
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn artificial photosynthetic system for photoaccumulation of two electrons on a fused dipyridophenazine (dppz)–pyridoquinolinone ligand†
Jean-François
Lefebvre
ab,
Julian
Schindler
 cd,
Philipp
Traber
c,
Ying
Zhang
cd,
Stephan
Kupfer
c,
Stefanie
Gräfe
c,
Isabelle
Baussanne
b,
Martine
Demeunynck
b,
Jean-Marie
Mouesca
f,
Serge
Gambarelli
f,
Vincent
Artero
a,
Benjamin
Dietzek
*cde and
Murielle
Chavarot-Kerlidou
*a
cd,
Philipp
Traber
c,
Ying
Zhang
cd,
Stephan
Kupfer
c,
Stefanie
Gräfe
c,
Isabelle
Baussanne
b,
Martine
Demeunynck
b,
Jean-Marie
Mouesca
f,
Serge
Gambarelli
f,
Vincent
Artero
a,
Benjamin
Dietzek
*cde and
Murielle
Chavarot-Kerlidou
*a
aLaboratoire de Chimie et Biologie des Métaux, Univ. Grenoble Alpes, CNRS, CEA, 38000 Grenoble, France. E-mail: murielle.chavarot-kerlidou@cea.fr
bUniv. Grenoble Alpes, CNRS, DPM, 38000 Grenoble, France
cInstitute of Physical Chemistry, Abbe Center of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
dDepartment Functional Interfaces, Leibniz Institute of Photonic Technology Jena (IPHT), Albert-Einstein-Straße 9, 07745 Jena, Germany. E-mail: benjamin.dietzek@leibniz-ipht.de
eCenter for Energy and Environmental Chemistry, Friedrich Schiller University Jena, Philosophenweg 8, 07743 Jena, Germany
fUniv. Grenoble Alpes, CEA, CNRS, INAC-SyMMES, 38000 Grenoble, France
First published on 2nd April 2018
Abstract
Increasing the efficiency of molecular artificial photosynthetic systems is mandatory for the construction of functional devices for solar fuel production. Decoupling the light-induced charge separation steps from the catalytic process is a promising strategy, which can be achieved thanks to the introduction of suitable electron relay units performing charge accumulation. We report here on a novel ruthenium tris-diimine complex able to temporarily store two electrons on a fused dipyridophenazine–pyridoquinolinone π-extended ligand upon visible-light irradiation in the presence of a sacrificial electron donor. Full characterization of this compound and of its singly and doubly reduced derivatives thanks to resonance Raman, EPR and (TD)DFT studies allowed us to localize the two electron-storage sites and to relate charge photoaccumulation with proton-coupled electron transfer processes.
Introduction
The development of solar-driven chemistry exploiting sunlight to produce fuels and commodity chemicals (ammonia, hydrogen peroxide…) from readily available feedstocks (water, carbon dioxide, atmospheric nitrogen and oxygen…) is a top research priority for the 21st century.1–4 Natural photosynthesis is an inspiration in that field,5–7 and the first functional molecular-based photocathodes for hydrogen production and CO2 reduction recently appeared in the literature,8–12 combining molecular photosensitizers as a surrogate for photosystems and catalytic centers mimicking enzymes involved in bioenergetic processes. These artificial photosynthetic systems however display low energy conversion efficiencies because the light-induced charge separation process is a single-electron event that occurs on the fs–ns timescale, i.e. several orders of magnitude faster than multielectron catalysis. Consequently, charge recombination and/or degradation processes most of the time take place before catalysis can proceed. A solution to overcome this limitation therefore requires decoupling these two processes and here again, inspiration can be found in nature. For example, reversible quinone/hydroquinone couples are employed as electron relays in the photosynthetic machinery:13 after two consecutive photon absorption – charge separation steps at photosystem II (PS II), two electrons are transported by plastoquinone QB (among other electron transport cofactors), thanks to proton-coupled electron transfer (PCET) processes. Moreover, the photosynthetic process generates NAD(P)H as a key biochemical intermediate via the addition of two electrons and one proton to NAD(P)+. NAD(P)H further acts as a versatile reductant to drive reductive enzymatic reactions in the dark, such as CO2 fixation (in the Calvin cycle).13 Hence, the integration of charge accumulation sites in the molecular design of the photocatalytic system and the implementation of PCET processes is an appealing strategy towards the temporary storage of photogenerated electrons before delivery to the catalyst.Since the initial report by Wasielewski and co-workers in 1992,14 a few molecular systems have been reported that photoaccumulate reducing equivalents.15–17 The electron storage sites range from organic structures such as naphthalene or perylene bisimides,14,18 anthraquinone,19 and viologen derivatives,20,21 to iridium(III)22 or rhodium(III)23 complexes and polyoxometalates.24,25 Ruthenium complexes containing NAD(P)+/NAD(P)H mimics were also elegantly developed by the group of Tanaka.26–28 Of particular interest is the work of MacDonnell and co-workers on dinuclear ruthenium complexes bearing extended poly-N-heterocyclic bridging ligands29–32 with, in particular, a quinone-containing bridging ligand undergoing a light-driven proton-assisted four-electron reduction process.29 These authors also clearly showed that a bent bridging ligand can accumulate two electrons at a potential suitable to drive proton reduction.32
Implementing such a strategy in fuel-producing photocathodes however puts strong topological constraints on the molecular design. Indeed, unidirectional electron transfer from the electrode substrate to the accumulation site and through the photosensitizer is required. In addition, the photoaccumulation site should be remote from the electrode surface in order to limit the recombination processes.33 This precludes the use of previously reported systems in which two photosensitizing units straddle the accumulation site.14,18,19,24,29,30,32 Conversely, systems based on a single light-harvesting center such as iminoimidazole-containing ruthenium photosensitizers, whose properties have been studied by two of us,34,35 and disubstituted dipyrido-[3,2-a:2′,3′-c]phenazine (dppz)-based ruthenium complexes36 are instrumental to the aim of designing molecular photoelectrodes with enhanced efficiency. We therefore designed a novel ruthenium tris-diimine complex, [1](PF6)2 (Scheme 1), that features an original bent-shaped polyazaaromatic ligand, namely a pyridoquinolinone moiety fused to a dppz-chelating unit. We report herein its full steady state spectroscopic and electrochemical characterization and show how the pyridoquinolinone subunit drastically modulates the electronic properties of the complex toward charge accumulation. Photolysis experiments unambiguously established the ability of [1](PF6)2 to accumulate two electrons upon visible light irradiation in the presence of a sacrificial electron donor, and to further stabilize the photogenerated two-electron reduced state thanks to a PCET process. The electron storage sites on the π-extended ligand were identified at the (time-dependent) density functional level of theory ((TD)DFT) and by EPR and resonance Raman studies on independently prepared singly and doubly reduced derivatives.
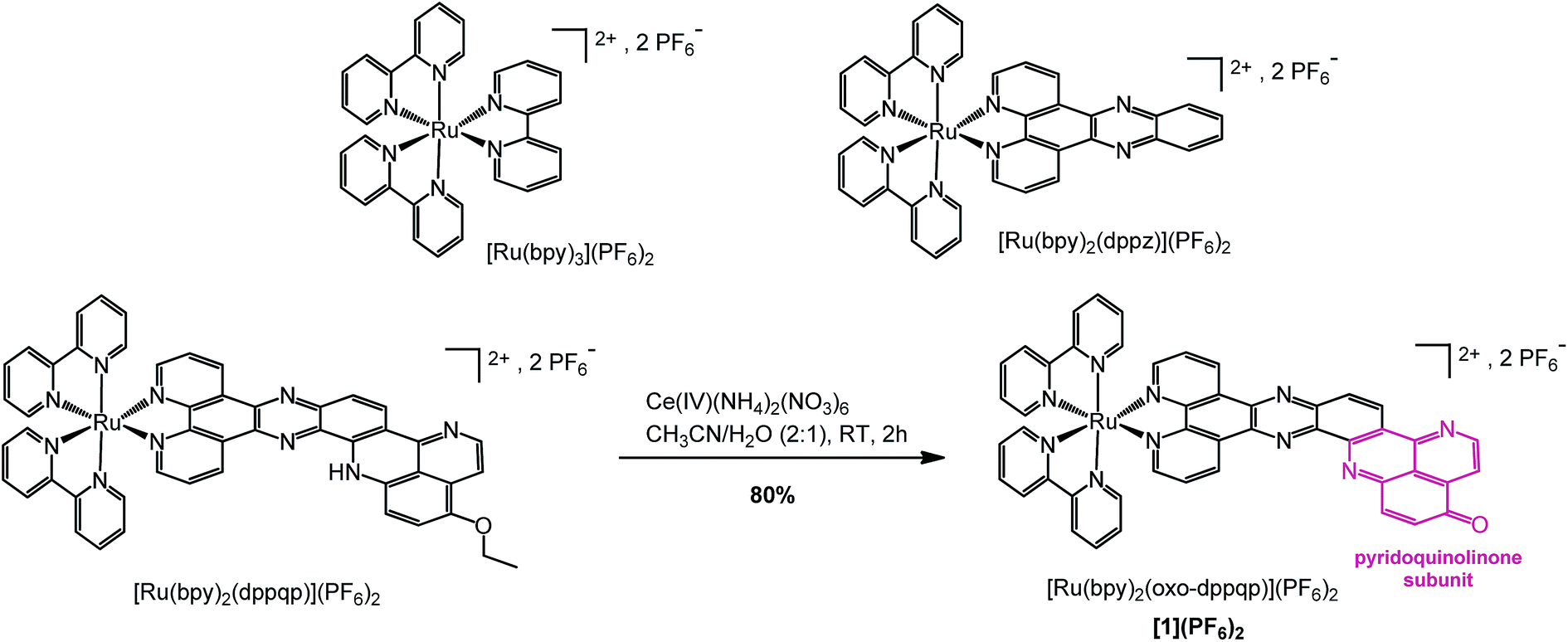 | ||
| Scheme 1 Synthesis of [Ru(bpy)2(oxo-dppqp)](PF6)2 ([1](PF6)2) from [Ru(bpy)2(dppqp)](PF6)2 and chemical structures of reference complexes [Ru(bpy)3](PF6)2 and [Ru(bpy)2(dppz)](PF6)2. | ||
Results and discussion
We previously reported an efficient “chemistry on the complex” strategy to synthesize variously substituted ruthenium tris-diimine complexes containing π-extended ligands, such as [Ru(bpy)2(dppqp)](PF6)2 (dppqp = dipyrido[3,2-a;2′,3′-c]pyrido[2′′,3′′-4,5]quinolino[2,3-h]phenazine, Scheme 1).37 Following previously published methods,38,39 deethylation by cerium ammonium nitrate (CAN, Ce(IV)(NH4)2(NO3)6) treatment, followed by spontaneous air oxidation of the latter afforded [Ru(bpy)2(oxo-dppqp)](PF6)2 ([1](PF6)2), featuring a fused dipyridophenazine (dppz)–pyridoquinolinone ligand (oxo-dppqp = dipyrido[3,2-a:2′,3′-c]pyrido[2′′,3′′-4,5]quinolino[2,3-h]phenazin-15-one) in 80% yield after purification.Aggregation through π-stacking is commonly observed for planar polyazaaromatic ruthenium complexes.37,40 Such a π-stacking behavior is supported by quantum chemical simulations for [1](PF6)2 (Fig. S3 and Table S1†) and was evaluated by a concentration-dependent 1H NMR study in CD3CN (Fig. S1 & S2†). The threshold concentration below which the monomer is predominant in solution was determined to be 5 × 10−4 M (see details in the ESI†), in line with a previous study on related Ru complexes;37 accordingly, the following spectroscopic and electrochemical measurements were performed at lower concentrations of [1](PF6)2.
The UV/vis spectrum of [1](PF6)2 displays typical features of ruthenium tris-diimine complexes (Fig. 2 and S4†), with an intense ligand-centered (LC) transition at 290 nm, characteristic of the bipyridine ligands, and metal-to-ligand charge-transfer (MLCT) bands observed between 400 and 500 nm. The high molar absorptivity in the visible range compared to the reference complex [Ru(bpy)2(dppz)](PF6)2 (Fig. S4†) suggests some contribution from the π-extended oxo-dppqp ligand. This is in agreement with TDDFT calculations predicting a strongly absorbing ligand-centered 1ππ* state on the π-extended ligand (see S8 in Fig. S5†).41
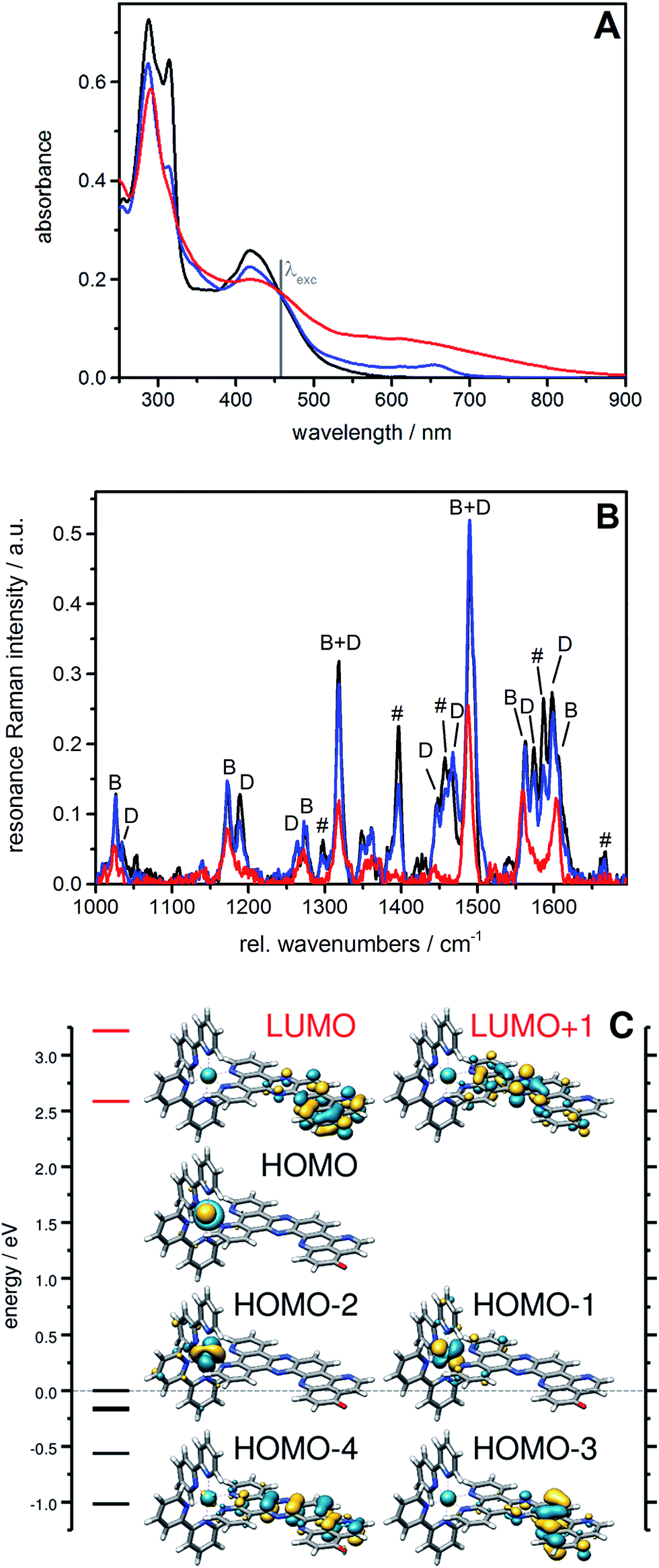 | ||
| Fig. 1 UV/vis (A) and RR spectra (B) of [1]2+ (black) in comparison with the spectra collected during controlled potential electrolysis: first (blue; electrolysis: 1 min at −0.5 V vs. Ag/AgCl) and second reduction (red; electrolysis: 1 min at −0.85 V vs. Ag/AgCl) in 0.1 M tetrabutylammonium tetrafluoroborate/acetonitrile. The RR spectrum was obtained upon excitation at 458 nm (solvent bands subtracted). The RR bands characteristic of vibrations of the bpy ligand (symbol B) as well as of the dppz moiety (symbol D) and the iminobenzoquinone moiety (symbol #) of the oxo-dppqp ligand are indicated. (C) MO diagram for [1]2+ including occupied (black) and virtual (red) frontier orbitals; orbital energies are given with respect to the energy of the HOMO. | ||
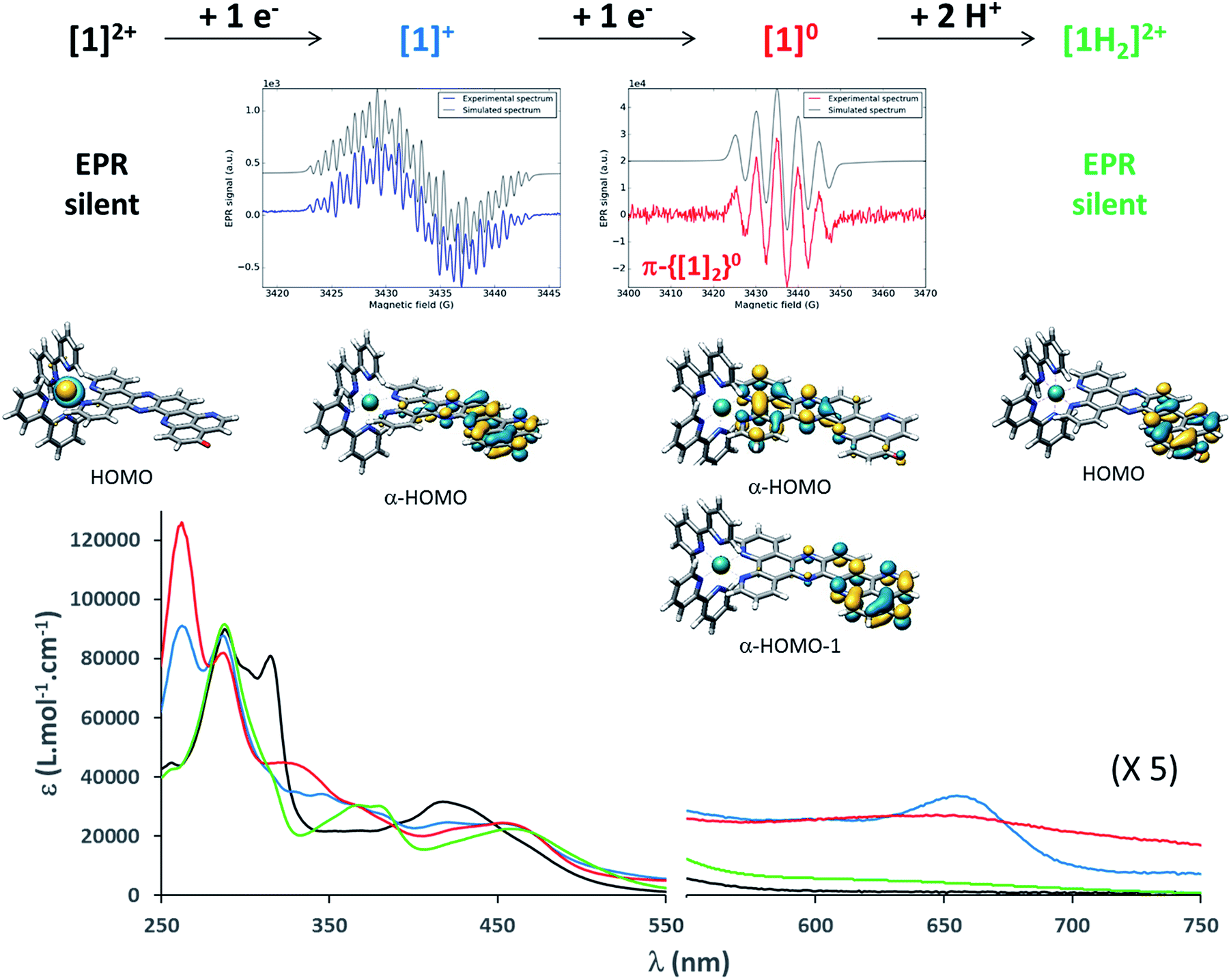 | ||
| Fig. 2 From top to bottom: summary of the chemical reduction process, EPR spectra, calculated structures and frontier molecular orbitals carrying excess charge, and UV/vis absorption spectra of [1]2+, [1]+, [1]0 and [1H2]2+ (see the ESI† for experimental details). | ||
The redox properties of [1]2+ were determined by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) (Fig. S6†) and are reported in Table 1. Oxidation of the Ru center is observed at +0.86 V vs. Fc+/Fc, which is slightly more anodic than the RuIII/RuII processes in [Ru(bpy)3](PF6)2 and [Ru(bpy)2(dppz)](PF6)2 and most likely reflects an increase of the electron-accepting properties of the π-extended ligand. In addition, [1]2+ displays five successive reduction processes; the first one at −0.87 V vs. Fc+/Fc is not observed for any of the reference complexes (Fig. S7 & S8†) and is tentatively assigned to a reduction on the pyridoquinolinone moiety. The second cathodic process occurs at a potential very close to the one obtained for the parent [Ru(bpy)2(dppz)](PF6)2 complex (Fig. S7†) which indicates a reduction located on the dppz moiety of the ligand. The following reductions are attributed to the three diimine ligands, by comparison to the reference complexes (Fig. S7 & S8†).
| RuIII/II | Red1 | Red2 | Red3 | Red4 | Red5 | |
|---|---|---|---|---|---|---|
| Ru(bpy)32+ | +0.82 | — | — | −1.73 | −1.91 | −2.18 |
| Ru(bpy)2(dppz)2+ | +0.84 | — | −1.36 | −1.79 | −2.01 | −2.49 |
| [1]2+ | +0.86 | −0.87 | −1.32 | −1.81 | −2.02 | −2.43 |
To gain more insights into the structures of the reduced compounds, the (electro)chemically reduced forms of [1]2+ (first and second reduction, generating [1]+ and [1]0, respectively) were investigated by UV/vis absorption spectroscopy (Fig. 2, S9 and S10†) and UV/vis and resonance Raman (RR) spectroelectrochemistry (SEC; Fig. 1 and Fig. S11†), in combination with TDDFT calculations (see the ESI† for details).
A 15 μM solution of [1]2+ in CH3CN was chemically reduced with cobaltocene32 under rigorously anaerobic conditions. The formation of the singly reduced derivative [1]+ is accompanied by significant changes in the UV/vis spectrum, with the growth of a very characteristic absorption feature at 653 nm (Fig. 2). In addition, three isosbestic points are observed at 326, 394 and 445 nm, associated with the disappearance of the absorption band at 315 nm and the growth of a new one at 348 nm (Fig. S9†). In contrast, the absorption of the doubly reduced species [1]0, generated by the addition of a second reducing equivalent to the solution, extends above 700 nm and is almost featureless (Fig. 2 and S10†). Such broad absorption features above 500 nm typically originate from ligand-centered states of ππ* nature of the reduced ligand.42 Similar spectral changes were also observed for electrochemically generated [1]+ and [1]0 (Fig. 1A).43 In the case of [Ru(bpy)2(dppz)]2+,42,44 the first reduction is centered on the phenazine moiety and is characterized by a broad absorption band from 500 to above 800 nm with notable absorption contributions beyond 700 nm (Fig. S11†), thus resembling the spectral features of doubly reduced [1]0. These data support the above assignment from cyclic voltammetry with the first reduction of [1]2+ centered at the pyridoquinolinone fragment and the second reduction localized on the phenazine fragment.
The RR spectrum of [1]2+ (collected upon excitation at 458 nm) consists of bands characteristic of vibrations of the bpy ligand (marked with B, Fig. 1B) as well as of the dppz moiety (marked with D) and the iminobenzoquinone moiety of the oxo-dppqp ligand (marked with #). These bands were assigned by comparison with the RR spectra of [Ru(bpy)2(dppz)]2+, [Ru(bpy)3]2+ (Fig. S12†) and [Ru(dppz)3]2+.45 In the first reduction process, a decrease of the bands attributed to the iminobenzoquinone moiety is observed, whereas MLCT transitions to the bpy ligands and the dppz moiety are sparsely affected. This again indicates that the additional charge is likely to be localized at the distal pyridoquinolinone moiety. In the second reduction process, the bands characteristic of the dppz moiety (symbol D) vanish, while the bands arising from bpy vibrations (symbol B) are still observed. Such an effect is also observed during the first reduction of the reference [Ru(bpy)2(dppz)]2+ (Fig. S13†). Thus, for [1]0, MLCT transitions to the dppz moiety do not take place anymore, in agreement with a second reduction localized on this part of the ligand.
To support these UV/vis and RR studies, quantum chemical calculations were performed at the DFT and TDDFT level of theory on [1]2+, as well as on the singly and doubly reduced species [1]+ and [1]0. [1]+ is clearly of doublet multiplicity; TDDFT allows us to assign the absorption feature at 653 nm to the bright intraligand states (localized on the oxo-dppqp ligand) D7 and D8, calculated at 605 and 576 nm respectively (Fig. S5†), of the singly reduced doublet [1]+. The doubly reduced complex, [1]0, may be either of singlet or triplet character (Fig. S14†). The DFT calculations reveal a near degeneracy of 1[1]0 and 3[1]0, while the triplet is slightly favored by 0.1 eV. Such an energy difference value is below the margin of error of DFT and thus no unambiguous assignment of the spin state of [1]0 is feasible. However, the simulated UV/vis absorption spectrum of the doubly reduced triplet species can be clearly associated with the experimental absorption spectra of [1]0 (Fig. S15†). In particular, the broad absorption band between 550 and 800 nm is unambiguously assigned to three bright oxo-dppqp-centered intraligand states (T12, T15 and T17) calculated at 759, 588 and 574 nm. On the other hand, no agreement is evident for the simulated 1[1]0 (Fig. S15†). Thus, the TDDFT calculations suggest that [1]0 exists in the triplet state. This is further supported below by electronic paramagnetic resonance experiments (EPR, vide infra). In addition, from the performed quantum chemical simulations, the first one-electron reduction is found to be localized on the iminobenzoquinone moiety (α-HOMO of [1]+), while the excess charges in doubly reduced 3[1]0 (triplet state)—compared to non-reduced [1]2+—are localized on the iminobenzoquinone moiety (α-HOMO-1) and on the dppz moiety (α-HOMO) as illustrated in Fig. 2. Further details with respect to the quantum chemical simulations are presented in the ESI.†
EPR is the spectroscopic technique of choice to probe the chemical environment of radical species. The chemical reduction procedure was thus adapted to record the X-band EPR spectra of [1]+ and [1]0 (200 μM in CH3CN/propionitrile; see UV/vis monitoring in Fig. S16†). The X-band EPR spectrum of the former, recorded in deoxygenated solution at room temperature, is characteristic of an organic radical species (g = 2.003), with a very slow relaxation rate (Fig. 2). It displays numerous, easily saturated, very narrow overlapping lines (0.3 G peak to peak) and could be simulated as a monoradical coupled to 3 nitrogen atoms (spin 1 nucleus; hyperfine coupling constants of 7.3, 4.8, and 3.9 MHz) and 6 hydrogen atoms (hyperfine coupling constants of 6.3, 5.4, 4.2, 3.7, 2.0, and 1.8 MHz). This finding is in agreement with the spin density of [1]+, which is predominantly localized on the pyridoquinolinone moiety (see Fig. S17†). [1]0 is also EPR active, which unambiguously confirms that this species exists in a triplet spin state. Its spectrum displays five equidistant broad lines (2.2 G peak to peak) with relative intensity 1–2–3–2–1 (Fig. 2), attributed to a hyperfine coupling with two equivalent N atoms with a coupling constant of 13.8 MHz (4.9 G for g = 2). It appeared to be strongly similar to the spectrum of singly reduced [Ru(bpy)2(dppz)](PF6)2 (Fig. S18†), which displayed a similar broad quintet with a hyperfine coupling constant of 14.2 MHz (5.05 G for g = 2). The only significant difference is a smaller hydrogen hyperfine coupling observed for the latter.42 Due to the similarity between the EPR spectra of the doubly reduced species [1]0 and the singly reduced species of [Ru(bpy)2(dppz)](PF6)2, we propose the formation of the dimer π-{[1]2}0, through π-stacking of two [1]0 complexes, occurring under the EPR analysis conditions. In such a structure, pairing of the electrons located on each pyridoquinolinone moiety would take place (thus precluding their EPR detection); on the other side, the electrons located on each pyrazine ring are too far from each other to interact, leading to the observed signature, identical to the one from a single dppz radical species. Indeed, an increase of the π-stacking propensity of these complexes is likely to occur upon ligand reduction,46 in agreement with the higher binding energy values calculated for the dimers of [1]1+ and [1]0 (Fig. S3 and Table S1†), compared to [1]2+. Bonding energies – and thus the tendency to aggregate – were calculated for {[1]2}0 and compared to those of {[1]2}4+ (dimer of [1]2+) and the intermediate {[1]2}2+ (dimer of [1]+) using DFT. The most stable combinations of the respective monomers are presented in Table S1† along with the calculated bonding energies and stacking distances. Interestingly, an increased bonding energy of −251 kJ mol−1 was observed for {3[1]2}0 (formed by two doubly reduced triplets), with respect to merely −132 kJ mol−1 for {[1]2}4+. This finding clearly demonstrates that the system exhibits an enhanced tendency to aggregate upon (double) reduction.
This set of experimental and theoretical data unambiguously confirmed that the first two reduction processes are both located on the fused dppz-pyridoquinolinone π-extended ligand of [1](PF6)2. This property is a prerequisite to achieve unidirectional (multi)charge photoaccumulation on a single site. From the point of view of the potential, the values of −0.87 V vs. Fc+/Fc (−0.30 V vs. NHE) and −1.32 V vs. Fc+/Fc (−0.75 V vs. NHE) are in the same range as that reported for the best performing complexes in the literature.19,32,36 Moreover, after the first reduction, this complex preserves its ability to generate a reactive MLCT state, which is mandatory to drive a second reduction process under visible light irradiation that leads to charge accumulation. Indeed, the broad absorption band between 400 and 500 nm is only slightly affected by the first reduction, with a small decrease of the intensity at around 420 nm and the appearance of a shoulder at 455 nm (Fig. 2). This is further supported by the RR study – the MLCT transitions to the bpy ligands and the dppz moiety being only sparsely affected by single reduction (Fig. 1) – and by TDDFT, where the excited state properties, such as the excitation energies and oscillator strengths (i.e. D28, D38, D40 and D44 calculated at 444, 424, 421 and 405 nm – see Fig. S5†) are preserved for [1]+.
All these observations thus validated the design of [1](PF6)2 to achieve light-driven electron storage and prompted us to assess its behavior under photolysis conditions. A very fast photochemical reaction occurred when complex [1](PF6)2 was irradiated at 450 nm in the presence of triethylamine (TEA) as a sacrificial electron donor. The spectral signature of [1]+ was transiently observed during the first minute of photolysis (Fig. 3); the absorption at 655 nm reached a maximum after 10 s of irradiation, while [1]2+ was still present in solution (observed at 315 nm, see Fig. S19†). Subsequently, the spectra rapidly evolved toward a new spectral feature, strikingly different from the signature of [1]0, and the reaction was completed after 5 minutes (Fig. 3). As it turns out, the newly formed species is EPR silent (see details of EPR measurements in the ESI†) and its absorption spectrum perfectly matches the one of the EPR silent doubly reduced doubly protonated derivative [1H2]2+, independently prepared by the addition of trifluoroacetic acid (TFA) to [1]0 (Fig. 3). A quantum yield of 66% (based on the amount of absorbed photons) was measured, which is very high for a reaction involving two successive light-driven electron transfers (see details in ESI, Fig. S20†). In our photolysis experiment, conducted in dry CH3CN, the source of protons is most likely the radical cation TEA˙+. The latter is generated upon reductive quenching of the Ru excited state by TEA, and is known to decompose with the release of one equivalent of H+.47 Similar proton transfers were characterized under photolysis conditions by MacDonnell and co-workers.29,36
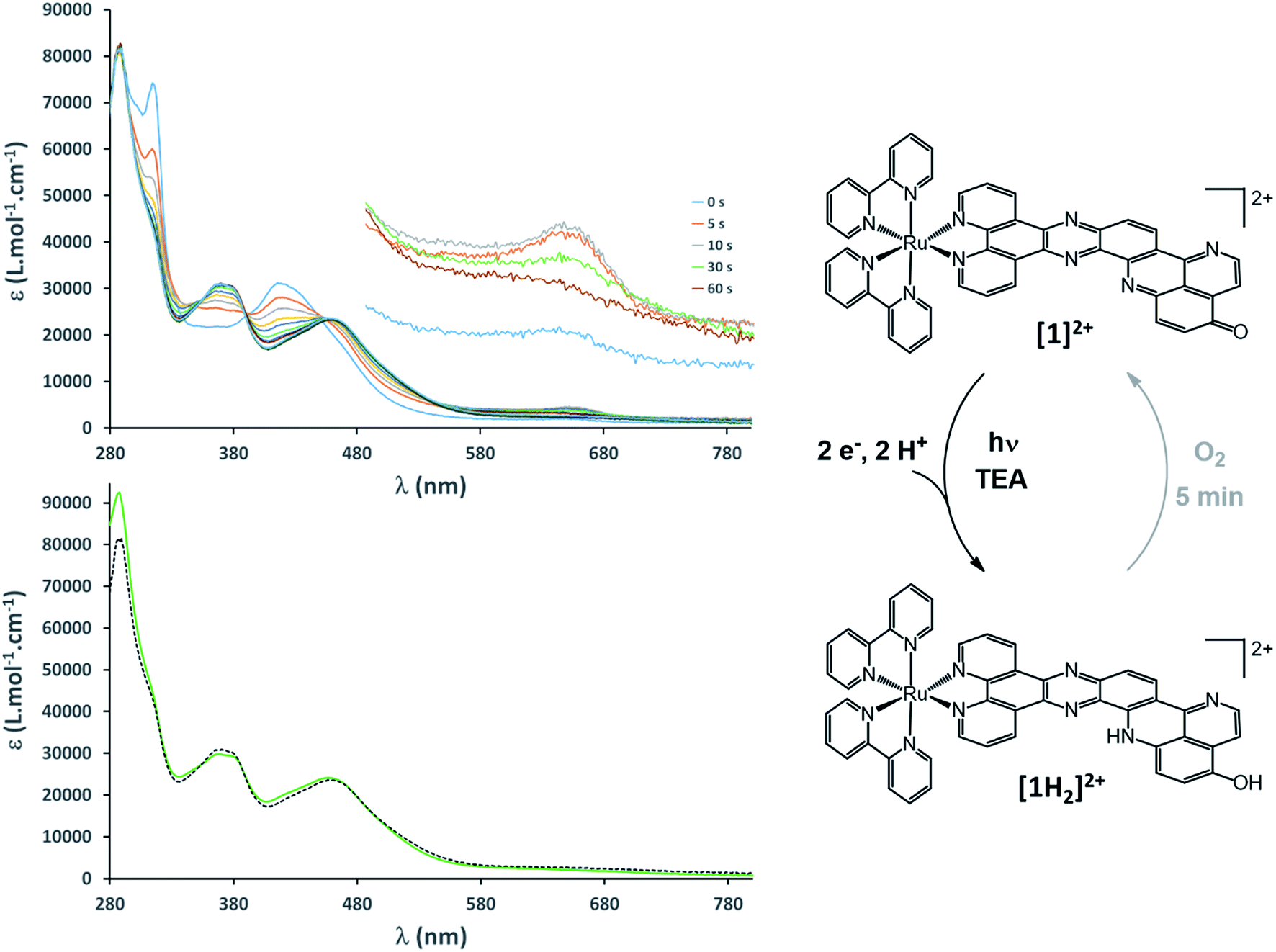 | ||
| Fig. 3 Top: UV/vis monitoring of a continuous photolysis experiment (λexc = 450 nm; 15 μM in CH3CN in the presence of 0.15 M TEA) (inset: zoom-in of the 600–700 nm area where the formation of [1]+ is observed). Bottom: comparison of the photolysis experiment (t = 5 min; dotted line) with [1H2]2+ (green line). Right: summary scheme of the reversible light-driven charge accumulation process. | ||
The equilibrium structure of [1H2]2+ was investigated at the DFT level of theory; the most stable doubly protonated doubly reduced species is an aminophenol derivative (Fig. 2 and 3), suggesting that electronic redistribution takes place within the doubly reduced π-extended ligand upon protonation. This structure was further confirmed by 1H NMR analysis of the photolysis experiment (see ESI, Fig. S21 and Tables S2 and S3†). Charge compensation and stabilization through proton-coupled electron transfer thus takes place during light-driven reduction of [1](PF6)2. Although occurring to the detriment of the reducing power of the system (see Fig. S22†), such a stabilization process was also encountered with various biological cofactors, plastoquinone QB typically, and proved essential for their electron relay activity.
Very few studies have addressed the stability of the system under photoaccumulation conditions. Importantly, we found that fast reoxidation of [1H2]2+ to the initial [1]2+ complex occurs upon exposing the photolysis solution to air (Fig. S23†). The quasi-reversibility of proton-coupled electron accumulation on [1](PF6)2 is also attested by the cyclic voltammogram of [1]2+ measured in the presence (see Fig. S22†). This contrasts with a previously reported dinuclear Ru complex with a bent tetraazatetrapyridopentacene bridging ligand, which is able to store two electrons at slightly more negative potentials; regeneration of this complex upon exposure to air takes up to 12 hours, owing to a detrimental radical-anion dimerization process.32
Conclusion
Appending a pyridoquinolinone subunit to the archetype [Ru(bpy)2(dppz)](PF6)2 complex successfully endowed the novel ruthenium trisdiimine photosensitizer [1](PF6)2 with multiple charge photoaccumulation properties. The latter, together with its singly and doubly reduced derivatives, was spectroscopically and electrochemically characterized. The combination of resonance Raman and EPR spectroscopies with DFT calculations proved instrumental to accurately localize the electron storage sites on the π-extended ligand. Remarkably, this system reproduces the 2e−/2H+ mechanism at work for both the light-driven reduction and dark oxidation of the plastoquinone photosynthetic cofactor: under visible light irradiation in the presence of a sacrificial electron donor, reversible photoaccumulation of two electrons, coupled to protonation steps, was achieved. This study thus represents an important breakthrough for the design of next generation artificial photosynthetic devices relying on a rational decoupling of light-driven single-electron events and targeted multielectron redox catalysis. Although the redox potential of the [1H2]2+ species in DMF is too high to warrant hydrogen production, it is still in line with other multi-electron reduction processes such as O2 reduction to H2O2,48 the latter being another relevant target in the context of solar-driven chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Fonds der Chemischen Industrie (B. D. and J. S.), the French National Research Agency (Labex Program, ARCANE, ANR-11-LABX-0003-01) and the CEA DRF Impulsion 2017 Program (Project “Light-Acc-Cat”). All calculations have been performed at the Universitätsrechenzentrum of the Friedrich Schiller University Jena. We acknowledge the COST Action CM1202, PERSPECT-H2O: Supramolecular Photocatalytic Water Splitting.Notes and references
- http://www.euchems.eu/solar-driven-chemistry/ .
- Solar Fuels and Artificial Photosynthesis: Science and innovation to change our future energy options, RSC report, January 2012 Search PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695 Search PubMed.
- A. Thapper, S. Styring, G. Saracco, A. W. Rutherford, B. Robert, A. Magnuson, W. Lubitz, A. Llobet, P. Kurz, A. Holzwarth, S. Fiechter, H. de Groot, S. Campagna, A. Braun, H. Bercegol and V. Artero, Green, 2013, 3, 43 CrossRef.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC.
- W. Song, Z. Chen, M. K. Brennaman, J. J. Concepcion, A. O. T. Patrocinio, N. Y. M. Iha and T. J. Meyer, Pure Appl. Chem., 2011, 83, 749 CrossRef CAS.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946 CrossRef CAS PubMed.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760 CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085 CrossRef CAS PubMed.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90 CrossRef CAS.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308 CrossRef CAS PubMed.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC.
- M. P. O'Neil, M. P. Niemczyk, W. A. Svec, D. Gosztola, G. L. Gaines and M. R. Wasielewski, Science, 1992, 257, 63 Search PubMed.
- Y. Pellegrin and F. Odobel, Coord. Chem. Rev., 2011, 255, 2578 CrossRef CAS.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840 CrossRef PubMed.
- A. G. Bonn and O. S. Wenger, Chimia, 2015, 69, 17 CrossRef CAS PubMed.
- M. Skaisgirski, X. Guo and O. S. Wenger, Inorg. Chem., 2017, 56, 2432 CrossRef CAS PubMed.
- M. Orazietti, M. Kuss-Petermann, P. Hamm and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 9407 CrossRef CAS PubMed.
- K. Kitamoto, M. Ogawa, G. Ajayakumar, S. Masaoka, H.-B. Kraatz and K. Sakai, Inorg. Chem. Front., 2016, 3, 671 RSC.
- R. M. Young, S. C. Jensen, K. Edme, Y. Wu, M. D. Krzyaniak, N. A. Vermeulen, E. J. Dale, J. F. Stoddart, E. A. Weiss, M. R. Wasielewski and D. T. Co, J. Am. Chem. Soc., 2016, 138, 6163 CrossRef CAS PubMed.
- S. M. Molnar, G. Nallas, J. S. Bridgewater and K. J. Brewer, J. Am. Chem. Soc., 1994, 116, 5206 CrossRef CAS.
- M. Elvington and K. J. Brewer, Inorg. Chem., 2006, 45, 5242 CrossRef CAS PubMed.
- K. J. Elliott, A. Harriman, L. Le Pleux, Y. Pellegrin, E. Blart, C. R. Mayer and F. Odobel, Phys. Chem. Chem. Phys., 2009, 11, 8767 RSC.
- B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504 CAS.
- T.-a. Koizumi and K. Tanaka, Angew. Chem., Int. Ed., 2005, 44, 5891 CrossRef CAS PubMed.
- T. Fukushima, T. Wada, H. Ohtsu and K. Tanaka, Dalton Trans., 2010, 39, 11526 RSC.
- K. Kobayashi, H. Ohtsu, K. Nozaki, S. Kitagawa and K. Tanaka, Inorg. Chem., 2016, 55, 2076 CrossRef CAS PubMed.
- R. Konduri, H. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185 CrossRef CAS PubMed.
- R. Konduri, N. R. de Tacconi, K. Rajeshwar and F. M. MacDonnell, J. Am. Chem. Soc., 2004, 126, 11621 CrossRef CAS PubMed.
- N. R. de Tacconi, R. O. Lezna, R. Konduri, F. Ongeri, K. Rajeshwar and F. M. MacDonnell, Chem.–Eur. J., 2005, 11, 4327 CrossRef CAS PubMed.
- S. Singh, N. R. de Tacconi, N. R. G. Diaz, R. O. Lezna, J. Munoz Zuniga, K. Abayan and F. M. MacDonnell, Inorg. Chem., 2011, 50, 9318 CrossRef CAS PubMed.
- N. Queyriaux, R. A. Wahyuono, J. Fize, C. Gablin, M. Wächtler, E. Martinez, D. Léonard, B. Dietzek, V. Artero and M. Chavarot-Kerlidou, J. Phys. Chem. C, 2017, 121, 5891 CAS.
- L. Zedler, S. Kupfer, I. R. de Moraes, M. Wächtler, R. Beckert, M. Schmitt, J. Popp, S. Rau and B. Dietzek, Chem.–Eur. J., 2014, 20, 3793 CrossRef CAS PubMed.
- S. Kupfer, Phys. Chem. Chem. Phys., 2016, 18, 13357 RSC.
- J. M. Aslan, D. J. Boston and F. M. MacDonnell, Chem.–Eur. J., 2015, 21, 17314 CrossRef CAS PubMed.
- J.-F. Lefebvre, D. Saadallah, P. Traber, S. Kupfer, S. Grafe, B. Dietzek, I. Baussanne, J. De Winter, P. Gerbaux, C. Moucheron, M. Chavarot-Kerlidou and M. Demeunynck, Dalton Trans., 2016, 45, 16298 RSC.
- L. Bouffier, M. Demeunynck, A. Milet and P. Dumy, J. Org. Chem., 2004, 69, 8144 CrossRef CAS PubMed.
- L. Bouffier, B. Baldeyrou, M.-P. Hildebrand, A. Lansiaux, M.-H. David-Cordonnier, D. Carrez, A. Croisy, O. Renaudet, P. Dumy and M. Demeunynck, Bioorg. Med. Chem., 2006, 14, 7520 CrossRef CAS PubMed.
- K. Ritter, C. Pehlken, D. Sorsche and S. Rau, Dalton Trans., 2015, 44, 8889 RSC.
- J. Schindler, Y. Zhang, P. Traber, J.-F. Lefebvre, S. Kupfer, M. Demeunynck, S. Gräfe, M. Chavarot-Kerlidou and B. Dietzek, J. Phys. Chem. C, 2018, 122, 83 CAS.
- J. Fees, W. Kaim, M. Moscherosch, W. Matheis, J. Klima, M. Krejcik and S. Zalis, Inorg. Chem., 1993, 32, 166 CrossRef CAS.
- A much more pronounced absorbance was measured above 500 nm for electrochemically generated [1]0 (Fig. 1). The higher complex concentration employed for this measurement, and additionally the presence of an electrolyte, most probably modify the pi-stacking equilibrium for [1]0 and could account for this observation.
- L. Zedler, J. Guthmuller, I. Rabelo de Moraes, S. Kupfer, S. Krieck, M. Schmitt, J. Popp, S. Rau and B. Dietzek, Chem. Commun., 2014, 50, 5227 RSC.
- C. G. Coates, L. Jacquet, J. J. McGarvey, S. E. J. Bell, A. H. R. Al-Obaidi and J. M. Kelly, J. Am. Chem. Soc., 1997, 119, 7130 CrossRef CAS.
- N. R. de Tacconi, R. Chitakunye, F. M. MacDonnell and R. O. Lezna, J. Phys. Chem. A, 2008, 112, 497 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Chem.–Eur. J. DOI:10.1002/chem.201704512.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04348a |
| This journal is © The Royal Society of Chemistry 2018 |