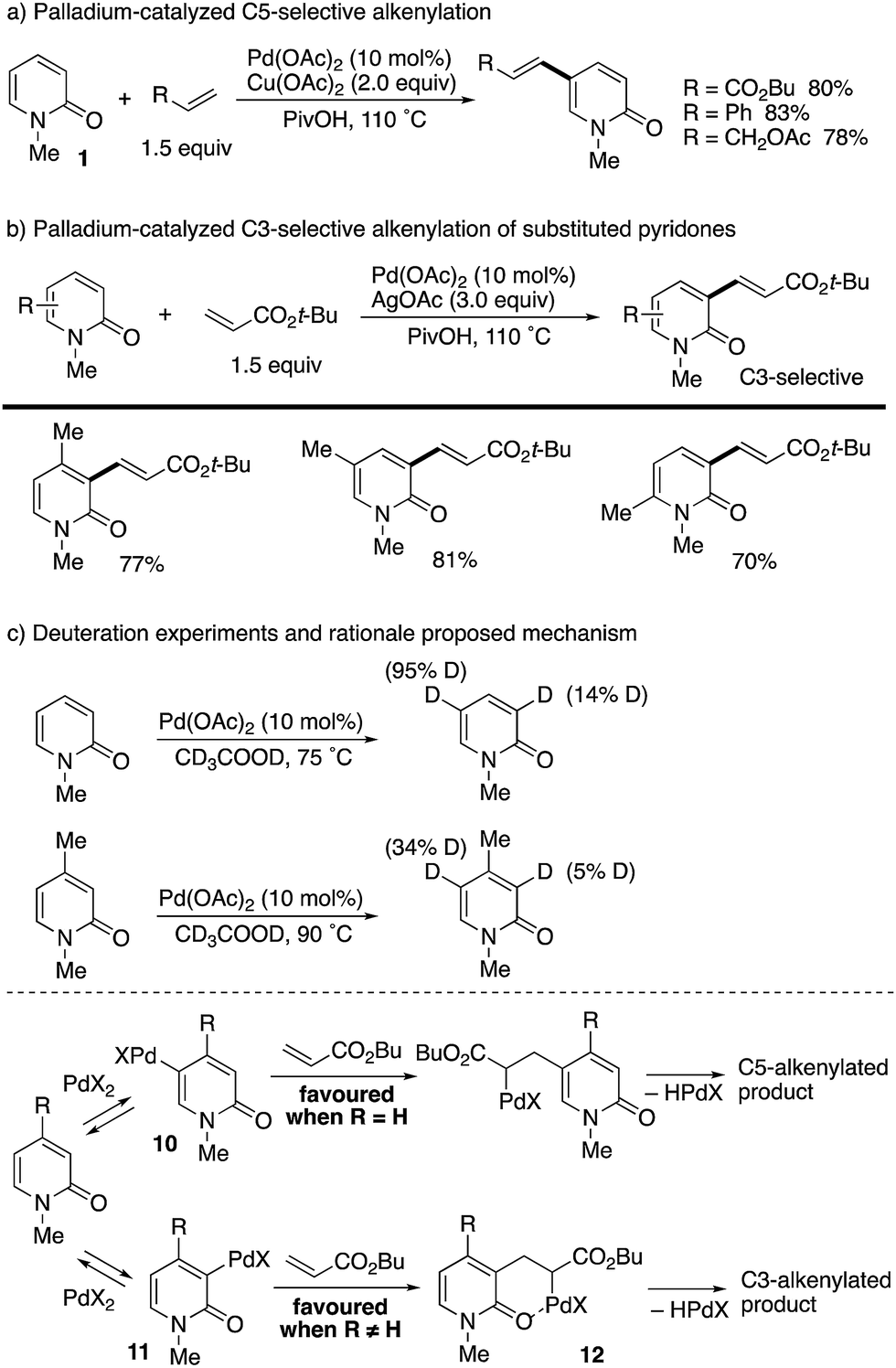
After about 30 years, a catalytic variant was developed with the aid of Cu(OAc)2 terminal oxidant by Li and co-workers (Scheme 10).18 Styrene and allyl acetate as well as acrylates could be employed for coupling with unsubstituted 1 at the C5 position (Scheme 10a). Interestingly, when substituents were introduced at any positions, the reaction site was completely switched to the C3 position (Scheme 10b). In these cases, Ag-based oxidants gave better isolated yields. The above intriguing phenomena can be reasonably explained by the deuterium incorporation experiment (Scheme 10c); C–H palladation reversibly occurred mainly at the most electron-rich C5 position but also at the C3 position, and thus the site selectivity can be determined by the insertion step of the olefin to the Pd–C (pyridone) bond. Without any substituents on the pyridone ring (R = H), the kinetically favoured C5-palladated species 10 undergoes the insertion predominantly. On the other hand, the substituent (R ≠ H) can accelerate the insertion of olefin into the C3-palladated pyridone 11 because the formation of the six-membered palladacycle intermediate 12 is driven by positive Thorpe–Ingold effects.
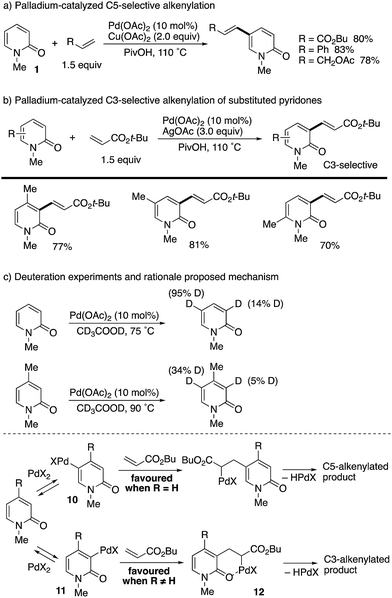 |
| | Scheme 10 Palladium-catalysed C5- and C3-alkenylation of 2-pyridones. | |
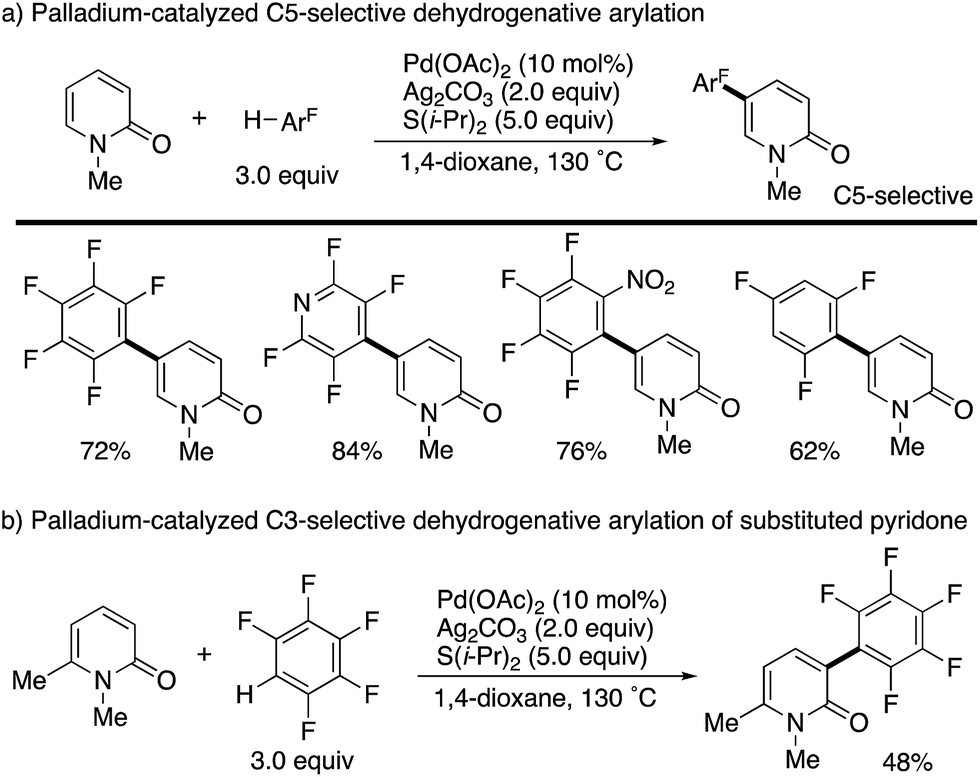
Additionally, Li succeeded in the palladium-catalysed dehydrogenative C5-arylation with polyfluoroarenes (Scheme 11a). The use of Ag2CO3 oxidant and S(i-Pr)2 additive efficiently promoted the reaction. As in Scheme 10, substituent-dependent site-selectivity switching was observed (Scheme 11b).
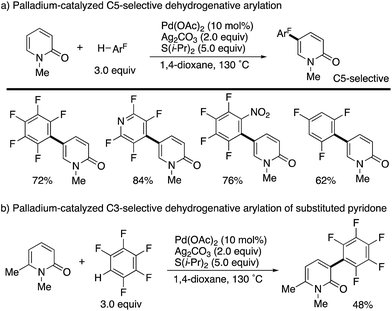 |
| | Scheme 11 Palladium-catalysed dehydrogenative C5- and C3-arylation of 2-pyridones with polyfluoroarenes. | |
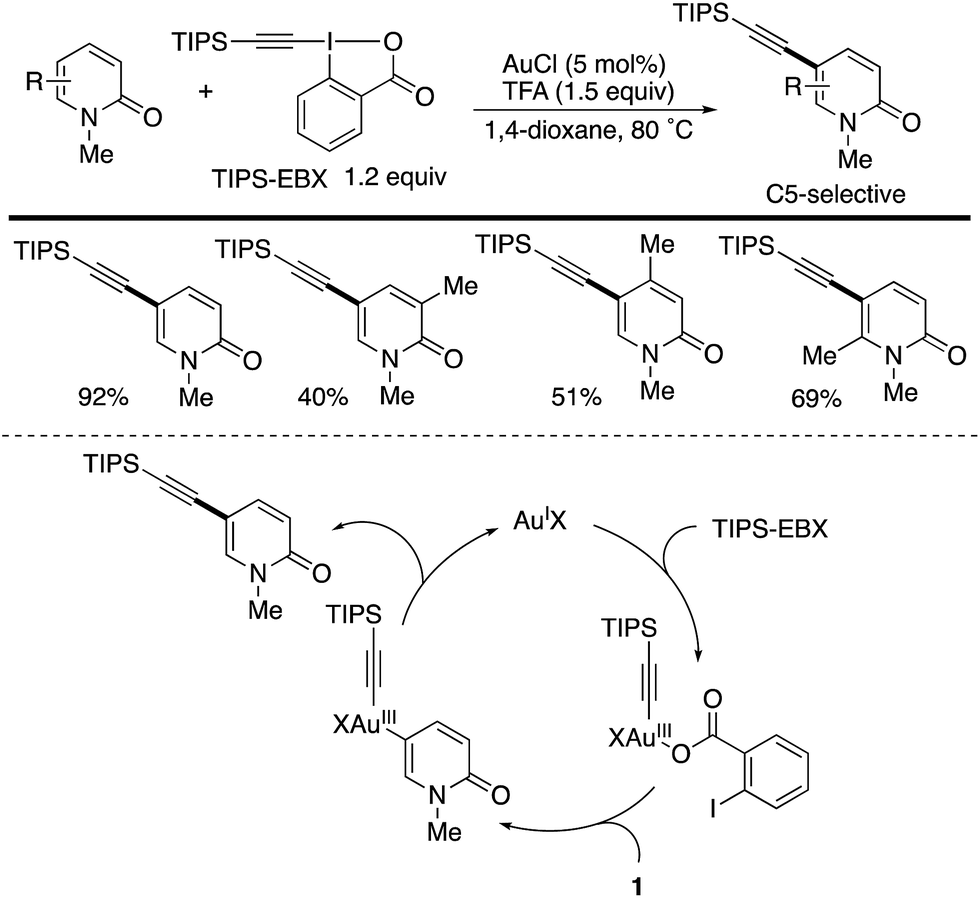
Another C5-selective functionalization is gold-catalysed direct alkynylation with the hypervalent I(III) reagent TIPS-EBX (Scheme 12).19 Different from the palladium catalysis (Schemes 10 and 11), uniformly high C5-selectivity was achieved irrespective of the substitution pattern. Several control experiments suggest that the mechanism involves electrophilic auration on an Au(III) species generated from the reaction of the starting Au(I) and TIPS-EBX, which occurs selectively at the most electron-rich C5 position. However, the authors cannot completely exclude the possibility of an alternative mechanism that includes the nucleophilic attack of pyridone by the alkyne moiety of TIPS-EBX, activated by gold (alkyne π-activation mechanism).
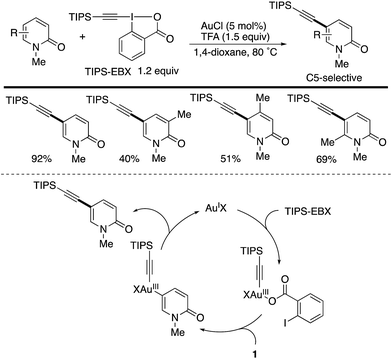 |
| | Scheme 12 Gold-catalysed C5-selective alkynylation of 2-pyridones with TIPS-EBX. | |
C6-Selective functionalization
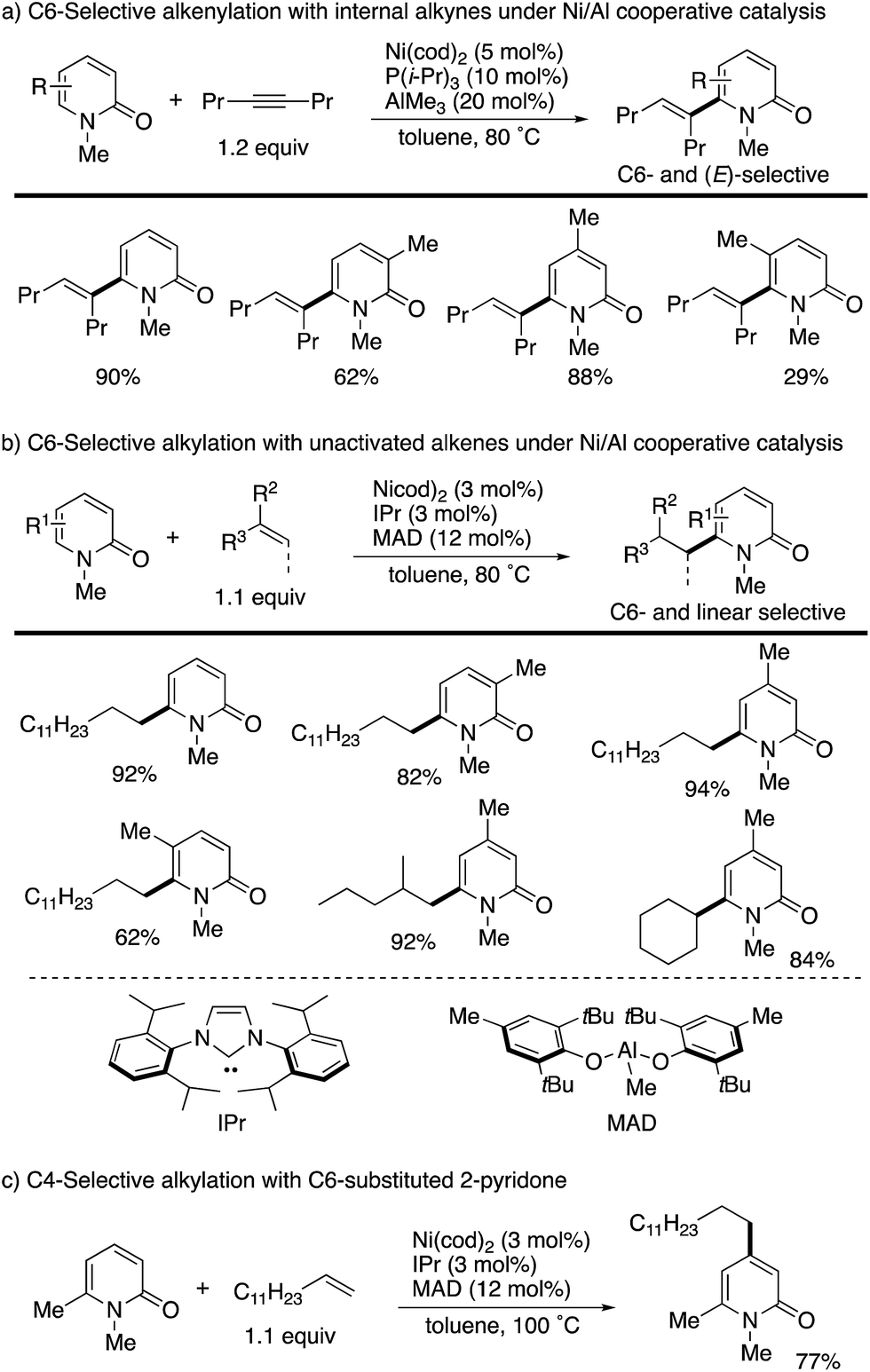
The C6 position is the most electron-deficient site of the 2-pyridone, and its selective C–H functionalization thus requires completely different strategies to those for C3- and C5-selective C–H activation. In 2009, Nakao and Hiyama reported the first successful example of C6-selective alkenylation with internal alkynes under elegantly designed Ni/Al cooperative catalysis (Scheme 13a).20 By using Ni(cod)2/P(i-Pr)3 and AlMe3 catalysts, a variety of 2-pyridones could be directly coupled with alkynes to form the corresponding C6-alkenylated products with high (E)-selectivity. A small amount of 4,6-dialkenylated products (∼5%) was also formed, but substantially high C6-selectivity was generally observed. Subsequently, the same research group succeeded in direct alkylation with unactivated olefins (Scheme 13b).21 The simple change of ligand and aluminum species to IPr and MAD allowed for efficient and linear selective coupling with terminal alkenes, 1,1-disubstituted alkenes, and the even more challenging internal alkene. If the most reactive C6 position was blocked by the substituent, the secondary electron-deficient C4 C–H was selectively alkylated (Scheme 13c). Such a reactivity trend is in marked contrast to that observed in the above radical and electrophilic metalation processes.
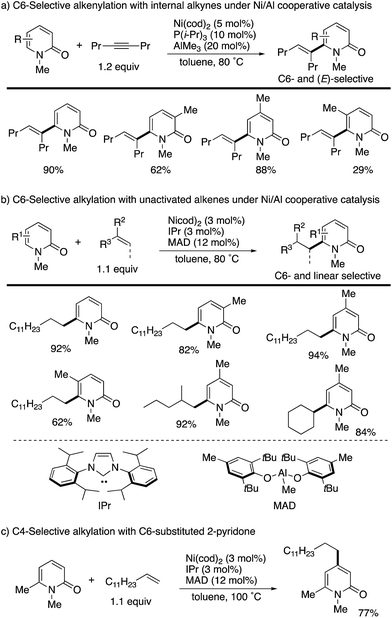 |
| | Scheme 13 Ni/Al-co-catalysed C6-selective alkenylation and alkylation. | |
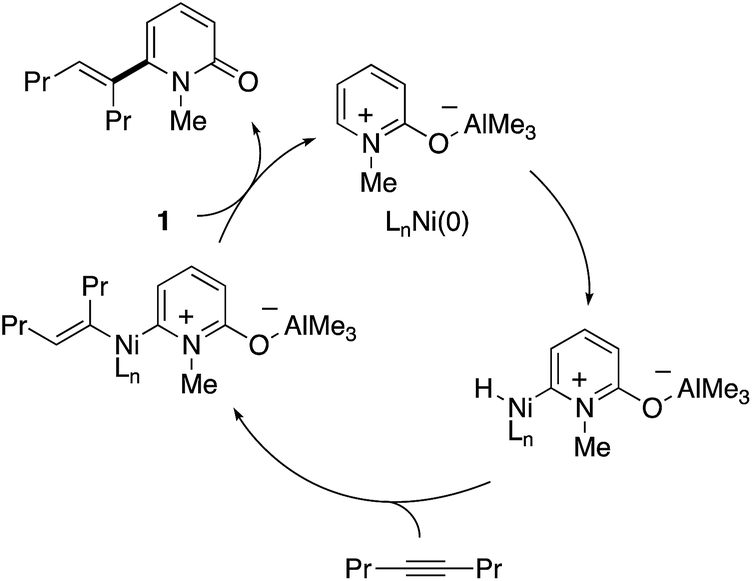
The initially proposed reaction mechanism is shown in Scheme 14. The carbonyl of 2-pyridone 1 is coordinated to the Lewis acidic Al center to increase the electrophilicity of the C6 position. The activated C6 C–H bond oxidatively adds to the electron-rich Ni(0) species (oxidative addition) to form a Ni–H intermediate. The formation of the C6-alkenylated product then follows from alkyne insertion and reductive elimination. However, recent computational studies based on DFT suggest that a ligand-to-ligand H transfer mechanism without formation of the Ni–H species is more likely.22
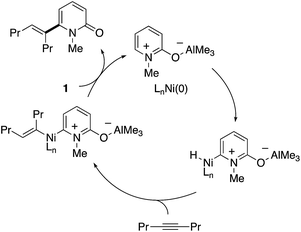 |
| | Scheme 14 The initially proposed mechanism for Ni/Al-co-catalysed C6-selective alkenylation with alkynes. | |
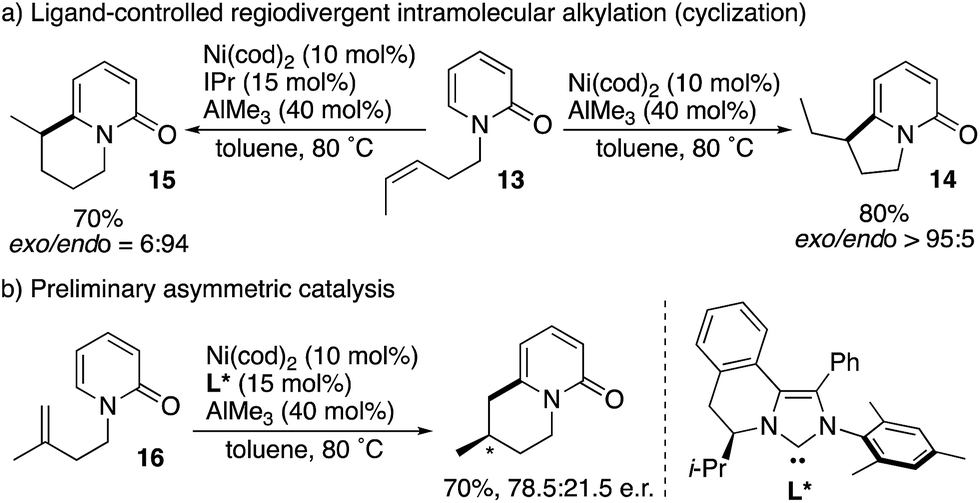
Cramer subsequently developed intramolecular versions of the nickel-catalysed C6-selective alkylation reaction (Scheme 15).23 Notably, ligand-controlled regiodivergency was observed; from the single starting substrate 13, the combination of the cod ligand and AlMe3 afforded the corresponding exo-cyclization product 14 whereas the endo-cyclization product 15 was selectively formed in the presence of the IPr ligand and AlMe3 (Scheme 15a). Moreover, enantioselective endo-cyclization of 16 was possible by using the chiral NHC ligand L*, while still preliminary (Scheme 15b).
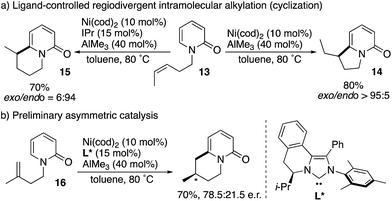 |
| | Scheme 15 Ligand-controlled regiodivergent Ni-catalysed intramolecular C6-selective alkylation. | |
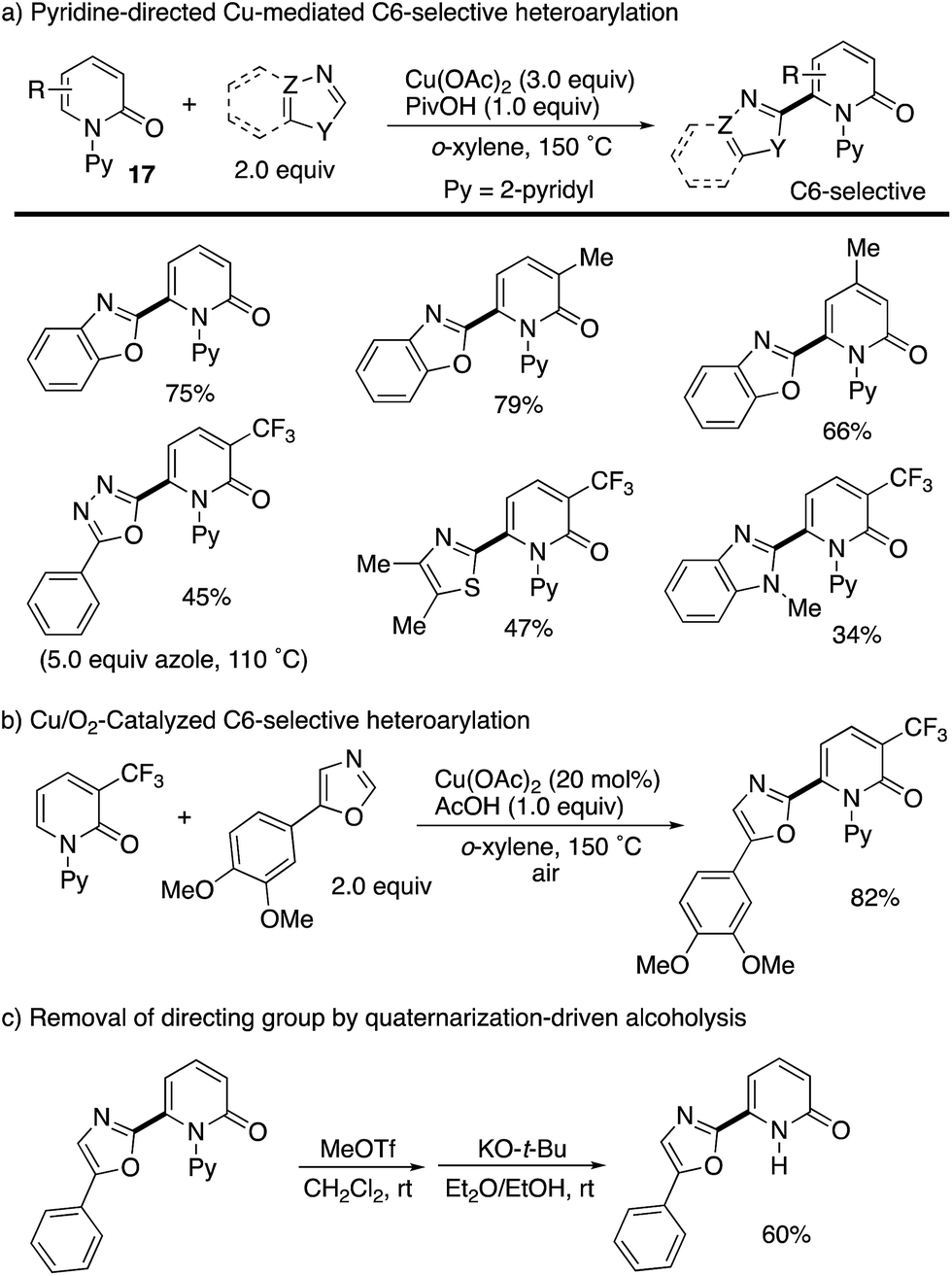
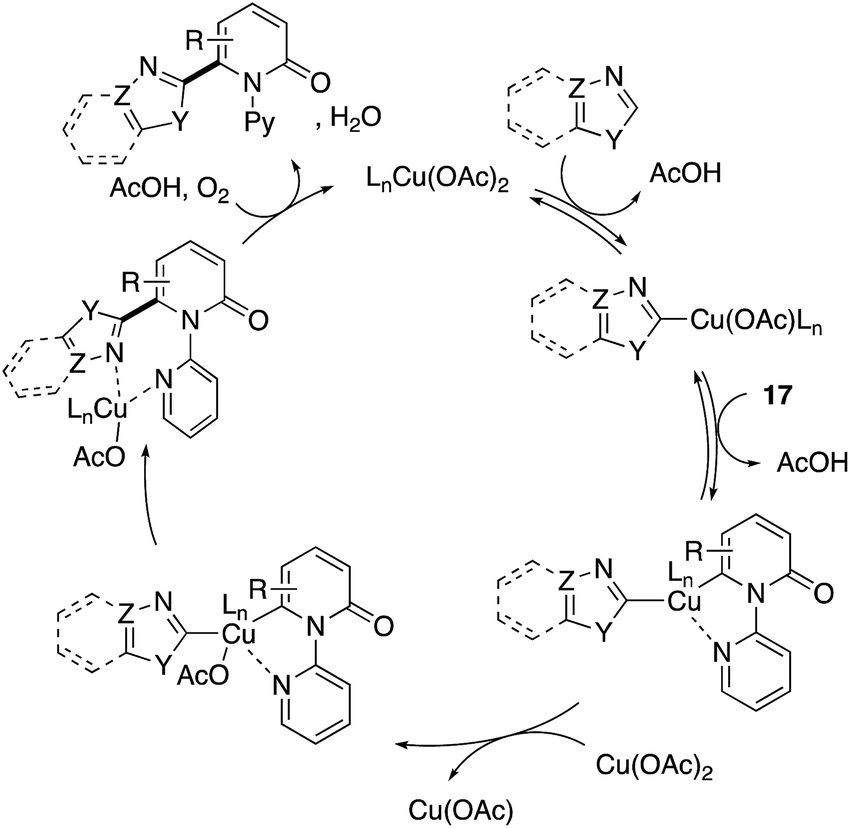
The more robust C6-selectivity is induced by a suitable directing group. In 2014, Hirano and Miura designed N-(2-pyridyl)-2-pyridones 17 and successfully performed copper-mediated C6-selective dehydrogenative heteroarylation with 1,3-azoles (Scheme 16a).24 The reaction system was compatible with oxazole, thiazole, and imidazole, and C6-arylated 2-pyridones were exclusively formed without any contamination of regioisomers. Additionally, the use of molecular oxygen rendered the reaction catalytic in copper (Scheme 16b). The 2-pyridyl directing group was essential for obtaining the reactivity as well as C6-selectivity, but could be readily removed by quaternarization-driven alcoholysis after the coupling event (Scheme 16c). The plausible mechanism includes: (1) initial cupration of the more acidic 1,3-azole C–H, (2) second cupration of the pyridone C6 C–H assisted by the coordination of a pyridyl group, (3) oxidation into a Cu(III) species with additional Cu(II) salt (disproportionation), (4) productive reductive elimination and (5) dissociation of the C6-arylated product and reoxidation to the starting Cu(II) by molecular oxygen and AcOH (Scheme 17). Both of the C–H cupration steps are reversible, and the cross-coupling selectivity is thus determined in the oxidation or reductive elimination step.
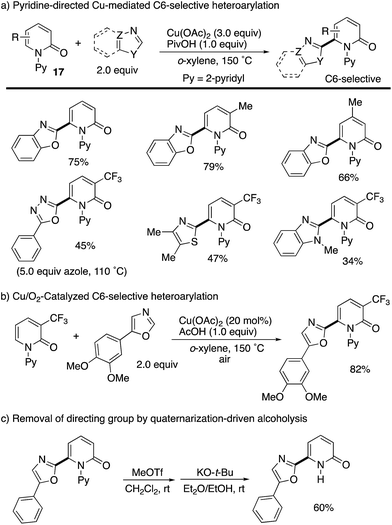 |
| | Scheme 16 Pyridine-directed, Cu(II)-promoted C6-selective heteroarylation and removal of the pyridine directing group. | |
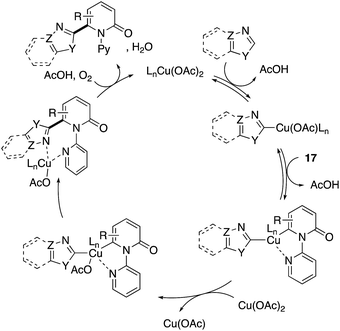 |
| | Scheme 17 Plausible mechanism for Cu(II)-catalysed directed C6-heteroarylation. | |
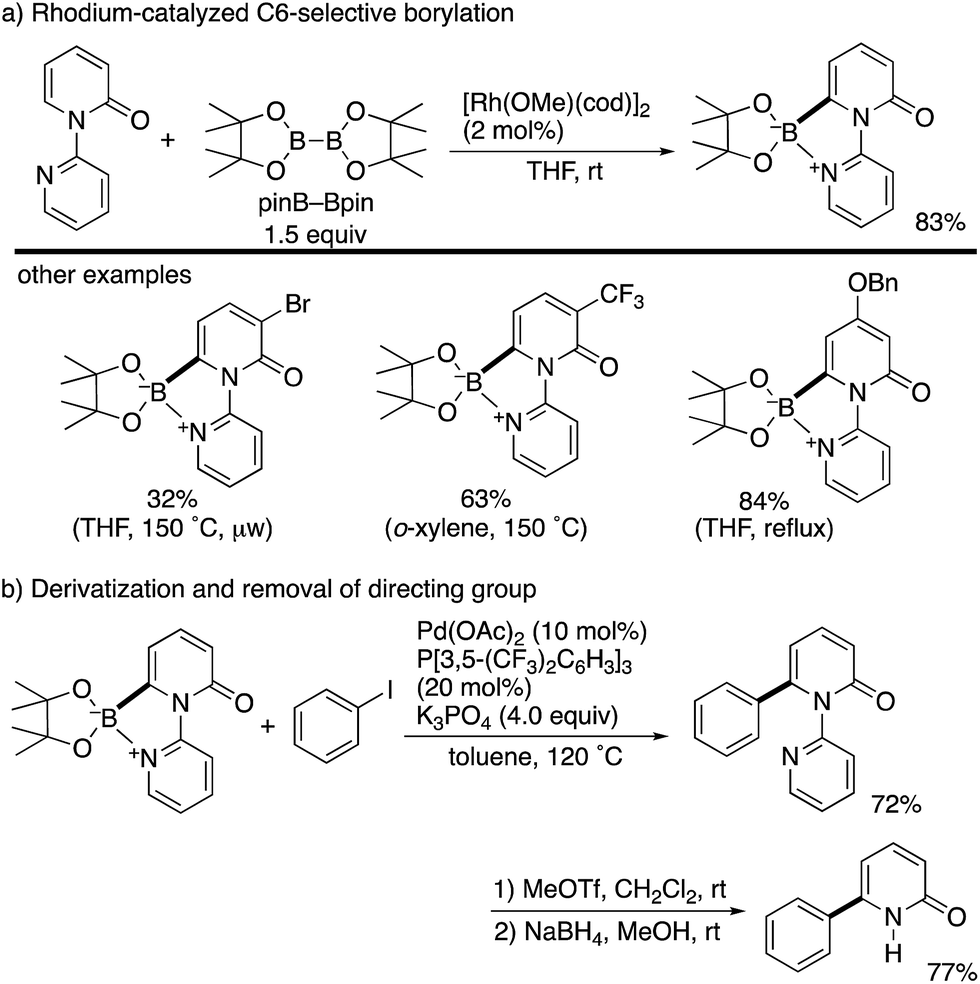
By using the attachable and detachable directing group strategy, Hirano and Miura then developed a related C6-selective C–H functionalization including rhodium-catalysed borylation (Scheme 18)25 and nickel-catalysed alkylation (Scheme 19)26 with bis(pinacolato)diboron (pinB–Bpin) and dienes/activated alkenes, respectively. The former rhodium catalysis provided various C6-borylated pyridones in moderate to good yields, although the reactivity was highly dependent on the steric and electronic nature of the substituents (Scheme 18a). The Suzuki–Miyaura cross-coupling reaction was followed by the removal of the directing group to form the 6-phenyl-NH-pyridone in a good overall yield (Scheme 18b). The latter nickel catalysis promoted unique ring-contracting C–H coupling with 1,8-cyclooctadiene (cod), which was accelerated by the addition of K3PO4 (Scheme 19).
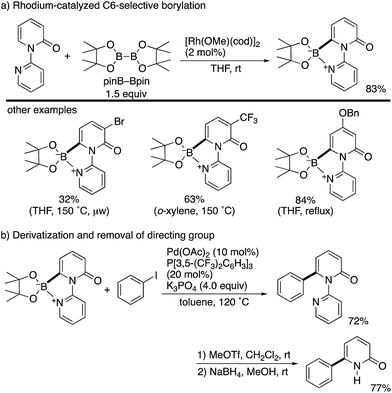 |
| | Scheme 18 Rh-Catalysed directed C6-borylation and application to Suzuki–Miyaura coupling. | |
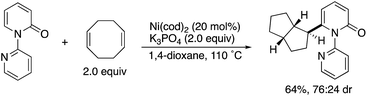 |
| | Scheme 19 Ni-Catalysed directed C6-alkylation. | |
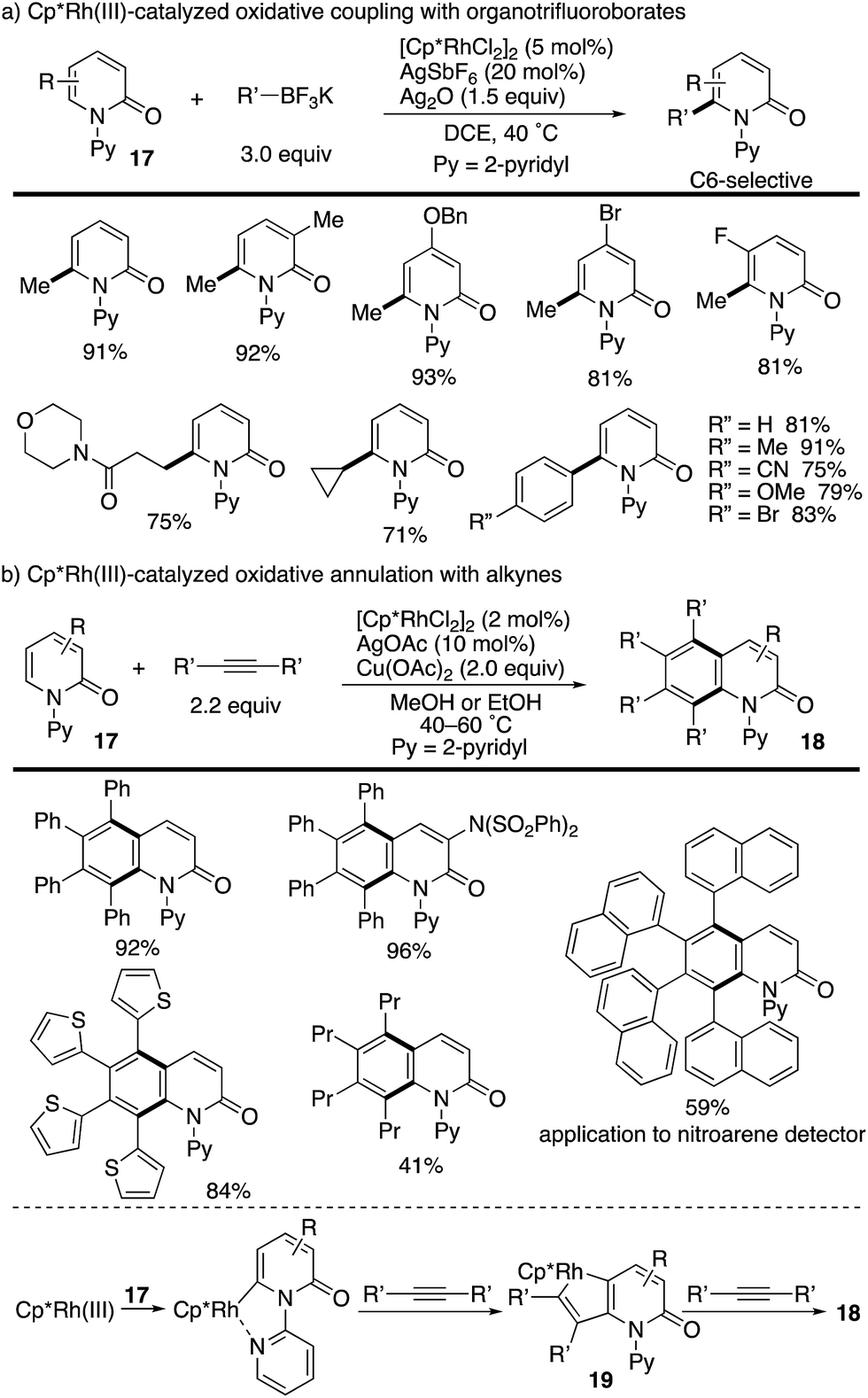
The above seminal work by Hirano and Miura prompted several groups to join this research field. Liu and co-workers reported the Cp*Rh(III)-catalysed oxidative coupling of N-(2-pyridyl)-2-pyridones 17 with organotrifluoroborates (Scheme 20a).27 The Cp*Rh(III)/Ag2O catalysis was tolerated with not only arylborons but also alkylborons to deliver various C6-functionalized 2-pyridones in good yields. More recently, a related annulation reaction with two alkyne molecules was developed by Patra, leading to multi-substituted isoquinolinone derivatives 18 (Scheme 20b).28 The reaction is considered to proceed via (1) a pyridine-directed first C–H rhodation at the C6 position, (2) insertion of one alkyne, (3) metalacycle 19 formation via a second C–H rhodation at the C5 position, (4) insertion of another alkyne and (5) reductive elimination. Additionally, the naphthalene-substituted highly π-extended product can be applied to a nitroarene detector.
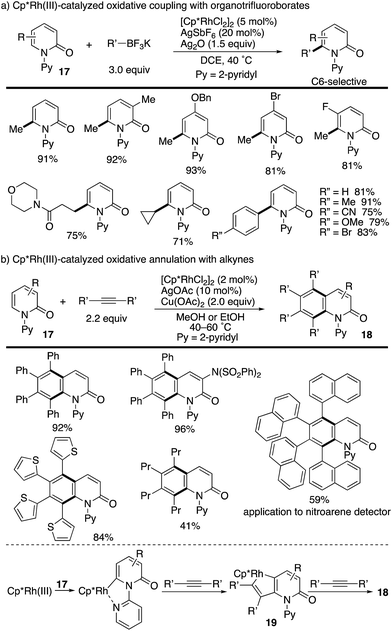 |
| | Scheme 20 Cp*Rh(III)-Catalysed directed oxidative coupling at the C6 position. | |
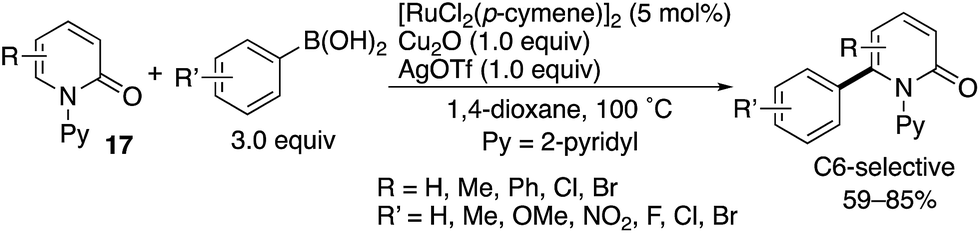
Das also developed a similar oxidative arylation reaction by using a Ru(II) catalyst combined with Cu- and Ag-based oxidants (Scheme 21).29
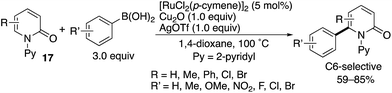 |
| | Scheme 21 Ru(II)-Catalysed directed C6-arylation. | |
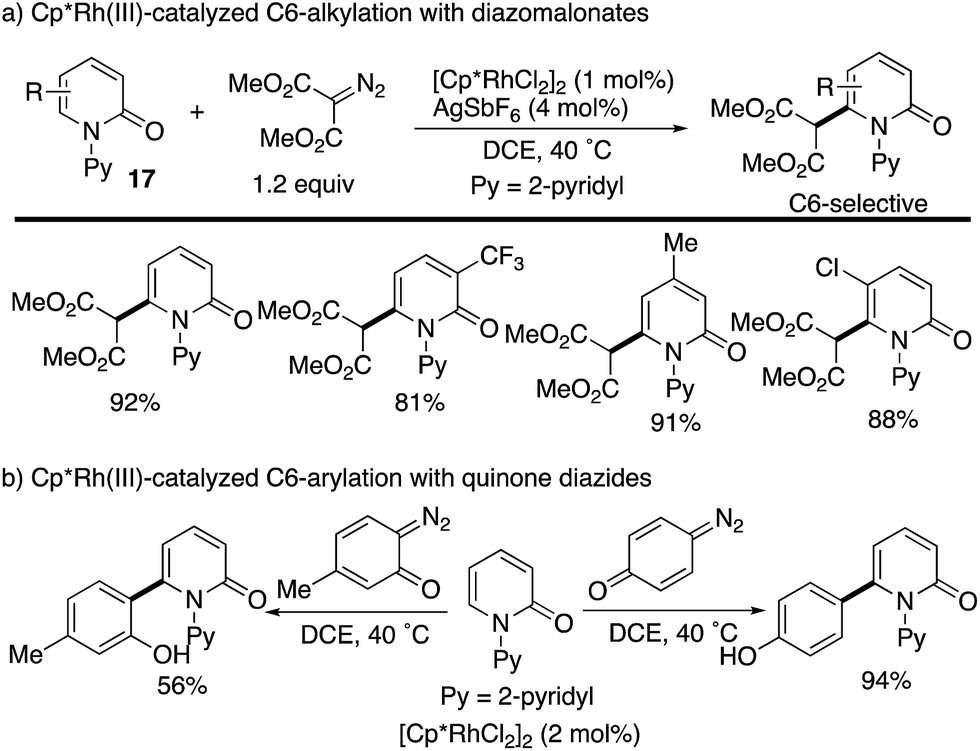
The Cp*Rh complexes also catalysed the redox-neutral C–H coupling with diazo compounds (Scheme 22). Samanta reported C6-selective alkylation with diazomalonates (Scheme 22a).30 The same authors subsequently developed a related coupling with quinone diazides to introduce the phenol substituents at the C6 position (Scheme 22b).31 Also in these cases, the C6 selectivity was high independent of the substitution pattern on the pyridone ring.
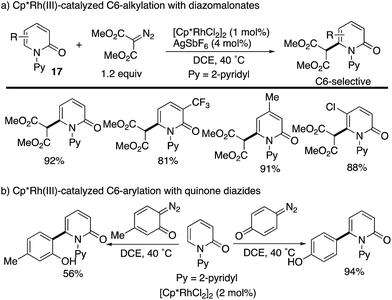 |
| | Scheme 22 Cp*Rh(III)-Catalysed directed coupling with diazo compounds at the C6 position. | |
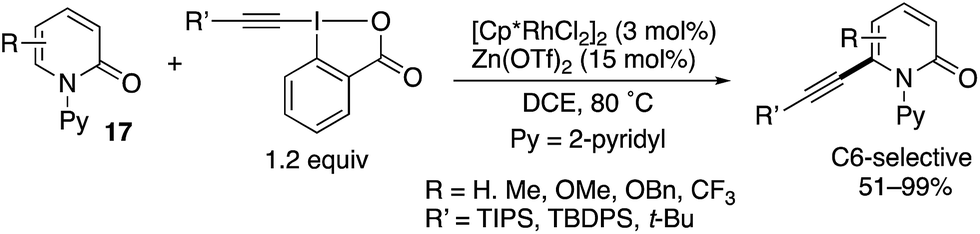
Directed alkynylation was also possible. With TIPS-EBX, which is the same as was used in Scheme 12, the N-(2-pyridyl)-2-pyridones 17 underwent C6-alkynylation in the presence of the [Cp*RhCl2]2 catalyst and Zn(OTf)2 additive (Scheme 23).19 Also note that TBDPS- and t-Bu-substituted EBXs could also be employed.
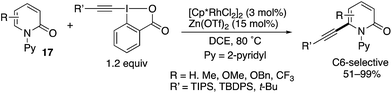 |
| | Scheme 23 Cp*Rh(III)-Catalysed directed C6-alkynylation with EBX reagents. | |
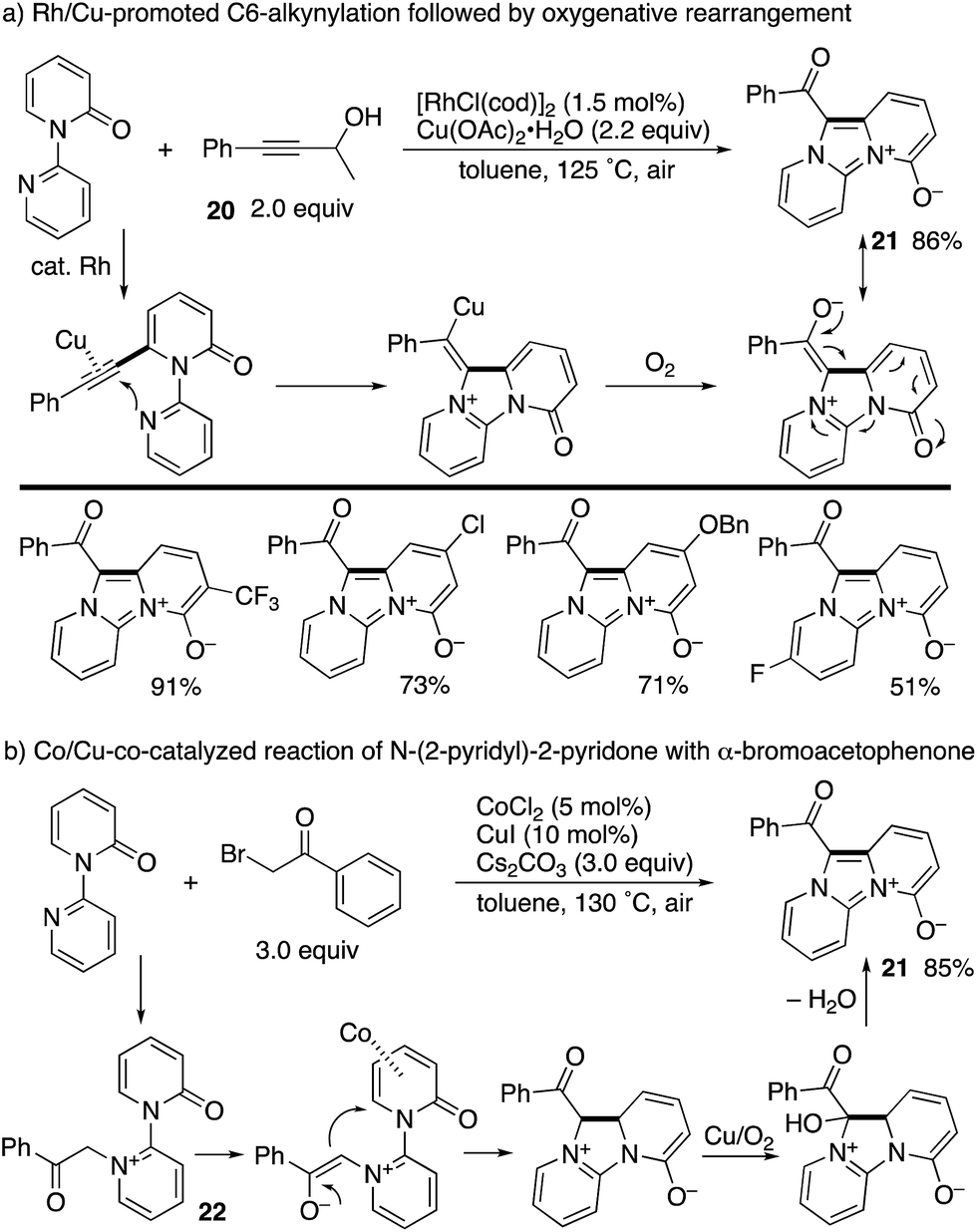
A Rh/Cu-promoted domino reaction including C6-alkynylation was developed by Li and co-workers (Scheme 24a).32 Rhodium-catalysed oxidative alkynylation with the propargyl alcohol 20, as the masked terminal alkyne, was followed by copper-mediated oxygenative rearrangement to furnish imidazodipyridinium-4-olates 21 in good yields. Very recently, the same research group reported that a similar imidazodipyridinium 21 could be synthesized from N-(2-pyridyl)-2-pyridones and α-bromoacetophenones under aerobic Co/Cu cooperative catalysis (Scheme 24b).33 The reaction pathway includes the N-alkylated intermediate 22, and subsequent Co-promoted intramolecular conjugated addition and Cu/O2-mediated oxidative aromatization could occur.
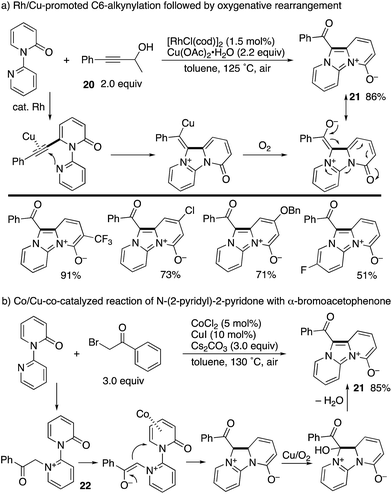 |
| | Scheme 24 Rh/Cu-co-catalysed domino alkynylation/oxygenative rearrangement and related Co/Cu aerobic catalysis. | |
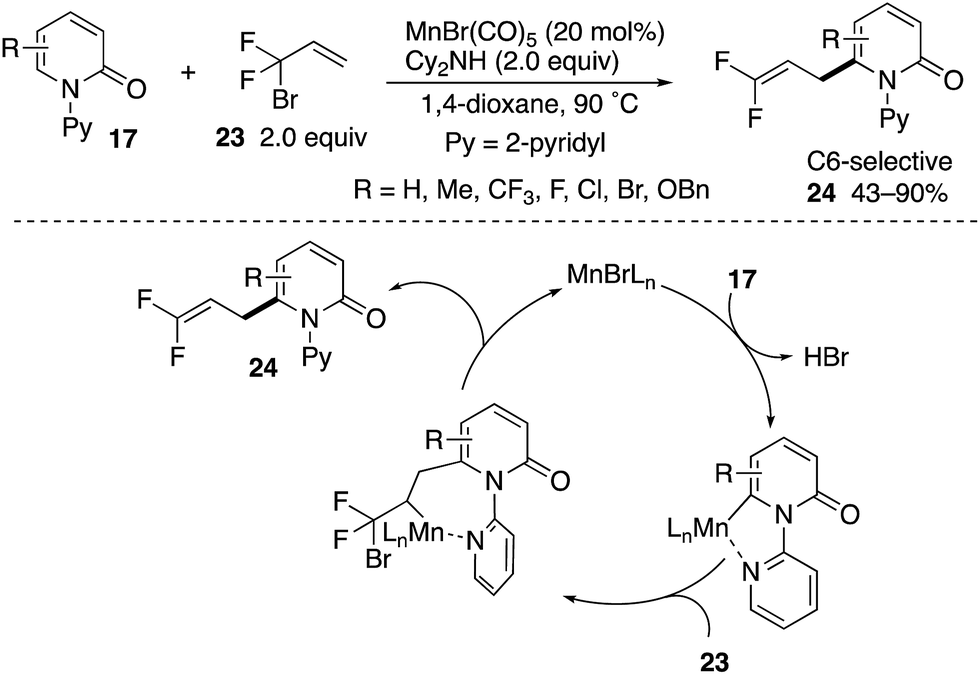
A Mn(I)-catalysed C6-allylation with 3-bromo-2,2-difluoropropene (23) was also developed by Zhang (Scheme 25).34 The reaction proceeded site-selectively and regioselectively to deliver the 3,3-difluoroallylation products 24 in good yields. The successful introduction of a difluoromethylene unit deserves much attention from the pharmaceutical point of view because of the unique steric and electronic nature of fluorine in medicinal applications.35 The reaction occurs through (1) coordination-assisted C6–C–H manganesation, (2) regioselective insertion of olefin and (3) β-Br elimination.
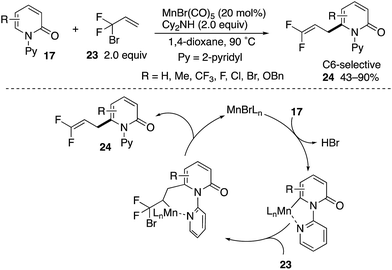 |
| | Scheme 25 Mn(I)-Catalysed directed C6-3,3-difluoroallylation. | |
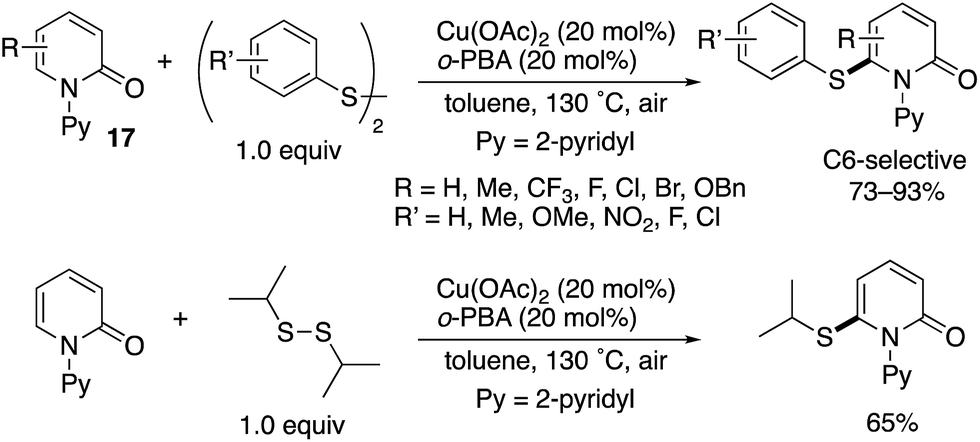
Although chemists have mainly focused on the development of C–C forming reactions at the C6 position, C6-selective heteroatom-functionalization is also possible. Jiang and Liu reported a Cu(II)-catalysed thiolation with disulfides (Scheme 26).36 Both diaryl- and dialkyldisulfides participated in the reaction, and the corresponding C6-thiolated pyridones were obtained in good to high yields. Similar to in Scheme 16, the acidic additives had positive effects on the reaction, with 2-biphenylcarboxylic acid (o-PBA) proving to be optimal in the C–H thiolation.
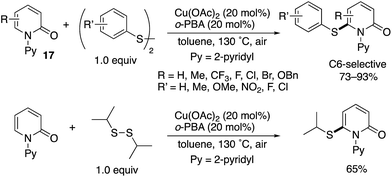 |
| | Scheme 26 Cu(II)-Catalysed directed C6-thiolation. | |
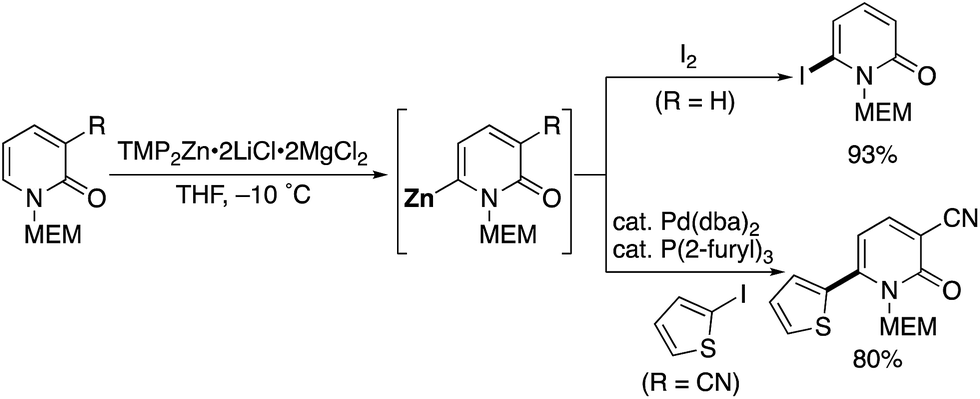
Very recently, Knochel reported a methoxyethoxymethyl (MEM)-directed C6-selective zincation with TMP2Zn·2LiCl·2MgCl2 (Scheme 27).37 The zincation reaction proceeded smoothly under mild conditions, and subsequent trapping with I2 afforded 6-iodo-2-pyridone, which is a useful building block for further elaboration. Negishi cross-coupling of zincated pyridone was also possible.
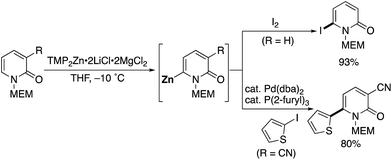 |
| | Scheme 27 MEM-Directed C6-selective zincation. | |
Sterically controlled site-selective C–H borylation
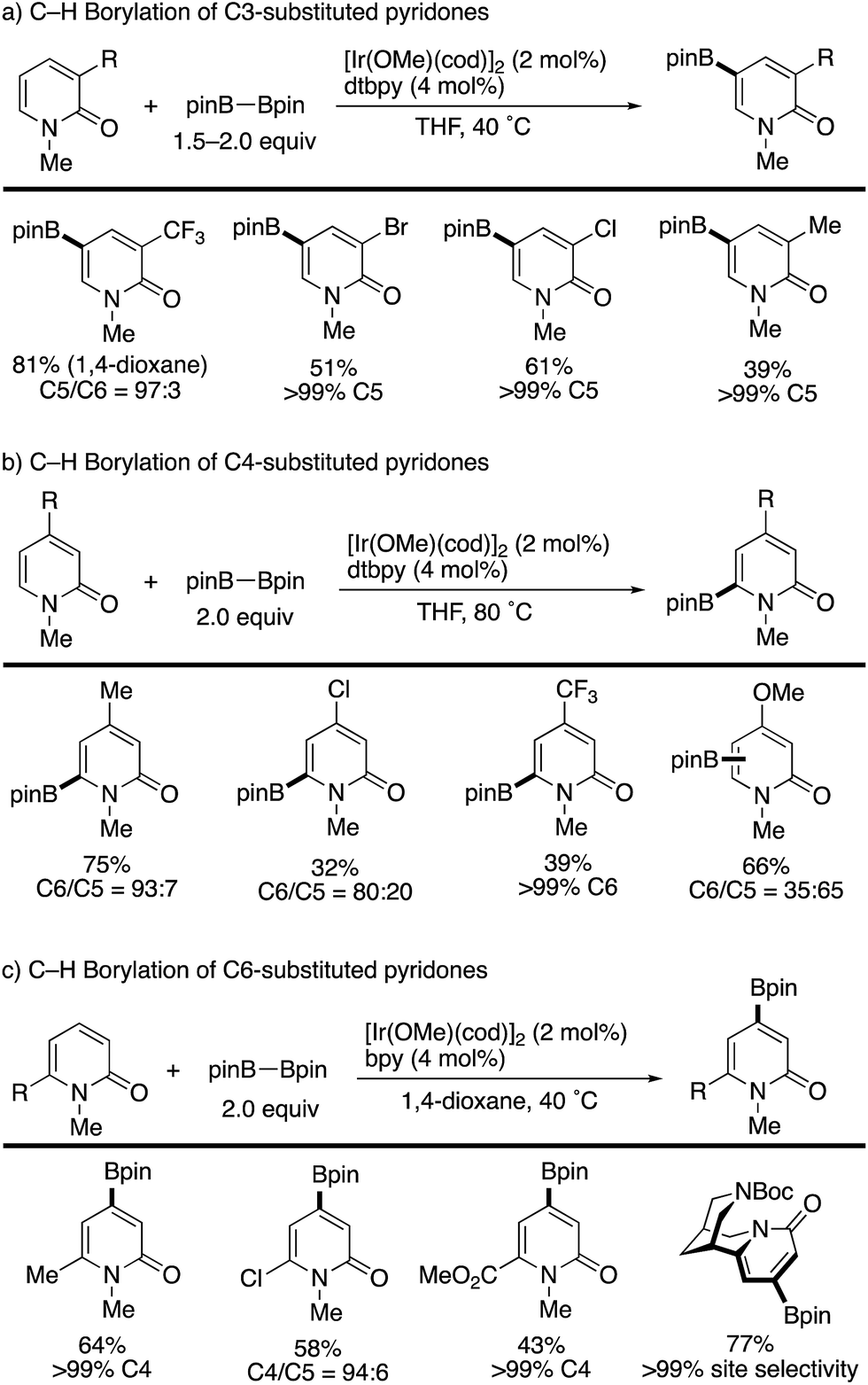
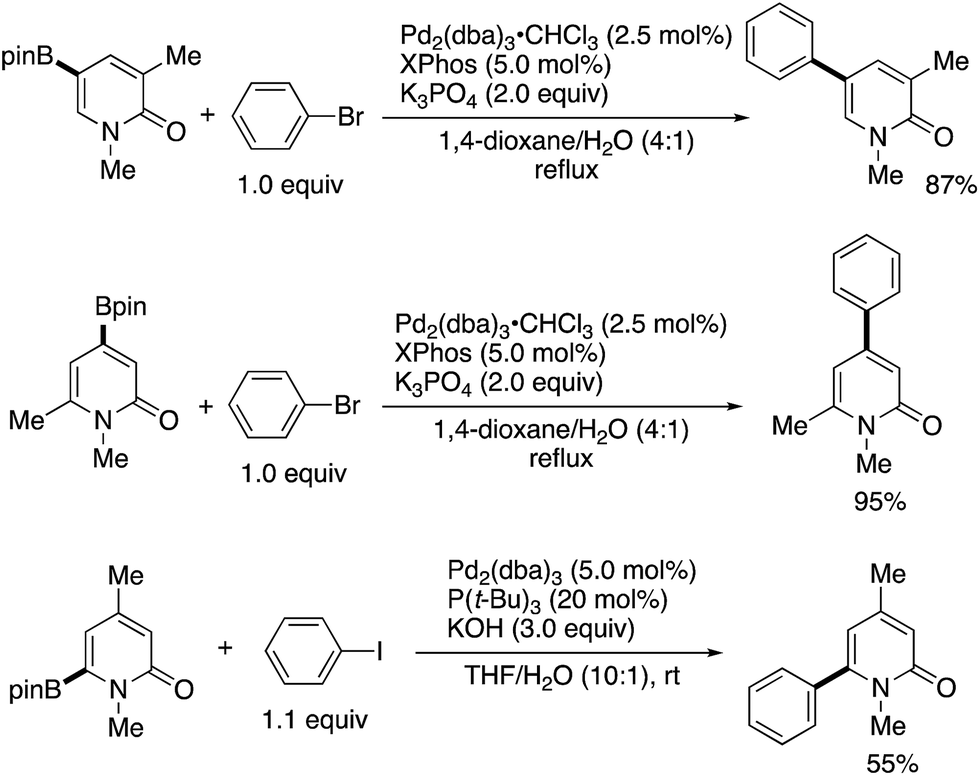
Iridium-catalysed nondirected C–H borylation is now recognized to be one of the most powerful and reliable strategies for the introduction of functional groups to various aromatic and heteroaromatic compounds.38 However, the site-selective C–H borylation of 2-pyridones remained underdeveloped until the seminal work by Hirano and Miura (Scheme 28) was reported.39 By a judicious choice of catalyst and solvent, the C3-, C4-, and C6-substituted pyridones were borylated, and the corresponding C5-, C6-, and C4-borylated products were formed with high site-selectivity (dtbpy = 4,4,-di(tert-butyl)-2,2′-bipyridine, bpy = 2,2′-bipyridine). Particularly notable is the successful C–H -borylation at the C4 position (Scheme 28c), which is otherwise difficult to access by other C–H activation strategies; just a single example under Ni/Al cooperative catalysis was reported, mentioned in Scheme 13c. Additionally, Boc-protected (−)-cytisine was also successfully borylated, giving a single isomer. The obtained borylated products were readily manipulated by Suzuki–Miyaura coupling to deliver the corresponding arylated 2-pyridones in good yields (Scheme 29).
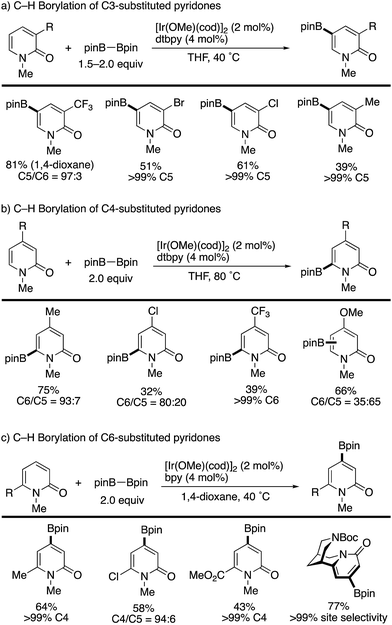 |
| | Scheme 28 Ir-Catalysed nondirected site-selective C–H borylation. | |
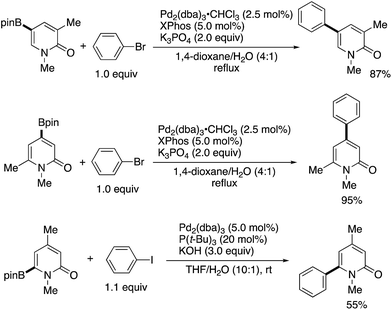 |
| | Scheme 29 The Suzuki–Miyaura cross-coupling of borylated 2-pyridones. | |
The site-selectivity was mainly controlled by steric factors, and most sterically accessible positions were selectively borylated. However, the reaction of the C5-substituted pyridones and unsubstituted pyridones resulted in much lower conversion and/or nonselective multi borylation. Further developments are still awaited.
Conclusions
In this minireview, we have demonstrated the recent progress of site-selective C–H functionalization of 2-pyridones, which are privileged structural motifs in many important bioactive molecules. Due to its conjugated enone-like structure, the pyridone ring has large electronic biases, and thus site selectivity can be controlled by optimization of the electronic nature of the catalyst (organometallic control). However, some reaction sites are often competitive (e.g. C3 vs. C5, see Scheme 10). In such a case more robust strategies, namely those involving radicals or directing groups, are still essential. The steric control protocol is also powerful in specific cases. However, we currently have no solution for the substituent-independent C4-selective C–H functionalization of general 2-pyridones, involving parent unsubstituted ones. Additionally, applications to complex molecule synthesis still remains elusive. We believe that the development of completely new methodologies and/or concepts addresses the current problems and enables the truly useful, productive, and reliable synthesis of multi-substituted, highly functionalized pyridones, which can open a door to new drug molecules based on 2-pyridone.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant no. JP 15H05485 (Grant-in-Aid for Young Scientists (A)) to K. H. and JP 17H06092 (Grant-in-Aid for Specially Promoted Research) to M. M.
Notes and references
-
(a) M. Torres, S. Gil and M. Parra, Curr. Org. Chem., 2005, 9, 1757 CrossRef CAS
 ;
(b) I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1 CrossRef CAS PubMed ;
(c) S. Hibi, K. Ueno, S. Nagato, K. Kawano, K. Ito, Y. Norimine, O. Takenaka, T. Hanada and M. Yonaga, J. Med. Chem., 2012, 55, 10584 CrossRef CAS PubMed ;
(d) P. Hajek, H. McRobbie and K. Myers, Thorax, 2013, 68, 1037 CrossRef PubMed .
;
(b) I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1 CrossRef CAS PubMed ;
(c) S. Hibi, K. Ueno, S. Nagato, K. Kawano, K. Ito, Y. Norimine, O. Takenaka, T. Hanada and M. Yonaga, J. Med. Chem., 2012, 55, 10584 CrossRef CAS PubMed ;
(d) P. Hajek, H. McRobbie and K. Myers, Thorax, 2013, 68, 1037 CrossRef PubMed .
- For recent selected reviews, see:
(a) F. Kakiuchi and T. Kochi, Synthesis, 2008, 2008, 3013 CrossRef ;
(b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed ;
(c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed ;
(d) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed ;
(e) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC ;
(f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed ;
(g) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096 CrossRef CAS PubMed ;
(h) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed ;
(i) L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC ;
(j) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed ;
(k) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed ;
(l) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed ;
(m) J. Yuan, C. Liu and A. Lei, Chem. Commun., 2015, 51, 1394 RSC ;
(n) K. Hirano and M. Miura, Chem. Lett., 2015, 44, 868 CrossRef CAS ;
(o) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894 CrossRef CAS PubMed ;
(p) F. Wang, S. Yu and X. Li, Chem. Soc. Rev., 2016, 45, 6462 RSC ;
(q) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed ;
(r) M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000 CrossRef PubMed ;
(s) M. P. Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654 CrossRef PubMed ;
(t) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed .
- T. Kino, Y. Nagase, Y. Ohtsuka, K. Yamamoto, D. Uraguchi, K. Tokuhisa and T. Yamakawa, J. Fluorine Chem., 2010, 131, 98 CrossRef CAS .
- A. Nakatani, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2013, 19, 7691 CrossRef CAS PubMed .
- For selected examples of Mn(III)-mediated radical reactions, see:
(a) B. B. Snider, Chem. Rev., 1996, 96, 339 CrossRef CAS PubMed ;
(b) A. W. J. Logan, J. S. Parker, M. S. Hallside and J. W. Burton, Org. Lett., 2012, 14, 2940 CrossRef CAS PubMed ;
(c) J. Yin, C. Wang, L. Kong, S. Cai and S. Gao, Angew. Chem., Int. Ed., 2012, 51, 7786 CrossRef CAS PubMed ;
(d) X. Wang, X. Wang, X. Tan, J. Lu, K. W. Cormier, Z. Ma and C. Chen, J. Am. Chem. Soc., 2012, 134, 18834 CrossRef CAS PubMed ;
(e) S. K. Guchhait, M. Kashyap and S. Saraf, Synthesis, 2010, 1166 CrossRef CAS ;
(f) A. Dickschat and A. Studer, Org. Lett., 2010, 12, 3972 CrossRef CAS PubMed ;
(g) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed .
- A. Nakatani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 1377 CrossRef CAS PubMed .
-
(a) Y. M. Osornio, R. Cruz-Almanza, V. Jiménez-Montaño and L. D. Miranda, Chem. Commun., 2003, 2316 RSC ;
(b) O. Guadarrama-Morales, F. Méndez and L. D. Miranda, Tetrahedron Lett., 2007, 48, 4515 CrossRef CAS .
-
(a) G. A. Russell and R. F. Bridger, Tetrahedron Lett., 1963, 4, 737 CrossRef ;
(b) T. Suehiro, A. Suzuki, Y. Tsuchida and J. Yamazaki, Bull. Chem. Soc. Jpn., 1977, 50, 3324 CrossRef CAS .
-
(a)
A. Studer, in Radicals in Organic Synthesis, ed. P. Renaud and M. Sibi, Wiley-VCH, Weinheim, 1st edn, 2001, vol. 2, p. 44 Search PubMed ;
(b)
S. E. Vaillard, B. Schulte and A. Studer, in Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH, Weinheim, 2009, p. 475 Search PubMed ;
(c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed .
- For selected reviews on the photoredox catalysis, see:
(a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed ;
(b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC ;
(c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed ;
(d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed ;
(e) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS PubMed .
- A. Najib, S. Tabuchi, K. Hirano and M. Miura, Heterocycles, 2016, 92, 1187 CrossRef CAS .
- A. Modak, S. Rana and D. Maiti, J. Org. Chem., 2015, 80, 296 CrossRef CAS PubMed .
- P. Chauhan, M. Ravi, S. Singh, P. Prajapati and P. P. Yadav, RSC Adv., 2016, 6, 109 RSC .
- E. E. Anagnostaki, A. D. Fotisdou, V. Demertzidou and A. L. Zografos, Chem. Commun., 2014, 50, 6879 RSC .
- J. Buck, J. P. Madelay and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1992, 67 RSC .
- T. Itahara and F. Ouseto, Synthesis, 1984, 488 CrossRef CAS .
- C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS PubMed .
- Y. Chen, F. Wang, A. Jia and X. Li, Chem. Sci., 2012, 3, 3231 RSC .
- Y. Li, F. Xie and X. Li, J. Org. Chem., 2016, 81, 715 CrossRef CAS PubMed .
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 15996 CrossRef CAS PubMed .
- R. Tamura, Y. Yamada, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2012, 51, 5679 CrossRef CAS PubMed .
-
(a) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef CAS PubMed ;
(b) S. Tang, O. Eisenstein, Y. Nakao and S. Sakaki, Organometallics, 2017, 36, 2761 CrossRef CAS .
- P. A. Dones and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 633 Search PubMed .
- R. Odani, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2014, 53, 10784 CrossRef CAS PubMed .
- W. Miura, K. Hirano and M. Miura, Org. Lett., 2016, 18, 3742 CrossRef CAS PubMed .
- W. Miura, K. Hirano and M. Miura, J. Org. Chem., 2017, 82, 5337 CrossRef CAS PubMed .
- P. Peng, J. Wang, H. Jiang and H. Liu, Org. Lett., 2016, 18, 5376 CrossRef CAS PubMed .
- A. Biswas, D. Giri, D. Das, A. De, S. K. Patra and R. Samanta, J. Org. Chem., 2017, 82, 10989 CrossRef CAS PubMed .
- K. A. Kumar, P. Kannaboina and P. Das, Org. Biomol. Chem., 2017, 15, 5457 Search PubMed .
- D. Das, A. Biswas, U. Karmakar, S. Chand and R. Samanta, J. Org. Chem., 2016, 81, 842 CrossRef CAS PubMed .
- D. Das, P. Poddar, S. Maity and R. Samanta, J. Org. Chem., 2017, 82, 3612 CrossRef CAS PubMed .
- T. Li, Z. Wang, K. Xu, W. Liu, X. Zhang, W. Mao, Y. Guo, X. Ge and F. Pan, Org. Lett., 2016, 18, 1064 CrossRef CAS PubMed .
- T. Li, C. Fu, Q. Ma, Z. Ma, Y. Yang, H. Yang, R. Lv and B. Li, J. Org. Chem., 2017, 82, 10263 CrossRef CAS PubMed .
- J. Ni, H. Zhao and A. Zhang, Org. Lett., 2017, 19, 3159 CrossRef CAS PubMed .
-
(a) J.-M. Altenburger, G. Y. Lassalle, M. Matrougui, D. Galtier, J.-C. Jetha, Z. Bocskei, C. N. Berry, C. Lunven, J. Lorrain, J.-P. Herault, P. Schaeffer, S. E. O'Connor and J.-M. Herbert, Bioorg. Med. Chem., 2004, 12, 1713 CrossRef CAS PubMed ;
(b) Y. Pan, J. Qiu and R. B. Silverman, J. Med. Chem., 2003, 46, 5292 CrossRef CAS PubMed ;
(c) P. M. Weintraub, A. K. Holland, C. A. Gates, W. R. Moore, R. J. Resvick, P. Bey and N. P. Peet, Bioorg. Med. Chem., 2003, 11, 427 CrossRef CAS PubMed .
- P. Peng, J. Wang, C. Li, W. Zhu, H. Jiang and H. Liu, RSC Adv., 2016, 6, 57441 RSC .
- D. S. Ziegler, R. Greiner, H. Laura, K. Karaghiosoff and P. Knochel, Org. Lett., 2017, 19, 5760 CrossRef CAS PubMed .
- Selected seminal works and reviews:
(a) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS ;
(b) G. A. Chotana, M. A. Rak and M. R. Smith III, J. Am. Chem. Soc., 2005, 127, 10539 CrossRef CAS PubMed ;
(c) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed ;
(d) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC .
- W. Miura, K. Hirano and M. Miura, Synthesis, 2017, 49, 4745 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Masahiro
Miura
* and
Masahiro
Miura