
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of plasma-assisted catalytic conversion of gaseous carbon dioxide and methane into value-added platform chemicals and fuels
Harinarayanan Puliyalil
 ,
Damjan Lašič Jurković
*,
Venkata D. B. C. Dasireddy
and
Blaž Likozar
,
Damjan Lašič Jurković
*,
Venkata D. B. C. Dasireddy
and
Blaž Likozar
Department of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova 19, 1001 Ljubljana, Slovenia. E-mail: damjan.lasic@ki.si
First published on 2nd August 2018
Abstract
CO2 and CH4 contribute to greenhouse gas emissions, while the production of industrial base chemicals from natural gas resources is emerging as well. Such conversion processes, however, are energy-intensive and introducing a renewable and sustainable electric activation seems optimal, at least for intermediate-scale modular operation. The review thus analyses such valorisation by plasma reactor technologies and heterogeneous catalysis application, largely into higher hydrocarbon molecules, that is ethane, ethylene, acetylene, propane, etc., and organic oxygenated compounds, i.e. methanol, formaldehyde, formic acid and dimethyl ether. Focus is given to reaction pathway mechanisms, related to the partial oxidation steps of CH4 with O2, H2O and CO2, CO2 reduction with H2, CH4 or other paraffin species, and to a lesser extent, to mixtures' dry reforming to syngas. Dielectric barrier discharge, corona, spark and gliding arc sources are considered, combined with (noble) metal materials. Carbon (C), silica (SiO2) and alumina (Al2O3) as well as various catalytic supports are examined as precious critical raw materials (e.g. platinum, palladium and rhodium) or transition metal (e.g. manganese, iron, cobalt, nickel and copper) substrates. These are applied for turnover, such as that pertinent to reformer, (reverse) water–gas shift (WGS or RWGS) and CH3OH synthesis. Time-on-stream catalyst deactivation or reactivation is also overviewed from the viewpoint of individual transient moieties and their adsorption or desorption characteristics, as well as reactivity.
 Harinarayanan Puliyalil | Dr Harinarayanan Puliyalil completed his master's degree in chemistry from Mahatma Gandhi University, Kerala, India in 2012. Later he joined Jozef Stefan Institute, Slovenia for his doctoral studies under the supervision of Prof. Uros Cvelbar. His research was mainly focused on plasma surface modification and plasma selective etching of polymeric materials. In 2016, he joined in the research group of Dr Blaz Likozar at the National Institute of Chemistry, Slovenia for his post doctoral studies, where we worked on atmospheric pressure plasma assisted CH4 and CO2 valorization process. Currently he is working as an R&D engineer at IMEC, Belgium. |
 Damjan Lašič Jurković | Damjan Lašič Jurković has finished his Bachelors and Masters degrees in Chemical Engineering at the University of Ljubljana, Faculty of Chemistry and Chemical Technology (FKKT). Afterwards, he was employed in 2015 at the Department of Catalysis and Chemical Reaction Engineering at National Institute of Chemistry in Ljubljana as a researcher, and is currently working there on his PhD degree in Chemical Engineering (FKKT). His work focuses mainly on plasma and plasma-catalytic activation of methane, as well as reaction kinetics and reactor modelling. |
 Venkata D. B. C. Dasireddy | Dr Venkata D. B. C. Dasireddy (male) (Research Associate, Department of Catalysis and Chemical Engineering) received his PhD in Applied Chemistry in 2013 at University of KwaZulu Natal, South Africa. He has been working at NIC for 3 years. He was also a postdoctoral researcher at University of KwaZulu Natal from 2013–2015. His expertise lies mainly in heterogeneous catalysis for green chemistry applications, chemical (process) engineering, reactor design and activating of C–H bonds through heterogeneous catalysis. He published over 53 scientific research papers (28 as first author) up to now and has a h-index of 11 (300 citations). |
 Blaž Likozar | Prof. Dr Blaž Likozar earned his PhD in Chemical Engineering in 2008 at the University of Ljubljana, Faculty of Chemistry and Chemical Technology. He has been a member of NIC since 2011, working senior research fellow, assistant professor and department head in the field of chemical engineering (Department of Catalysis and Chemical Reaction Engineering); leading the programme “Chemical Reaction Engineering”/several research projects. His expertise lies (among others) in heterogeneous catalyst materials, modelling, simulation and optimization of process fluid mechanics, transport phenomena and chemical kinetics. He worked in 2014–2015 at the University of Delaware, as a Fulbright Program researcher. |
1. Introduction
According to some recent reports, 80% of the global energy requirements are satisfied by fossil fuels like oil, natural gas and coal.1 Due to its vast availability and lower emissions compared to oil or coal, natural gas consumption has increased significantly over past few decades. Natural gas reserves including both conventional and unconventional resources are estimated to be about 6330 trillion m3.2 It is demonstrated that CO2 emissions from natural gas combustion are 25–45% lower compared to those from coal or oil.3 Besides, it emits much lower quantities of toxic components including various oxides (NOx, SOx and CO), aromatic hydrocarbons and genotoxic materials.1,4 Natural gas primarily contains CH4 (≤95%) and is conventionally obtained as a by-product of petroleum refining. The major unconventional source of natural gas is termed shale gas, where CH4 is trapped in the micro pores of sedimentary rocks (shales). The gas can be recovered by hydraulic fracturing combined with horizontal drilling of the shale. Challenges, potential opportunities and related environmental issues of hydraulic recovery of CH4 have been addressed elsewhere.5,6 Another potentially abundant methane source are natural gas hydrates.7CH4 is directly used as a feedstock chemical as well as a fuel for energy production in residential, electric power, industrial and transport sectors.8 Liquefaction of CH4 is particularly important since most of the natural gas deposits are located at remote areas and long distance transportation is imperative to make it available to the market. However, compression of CH4 into liquid form requires considerable amounts of energy, which are not economical for commercial purposes.9 Due to a very low boiling point (−161.6 °C at a pressure of 1 atm) and high flammability, transportation of CH4 from stranded sources is challenging and its uses are limited.10 For this reason, research on controlled oxidation of CH4 to liquid fuels like CH3OH or conversion to easily liquefiable hydrocarbons has been given a lot of attention in the past decades.11,12 A new advance in the technology for the cost effective conversion of CH4 to liquids would allow easy storage and transportation, and exploitation of CH4 on a much larger scale. For instance, the urge for CH3OH production is increasing since CH3OH can be used in fuel cells for energy production.13–15 The major challenges that confront CH4 activation are the high bond dissociation energy (104 kcal mol−1) and covalent nature of C–H bonds. CH4 cannot form efficient coordinate bonds with transition metal catalysts or other electron deficient centres due to a low basicity of the molecule. As reported by Olah et al., only strong super acids can generate methyl carbocation by hydride ion abstraction.16,17
Leakage of CH4 into the atmosphere due to its high volatility is a serious concern. It is estimated that around 8% of CH4 escapes to the atmosphere during shale gas recovery, transportation, storage and distribution.18 This number can be reduced if we can convert CH4 into less volatile or liquid forms. A report in 2010 reveals that the relative CH4 contribution to the greenhouse effect is the second highest (16%), preceded only by CO2 (76%).19 According to some rough estimation, CH4 emission into the atmosphere is expected to increase by up to 23% by 2020 (equivalent to 8 million metric tons of CO2). Nevertheless, both CH4 and CO2 emissions have increased drastically and have been given increased attention in past decades due to the extensive use of fossil fuels and exploitation of natural gas on a very large scale.
To control the global warming and related climate changes, conversion of CH4 and CO2 into other convenient forms is essential. The research on the catalytic conversion of CO2 and CH4 for sustainable development and carbon cycle fixation is blooming.20–22 In general, photochemical, electrochemical, biological, catalytic or plasma assisted conversion methods are used to convert CO2 into useful compounds including urea, alcohols, carboxylic acids, lactones, heterocyclic compounds and polymeric materials.20,23–26 Similarly, CH4 conversion with thermal or catalytic pyrolysis, oxidative coupling, biological processes or plasma activation techniques is efficiently used for the large scale production of valuable products including higher hydrocarbons, hydrogen, synthesis gas, alkanes, CH3OH, carboxylic acids, alkenes or aromatics.27–30 It is worth mentioning that most desired products are liquid organic oxygenates and C-5+ hydrocarbons. However, even C-2 to C-4 hydrocarbons are easily compressible compared to CH4, which is promising for cheaper transportation. However, some of these compounds have strictly regulated transport and storage. For example, acetylene can spontaneously explode when stored at high pressures, typically above 2 bar, which is why it is mostly transported and stored by dissolving it in acetone. Despite of such practical challenges, the conversion of CH4 to various chemicals (for example acetylene) is considered as a challenge for the scientific community from the perspective of C–H bond activation.
The greatest potential for a major advance in CH4 catalytic conversion technology is in the discovery of a route for the formation of oxygenates i.e., CH3OH and formaldehyde.28,31–34 Presumably, H2 liberated as a by-product of the dehydrogenation of CH4 reacts with O2 on the catalyst to form a surface peroxide species, which is responsible for the activation of CH4 to oxygenated compounds.31 There are some reports for oxidation of CH4 using oxygen over molybdenum based catalysts, in which very low conversions of CH4 (<10%) and a high selectivity (50%) towards formaldehyde were observed.35,36 Zeolite based catalysts are also explored in CH4 to CH3OH reaction and the ability of Cu-exchanged zeolites to convert CH4 to CH3OH at 130–200 °C using molecular oxygen as the oxidant was discovered.32,35,37
Nevertheless, it was found that CH4 conversion and rates of CH3OH formation drastically decreased due to several deactivation mechanisms, mainly coking and sintering. To overcome this deactivation, hydrogen was used as a co-feed (at temperatures above 400 °C), but it had a detrimental effect on CH4 conversion and CH3OH selectivity. In recent studies, a Fe-based zeolite showed catalytic formation of CH3OH from CH4 in aqueous hydrogen peroxide.37,38 Cu–Fe/ZSM-5 catalyst was reported as an active catalyst for the oxidation of CH4 to CH3OH. However, due to the high temperatures required for the reaction, reasonable selectivity was obtained only at insignificant conversions of 0.25% due to further oxidation.39
Similar to CH4, CO2 is a carbon resource that can be transformed into useful chemicals such as CH3OH, dimethyl ether, etc. by thermal catalysis40,41 Although most of the research focuses on CO2 hydrogenation to CH3OH, CO2 conversions remain low (<20%) due to difficulties in activating CO2.42,43 Industrial catalysts that are effective for CO-rich feed are not as effective for CO2-rich feeds under similar operation conditions.44 It is well established, however, that copper-based catalysts are typically used for CH3OH synthesis (at 240–260 °C and 40–50 bar) from CO2.43,45 Also due to feedstock differences, Cu/ZnO-based catalysts have been widely investigated and modified with various metal oxides for the hydrogenation of CO2.46–48
Although some superior catalysts for CO2 hydrogenation have been reported, the majority of new catalysts still contains copper as the main component, together with various modifiers and supports.43,49,50 In addition, several detailed studies were devoted to the influence of Ga and Pd and it was found that the catalytic performance of the Cu/Zn-based catalyst was improved when even little amounts of Ga and Pd were added.51,52 Nevertheless, the high cost of these additives hindered their practical application. However, the CH3OH yield in most experiments is typically below 30%. In addition to this, the hydrogenation of CO2 to CH3OH process requires a high quantity of H2 and has low CH3OH selectivity. Thus, CH3OH production using this method is still far from an economical point of view.41
As addressed before, catalytic conversion of CH4 and CO2 is associated with numerous issues such as high temperature operation, coking, sintering and low conversion rate and product selectivity. A big limitation of any thermal activation reaction of CH4 to liquid products is that most such products start decomposing at temperatures required for meaningful CH4 conversion, which makes it practically impossible to achieve high conversions and selectivities simultaneously, preventing high single-pass product yields. Recent developments in the field of plasma assisted conversion techniques provide a partial or complete solution to many of these issues. For this reason, considerable attention has been given to the research in the field of plasma assisted valorisation of CH4 and CO2.53–57 Inside the plasma, electron impact excitation, dissociation and ionization can generate numerous species including excited molecules, neutral atoms, atomic or molecular ions and metastable species at room temperature.58,59 Thus generated reactive species undergo recombination reactions to yield neutral molecules with upgraded product values.
Product formation inside plasma can be controlled by changing the density of the species generated in the system. This can be done with the regulation of various parameters such as gas flow rate, discharge power and feed gas composition. In many cases, by introducing suitable catalysts inside the discharge zone, the yield and selectivity of the product formation are improved.60 One of the roles of the catalyst inside plasma is to provide a suitable surface for efficient adsorption of various reactive species, which can modify the reaction pathways. In addition, the catalyst in the discharge zone can significantly modify the properties of the generated plasma by inducing effects such as surface discharge, improving the ionization and dissociation processes and many more. These influences are highly dependent on the physical and chemical characteristics of the packing material. The effects of catalyst on properties of plasma and vice versa are discussed in detail in the following sections. In this review, current state of art in the field of plasma assisted catalytic and non-catalytic processes used for CH4 and CO2 activation and conversion into valuable chemicals (especially higher hydrocarbons and organic oxygenates) and underlying mechanisms are provided. Important surface processes and role of plasma in both physical and chemical modification of various catalyst materials are also discussed.
2. Plasma: the fourth state of matter
Plasma is a state of matter that is essentially an ionized gas. Approximately 99% of our universe consists of plasma. An unambiguous definition of plasma cannot be easily determined, and plasma is usually defined as such when gas has certain properties, such as electrical conductivity, light emission, high gas dissociation levels etc. In plasma state, the gas contains a wide spectrum of different species like ions, electrons, atoms, radicals and neutral molecules, as well as their excited forms. Plasma is not charged overall; however, it contains free charge carriers and is therefore electrically conductive.612.1 Plasma generation
Plasma can be generated from neutral gas by different processes; thermal excitation being the most intuitive one. When gas gets heated to a high enough temperature, which is usually on the order of thousands of degrees, gas molecules have a high enough energy for spontaneous dissociation, excitation and ionization to take place. Thus, plasma is essentially formed because it's thermodynamically more favourable for the gas to be dissociated or ionized at that temperature. However, this methodology for plasma generation is not applauded due to high energy consumption and other technical issues associated with the operation.The second, kinetically more interesting and widely accepted approach for plasma generation is generation by means of electricity. A typical example is capacitively coupled plasma, where a high potential difference is applied between two electrodes. Due to the effects of electric field which causes an electrical discharge, the gas in between the electrodes is transformed into plasma.
The way discharge plasma generation works is that with a high enough voltage, termed breakdown voltage, current will start to flow through the otherwise non-conductive gas. The voltage can be on the order of thousands of volts, and for gas at atmospheric pressure, it can be very roughly estimated to about 3 kV for a 1 mm distance of gas between electrodes.62 Therefore, the strength of electric field in the gas is of a very high magnitude. This causes the electrons to have a high acceleration rate going from one electrode towards the other, and they can develop very high energies. Generally, the electrons will be accelerated until they interact with another particle, like a gas molecule. Therefore, at lower pressures, it is expected that the electrons will develop higher energies, because the mean free collision path with molecules is longer.
Accelerated electrons interact with atoms and molecules in two different ways, the first being so-called elastic collisions. In this type of interaction, electrons will transfer some of their energy to the molecule in form of kinetic energy, e.g. heat. This is because the energy transfer during the collision is not sufficient to cause any chemical changes in the molecule and just raises its temperature. The other type of interactions is termed inelastic collisions. In this case, when an electron of appropriate energy collides with an atom or molecule, the electron energy transfer is sufficient to induce phenomena such as excitation, ionization or dissociation. This generally supplies significantly less heat to the system, as most of the electron energy is used for state changing in order to overcome some of the associated energy barriers.
Both low pressure and atmospheric pressure plasmas have proved their vital roles in various fields of research including physics, chemistry, biology, medicine and material science.63–68 However, in chemical conversion routes, operational pressure close to atmospheric pressure is vastly accepted. The reason for this is that using low pressure is both expensive and harder to manage, and is also not favourable for reactions from a kinetic approach. Indeed, high pressure is generally used in those industrial catalytic processes where the thermodynamics doesn't restrict it, to speed up the reaction. While it's possible to use plasma at higher pressures, it is usually not preferred because increasingly high voltages have to be used for the discharge. The voltage limitations may be overcome by reducing the discharge gap. However, the gap might be too narrow for any potential catalyst incorporation in the discharge zone. Therefore, it comes to no surprise that in CO2 and CH4 activation chemistry, most of the research is done using atmospheric pressure plasmas.
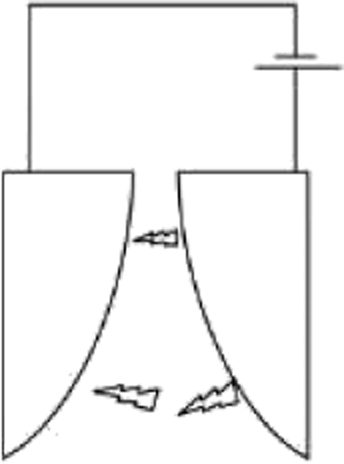
The setups to generate discharge plasma are many. The ones mainly used in CH4 and CO2 valorisation processes are briefly described below. The first obvious example is a spark plasma reactor, in which, two electrodes are placed at a distance, usually on the mm-cm order, and then AC voltage is applied to one of the electrodes, forming “sparks” between them. The electrode can be either narrow in shape, or one of them can be wider. Narrow electrodes produce a linear, spark-like plasma volume, whether the latter setup forms plasma in a conical shape.69
Another common setup for plasma generation is the gliding arc reactor, in which the electrodes are placed at an angle, so the distance between them increases in the flow direction of the gas. An arc is ignited at the shortest distance between electrodes, and then travels with the gas until it dies out because it cannot be sustained anymore at increased distance. For more information on gliding arc and their applications, the reader is referred to.62 Another of widely used plasma setup is rotating arc or vortex flow gliding arc, which is inherently very similar. It's different in a way that it has 3D geometry using a tube an inner cone as the electrode. Gas is introduced in a swirling motion and the reactor is capable of producing plasma on a larger volume.70
Dielectric barrier discharge (DBD) is a somewhat different plasma setup. It differs from the ones mentioned above in the fact that it has a layer of dielectric material between the gas and the electrodes. This dielectric material can pass a limited small amount of current in one direction, called the displacement current. However, it can conduct AC electricity. When the electricity is applied, the charge accumulation on the dielectric material can generate plasma as short lived streamers. The main advantage of using a dielectric in the way of the discharge is that it will stop the discharge on one spot after a streamer is formed, and thus forcing the discharge to happen on another spot on its surface, so plasma is generated as short lived streamers.71 This assures that the discharge forms all over the dielectric/electrode surface. On the other hand, in the reactor types discussed above, plasma is sustained in a single channel. Characteristics of various plasma systems mentioned above are presented in Table 1. Note that only the reactor setups used for CH4 and CO2 valorisation process are shown here, and arc plasma is added for comparison as it's a typical example of thermal plasma. Therefore, some widely used plasma generation systems such as microwave plasma or glow discharge have been excluded. Another important parameter is whether a pulsed plasma source is used, which is essentially a very fast turn-on/off switch.
| Reactor type | DBD | Spark | Corona | Gliding arc | Arc |
|---|---|---|---|---|---|
| Scheme |  |
 |
 |
 |
 |
| Electron energy [eV] | 1–30 | — | ∼5 | 1.4–2.1 | 1–10 |
| Electron density [cm−3] | 1012–1015 | 1014–1015 | 109–1013 | 1014–1015 | 1015–1019 |
| Current [A] | 1–50 | 20–30 | ∼10−5 | 0.1–50 | 30–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Gas temperature [K] | 300–500 | 400–1000 | ∼400 | 1000–3000 | 5 × 103–104 |
| Breakdown voltage [kV] | 5–25 | 5–15 | 10–50 | 0.5–4 | 10–100 |
The above mentioned setups, along with different parameters such as current, frequency, discharge gaps and voltage, can give a vast variety of plasma properties. One of the most important of these properties is the warmness of the plasma, which can be explained as a characteristic depending on the type of electron collision interactions in specific conditions. More details on warm and cold plasmas are illustrated in the following section.
2.2 Thermal and non-thermal discharge plasmas
The aforementioned electron collision types affect greatly the thermal properties of the plasma in question. Regarding those properties, we usually divide plasmas into thermal and non-thermal (cold) ones. Generally, in thermal plasmas, the gas temperature is relatively high and is comparable to electron temperature – the electrons and other species are in thermodynamic equilibrium. Non-thermal plasma temperature can be very low, even around room temperature, but it still contains high temperature electrons and excited and ionized species.The two main parameters that mostly affect the “warmness” of the plasma are electron density and pressure. Plasmas with high electron density, such as arc plasma, will be very warm due to the increased amount of elastic collisions, which heat up the gas. High electron density mainly increases the temperature because the energy input per volume is higher. In setups such as DBD, the electron density is low, and the plasma is relatively cold. Table 1 presented above lists some common plasma setups with accompanying characteristics.
It should be noted that all plasma sources discussed in the paper are inherently non-thermal. However, they can still differ greatly in the gas temperature, and are often referred to as cold and warm plasmas, to signify this difference. Herein, we consider DBD and corona as cold plasmas, and other systems such as spark and gliding arc as warm plasmas.
3. Plasma as a reaction medium
High concentrations and potential energies of radicals, ions and excited species in plasma mean that many reactions between said species will occur. Therefore, plasma is a highly reactive medium and exhibits a high potential for many chemical processes. Non-thermal plasma seems especially promising because of its inherent low temperature, which might be favourable for certain processes due to thermodynamics. An example of such a reaction is formation of organic oxygenates, that might be unstable at high temperatures, especially where oxidants are present in the system, as in the case of CO2 and CH4 reactions to liquid products. Therefore, plasma seems like a potential candidate for the conversion of CH4 and CO2 to liquid chemicals, fuels, and other value-added chemicals. In the following sections, a more detailed insight into plasma chemistry of CH4 and CO2 is given.3.1 Activation of CH4 and CO2 in plasma
CH4 and CO2 are generally considered to be relatively stable molecules that are hard to activate at low temperatures. Therefore, in such processes where these two molecules have to convert to useful products, high temperatures or aggressive co-reagents are required, as stated in the introduction section. In plasma, CH4 and CO2 can be activated or dissociated by means of high energy electron impact, which can be achieved even at room temperature. In this section, the mechanisms of CH4 and CO2 activation in plasma are discussed, whereas the mechanistic routes for the recombination of various activated plasma reactive species to valuable products are focused in the following sections. Some of the very elementary reactions and associated electron energies are presented in Table 2. Since H2, O2 and H2O are also used as co-reagents in the plasma valorisation of CH4 and CO2 that some of the elementary reactions for the excitation of these molecules are also presented in the table. More details on plasma excitation mechanisms of the above mentioned molecules can be found elsewhere.72,73| Reagents | Products | Interaction type | Electron energy required [eV] |
|---|---|---|---|
| CH4 + e | CH3˙ + H + e | Dissociation | 8.8 (ref. 72), 9 (ref. 64) |
| CH4 + e | CH2˙+ H2 + e | Dissociation | 9.4 (ref. 72), 10 (ref. 64) |
| CH4 + e | CH˙ + H2 + H + e | Dissociation | 12.5 (ref. 72), 11 (ref. 64) |
| CH4 + e | C˙ + 2H2 + e | Dissociation | 14.0 (ref. 72), 12 (ref. 64) |
| CH4 + e | CH4+ + 2e | Ionization | 12.6 (ref. 72) |
| CO2 + e | CO + O + e | Dissociation | 5.5 (ref. 70 and 71) |
| H2O + e | OH + O + e | Dissociation | 5.1 (ref. 73), 7 (ref. 74) |
| H2 + e | H + H + e | Dissociation | 4.5 (ref. 70) |
| O2 + e | O + O + e | Dissociation | 5.1 (ref. 70) |
| O2 + e | O2+ + 2e | Ionization | 12.5 (ref. 70) |
It can be seen from the Table 2 that CH4 gets mostly activated by dissociation, where one or more hydrogen atoms get removed.74–77 Higher electron energies are required for the removal of more than one hydrogen, so the extent to which each of these processes will take place is heavily dependent on electron energy distribution in a given system.76,77 In typical non-equilibrium plasmas, electron energies on the order of 5–10 eV are expected, as previously presented in Table 1. It can be seen that as CH4 has a high dissociation energy (9 eV), most of the dissociation will produce CH3 radicals rather than CH2 and CH ones. It is also evident that typical oxidants used alongside CH4 have much lower dissociation energy (5.5 eV for CO2 and 5.1 eV for O2). Therefore, it is possible to deduce that a significant portion of CH4 can get activated by excited oxidant species rather than dissociation. It should be noted that other activation mechanisms can occur in plasma, such as vibrational excitation, which can cause chemical reactions as well. For example, these kinds of mechanisms are highly pronounced in microwave plasmas. However, most of the processes dealing with CO2 and CH4 conversions to liquid chemicals use other plasma sources where dissociation and ionization are indeed the main activation mechanisms.
Besides electron collision, neutral molecules can be activated in other manners. One is thermal activation, which we can expect in warm plasmas, but is in most cases negligible in cold plasmas. By thermal activation, for example, CH4 can gradually lose each of its hydrogen atoms. Note that relatively high temperatures are required for this reaction, usually above 300 °C with a catalyst78,79 and 600 °C or above without one.80 However, even at those temperatures the conversions are negligible, so often temperatures above 1000 °C are used.81 In plasma, the activation can also proceed through phenomena such as charge or radical transfer, energy transfer, excitation by another molecule etc. These kinds of reactions play a very important role in many reaction pathways, and can be studied separately from electron collision process in afterglow plasma.82
Some of these previously mentioned activated species need to react among each other in order to form products. Obviously, even more reaction pathways are possible in this case. Possible reactions include different types of interactions, recombination being the most obvious. In this process, two species like radicals react to form a stable product. In general, two highly energetic species cannot recombine in an exothermic manner, because the resulting product would be unstable at the elevated temperature. In such cases, it is necessary to remove excess energy into the environment, e.g. to a passer-by spectator molecule, usually a neutral molecule. These kinds of reactions can therefore only take place in so called three body collisions.
4. Plasma valorisation of CH4 and CO2 into valuable chemicals
Conversion of the aforementioned greenhouse gases in the presence of plasma is extensively studied. Valorisation of CH4 is achieved mainly by plasma non-oxidative coupling to higher hydrocarbons and oxidative coupling to organic oxygenates. In the case of CO2, valorisation process involves partial or complete reduction with reducing agents such as H2 or CH4. This section provides an outlook into plasma valorisation of CH4 and CO2 and the influential factors associated with it. The results from the previous reports on CH4 and CO2 valorisation in various plasma reactors are summarised in Table 3.| Feed gas ratio | Reactor setup | SEI [kJ mol−1] | Plasma temperature [K] | Conversion [%] | Products (selectivities [%]) | Reference |
|---|---|---|---|---|---|---|
| Non-oxidative methane coupling | ||||||
| 5% CH4, 95% N2 | DBD AC | 1094.4 | 500 | CH4 – 14.8% | C2H6 (10.6%) | 77 |
| C2H4 (0.7%) | ||||||
| C2H2 (0.8%) | ||||||
| C-3 (2%) | ||||||
| 5% CH4, 95% N2 | DBD pulsed | 224.64 | 500 | CH4 – 12.4% | C2H6 (6.5%) | 77 |
| C2H4 (0.3%) | ||||||
| C2H2 (0.3%) | ||||||
| C-3 (0.9%) | ||||||
| 5% CH4, 95% N2 | Spark AC | 172.8 | 1000 | CH4 – 49.4% | C2H4 (0.3%) | 77 |
| C2H2 (86%) | ||||||
| 5% CH4, 95% N2 | Spark pulsed | 322.56 | 1000 | CH4 – 83.0% | C2H6 (2.6%) | 77 |
| C2H4 (3.1%) | ||||||
| C2H2 (45.9%) | ||||||
| C-3 (0.7%) | ||||||
| 5% CH4, 95% N2 | Rotating arc | 61.92 | 1000 | CH4 – 25.8% | C2H6 (0.2%) | 77 |
| C2H4 (1.1%) | ||||||
| C2H2 (42%) | ||||||
| 5% CH4, 95% N2 | Gliding arc | 72 | 3000 | CH4 – 23.7% | C2H2 (27.2%) | 77 |
| 5% CH4, 95% N2 | Hollow cathode | 28.8 | 2000 | CH4 – 42.2% | C2H6 (1.4%) | 77 |
| C2H4 (1.4%) | ||||||
| C2H2 (27%) | ||||||
| C-3 (0.3%) | ||||||
| 21.2% CH4, 78.8% Ar | Gliding arc, 80 mm length | 2090 | — | CH4 – 43.4% | C-2 (87.2%) | 87 |
| 100% CH4 | Gliding arc, 20 kHz, 150 mm length | 273.6 | — | CH4 – 47% | C2H2 (22%) | 88 |
| 15% CH4, 85% Ar | Gliding arc, 20 kHz, 150 mm length | 165.6 | — | CH4 – 65% | C2H2 (7%) | 88 |
| 100% CH4 | DBD, 75 kHz, 1 mm gap, 40 mm length | 867 | — | CH4 – 18% | C2H6 (30%) | 90 |
| C2H4 (3%) | ||||||
| C2H2 (3%) | ||||||
| C-3 to C-5 (27%) | ||||||
| 100% CH4 | Spark, 5 mm gap, 50 Hz DC, 5 kV, pulsed | 1059 | 440 | CH4 – 65% | C2H4 (5%) | 90 |
| C2H2 (75%) | ||||||
| C-3 to C-5 (5%) | ||||||
| 100% CH4 | Microwave, 1 kHz pulses of 60 μs, 30 mbar | 963.5 | 1500–2500 | CH4 – 90% | C2H2 (80%) | 93 |
| 100% CH4 | Corona, 1–2 kHz | 3854.1 | — | CH4 – 72% | C2H2 (56%) | 94 |
| C4H2 (8%) | ||||||
| C2H4 (3%) | ||||||
| 100% CH4 | DBD | 4624.9 | — | CH4 – 38% | C4H10 (5%) | 94 |
| C2H2 (4%) | ||||||
| C2H6 (25%) | ||||||
| C3H8 (10%) | ||||||
| 50% He, 50% CH4 | DBD, 1.2 mm gap, 120 mm length, 3 kHz | 10350 |
∼373 | CH4 – 18.4% | C2H6 (80.7%) | 95 |
| C2H4 (6.3%) | ||||||
| C2H2 (1.3%) | ||||||
| C3H8 (5.3%) | ||||||
| C-4+ (6.5%) | ||||||
| 10% CH4, 90% Ar | DBD, 3 mm gap, 4 mL volume, 10 kHz, 3–6 kV | 60 | — | CH4 – 13% | — | 76 |
| 10% CH4, 90% Kr | DBD, 3 mm gap, 4 mL volume, 10 kHz, 3–6 kV | 68.57 | — | CH4 – 23% | C2H6 (32%) | 247 |
| C2H4 (4%) | ||||||
| C2H2 (4%) | ||||||
| 100% CH4 | DBD, 8.8 mL volume, 10 kV, 20 kHz | — | — | CH4 – 55.0% | C2H6 (20.89%) | 97 |
| C2H6 (2.01%) | ||||||
| C3H6 (12.4%) | ||||||
| C-4 (11.54%) | ||||||
| C2H2 (4.85%) | ||||||
| 100% CH4 | DBD, 20 kHz, 3 mm gap, 13.6 mL volume, 40 kV, 20–50 kHz | 1296 | — | CH4 – 25.2% | C3H8 (4%) | 248 |
| C2H2 + C2H4 (12%) | ||||||
| C4H10 (19%) | ||||||
| C2H6 (34%) | ||||||
| 100% CH4 | DBD, 0.4 mm gap, 200 mm length, 6.4–8.6 kV | 3342 | 448 (wall) | CH4 – 25.1% | C-2 and C-3 (80.27%) | 98 |
| 100% CH4 | DBD, electrode with disks 5 mm apart | 7372.8 | — | CH4 – 10.2% | C2H6 (45%) | 99 |
| C3H8 (20%) | ||||||
| C2H4 (3%) | ||||||
| C2H2 (3%) | ||||||
| C-4 (10%) | ||||||
| C-5+ (12%) | ||||||
|
||||||
| Methane partial oxidation with O2, N2O or H2O | ||||||
| 20% O2, 80% CH4 | DBD, 1 mm gap, 50 mL volume, 20 kV, 30 kHz, 2 bar | 530 | 353 (wall) | CH4 – 15% | CH3OH (22%) | 108 |
| 50% H2, 50% O2 | DBD, double dielectric barrier | 633.6 | — | O2 – 90.8% | H2O2 (32.2%) | 112 |
| CH4 – 66.4% | H2O (18.5%) | |||||
| 50% Ar, 42.5% CH4 7.5% O2 | DBD, 3.5 mm gap, 17.3 mL volume, 10 ns pulses, 440 Hz, 25 kV | 112 | — | CH4 – 30% | CH3OH (18%) | 113 |
| O2 – 96% | HCHO (2%) | |||||
| C-2 (20%) | ||||||
| 5% CH4, 5% N2O, 90% Ar | DBD, 2 mL volume, 1 mm gap | 1029 | 330 (wall) | CH4 – 32.2% | CH3OH (10%) | 114 |
| N2O – 53.8% | HCHO (25%) | |||||
| C2H6 (10%) | ||||||
| 75% CH4, 25% O2 | DBD, 4 mm, 688 cm2 electrode surface | 849.6 | 301 (cooling fluid) | CH4 – 24% | CH3OH (17%) | 115 |
| O2 – 74% | Methyl formate (5%) | |||||
| HCOOH (16%) | ||||||
| HCHO (13%) | ||||||
| C2H5OH (1%) | ||||||
| 80% N2, 10% CH4, 10% O2 | DBD, cooled, 1 mm ID, twisted metallic electrode, 75 kHz | 672 | 298 (cooling fluid) | CH4 – 45% | CH3OH (17%) | 117 |
| O2 – 83% | HCHO (3%) | |||||
| HCOOH (9%) | ||||||
| 50%CH4, 50% O2 | DBD, cooled, 1.5 mm ID, twisted metallic electrode, 10 kHz | — | 283 (cooling fluid) | CH4 – 12% | CH3OH (10%) | 118 |
| HCHO (15%) | ||||||
| HCOOH (14%) | ||||||
| 50% CH4, 50% air | DBD, 10 kV, 10 kHz, 0.5 mm gap, 600 mm winding spiral ground | 864 | — | CH4 – 30% | CH3OH (9%) | 107 |
| 16% CH4, 84% H2O | Capacitively coupled plasma, DC, 133–1333 Pa | 345.6 | — | CH4 – 5% | CH3OH (20%) | 121 |
| HCHO (6%) | ||||||
| C2H6 (19%) | ||||||
| 50% CH4, 50% H2O | DBD, 2–3 kV, 250–2000 Hz, 1.8 mm ID, 500 Hz pulses of 400 ns | 246.4 | — | CH4 – 10% | CH3OH (7.5%) | 122 |
| H2O – 5% | ||||||
|
||||||
| CO2 activation with H2, CH4 or H2O | ||||||
| 67.4% CH4, 32.6% CO2 | DBD, 1.8 mm gap | 3600 | 338 (cooling fluid) | CH4 – 35% | Alcohols (5%) | 133 |
| CO2 – 20% | Acids (5%) | |||||
| C2H6 (19%) | ||||||
| C3H8 (9.3%) | ||||||
| C-4+ (9%) | ||||||
| 67.4% CH4, 32.6% CO2 | DBD, 1.1 mm gap, electrode with spacing | 3600 | 338 (cooling fluid) | CH4 – 55% | Alcohols (3%) | 133 |
| CO2 – 37% | Acids (8%) | |||||
| C2H6 (14%) | ||||||
| C3H8 (7.5%) | ||||||
| C-4+ (8%) | ||||||
| 66.8% CH4, 33.2% CO2 | DBD, 1 mm gap, 200 mm length, 25 kHz | 2400 | 333 (thermocouple in plasma) | CH4 – 64.3% | CH3COOH (5.2%) | 128 |
| CO2 – 43.1% | Propanoic acid (1%) | |||||
| CH3OH (0.3%) | ||||||
| C2H5OH (1.8%) | ||||||
| 50% CO2, 50% H2 | Surface discharge, 11 kV, 7 kHz | 518.4 | — | CO2 – 15% | DME (5%) | 134 |
| 50% CO2, 50% H2O | Negative corona plasma, 15 kV, 10.2 kHz | 6652.8 | 378 (thermostat) | CO2 – 18% | CH3OH (21%) | 136 |
| H2O – 14% | C2H5OH (13%) | |||||
4.1 Non-oxidative CH4 conversion
The simplest and most obvious CH4 plasma valorisation technique is the so-called non-oxidative CH4 conversion. In the process, pure CH4 is fed into a plasma zone, where reaction takes place. Two main mechanism are prevalent here, activation by electron collision and thermal activation. In both mechanisms, hydrogen is stripped from CH4 resulting in activated species which can react with another one to form stable products. As described by the studies mentioned later in the review, main products of this process are C2H6 and C2H2 with accompanying hydrogen. Higher hydrocarbons are also formed, but are increasingly scarcer with increasing carbon number. Although C-5+ hydrocarbons have been detected in the product mixtures of this reaction, high yields of such products were not obtained. An unwanted by-product of the process is pure carbon or coke, which can adversely affect the energy efficiency. In cases where CH4 needs to be valorised because of its remoteness and expensive/inefficient transport, even production of C2H6 C2H4 and C2H2 can be considered a sort of valorisation, however, safety issues need to be addressed as mentioned beforehand, especially for the case of acetylene. Firstly, these gases are much more easily compressible, making their transportation cheaper. Secondly, they can be transformed to liquids or other valuable products in a subsequent process. In some cases, even pure carbon products can be considered valuable in some forms, e.g. graphene nano-flakes83 and sheets.84 For a much more in-depth review of this particular process, the reader is referred to an extensive recent review by Scapinello et al.85The product selectivity of the process depends greatly on the gas temperature of the plasma, many times referred to as the “plasma warmness”. This can be seen in many studies discussed below, and is directly demonstrated by Lee et al. in a rather extensive experimental study.77 The authors showed a clear distinction between cold plasmas such as DBD and warmer plasmas such as spark, gliding arc, hollow cathode and rotating arc in terms of product distribution. Most of the product consists of either C2H6 or C2H4 and C2H2. C2H6 can be formed by the recombination of CH3 radical species, while C2H2 and C2H4 can be formed respectively from CH and CH2 intermediates, and also by dehydrogenation of C2H6.77 However, much higher electron energy is required for the dissociation of CH4 to CH and CH2. There are indeed some electrons present in the system with such high energies (>10 eV), but they are at the higher end of electron distribution and their concentration being very low, most of dissociation produces CH3. The main source of CH and CH2 species in the presented example is therefore assumed to be thermal dehydrogenation. Nevertheless, thermal gas phase dehydrogenation of the formed hydrocarbons is also possible resulting in consequent conversion of C2H6 to C2H4 and C2H4 to C2H2. Therefore, C2H6 is the main product in cold plasma systems due to a lack of further dehydrogenation mechanism. In warmer plasmas, C2H2 and C2H4 are more common. The study also demonstrates that warmer plasmas can convert CH4 into value added hydrocarbons at lower energy consumption. The efficiency of CH4 or CO2 activation in plasma is usually expressed in terms of specific energy input (SEI), which is stated as the ratio between the plasma power and the total gas flow rate.86 Conversion rate and energy efficiency for CH4 conversion in various atmospheric pressure plasma reactor types is compared in Fig. 1.
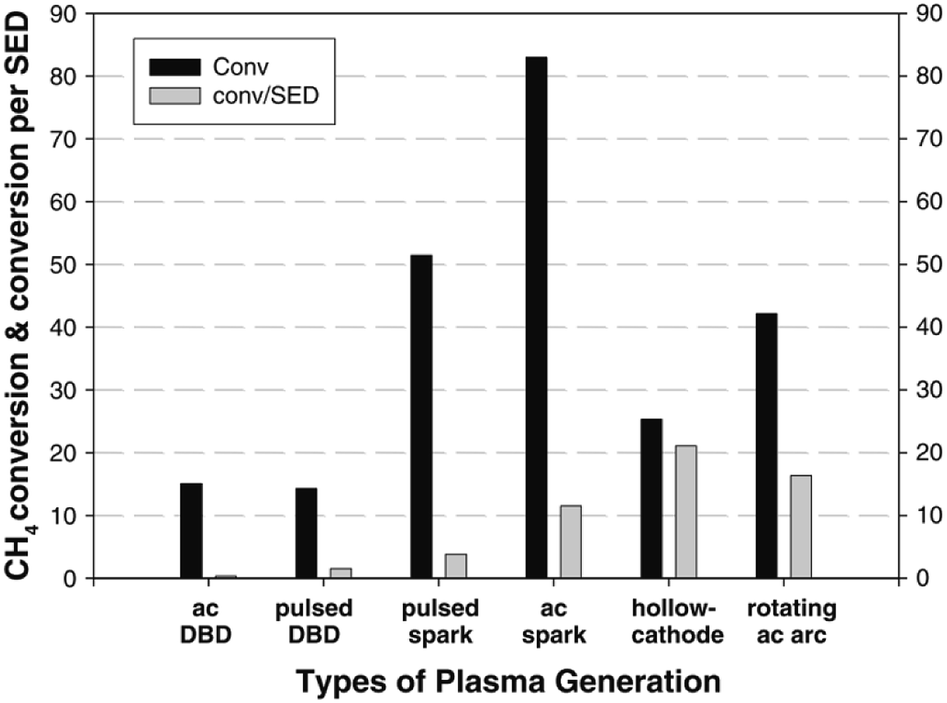 | ||
| Fig. 1 Comparison of CH4 conversion rate and associated energy efficiencies for various plasma reactor types. Reproduced from ref. 77 with permission from Springer Nature, copyright 2013. | ||
Non-oxidative CH4 coupling has been performed in multiple systems, gliding arc being one of them.87,88 In ref. 88, approximately 40% CH4 conversion was achieved with 20% selectivity towards C2H2 and 40% towards H2, which were the main products besides pure carbon and power was found to have little effect on the product distribution. When nitrogen was used alongside CH4, a maximum of 65% conversion of CH4 was achieved at 20% nitrogen with CH4. Regarding C2H2, maximum selectivity of 60% was achieved at a 50:50 N2:CH4 ratio. The reason for higher conversion in the presence of added N2 was that the vibrational excitation of N2 to its meta stable states [(N2(A) and N2(a′))] will help with the activation of CH4. Even though N2 has relatively higher dissociation energy, the above mentioned meta stable species can increase the rate of CH4 dissociation to various CHx radical species by transferring the potential energy to CH4 and thereby reduce the energy consumption. In another study, at a rather high specific energy input (SEI) of 2.09 MJ mol−1, 43% CH4 conversion was achieved with C-2 selectivity of 87% inside a gliding arc reactor.87 Influence of Ar addition on CH4 conversion was also studied, which determined that Ar addition into CH4 plasma reduced the energy input. It was revealed that a suitable percentage of Ar in the gas feed can increase the CH4 conversion by increasing the electron density in the reaction medium. However, both an increase,88 and decrease87 of C-2 hydrocarbon selectivity with Ar addition were reported. Nevertheless, no liquid products were detected in both cases.
Similar products (C2H2, H2 and C) were reported in studies related to non-oxidative CH4 coupling using a spark reactor.77,89,90 In a spark plasma channel with gas temperature in the range of 420–460 K was observed that lower reactor diameter and longer residence times increased the CH4 conversion but also the selectivity towards graphitic carbon deposition. Furthermore, the energy cost of C2H2 was lower at shorter residence time, which was about 12.1 kW h kg−1 under the optimized reaction conditions.90 The high selectivity towards C2H2 in plasma CH4 conversion is explained by a model considering more than 50 reactions and thermal balance and can be found elsewhere.91 It was observed that using gliding arc plasma could provide up to 35% improvement in conversion and energy efficiency when compared to pulsed plasma with parallel electrodes, most likely due to the distribution of the discharge over a larger gas volume.92
Pulsed microwave plasma (with gas temperature 1500–2500 K) was also used for C2H2 production, where the authors emphasized the importance of pulse length for conversion and selectivity.93 When pulse length was optimized at 60 microseconds at 1 kHz, CH4 conversion of 90% with 80% C2H2 selectivity was obtained. It was experimentally validated that by controlling the pulse duration, the densities of CH species and atomic H species in the system could be controlled which indeed influence the reaction pathways. Another study revealed that corona discharge can provide better C2H2 yield and higher CH4 conversion compared to that of DBD reactors at a given discharge power. This is ascribed to higher electron energy in the case of the corona reactor (10–20 eV) than in the case of the DBD reactor (1–10 eV).94
All of the aforementioned systems had somewhat thermal characteristics, and not much liquid products were detected. Experiments done in DBD were more promising in this regard, because a partial selectivity shift from C-2 to higher hydrocarbons can be observed when using colder plasmas. Yang et al. detected significant amount of propane and butane in DBD, and noticed a decrease in higher hydrocarbons formation as the SEI was increased.94 A screening of temperature and residence time on product distribution was presented in95 and it seemed that formation of C-5+ hydrocarbons peaked at the temperature of 200 °C. However, the major products were still C-2 hydrocarbons with a selectivity of over 70%. The authors also reported an enhanced conversion by 22% when CH4 was diluted with He, where CH4/He ratio equalled unity. This was attributed to an improved charge and energy transfer by added He into the system.
The effects of noble gas addition on CH4 conversion were further extended by Jo et al.76 Significant effect of diluent gases (He, Ne and Ar) on the conversion of CH4 was reported at a CH4 concentration of 10%. Noble gas addition did not affect the electron density, but it greatly affected the electron temperature, which explains higher CH4 conversion. Upon adding Ar, the conversion was the highest and almost double compared to that of He addition. In their next study,96 the effects of Kr and Xe were also compared and the study revealed that these gases had a significant effect on CH4 conversion. Kr addition resulted in the highest CH4 conversion, followed by Xe. On the other hand, He addition exhibited the highest selectivity towards C-3+ hydrocarbons, but the conversion was low. The difference in the selectivity and conversion depending on the noble gases in used in the system could be ascribed to their difference in the ionization cross section. Significant selectivity towards C-4+ hydrocarbons (∼10%) and propane (10–15%) were reported by Indarto et al.97 In this study, the C-2 selectivity was between 25 and 35%. It was noted that a high residence time favours the formation of shorter hydrocarbons (like C-2 or even pure carbon) and hydrogen. Another study dealt mainly with the effects of power, residence time of the gas in the reactor and discharge frequency.75 Both residence time as well as discharge power had lowered the energy efficiency as they were increased. Discharge frequency did not have much effect on either the energy efficiency or product selectivity. However, it affected the CH4 conversion and product yield, the highest being observed at 20 kHz. At this frequency, the maximum conversion of CH4 (25.2%) was observed at a CH4 flow rate of 50 mL min−1 and 45 W discharge power. In another study, residence time showed a big effect on C-2 and C-3 selectivity. C-3 selectivity increased with a longer residence time, while C-2 selectivity decreased, thus it is possible to control the product distribution merely by regulating residence time.98
Among many factors, discharge gap (the distance between the electrodes) is another crucial parameter that significantly influences the product formation in plasma assisted CH4 conversion processes. For instance, by changing the discharge gap, the rate of carbon deposition has been successfully controlled – it increased as the discharge gap increased.98 At the optimal discharge gap of 0.4 mm, combined selectivity towards C-2 and C-3 products of 80% with 25% CH4 conversion was reported at discharge power of 25 W. A study reports a modified DBD system with an electrode with disks attached at 5 mm intervals, effectively prolonging the discharge zone but decreasing the plasma density.99 The authors found out that with the same power but higher plasma density, the product selectivity was mainly in favour of C2H6 in a normal DBD, while the configuration with disks resulted in more C-3+ hydrocarbons.
A relatively simple kinetic model consisting of 8 species was developed,100 and was successfully fitted to the experimental data for non-oxidative CH4 coupling. A neural network model was also developed to determine the importance of different parameters on the product formation,101 by “teaching” the network using numerous data from multiple experimental studies. Using the model, it was found out that discharge power and gas flow were the most crucial parameters for the process, and that discharge frequency had next to no effect on the product distribution and conversion. While most other studies confirmed this predicted effects of flow and power,94,98 some studies reported a small but still significant effect of frequency.88 Note that this model was only fitted to experiments performed in non-oxidative CH4 coupling, while for example in oxidative coupling, it was shown that frequency can play and extremely important role.102 Models such as this one in the reference could be a very important part in further experiment designs. It has to be noted, however, that there were still many discrepancies between various models and experimental studies.
4.2 Partial oxidation of CH4
The partial oxidation of CH4 in plasma seems like a promising way of forming oxygenates. The reaction is initiated by electron collision with the neutral CH4/O2 molecules, forming both active oxygen derived and CH4 derived species. Major dissociation intermediates in this process are CH3 radical and O atom species. Articles discussing CH4 oxidation modelling103–105 go into greater depths of the mechanisms. It is of great importance to consider avoiding total combustion routes which form water and CO2, wasting both reagents. In gliding arc, for example,70 almost 100% selectivity towards syngas was achieved at O2/CH4 ratio of 0.6. While syngas can also be converted to liquids in subsequent processes, the focus of this review is more on the direct approaches. Therefore, thermal plasma characteristics should be avoided for this process and it should come to no surprise that most works published on the topic made use of DBD plasma.Another factor to consider is the decomposition of CH3OH in plasma, and the shifting selectivity towards total combustion products as the residence time increases, as described in detail in a review by Indarto.106 It is noted that the optimal residence time should be found so that the yield of CH3OH or other oxygenates is maximized, as there is a compromise between CH4 conversion and organic oxygenate selectivity in regards to the specific energy input (SEI).107 The SEI can be adjusted by controlling the total gas flow rate at the same power, as illustrated in Fig. 2.
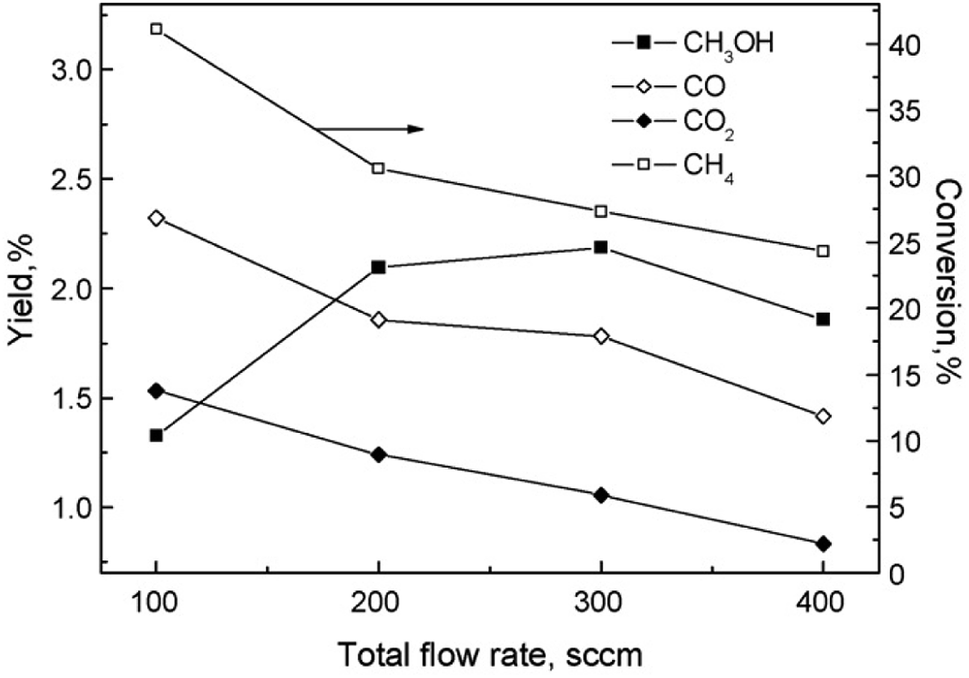 | ||
| Fig. 2 The effect of flow rate on product distribution (140 W, 7 kHz, 0.5 mm gap distance, ambient temperature, 1 bar, 1:1 CH4:O2). Reproduced from ref. 107 with permission from Elsevier, copyright 2009. | ||
For this process, the ratio of CH4 and oxygen is extremely important. It was proposed in108 that an oxygen concentration of 15% is optimal, because formation of CO and CO2 was favoured as the oxygen content increased further. At this ratio, maximum CH3OH yield of 3% was reported, with its selectivity being 30%. Using air instead of oxygen, a reduced yield of 2% was obtained. C2H6, C2H4, propane and ethanol were also detected in the products mixture, although at lower concentrations. The addition of inert gases, namely He and Ar, to the CH4/O2 mixture was also studied.109 A 2.5 times increase in CH3OH yield at 2.5 dilution ratio was observed. With further dilution, CH3OH selectivity decreased and C2H6 selectivity increased. As the Ar content in the gas feed increased, higher electron density was measured, which was responsible for the improved conversion rate and product selectivity. The effects of noble gases on the plasma properties and associated plasma chemistry were already discussed along with non-oxidative coupling reaction. Aghamir et al. found that at their operating conditions, C2H4 was the major product when the voltage is below 12 kV.110,111 As the voltage was increased to 18 kV, the selectivity to CH3OH could rise to up to 60%. At voltages higher than 14 kV, there was three times as much CH3OH in the product compared to C2H4. It should be noted that the increase in applied voltage does not induce any significant change in the internal electric field due to the formation of space charge in the region between the electrodes. Instead, it can increase the number of available electrons in the system and influence the gas phase reactions.
Hydrogen peroxide is also considered as a wanted product besides liquid organic oxygenates.112 A yield of 29.2% H2O2 was reported for CH4 partial oxidation in plasma. The yield was similar as in the case of H2/O2 plasma reaction, but the SEI was ∼10 times higher in the case of CH4. Also, up to 57.3% selectivity towards liquid oxygenates was reported, which mainly consisted of CH3OH, formaldehyde and formic acid. A dependence of the selectivity on SEI was also made in another study using a CH4/O2 mixture.113 It was found that there is a maximum to CH3OH and HCHO selectivities at 8 kJ mol−1, with 64% combined selectivity at this rather low SEI. In the same study, the reaction of CH4 with iodine in plasma for the synthesis of methyl iodide was reported, the selectivity being about 95%. Methyl iodide is a useful intermediate for further processing as it can be easily hydrolysed into methanol in alkaline media. As an alternative to O2, air, halogens and nitrous oxide were also used as efficient oxidizing agents.114 With an inlet of 90% Ar, 5% CH4 and 5% N2O, a total 10% yield of CH3OH and HCHO was measured with 40% combined selectivity. However, it is more convenient to use oxygen or air as the oxidizing agent in terms of commercial point of view.
In the research by Larkin et al., the previously addressed problem associated with the decomposition of liquid organic oxygenates within the plasma discharge zone was partially solved by using either in situ condensation by cooling the reactor115 or by using shorter residence time with condensation and recycle.116 In both cases, an increase in the yield of liquids, such as CH3OH, methyl formate, formic acid, formaldehyde and ethanol, was observed along with a total selectivity towards organic liquid products ranging from 24 to 52% depending on the reaction temperature. A similar approach can be found in the works of Nozaki, Okazaki and their colleagues.57,117,118 The authors used a cooled reactor (≤10 °C) to achieve in situ condensation of liquid products such as CH3OH, DME and formaldehyde. As these products condensed and were removed faster from the active plasma zone, there was a lower possibility of further decomposition. Therein, 70% and 30% combined selectivities of liquid organic oxygenates were observed when the yields were 5% and 20%, respectively, with the selectivity to syngas being about 40%. The authors remarked that a possibility of 30% one-pass yield with 80% selectivity might be possible if a syngas-to-DME reaction stage is added downstream. A kinetic modelling study of CH4 plasma oxidation was also performed to confirm the improved selectivity at lower temperatures.119 Simulations on CH4 partial oxidation in plasma, which were consistent with several experimental works, can be found elsewhere.104,120 This agreement seems promising for a better understanding of the process and future experiments in order to ensure a maximum selectivity and yield at minimum energy consumption.
4.3 Valorisation of CH4 by using water
Water being an abundant reagent, a process using it seems like a promising CH4 valorisation route. CH3OH synthesis from CH4 and water at lower pressures (133–133.3 × 102 Pa) in a capacitively coupled plasma reactor was performed.121 Formation of C2H6, C2H4, C2H2, and CH3OH was observed along with carbon monoxide as the major product. Among the carbon containing species, CH3OH selectivity was heavily dependent on the reagent mixture ratio. A maximum selectivity of 20% was observed at the CH4:H2O ratio of 1:5.
However, following studies mainly focused on the conversion at atmospheric pressure due to an ease of operation and scaling up. As an example, the process was performed in DBD122 where a CH3OH yield of about 1% was reported. By adding Kr (37.5%) to the reagent mixture, it was possible to enhance the conversions and CH3OH yield. Using a CH4:H2O:Kr ratio of 3:1:2.4, the conversion was increased by ∼30% and selectivities increased from ∼30 to almost double for CH3OH, C2H6 and butane. The authors attributed these effects due to the possibility that Kr and Ar have very high energies of their lowest metastable levels (10 and 11.5 eV), so they can cause the dissociation of CH4 and H2O by energy transfer to those molecules. Kr and Ar also have deep valleys in momentum-transfer cross section at the electron energies of 0.5 and 0.2 eV, respectively, which can vibrationally excite CH4 and H2O. No such effects were observed with the addition of He, which is missing a valley in its momentum-transfer cross-section profile.123 As an alternative explanation, it is well established that the addition of noble gases into the plasma can significantly increase the electron density in the system which in turn enhances the plasma ionization and dissociation rates.124,125
4.4 Plasma coupling of CH4 and CO2
While most research on simultaneous conversion of CH4 and CO2 was done on the process of dry reforming,126–129 some works focusing on liquid chemicals were also carried out. While dry reforming was mainly achieved in warmer plasmas, such as spark or gliding arc, the research aiming for liquids was mainly performed in DBD. In one of such studies,130 CH3OH and ethanol, along with various carboxylic acids such as formic, acetic, propanoic and butanoic acids were detected in the liquid products. Among the gaseous products formed, syngas was prominent (89%) along with lower amounts of C-2 (8.5%) and C-3 hydrocarbons (1.5%). The authors proposed that plasma dissociation of a CH4–CO2 mixture mainly produces CH3, H, CO and O species, which were converted into products. Additionally, possible mechanisms were proposed by means of DFT calculations. For example, it's possible for acetic acid to be produced by either subsequent additions of CO and OH to methyl radical, or an addition of COOH. It is speculated that COOH is mainly formed by recombination of CO and OH radicals.The process was also performed using stationary gas phase and high SEI (1400 kJ mol−1) in ref. 131. The authors observed the formation of long branched hydrocarbons, as at the longer reaction time at tested conditions, polymerization analogue took place. This could be explained as the addition of CHx species on to the long chain hydrocarbon molecules within the reaction media. During the experiments, approximately 20% of the products obtained were non-volatile liquids whereas 1% of the liquid fraction also consisted of oxygenates, such as alcohols, ketones, esters and acrylic acids. In another study,132 methane dry reforming was integrated with a coal pyrolysis process. The authors noted a significant increase in yield of tar (13.3%), which was ∼1.5 higher than the yield when no plasma was used.
The selectivities between oxygenates can be shifted by varying the discharge gap inside the DBD reactor, as observed in the case of non-oxidative coupling. When a gap of 1.1 mm was used, the conversion of CH4 and CO2 was higher, and the formation of acids and liquid hydrocarbons was favoured. When this gap was increased to 1.8 mm higher amounts of ethanol and CH3OH were formed.133 It is evident from these and other reported results that the discharge gap can influence the selectivities, however, the exact changes in the conditions are not well elucidated. For example, it is possible that the power density increases when the electrodes are at a shorter distance. In another work,128 a 5.2% selectivity towards acetic acid was observed at CH4:CO2 ratio of 2:1, with 64% and 43% of their respective conversions. Low amounts of ethanol (∼1.8%) and propanoic acid (∼1%) were also measured. In general, the overall selectivities towards liquids were low in the presented examples, and the major product in most cases was syngas. It is possible to obtain large amounts of CO in the gaseous product mixture as a result of CO2 dissociation, which would then be released as a major product.
4.5 Other CO2 conversion processes
Apart from CH4, hydrogen was used for CO2 reduction in several studies.134,135 A 85% CO2 conversion with 6% energy efficiency was reported at a H2:CO2 feeding ratio of 3:1.135 The study focused mainly on the reverse water-gas shift process, and thus the product was syngas. Although the production of syngas is not negligible in the gas to liquid agenda, as discussed below, a more direct conversion was investigated.134 A surface discharge reactor was used to convert a mixture of 50% CO2 in H2. Among the products, CO, DME and CH4 were formed in approximately equal concentrations. Reactions of CO2 with water inside various plasma reactors were also studied in detail.136,137
Oxalic acid and H2O2 were detected from a mixture of CO2 and H2O in microwave plasma reactor.138 In another study by the same group, CH3OH was also formed – both during the reaction of H2O and CO2 and also during water plasma cleaning of the deposited organic molecules in the reactor.137 The authors proposed that pressure is a very important parameter which affects CH3OH selectivity. It was observed that the CH3OH yield increased by a factor of about 3.5 when pressure was raised from 240 to 400 Pa. A negative corona discharge was used in water and CO2 to produce ethanol and CH3OH in approximately 3:1 ratio.136 At the pressure of 1 atm and the temperature of 105 °C, their combined yield reached 4.7%. As the pressure was increased to 4 atm, a yield of 11.9% was observed. The authors speculated that the above mentioned alcohols formed from anionic species generated by the electron attachment to water and CO2 (H2O− and CO2− respectively).
Overall, it is evident that non-oxidative CH4 conversion process yields mainly C-2 and C-3 hydrocarbons. It also has the disadvantage of coke production. In terms of useful liquid chemicals, it seems that this process might be most useful in a two stage systems where the resulting C2H2 or C2H6 could be converted downstream into liquid chemicals. Regarding direct conversion to liquids, using different oxidants in order to get organic oxygenates such as alcohols, aldehydes and carboxylic acids seems to be more promising. Using CO2 as the oxidant seems to produce mainly syngas and is not the most effective in this agenda. It should be noted that syngas can indeed be converted to liquids in a downstream process, but this presents additional investment and operational costs, and thus direct conversion methods are more welcome. Naturally, there are additional costs associated with direct methods as well, namely the need to perform recycling and other drawbacks when not being able to achieve high single-pass yields. Using water with both CO2 and CH4 seems to produce significant yields of oxygenates. However, more research needs to be done in this field. The most promising process seems to be the partial oxidation of CH4 using oxygen, where high yields of oxygenates were reported. It should be noted that many discrepancies between different experimental works have not yet been fully elucidated, so a better understanding of plasma mechanisms and pathways is also needed to optimize the experimental work. All the above processes can be performed in coupled plasma-catalytic systems as well, which may further increase the yields and energy efficiencies. Works dealing with plasma-catalytic conversion of either CO2 or CH4 are discussed further in the review.
5. Plasma-surface interactions
A material placed in plasma experiences a large number of complex interactions with various excited species generated therein. Such physical and chemical interactions between plasma and the catalyst embedded in the reactor can significantly affect the yield and selectivity of product formation. Thus, prior to the effects of a catalyst in plasma, it is worth mentioning the possible elementary plasma–surface interactions from the perspective of plasma catalysis.5.1 Adsorption
Adsorption is one of the most important surface processes in heterogeneous catalysis. The adsorption of plasma reactive species onto the surface of the interacting materials generally occurs either via physical adsorption (physisorption) or via chemical adsorption (chemisorption). Physisorption is facilitated by weak van der Waal's forces between the impinging particle and the interacting surface. Physisorption is a very weak and exothermic (ΔH = 1–25 kJ mol−1) interaction, which weakly binds the molecule on the surface.139 Even though the associated interactions are very weak, physisorption can sufficiently increase the lifetime of the incoming species in the proximity of the catalyst surface. This can significantly improve the performance level in plasma assisted conversions.Contrary to physisorption, the energy associated with chemisorption is much higher (ΔH = 40–400 kJ mol−1). When neutral plasma species reach the vicinity of the surface, electronic interactions are possible. Theoretical calculations show that the chemical interactions between valence states of the surface atoms with the incoming species are the primary reason for the chemical adsorption on the surface. This observation justifies the difference in the catalytic activity and selectivity of various transition metals in catalytic or hybrid plasma catalytic systems.140,141 Furthermore, chemisorption of vibrationally excited molecular species on the catalyst bed is one of the major reasons for the increased reaction kinetics in hybrid plasma catalyst systems.80 Such phenomena are experimentally validated through optical emission spectroscopic analysis.142
In addition to neutral atoms and excited molecules, plasma contains charged species including ions and electrons. Due to the lower mass, the mobility of the electrons in the system is much higher compared to that of positive ions. Thus a material floating in plasma acquires a negative potential. Report by Li et al. reveals that the negative charge accumulated on the electrode surface during the discharge remains for several minutes even after the plasma is turned off.143 This is an after effect of trapping of adsorbed electrons on the material surface. These trapped electrons are able to act as an electron reservoir and generate surface streamers. Such surface streamers can significantly improve the conversion rate in plasma catalysis, which is discussed in the following sections. On the other hand, due to lower energies, the influence of ions is usually disregarded in reactors operated at atmospheric pressure.
5.2 Recombination
Plasma gains its potential energy by means of inelastic collisions, leading to various processes including excitation, ionization or dissociation. This excess energy will be released back into the system in the form of radiation by the successful recombination of various energetic species. For instance, the energy released during the recombination of 2 atomic oxygen to form molecular oxygen is estimated to be around 5 eV.144 Recombination of various species in the gaseous phase is well presented in literature and is of greater importance in catalyst free activation of CH4 and CO2.113,139,145,146 However, the recombination on the surface of a solid material is largely influenced by its surface recombination coefficient (γ). Surface recombination coefficient for a solid material depends on its chemical composition, impinging plasma species and the surface temperature.147,148Three different mechanisms are commonly used to explain such reactions on the surface namely, Langmuir–Hinshelwood (LH) mechanism, Eley–Rideal (ER) mechanism and Mars–van Krevelen (MvK) mechanism. In the case of LH mechanism, the reaction occurs between two species which are in adsorbed state. Contrary to this, in ER mechanism a preadsorbed species reacts with an impinging species in the gaseous phase. LH mechanism prevails at medium temperature. Low surface diffusion coefficients and the high surface site coverage restrict LH mechanism at lower temperature.149 At elevated temperature, a shift from LH to ER mechanism can be observed due to higher desorption rate.150 The MvK mechanism is described by the incorporation of constituents from the catalyst lattice into the reaction by-products. However, due to the lack of consistency and accuracy of this mechanism, attention has been given predominantly to LH and ER mechanisms in heterogeneous catalysis.151,152
5.3 Ion implantation and sputtering
Ion implantation is the process in which high energy ions in the plasma penetrate into the bulk of a material. Such phenomena can alter the physical, chemical or morphological characteristics of the catalyst embedded in the discharge chamber. However, this process requires very high ion kinetic energy (a few keV).153,154 Sputtering is strikingly different from ion implantation processes, where the surface atoms or molecular species are knocked out by high kinetic energy of the incident ions. For sputtering to happen from a solid surface, the incident ion should have a kinetic energy above the binding energy of the surface atoms. However, both ion implantation and sputtering are very unlikely to happen in generally used atmospheric pressure plasmas for CH4 and CO2 conversions.1515.4 Deposition and etching
Plasma enhanced deposition techniques are well known for large scale production of various carbonaceous structures.155–157 However, carbon deposition during plasma assisted conversion of CH4 is a serious issue that decreases the energy efficiency and catalyst deactivation. The problems associated carbon deposition during both catalytic and plasma assisted catalytic conversion of CH4 is addressed elsewhere.158–160 It is considered that the dissociation of CH4 over the catalyst surface yields active monoatomic carbon.161 This carbon is generally removed into CO or CO2 by reacting with oxygen in the system. If the rate of carbon removal is less than the rate of its formation, carbon will polymerize on the catalyst surface. This process of carbon deposition, so-called coking, can adversely affect the activity and efficiency of the catalyst.One of the possible methods to reduce coke formation is to add small amounts of sulphur on the catalyst surface. However, the supply of H2S onto the catalyst surface can permanently deactivate the catalyst. Another approach is to alloy the catalyst surface with tetra- or penta-valent p metals such as Ge, Sn, Pb, As, Sb or Bi to avoid the metal-carbide formation on the surface, which is the precursor for the coke formation.162 Nevertheless, alloying can adversely affect the activity of the catalyst and reduce the conversion rate and selectivity. The most acceptable way to avoid coking is to use metals which are resistant towards metal–carbide bond formation.162–164
Contrary to deposition, etching is a process by which solid atoms or molecules are converted into small volatile molecules by reactive plasma species.165 Plasma etching can be used for the removal of carbon deposited on the catalyst surface, in order to recover the catalytic activity. This can be achieved in situ by leaking sufficient amount of O2 into the system. Nevertheless, etching of active metals or inorganic supports used in plasma conversion does not happen as these materials form only solid by-products on reacting with the plasma reactive species.
The major challenge of the plasma research field is to elucidate the specific roles of each type of interaction and distinguish them on the basis of numerical values. For example, the prediction of various constants such as adsorption and recombination coefficients, ionization and dissociation constants in the presence of a catalyst within the discharge zone, and how the physical and chemical properties of the catalysts influence these values. Since plasma is a complex mixture of various species and the catalyst used for a specific reaction can be very unique in terms of substrate, specific surface area, pore size, type of metal, size of metal particles, etc., this remains a great challenge faced by the plasma community.
6. Plasma pretreatment of the catalyst
Plasma technology has been established as a key tool for the surface modification of various catalytic materials. Plasma treatment has proven to be a fast and efficient technique to replace calcination process, which is one of the most important steps in catalyst synthesis. Indeed, plasma treated catalysts exhibit smaller particle size and lower extension of undesired surface contaminates.166 Alongside, plasma assisted doping can be an additional benefit. Thus plasma processing is largely used for the pretreatment of various catalysts to induce physical or chemical modifications to improve their activity. Through various physical and chemical interactions, plasma can modify the surface morphology and chemical structure of the interacting material.The physical modification is mainly associated with the changes in specific surface area or surface morphology, which in turn modifies the electronic properties of the material.167 The increase in specific surface area can originate either from improved dispersion of the active catalyst on the support or by creating wrinkled structures on the surface.168,169 However, plasma treatments have different effects on specific surface area for different catalysts.
One of the major applications of plasma physical modification of catalyst materials is to reduce coke formation and subsequent deactivation of the catalysts during the dry reforming reaction, which is speculated to occur due to the flattening of the catalyst after plasma exposure (Fig. 3).161 This is a cost-effective strategy to avoid the use of expensive noble metals for the prevention of coke formation. Other beneficial examples of plasma physical modification of the catalyst surface are provided in literature for various other processes as well.170–176
 | ||
| Fig. 3 Schematic representation of CO2/CH4 reforming over (a and c) NiAl–C (calcinated); and (b and d) NiAl-PC (plasma treated prior to calcination) samples. Reproduced from ref. 161 with permission from Elsevier, copyright 2008. | ||
The underlying mechanisms on plasma induced morphological changes on various catalytic materials are studied in detail. It is assumed that trapping of electrons from plasma on the metallic particles creates a thin plasma sheath around it.66 Due to this, these particles will experience strong electrostatic repulsive forces from the flow of electrons from the surrounding plasma. This coulombic force of repulsion can cause elongation or distortion of the metallic particles on the surface.177 Alongside, plasma electrons can persuade faster nucleation and slower crystal growth of the metallic particles which in turn reduces the particle size.
As a chemical modification approach, plasma treatment is used as an appropriate technique for various oxidation and reduction reactions. In the case of catalyst design and fabrication, H2 plasma reduction of P2O5 in the presence of metal oxide precursors has been found to be efficient for the synthesis of metal phosphides used for hydrodesulphurization catalysis.178
In the context of CH4 reforming, reduction of NiO to active metal is well known.179,180 In a recent report, Rafik et al. presented a low temperature H2 plasma pretreatment of a Ce–Zr promoted Ni catalyst with the intention of improving its activity towards CO2 methanation.181 H2 plasma pretreatment can successfully replace the conventional thermal pretreatment and allows the reduction reaction to take place at ambient temperatures with lower hydrogen consumption.181 In another example, Ar plasma pretreatment of Ir/Al2O3 catalyst was used for the reduction of Ir ions into metallic Ir, which improved the catalytic activity towards CO2 reforming over CH4.182 However, plasma pretreatments may not necessarily induce the same chemical effects to different metallic species. For example, after a Co–MnOx catalyst was exposed to DBD discharge, the relative ratio of M4+/Mn3+ and Co2+/Co3+ showed a notable increase.183 Thus induced chemical changes increased the electron transfer efficiency between Mn and Co species and thereby improved the NO catalytic oxidation activity.
In addition to the changes in oxidation states, plasma treatment can increase the concentration of chemisorbed O2, which can improve the activity of the catalyst. Further, heteroatom incorporation can be employed in order to improve the catalyst activity for applications such as oxidation, reduction, molecular abatement and reactions in fuel cells.184–186 Chemical reactions between the plasma reactive species and the catalyst material create surface defects and the incorporation of heteroatoms (doping), which can in turn effectively modify the electronic band structures and surface states of the material.167,187 Numerous other examples of chemical catalyst modifications using plasma can be found in literature.188–193
All the above mentioned processes are extremely important to research, as they take place during plasma catalytic processes for chemical synthesis. A good understanding of these mechanisms is extremely important to enable good experimental performance.
7. Synergistic effects in plasma assisted catalytic conversion
Possible interactions between plasma and a material placed inside the discharge zone have been discussed already. Indeed, when a catalyst is introduced into the plasma chamber, it can slightly or significantly change the properties of plasma. Nevertheless, the influence of a catalyst inside a hybrid plasma catalytic chamber originates from the interplay between numerous complex surface reactions. The synergistic effects inside the hybrid plasma catalytic system can be defined as the ratio of the degree of conversion in the presence of catalyst along with the plasma to the sum of conversions with the catalyst and plasma measured separately.194 This synergy can evidently influence the reaction kinetics, product yield and selectivity by providing an alternative reaction pathway. It should be noted that the presence of a catalyst does not necessarily induce a positive impact on the product yield or selectivity. Related examples are discussed later in this section.In general, the role of a catalyst in thermal catalysis is to provide new reaction pathways, which might successfully reduce the energy of activation for the reaction. However, the effects a catalyst has on plasma are somewhat different compared to those it has on a thermal catalytic reaction. In general, catalyst is active at lower temperatures in hybrid plasma systems. Plasma is often used to achieve catalytic reaction even at room temperature. Dissociated atoms in plasma are highly electrophilic and easily get adsorbed at the electron rich centres on the catalyst surface, react among each other and desorb away.195 Also the chemisorption of plasma excited species is found to be faster and more efficient than ground state molecules. Another plausible reaction could be between the chemisorbed species on the catalyst surface and the dissociated atoms, radicals or excited species in the gaseous phase through previously described ER mechanism. In another sense, the surface reactions happening inside hybrid plasma catalyst chamber are more complex than conventional thermal catalysis and are still in a need of thorough investigation. A comparison of thermal and plasma catalytic process are schematically presented in Fig. 4.195
 | ||
| Fig. 4 Schematic comparison of thermal and plasma catalytic surface processes. Reproduced from ref. 195 with permission from Springer Nature, copyright 2015. | ||
The dissociation mechanism on the surface of an active material is explained differently in the presence and absence of plasma. When a CO2 molecule approaches the surface of NiO/TiO2 catalyst, it gets adsorbed at the oxygen vacancies and undergoes dissociative electron attachment (DEA).196 DEA is defined as the low energy electron induced formation of a negative metastable ion (in the present example: CO2−), which undergoes subsequent dissociation. On the other hand, when plasma is introduced along with the catalyst, the dissociation rate tends to increase.197 In one of the recent reports, the influence of various plasma pretreated NiO/TiO2 catalysts inside a microwave discharge for CO2 conversion clarifies the plasma-catalyst synergy.198 By introducing Ar plasma pretreated NiO/TiO2 inside the plasma chamber, the energy and conversion efficiencies were found to increase from 9.6% to 17.2% and 23% to 42% respectively. However, it should be noted that the efficiency of all the tested catalysts was not high enough to provide a positive impact as illustrated in Fig. 5. A significantly improved conversion under hybrid plasma catalytic system is assumed due to a higher threshold energy value for DEA cross section in gaseous phase (5–10 eV) compared to the one at the catalyst surface (1.7 eV).198 The effect could not be generalized for all the tested catalysts, as clear from the results presented in Fig. 5. This clarifies the complexity and lack of understanding in the grass root level in plasma catalysis.
 | ||
| Fig. 5 CO2 conversion and energy efficiencies, measured during the plasma-catalysis CO2 dissociation, are shown for the NiO/TiO2 catalysts prepared by plasma treatment with different gases (O2, Ar, CO2). Reproduced from ref. 198 with permission from Elsevier, copyright 2016. | ||
Plasma-catalyst synergistic effects during CH4 activation were well studied in the past. Nozaki et al. explained the effects of plasma activation of CH4 in a DBD reactor with and without a catalyst packing.80 In a Ni/SiO2 catalyst only process, there was no CH4 conversion at temperatures below 400 °C and a spontaneous increase in the conversion rate was observed at temperatures around 600 °C. When the catalyst was coupled with plasma, strong synergistic effects yielded very high conversion rates (roughly >50% increase) in the temperature range between 400 and 600 °C.80
In direct plasma conversion, a very large amount of vibrationally excited CH4 existed in the system, which stayed non-reactive in the absence of any active catalyst. However, such vibrationally excited molecules can get adsorbed easily on the catalyst surface and get converted into products.199 In a very recent study, Kim et al. compared various aspects of CH4 conversion by considering: (1) gas phase dissociation of CH4, (2) dissociation in the presence of porous Al2O3 and Ni/Al2O3 catalysts, (3) thermal effects due to plasma and (4) interactions between Ni catalyst and excited plasma species.60 It is clear that the effect of temperature is predominantly seen in the presence of an active Ni catalyst. Temperature dependence on conversion in various tested conditions is compared in Fig. 6,200 which describes the plasma-catalyst synergy as a function of temperature. Additionally, higher surface area of the incorporated material further facilitates the vibrational excitation and subsequent degradation of CH4 by inducing higher bond polarization.201
 | ||
| Fig. 6 (a) Reaction environments for the CH4 reforming and (b) profiles of CH4 conversions (XCH4) and H2 yields (XH2) at various reaction environments obtained via bulk temperature controls. Reproduced from ref. 200 with permission from American Chemical Society, copyright 2016. | ||
Incorporation of the catalyst inside the plasma reactor does not necessarily increase the conversion rate or product selectivity in all the cases. For example, in a fully packed reactor, the formation of filamentary micro discharges will be significantly reduced due to the decrease in discharge volume.202 As a result, efficiency of molecular conversion can be reduced to a large extend. Another interesting example can be found in the report by Sentek et al.203 A catalyst free DBD reactor showed higher CH4 conversion compared to the one embedded with Pd/Al2O3 catalyst. However, it's worth mentioning that incorporation of the catalyst significantly increased the product selectivity towards C2H6. Similar examples of a reduction in molecular conversion and an increased product selectivity in catalyst packed plasma reactors can be found elsewhere.202 It is indeed clear that the effects of plasma on the catalyst are not invariably synergistic. The sum of individual effects of plasma and catalyst taken separately can be superior or inferior to the effect using plasma and the catalyst, which can be reflected in multiple outcomes including changes in conversion rate, energy efficiency or product selectivity.
8. Catalyst selection strategy for plasma-hybrid reactors
To date, there are hundreds of articles on plasma catalytic activation of CH4 and CO2. The most widely accepted strategy is to use a catalyst that is found efficient for conventional thermal catalysis. For example, Mo, Cu or Ni based catalysts can be used in thermal catalysis for the conversion of CH4 or CO2 into liquid fuels like CH3OH.204,205 Catalysts derived from the same group of elements are also efficient in hybrid plasma catalytic systems.206,207 Similar catalyst selection strategy has been applied for many other plasma assisted conversions including dry reforming, CO oxidation, CH4 conversion to syngas and molecular abatement.202,208–210 Nevertheless, no general rule has been established yet for the catalyst selection for plasma assisted conversion reactions. The activity of different catalysts inside the plasma chamber is determined by multiple factors ranging from chemical composition to physical properties. This section focuses on various effects that catalyst properties have on plasma, and how these effects can influence conversion rates, selectivities and energy efficiencies.The position of the catalyst, embedded in the plasma discharge chamber, can largely influence the extent of various synergistic effects and thus the conversion rate. Based on the packing strategy, catalyst embedded plasma reactors can be classified into three classes, namely single stage, double stage and multistage plasma reactors.211 In a single stage reactor, the catalyst is partially or completely immersed in the discharge zone. In such a system, plasma particles directly interact with the catalyst and the catalyst can even change the discharge behaviour of the plasma.
In a double stage reactor, the catalyst bed is located downstream of the discharge zone. In many cases, single stage reactors were found better than double stage reactors for multiple purposes including dry reforming, volatile organic compound abatement and air purification.211–213 Certainly, this claim cannot be generalized. In contrast to single stage or double stage reactors, multistage plasma reactors are more interesting for industrial scale processing, which allows a stepwise activation or deactivation of various species.
In the case of atmospheric pressure plasmas, chemically reactive species have a very short lifetime, which is on the order of a few ns to several μs. For example, the estimated lifetime of an electron is as low as 10 ns whereas atomic O(1D) and OH radical species have lifetimes on the order of a few μs.214,215 O3 species having a much larger lifetime (a few minutes) can be attributed to their lower reactivity compared to that of atomic or radical species. Nevertheless, lifetimes of any species inside the plasma system are largely influenced by the medium surrounding the active species under examination.215 For an effective utilization of the reactive species in hybrid plasma catalyst systems, plasma should be as close as possible to the catalyst bed to enable direct plasma-catalyst interaction which allows efficient diffusion of the species onto the surface.211 The criteria for direct interaction between the plasma and catalyst can be expressed as,
| Λ = l/(LD + Lef) ≤ 1 |
In plasma assisted conversion, the catalyst bed usually consists of an active material coated on a suitable support. Such supports can be mainly classified as semiconductors (metal oxides), ferroelectric materials or zeolites. The electrical properties of the catalyst are very important in determining the electrical properties of the plasma. For example, highly conductive materials are not the best candidates for hybrid plasma reactors. When a dielectric material is introduced into the discharge zone, charge accumulation on the surface of the catalyst creates a non-uniform distribution of the electric field.216 Such a distribution indeed enhances the electric field inside the plasma.
When the catalyst is embedded in pellets, the effects are mainly influenced by the curvature, contact angle and dielectric constant. The enhancement of the electric field is much higher at sharp edges due to the electric edge effect.217 Furthermore, plasma parameters such as electron temperature and densities of radicals, ions and electrons show significant increase with the increase in dielectric constant of the packed material.218 As presented in Fig. 7, the increase in dielectric constant of the packing material and the applied voltage in an AC discharge favours an exponential increase in the power of the partial discharge.219 In some of the early studies, the molecular conversion rate in plasma was found to increase merely by incorporating a dielectric material in the discharge zone, which emphasizes the aforementioned influences.220
 | ||
| Fig. 7 Power of the partial discharge in the packed bed with different specific dielectric constant εs. Reproduced from ref. 219 with permission from Elsevier, copyright 1990. | ||
The shape and morphology of the embedded material is significantly important as it determines the properties of plasma and associated molecular conversion rate.221 Especially the influence of porous structures on the catalyst surface has to be addressed in this context. The formation of micro discharges inside the pores of a dielectric material placed in plasma is well known. Zhang et al. reported that inner regions of the pore are characterized by enhanced electric field strength, lower electron density and higher electron temperature, electron impact ionization rate and ion density and these factors can remarkably influence the plasma catalytic process.222 Nevertheless, pore size and discharge voltage were the key parameters that influenced the formation of micro discharges inside the pore.
Theoretical investigations on the influence of the material dielectric constant and pore diameter on the plasma properties give better insight into the related issues.223 It is postulated that plasma generation inside large pores is enhanced in a broader range of dielectric constants whereas inside smaller pores the enhancement is mostly limited to lower dielectric constants. Thus the most commonly used catalyst supports such as Al2O3, SiO2 or zeolites (εr > 11) allow micro discharges inside smaller pores. On the other hand, ferroelectric materials such as BaTiO3 (εr ∼ 10000) cannot yield plasma enhancement even inside pores up to a size of 100 μm.223 (Fig. 8) The paper gives an outlook into the formation of discharge inside the pores of a catalyst based on the pore size and material dielectric constant. Even though discharge inside the catalyst pores are still considered as one of the influential factor that determines the conversion rates and product selectivity, its role in plasma catalysis is not well defined yet. One major limitation of this paper is that the model is less applicable for many commonly used catalysts which are having nano porous structures.
 | ||
| Fig. 8 Distributions of the electron impact ionization rate, averaged over time in one AC cycle, for different dielectric constants: (a) εr = 25, (b) εr = 300, (c) εr = 400, and (d) εr = 1000, for a helium discharge sustained at 20 kV with a 100 μm pore. Reproduced from ref. 223 with permission from American Chemical Society, copyright 2016. | ||
The effects of metallic particles on plasma properties are studied extensively by many researchers. As reported by Jo et al., the incorporation of Pt metal into Al2O3 increased the formation of CH3 radicals compared to CH2 or CH ones during CH4 discharge.224 This was attributed to the reduction in the electric field in the voids between the catalyst pellets along with the increased electric field in the proximity of Pt. This effect can be efficiently utilized in order to achieve better control over the type of reactive species in the plasma as well as the product selectivity.
Kim et al. studied the effects of metallic nanoparticles on surface discharge formation in the case of zeolites.225 The VOC oxidation ability inside an Ag/Cu-zeolite catalyst loaded plasma reactor tended to increase with metal loading. Firstly, metal loading can increase the plasma-catalyst interaction cross section. Additionally, metal particles on the surface enhance the electric field and allow the expansion of plasma over the catalyst surface, resulting in surface streamers. It is well known that physical and chemical characteristics of surface streamers on metal–zeolite catalysts are largely correlated with the type of metal and Si/Al ratio. Contrary to this, when the catalyst support is derived from BaTiO3, it yields micro discharges restrained to a lower volume.226 Even though surface streamers are assumed to be a necessity for a better molecular conversion in plasma catalysis, their physical and chemical characteristics have not been fully elucidated yet, although lots of strong efforts have been made recently. In particular, the coupled modelling and experimental study by Wang et al.227 inspected the formation of discharge in a packed bed reactor utilising different dielectric materials. The study uncovers three different types of surface discharges, namely positive restrikes, filamentary microdischarges and surface ionization waves. They noticed a clear distinction between the surface discharge type depending on the dielectric constant of the packing material. At low dielectric constants (εr = 5), there is a prevalence of surface discharge, which can be spread over multiple adjacent beads, while spatially limited filamentary discharges at bead contact points are preferred at high dielectric constants (εr = 1000). The authors emphasize the importance of such knowledge when designing plasma compatible catalysts and processes.
Thus in conclusion, the physical effects of a catalyst inside plasma reactors seem to be mostly clear. These effects include the change in discharge behaviour, electric field enhancement and an enhanced electron energy distribution.167 However, the chemical effects of a catalyst inside plasma are not well understood yet. The primary reason is that it is impossible to precisely differentiate the physical and chemical effects when the catalyst is placed inside the discharge zone. The second is the lack of sufficient experimental techniques for a clear-cut understanding of the surface processes at the solid–gas interphase. CH3OH selectivity in the presence of various metal species (Pt, Fe2O3, Cu, Zn, CeO2) on dielectric supports (ceramic, Al2O3) in plasma is presented. However, a proper explanation on the chemical effects of the catalyst is not properly explained.107,228
It was already mentioned that the covalent nature of the chemical bonds is the major problem for getting CH4 coordinated to an active transition metal. However, this is not the case for CO2. As CO2 is a Lewis acid, its ability to react or coordinate with various basic compounds has been utilized for CO2 capture, fixation and activation.229 A representative example can also be seen in the case of plasma assisted conversion of CH4 with CO2 in a DBD reactor. Among Ni catalysts supported on various compounds (γ-Al2O3, MgO, SiO2, and TiO2), CO2 conversion rate decreased in the order Ni/γ-Al2O3 > Ni/MgO > Ni/SiO2 > Ni/TiO2 whereas plasma only conversion yielded an even lower conversion rate. One of the expected reasons for a higher CO2 conversion rate in the case of Ni/γ-Al2O3 was due to a greater number of strong basic sites on the catalyst surface, which increased the residence time of the molecule in the discharge zone and increased the extend of interaction with the reactive species. Furthermore, the catalyst showed a higher selectivity towards syngas and C-3 to C-4 hydrocarbons as well as a lower carbon deposition.160
For better active metal selection strategy for CH4 and CO2 valorisation, thorough understanding of the intermediate transition states and reaction pathways is necessary. Recently, Zhao et al. reported a density functional theory calculation for the efficient coupling of CH4 with CO2 over Zn doped Ce catalyst in plasma free atmosphere.230 The article nicely presented the formation of Zn–CH3 bonds by dissociative chemisorption of CH4. Later, the insertion of CO2 yielded a three centred metal–acetate transition state, which can regenerate the catalyst by means of CH3COOH elimination. Even though the study was reported for a plasma free catalytic conversion, the extension of such models into hybrid plasma systems would enable the experimentalist to choose a better catalyst for hybrid plasma catalytic systems.
In the case of plasma catalytic systems, such modelling can pose a significant challenge since plasma is a complex mixture of numerous excited species. Furthermore, while taking into account the influence of various plasma parameters and the influence of the catalyst on the properties of plasma, such modelling becomes even more complicated. Moreover, the influence of the catalyst on the properties of plasma is still unclear. Before concluding this section, it's worth quoting a question put forward by Prof. J. C. Whitehead, in one of his recent articles:194 ‘Will it be possible to design a catalyst that can be activated by plasma but is inactive thermally?’ Due to limitations in the fundamental understanding of the basic mechanisms, to achieve such a milestone would be a great challenge for the plasma community.
9. Plasma assisted catalytic conversion of CH4 and CO2 into valuable chemicals
The field of plasma assisted catalytic conversion of CH4 and CO2 has been given great attention in recent times. The results from recent publications are summarised in Table 4. One of the accepted approaches for CO2 utilization is to reduce it with H2 inside a catalyst embedded plasma reactor, producing liquid fuels such as CH3OH.206 By introducing a commercial CuO/ZnO/Al2O3 catalyst inside the discharge zone, CH3OH yield increased up to 10 times compared to the one obtained in catalyst free discharge at a gas temperature of 100 °C. Furthermore, the selectivity increased up to 20%. One of the addressed problems in CO2 reduction was a very low CH3OH selectivity (<1% in the presented example) and a large degree of H2 consumption during the process. To overcome the above mentioned issues of H2 consumption and low liquid product selectivity, widely accepted method is the CO2 reforming by CH4, which allows the large scale utilization of both gases. Furthermore, readily available hydrocarbons like C2H6 can also be used as reagents for CO2 reduction.| Feeding gas ratio | Reactor configuration | Conversion (CO2/HCs) | T (°C) | Packing material | Value added products and selectivity | Reference | ||
|---|---|---|---|---|---|---|---|---|
| a Catalyst in the afterglow. | ||||||||
| CO2 + H2 (1:3) |
DBD; discharge gap 1 mm, | 14%/— | 100 | CuO/ZnO/Al2O3 | CH3OH (7–10%) | 249 | ||
| CO2 + C2H6 | DBD | NA/100% | RT | VOx/Al2O3 | HCHO (11.4%) | 250 | ||
| CO2 + CH4 (1:3) |
DBD, 500W | C-2 | C-3 | 239 | ||||
| NA | Fleece | 11.3 | 8.7 | |||||
| NA | NaA | 13 | 10.3 | |||||
| NA | NaY | 12.3 | 10 | |||||
| NA | HY | 15.2 | 11 | |||||
| CH4 + CO2 + Heat flow of 14 + 1+ 65 mL min−1 | DBD, V = 18 kV, frequency: 300 Hz | C-2 | C-3 | C-4 | 241 | |||
| 23.3/19.5 | RT | La2O3/γ-Al2O3 | 42.9 | 12.4 | 5.6 | |||
| 32.0/20.9 | 200 | La2O3/γ-Al2O3 | 41.2 | 15.0 | 8.7 | |||
| 56.1/21.4 | 400 | La2O3/γ-Al2O3 | 39.5 | 11.7 | 5.3 | |||
| CO2 + CH4 (1:2) |
Corona discharge with hollow Cu ground electrode; power 30 W, feed flow rate 25 mL min−1 | C-2 | 240 | |||||
| 16.7/43.4 | RT | γ-Al2O3 | 30.6 | |||||
| 22.1/24.5 | RT | 5% La2O/γ-Al2O3 | 70.6 | |||||
| 22/23.8 | RT | 0.1% Pd–5% La2O/γ-Al2O3 | 70.4 | |||||
| CH4 + air (1:1) |
P = 140W; discharge frequency, 7 kHz; DBD discharge gap = 2.5 mm |
CH3OH | 251 | |||||
| —/25–26% | No effect | Plasma only | ∼7.6% | |||||
| 150 | Ceramic pellet (CP)a | >8% | ||||||
| —/25–26% | 150 | Pt/CPa | ∼9% | |||||
| —/25–26% | 150 | Fe2O3/CPa | 10.66% | |||||
| —/25–26% | 150 | CeO2/CPa | >8.5% | |||||
| CH4 + air (1:1) |
DBD discharge gap = 2.5 mm, feed flow rate = 300 sccm | CH3OH | 228 | |||||
| —/24.5–25.5% | 150 | CPa | ∼8.5% | |||||
| —/24.5–25.5% | 150 | CuO/CPa | 9% | |||||
| —/24.5–25.5% | 150 | Fe2O3/CPa | 10.1% | |||||
| —/24.5–25.5% | 150 | Fe2O3–CuO/CPa | 11.3% | |||||
| CH4 + air | P = 61 W; discharge frequency,7 kHz; DBD discharge | CH3OH | 252 | |||||
| —/35% | 150 | CuO/γ-Al2O3 | 2.5% | |||||
| —/36% | 150 | Mo–CuO/γ-Al2O3 | 3.5% | |||||
In a very recent report, plasma assisted catalytic conversion of CO2 by C2H6 is achieved. A V5+/Al2O3 catalyst, which is known to be one of the most efficient catalysts for oxidative dehydrogenation of C2H6, was used for the reaction.231 The catalyst was embedded in the discharge zone along with glass balls and BaTiO3. Major reaction by-products obtained include H2, CO, CH4, C3H6 and HCHO with a 100% C2H6 conversion and high HCHO selectivity (11.4%). The improvement in selectivity was attributed to the synergistic effects of active vanadium catalyst, ferroelectric BaTiO3 and plasma activation. The process allows a successful utilization of CO2 for the production of HCHO, a necessary chemical in industry used for wood processing, textile manufacturing and the production of formaldehyde resins, fertilizers, chelating agents and polyhydric alcohols.232
The ability of Cu to adsorb CO2 as COO− species on the surface, which will then reduce to a crucial intermediate HCOO−, is an important advantage of Cu the plasma catalytic conversion CO2 into alcohols. This was validated by Zhao et al. in the selective synthesis of ethanol from CO2 and water vapour with the assistance of commercially available Cu/ZnO/Al2O3 catalyst packed in a negative corona reactor. It is assumed that the active Cu species on the surface are partially oxidised in the plasma, which provides better selectivity towards ethanol compared to other competing products such as methanol.233
Oxidative CH4 coupling of CH4 with CO2 in hybrid plasma catalytic reactors significantly increases the CH4 conversion along with the inhibition of carbon deposits. During such reactions, the major product obtained in many reports is syngas.234–236 In addition to that, a large number of gaseous hydrocarbons (C2H6, C2H4, etc.), liquid hydrocarbons (pentane), and organic oxygenates could be also produced. It was presented that by introducing CO2 in the feeding gas, the conversion of CH4 over catalysts inside a DBD can be increased. The mechanism behind this is well explained in literature.234 Dissociation of CO2 can yield atomic oxygen species (e.g. O(1D)), which can easily pick up a hydrogen atom from CH4 to generate CH3 radicals. These hydrocarbon radicals will further react with other hydrocarbon radicals, atomic oxygen or OH radicals to yield higher hydrocarbons or liquid oxygenates.
It is well known that one can change the properties of plasma by changing the electrode material used for its generation.237 Using this principle, optimal plasma-catalyst synergy has been reached for the reduction of CO2 with CH4 inside DBD with various metallic electrodes.238 When the electrode material was changed from steel to Ni or Cu, the selectivity towards carboxylic acids were almost doubled. In this study, the authors gave much attention to the basic understanding of plasma-catalyst interactions rather than achieving a better product yield. Such research should be encouraged to cover the gap between plasma assisted conversion and the underlying mechanisms associated with it.
Zhang et al. compared the effects of various zeolite packings inside DBD on the selectivity towards higher hydrocarbons and liquid fuels.239 Incorporation of zeolite HY in the discharge zone significantly reduced both CH4 and CO2 conversion rates whereas selectivity towards C-4 hydrocarbons reached above 50%. On the other hand, the selectivity towards C-4 hydrocarbons was significantly lower in zeolite NaY packed reactors. The selectivity towards C-4 hydrocarbons has been found to decrease depending on the packing material in the order, zeolite HY > zeolite NaA > zeolite NaY > fleece. This study further supported the previous statement that a very small change in the chemical properties of the catalyst can significantly influence the reaction pathway or resulting products in a hybrid plasma reactor. The explanation for such observations is still a puzzle for the plasma catalysis community.
One of the most convenient ways to achieve higher hydrocarbon yield is to increase the discharge power. However, the yield of liquid products such as CH3OH or lower hydrocarbons tends to decrease with an increase in discharge power. It is assumed that lower hydrocarbons get activated and are converted into higher ones at elevated powers. However, these products are being degraded by plasma (and the elevated temperature at higher power) as well, so after some point, the increased plasma power will negatively affect the amounts of higher hydrocarbons in the product stream, and products such as H2, coke, and CO will be prevalent. It should be noted that the properties of plasma vary significantly with the changes in discharge power and thus the reaction pathways in plasma catalysis are completely modified. Thus it is very difficult to make a general conclusion on such influences of discharge parameters.
In CH4 conversion with CO2, sufficient amount of CO2 is necessary to provide an oxidizing atmosphere, which can reduce the carbon deposition and following catalyst deactivation. Zhang et al. presented the effect of CO2 concentration on product selectivity during C-2 hydrocarbon formation inside a hybrid pulse corona La2O3/γ-Al2O3 catalyst reactor.240 As CO2 content in the feed was increased by a factor of 4, C2H2 selectivity decreased by about 35% along with a significant increase in the formation of C2H6 and C2H4 (5 to 26% and 7 to 16% respectively). By increasing the CO2 concentration in the feed gas, it is possible to increase the CH4 conversion. However, for higher concentrations of CO2, the selectivity towards higher hydrocarbons or alcohols tends to decrease with a simultaneous increase in the selectivity towards CO or CO2 in the outlet. Furthermore, the influence of Pd doping on La2O3/γ-Al2O3 in the plasma catalytic conversion of CH4 over CO2 was studied. It was revealed that traces of Pd on La2O3/γ-Al2O3 increased the C-2 selectivity up to 70% and along with a high C2H4 (about 65%) in the product mixture. Whereas in the case of corresponding La2O3/γ-Al2O3 without Pd yielded very high C2H2 (76%) with C2H4 yield below 12%. The results revealed the efficiency of Pd–La2O3/γ-Al2O3 catalyst for C2H2 hydrogenation under pulse corona discharge. On the other hand, the yield towards C-2 HCs was lower inside La2O3/γ-Al2O3 hybrid DBD reactor at a discharge frequency of 300 Hz. However, inside the hybrid DBD reactor, there was significantly higher selectivity of C-3 and C-4 HCs (12.4 and 5.6% respectively) at room temperatures.241 Such differences in the yield and selectivity might be attributed to the difference in the plasma-catalyst interactions arose due to the distinct properties of generated plasmas in different reactor configurations. Another approach for the valorisation of CH4 is its controlled oxidation with atmospheric air or O2 due to cost effectiveness. In a related study, the effects of various catalysts deposited on ceramic particles (CP) in a two stage hybrid plasma catalyst reactor revealed that CH3OH formation was occurring in two stages.107 In the first stage, CHx radicals formed as a result of CH4 dissociation and then reacted with OH radicals and a sufficient number of atomic H to form CH3OH. Syngas was also formed as a by-product. In the second stage, this synthesis gas was converted on the active catalyst surface to yield CH3OH. At a moderate temperature of 150 °C, the CH3OH selectivity for various catalysts decreased in the order Fe2O3/CP > Pt/CP > CeO2/CP > CP > plasma only. The incorporation of a Fe2O3 catalyst increased the CH3OH selectivity by up to 50% compared to that of plasma only process.
In thermal catalysis, Cu is widely used as a promoter to improve the activity and selectivity of Fe2O3 catalysts towards alcohols.242 The same catalyst selection strategy has been applied in a two stage plasma catalytic conversion process.228 The comparison of catalyst performance in plasma revealed that CH3OH selectivity was 10.5% lower when Fe2O3/CP was used instead of the CuO modified catalyst. The improvement in the activity of the catalyst was ascribed to a reduced electron density around Fe3+ as well as a reduction in oxygen vacancy concentration after CuO doping. Furthermore, it was revealed that the CuO promoter didn't have much influence on CH4 conversion, but merely improved CH3OH selectivity. Similar effects of Mo doping of a CuO/Al2O3 catalyst on the selectivity towards CH3OH formation were also reported.207 The mechanism of CH3OH production is speculated as follows, where the influence of the oxygen vacancies is considered.
| CH4 + e → CH3 + H + e | (1) |
| O2 + e → O + O + e | (2) |
| H + O + CH3 + 2M–O → M–OCH3 + M–OH | (3) |
| M–OCH3 + M–OH + O → CH3OH + 2M–O | (4) |
| V0 + O = C → V0⋯O2−⋯C2+ | (5) |
| V0⋯O2−⋯C2+ + 2H2 → V0 + CH3OH | (6) |
Both single stage and two stage plasma catalytic systems are well known and widely exploited for various applications including molecular abatement, CH4 or CO2 activation, air purification and many more. However, the studies have to be extended into liquid fuel synthesis from CH4 to delve more deeply into plasma-catalyst interactions and resulting molecular conversion and product selectivity.
A study presented on the partial oxidation of CH4 with air over a Fe2O3–CuO/γ-Al2O3 catalyst using DBD compared the effects of various catalyst packing strategies.243 Inside the single stage plasma reactor, CH4 conversion rate was much higher and the yielded products indicated higher extension of total CH4 oxidation in a reactor packed solely with Al2O3. On the other hand, with a Fe2O3–CuO/γ-Al2O3 catalyst, CH3OH yield was 68% higher than that of pure Al2O3 at 200 °C. Whilst in the case of a two stage plasma reactor, the maximum CH3OH yield achieved with the Fe2O3–CuO/γ-Al2O3 catalyst was only 21% higher at a temperature of 150 °C. By introducing the catalyst in the plasma zone, it was possible to reduce the unwanted ozone emission in the outlet.19,244 However, the catalyst embedded in the discharge zone was more prone to carbon deposition and consequent deactivation.243
Some researchers currently focus on the efficiency of various plasma catalytic systems to oxidize CH4 into liquid fuels in the presence of oxides of nitrogen like N2O. Even though such processes are promising for new advance in the field, the conversion of CH4 is always preferred using cheap oxidants including O2, air or CO2 since most of the CH4 depositions are in remote regions and the usage of reagents which are readily available at the location is necessary.
10. Economic viability and industrialization of plasma assisted conversion
The energy efficiency, economic viability and ease of scale up are some of the important aspects of any process evaluation. One of the approaches would be to calculate the energy needed for the formation of unit mole of a particular compound. Some of the lowest electricity costs of methanol, produced by plasma oxidation of methane, that we found through our literature survey, were 69 kW h kg−1,117 87.7 kW h kg−1,116 142.6 kW h kg−1 (ref. 114) and 201.9 kW h kg−1.108 Among these results, the cheapest price of electricity per liter of methanol produced would be roughly 6.5$ per l, assuming an approximate electricity price of 12 cents per kW per h. This price is obviously not taking into account the price of the feedstock, other operational costs and transport. For comparison, current market methanol production costs are on the approximate level of 0.1–0.3$ per l.245As the research is done on laboratory-scale setups, the direct comparison with industrially produced methanol might not be the fairest, but it can give a very rough idea on the feasibility of the process. For smaller scale production, the costs associated with a plasma reactor may be significantly lower than that of a two-step steam-reforming to methanol synthesis plant. Compared to the thermal catalysis, fast turn on/off time and room temperature operation are significant advantageous of plasma assisted conversion processes as well, which can reduce the energy consumption to larger extend. Nevertheless, one can question the feasibility of plasma catalysis as the process is operated at atmospheric pressure with limited gas inlet flows. Such a drawback can be easily overcome by utilizing the parallel reactor concept for DBD as introduced by Kogelschatz for large scale ozone synthesis.246 The industrial scale reactor thus constructed consists of bundles of small scale reactors arranged in parallel, as presented in Fig. 9.246 Such reactors allow operations at higher gas inlet flows, incorporation of the catalyst and higher energy input.
 | ||
| Fig. 9 Assembly of smaller reactor bundles for the industrial scale plasma scale processing (top) and the enlarged view of smaller reactor (bottom). Reproduced from ref. 246 with permission from Springer Nature, copyright 2003. | ||
The major advantages of plasma operation are attributed to high reactivity and the possibility of low temperature operation. This is beneficial for surpassing certain thermodynamic limitations for the thermal process in cases such as production of organic oxygenates in a single-step process. However, it does not mean that the energy cost of the operation is lower, as plasma can be similarly energy intensive as heating the gas to a high temperature. Therefore, to determine the industrial viability of the process, the energy cost has to be given great attention.
11. Conclusions, challenges and future outlook
In this review, we presented an overview of both plasma and plasma assisted catalytic conversion of CH4 and CO2 into valuable chemicals, which are either liquids or gases with much lower energy for liquefaction compared to CH4. The inspiration behind this is based on large scale exploitation and easy storage of widely available natural gas resources with an additional benefit of carbon cycle balance and consequent control of greenhouse effects due to CO2 and CH4 emission.The articles presented in this review evidently support the future potential of plasma catalysis for efficient valorisation of greenhouse gases into liquids and other useful fuels on a larger scale. Plasma assisted conversion has multiple benefits such as fast switch on and off times, low temperature operation and reduced coking. Furthermore, synergistic effects inside plasma hybrid catalyst reactors are promising for the improvement of product selectivity and energy efficiency and could help to upgrade conventional thermal catalysis.
However, both the plasma and the catalytic community are still facing numerous challenges regarding the aforementioned technology. One is the over oxidation of the liquid products formed on the catalyst bed due to their high retention times. Extremely high reactivity of the plasma reactive species and micro discharges in the catalyst pores can facilitate this unwanted degradation. Thus it is very important to fabricate suitable catalysts that would enable sufficient micro discharges to enhance CO2 and CH4 conversions and allow easy diffusion of the desired products from the catalyst bed.
As mentioned before, low temperature operation is one of the key advantages of plasma hybrid catalyst reactors. Even though catalysts operate efficiently at lower temperatures (∼100 °C) when plasma is present, the primary challenge is to build up suitable plasma-catalyst systems that operate efficiently even at room temperatures. This would provide exceptionally good energy efficiency, one of the most important criteria for industrial scale production.
This issue can be overwhelmed by achieving a more precise understanding of the underlying mechanisms and by developing an advanced catalyst selection strategy. As mentioned before, the most commonly accepted strategy for catalyst selection for plasma hybrid reactors is to pick the ones which are efficient for conventional catalytic conversion. A better catalyst selection strategy can be achieved only by combining advanced level simulation on plasma, catalysis and plasma–surface interactions and validate them with dedicated experiments.
Furthermore, scrutiny on the interaction of various excited states (electronically and vibrationally) with the catalyst surface and their prominent role in the product formation is another challenge. Indeed, a development in experimental techniques that allow precise monitoring of the reactions at the plasma-catalyst interface is necessary. Overall, a much deeper fundamental understanding of the process is required in order to bring plasma catalysis closer to the industry, and focused research with strong interdisciplinary bonds is needed to achieve it.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the European Commission, as the work was partially established within the ADREM project. This project has received funding through a Sustainable Process Industry through Resources an Energy Efficiency (SPIRE) call under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 680777). The authors also acknowledge the financial support from the Slovenian Research Agency (ARRS) (research core funding No. P2–0152), and the project “Direct Conversion of Methane to Higher Hydrocarbons Using Superacid Catalysts” (J2-7319) which was financially supported by the Slovenian Research Agency.Notes and references
- D. Saha, H. A. Grappe, A. Chakraborty and G. Orkoulas, Chem. Rev., 2016, 116, 11436–11499 CrossRef PubMed.
- G. Iaquaniello, A. Salladini, E. Palo and G. Centi, ChemSusChem, 2015, 8, 717–725 CrossRef PubMed.
- R. Kerr, Science, 2010, 328, 1624–1626 CrossRef PubMed.
- K. Hayhoe, H. S. Kheshgi, A. K. Jain and D. J. Wuebbles, Clim. Change, 2002, 54, 107–139 CrossRef.
- V. M. Torres, J. Thomas, D. W. Sullivan, A. Hendler, S. C. Herndon, C. E. Kolb, M. P. Fraser, a. D. Hill, B. K. Lamb, R. F. Sawyer, J. H. Seinfeld, D. T. Allen and M. Harrison, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18023 Search PubMed.
- D. M. Kargbo, R. G. Wilhelm and D. J. Campbell, Environ. Sci. Technol., 2010, 44, 5679–5684 CrossRef PubMed.
- C.-G. Xu and X.-S. Li, RSC Adv., 2015, 5, 54672–54699 RSC.
- C. Hammond, S. Conrad and I. Hermans, ChemSusChem, 2012, 5, 1668–1686 CrossRef PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef PubMed.
- J. R. Webb, T. Bolaño and T. B. Gunnoe, ChemSusChem, 2011, 4, 37–49 CrossRef PubMed.
- U. P. M. Ashik, W. M. A. Wan Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 44, 221–256 CrossRef.
- E. V. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Catal. Sci. Technol., 2017, 7, 366–381 RSC.
- F. Achmad, S. K. Kamarudin, W. R. W. Daud and E. H. Majlan, Appl. Energy, 2011, 88, 1681–1689 CrossRef.
- P. Mierczynski, K. Vasilev, A. Mierczynska, W. Maniukiewicz and T. P. Maniecki, Appl. Catal., A, 2014, 479, 26–34 CrossRef.
- P. Mierczynski, K. Vasilev, A. Mierczynska, W. Maniukiewicz, R. Ciesielski, J. Rogowski, I. M. Szynkowska, A. Y. Trifonov, S. V. Dubkov, D. G. Gromov and T. P. Maniecki, Catal. Sci. Technol., 2016, 6, 4168–4183 RSC.
- G. A. Olah, G. Klopman and R. H. Schlosberg, J. Am. Chem. Soc., 1969, 91, 3261–3268 CrossRef.
- G. a. Olah and R. H. Schlosberg, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef.
- R. W. Howarth, R. Santoro and A. Ingraffea, Clim. Change, 2011, 106, 679–690 CrossRef.
- W. C. Chung and M. B. Chang, Renewable Sustainable Energy Rev., 2016, 62, 13–31 CrossRef.
- B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52–62 CrossRef PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef PubMed.
- T. V. Choudhary, E. Aksoylu and D. Wayne Goodman, Catal. Rev., 2003, 45, 151–203 CrossRef.
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef PubMed.
- A. J. Hunt, E. H. K. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306–322 CrossRef PubMed.
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef PubMed.
- M. V. Landau, R. Vidruk and M. Herskowitz, ChemSusChem, 2014, 7, 785–794 CrossRef PubMed.
- A. Holmen, Catal. Today, 2009, 142, 2–8 CrossRef.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15–23 CrossRef.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef PubMed.
- V. D. B. C. Dasireddy and B. Likozar, Energy Technol., 2017, 5, 1344–1355 CrossRef.
- A. V. Annapragada and E. Gulari, J. Catal., 1990, 123, 130–146 CrossRef.
- R. Horn, R. Schlögl and R. Schlögl, Catal. Lett., 2015, 145, 23–39 CrossRef.
- R. Palkovits, C. von Malotki, M. Baumgarten, K. Mullen, C. Baltes, M. Antonietti, P. Kuhn, J. Weber, A. Thomas and F. Schuth, ChemSusChem, 2010, 3, 277–282 CrossRef PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef PubMed.
- M. J. Da Silva, Fuel Process. Technol., 2016, 145, 42–61 CrossRef.
- Z. R. Ismagilov, E. V. Matus, M. A. Kerzhentsev, L. T. Tsikoza, I. Z. Ismagilov, K. D. Dosumov and A. G. Mustafin, Pet. Chem., 2011, 51, 174–186 CrossRef.
- C. Kalamaras, D. Palomas, R. Bos, A. Horton, M. Crimmin and K. Hellgardt, Catal. Lett., 2016, 146, 483–492 CrossRef.
- B. Puértolas, A. K. Hill, T. García, B. Solsona and L. Torrente-Murciano, Catal. Today, 2015, 248, 115–127 CrossRef.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. Hasbi Ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor, H. Hagen, E. E. Stangland, J. H. Kang, J. M. Moulijn, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 689–699 CrossRef.
- S. Specchia, Int. J. Hydrogen Energy, 2014, 39, 17953–17968 CrossRef.
- S. Perathoner and G. Centi, ChemSusChem, 2014, 7, 1274–1282 CrossRef PubMed.
- X. Liang, X. Dong, G. Lin and H. Zhang, Appl. Catal., B, 2009, 88, 315–322 CrossRef.
- Y. Zhang, L. Zhong, H. Wang, P. Gao, X. Li, S. Xiao, G. Ding, W. Wei and Y. Sun, J. CO2 Util., 2016, 15, 72–82 CrossRef.
- P. Gao, F. Li, F. Xiao, N. Zhao, W. Wei, L. Zhong and Y. Sun, Catal. Today, 2012, 194, 9–15 CrossRef.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef.
- P. Khirsariya and R. K. Mewada, Procedia Eng., 2013, 51, 409–415 CrossRef.
- I. H. Tseng, W. C. Chang and J. C. S. Wu, Appl. Catal., B, 2002, 37, 37–48 CrossRef.
- H. Lei, R. Nie, G. Wu and Z. Hou, Fuel, 2015, 154, 161–166 CrossRef.
- O. S. Joo, K. D. Jung, I. Moon, a Y. Rozovskii, G. I. Lin, S. H. Han and S. J. Uhm, Ind. Eng. Chem. Res., 1999, 38, 1808–1812 CrossRef.
- N. Ahmed, Y. Shibata, T. Taniguchi and Y. Izumi, J. Catal., 2011, 279, 123–135 CrossRef.
- Y. Song, X. Liu, L. Xiao, W. Wu, J. Zhang and X. Song, Catal. Lett., 2015, 145, 1272–1280 CrossRef.
- S. E. Collins, D. L. Chiavassa, A. L. Bonivardi and M. A. Baltanás, Catal. Lett., 2005, 103, 83–88 CrossRef.
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- A. Bogaerts, T. Kozák, K. Van Laer and R. Snoeckx, Faraday Discuss., 2015, 183, 217–232 RSC.
- A. Bogaerts and E. C. Neyts, ACS Energy Lett., 2018, 3, 1013–1027 CrossRef.
- A. Indarto, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 1038–1043 Search PubMed.
- T. Nozaki and K. Okazaki, Green Process. Synth., 2012, 1, 517–523 Search PubMed.
- H. Puliyalil and U. Cvelbar, Nanomaterials, 2016, 6, 108 CrossRef PubMed.
- G. Filipic, O. Baranov, M. Mozetic and U. Cvelbar, J. Appl. Phys., 2015, 117, 1–10 CrossRef.
- J. Kim, M. S. Abbott, D. B. Go and J. C. Hicks, ACS Energy Lett., 2016, 94–99 CrossRef.
- C. Tendero, C. Tixier, P. Tristant, J. Desmaison and P. Leprince, Spectrochim. Acta, Part B, 2006, 61, 2–30 CrossRef.
- A. Fridman, S. Nester, L. a. Kennedy, A. Saveliev and O. Mutaf-yardimci, Prog. Energy Combust. Sci., 1999, 25, 211–231 CrossRef.
- X. Q. Deng, J. L. Liu, X. S. Li, B. Zhu, X. Zhu and A. M. Zhu, Catal. Today, 2017, 281, 630–635 CrossRef.
- J. Gruenwald, J. Reynvaan, T. Eisenberg and P. Geistlinger, Contrib. Plasma Phys., 2015, 55, 337–346 CrossRef.
- K. Bazaka, M. V. Jacob, R. J. Crawford and E. P. Ivanova, Acta Biomater., 2011, 7, 2015–2028 CrossRef PubMed.
- G. Filipič and U. Cvelbar, Nanotechnology, 2012, 23, 194001 CrossRef PubMed.
- K. Bazaka, M. V. Jacob, W. Chrzanowski and K. Ostrikov, RSC Adv., 2015, 5, 48739–48759 RSC.
- K. Vasilev, S. S. Griesser and H. J. Griesser, Plasma Processes Polym., 2011, 8, 1010–1023 CrossRef.
- V. Shapoval, E. Marotta, C. Ceretta, N. Konjević, M. Ivković, M. Schiorlin and C. Paradisi, Plasma Processes Polym., 2014, 11, 787–797 CrossRef.
- D. H. Lee, K. T. Kim, M. S. Cha and Y. H. Song, Plasma Processes Polym., 2007, 31(II), 3343–3351 Search PubMed.
- U. Kogelschatz, Plasma Chem. Plasma Process., 2003, 23, 1–46 CrossRef.
- C. J. Liu, G. H. Xu and T. Wang, Fuel Process. Technol., 1999, 58, 119–134 CrossRef.
- T. Silva, N. Britun, T. Godfroid and R. Snyders, Plasma Processes Polym., 2014, 23, 25009 Search PubMed.
- S. Jo, D. H. Lee and Y. H. Song, Chem. Eng. Sci., 2015, 130, 101–108 CrossRef.
- C. Xu and X. Tu, J. Energy Chem., 2013, 22, 420–425 CrossRef.
- S. Jo, D. Hoon Lee, W. Seok Kang and Y.-H. Song, Phys. Plasmas, 2013, 20, 83509 CrossRef.
- D. H. Lee, Y. H. Song, K. T. Kim and J. O. Lee, Plasma Chem. Plasma Process., 2013, 33, 647–661 CrossRef.
- D. Soulivong, S. Norsic, M. Taoufik, C. Copéret, J. Thivolle-Cazat, S. Chakka and J. M. Basset, J. Am. Chem. Soc., 2008, 130, 5044–5045 CrossRef PubMed.
- L. Yuliati, T. Hattori and H. Yoshida, Phys. Chem. Chem. Phys., 2005, 7, 195 RSC.
- T. Nozaki, N. Muto, S. Kado and K. Okazaki, Catal. Today, 2004, 89, 57–65 CrossRef.
- Y. Xu and L. Lin, Appl. Catal., A, 1999, 188, 53–67 CrossRef.
- J.-C. Legrand, a.-M. Diamy, R. Hrach and V. Hrachová, Vacuum, 1999, 52, 27–32 CrossRef.
- A. Mohanta, B. Lanfant, M. Asfaha and M. Leparoux, Appl. Phys. Lett., 2017, 110, 093109-1–093109-5 Search PubMed.
- A. Wu, X. Li, J. Yan, J. Yang, C. Du, F. Zhu and J. Qian, Appl. Energy, 2017, 195, 67–79 CrossRef.
- M. Scapinello, E. Delikonstantis and G. D. Stefanidis, Chem. Eng. Process., 2017, 117, 120–140 CrossRef.
- M. Heintze and B. Pietruszka, Catal. Today, 2004, 89, 21–25 CrossRef.
- S. Hu, B. Wang, Y. Lv and W. Yan, Plasma Sci. Technol., 2013, 15, 555–561 CrossRef.
- A. Indarto, J. W. Choi, H. Lee and H. K. Song, Energy, 2006, 31, 2650–2659 CrossRef.
- S. Kado, Y. Sekine, K. Urasaki, K. Okazaki and T. Nozaki, Stud. Surf. Sci. Catal., 2004, 147, 577–582 CrossRef.
- S. Kado, Y. Sekine, T. Nozaki and K. Okazaki, Catal. Today, 2004, 89, 47–55 CrossRef.
- a. V. Kirikov, V. V. Ryzhov and a. I. Suslov, Tech. Phys. Lett., 1999, 25, 794–795 CrossRef.
- A. Majidi Bidgoli, A. Ghorbanzadeh, R. Lotfalipour, E. Roustaei and M. Zakavi, Energy, 2017, 125, 705–715 CrossRef.
- M. Heintze, M. Magureanu and M. Kettlitz, J. Appl. Phys., 2002, 92, 7022–7031 CrossRef.
- Y. Yang, Plasma Chem. Plasma Process., 2003, 23, 283–296 CrossRef.
- H. Tarverdi, Iran. J. Chem. Chem. Eng., 2005, 24, 63–71 Search PubMed.
- S. Jo, D. Hoon Lee and Y.-H. Song, Chem. Eng. Sci., 2015, 130, 101–108 CrossRef.
- A. Indarto, J.-W. Choi, H. Lee and H. K. Song, J. Nat. Gas Chem., 2006, 15, 87–92 CrossRef.
- B. Wang, W. Yan, W. Ge and X. Duan, J. Energy Chem., 2013, 22, 876–882 CrossRef.
- K. Konno, K. Onoe, Y. Takiguchi and T. Yamaguchi, Chem. Eng. Res. Des., 2015, 95, 144–149 CrossRef.
- A. Indarto, N. Coowanitwong, J. W. Choi, H. Lee and H. K. Song, Fuel Process. Technol., 2008, 89, 214–219 CrossRef.
- S. Y. Liu, D. H. Mei, Z. Shen and X. Tu, J. Phys. Chem. C, 2014, 118, 10686–10693 CrossRef.
- C. Liu, A. Marafee, B. Hill, G. Xu, R. Mallinson and L. Lobban, Ind. Eng. Chem. Res., 1996, 35, 3295–3301 CrossRef.
- V. Goujard, T. Nozaki, S. Yuzawa, A. Ağiral and K. Okazaki, J. Phys. D: Appl. Phys., 2011, 44, 274011 CrossRef.
- C. De Bie, J. Van Dijk and A. Bogaerts, J. Phys. Chem. C, 2015, 119, 22331–22350 CrossRef.
- I. Antonius, C. Jae-Wook, L. Hwaung and S. H. Keun, Chin. Sci. Bull., 2008, 53, 2783–2792 Search PubMed.
- A. Indarto, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 1038–1043 Search PubMed.
- L. Chen, X. W. Zhang, L. Huang and L. C. Lei, Chem. Eng. Process., 2009, 48, 1333–1340 CrossRef.
- L. Zhou, B. Xue, U. Kogelschatz and B. Eliasson, Plasma Chem. Plasma Process., 1998, 18, 375–393 CrossRef.
- M. Okumoto, Z. Su, S. Katsura and A. Mizuno, IEEE Trans. Ind. Appl., 1999, 35, 1205–1210 CrossRef.
- F. M. Aghamir, N. S. Matin, A. H. Jalili, M. H. Esfarayeni, M. A. Khodagholi and R. Ahmadi, Plasma Processes Polym., 2004, 13, 707–711 Search PubMed.
- F. M. Aghamir, N. S. Matin, A.-H. Jalili and M.-A. Esfarayeni, J. Plasma Fusion Res. SERIES, 2004, 6, 696–698 Search PubMed.
- J. Zhou, Y. Xu, X. Zhou, J. Gong, Y. Yin, H. Zheng and H. Guo, ChemSusChem, 2011, 4, 1095–1098 CrossRef PubMed.
- M. Okumoto and A. Mizuno, Catal. Today, 2001, 71, 211–217 CrossRef.
- H. Matsumoto, S. Tanabe, K. Okitsu, Y. Hayashi and S. L. Suib, J. Phys. Chem., 2001, 105, 5304–5308 CrossRef.
- D. W. Larkin, L. L. Lobban and R. G. Mallinson, Catal. Today, 2001, 71, 199–210 CrossRef.
- D. W. Larkin, L. L. Lobban and R. G. Mallinson, Ind. Eng. Chem. Res., 2001, 40, 1594–1601 CrossRef.
- T. Nozaki, A. Hattori and K. Okazaki, Catal. Today, 2004, 98, 607–616 CrossRef.
- T. Nozaki, A. Aĝiral, S. Yuzawa, J. G. E. Han Gardeniers and K. Okazaki, Chem. Eng. J., 2011, 166, 288–293 CrossRef.
- V. Goujard, T. Nozaki, S. Yuzawa, A. Ağiral and K. Okazaki, J. Phys. D: Appl. Phys., 2011, 44, 274011 CrossRef.
- A. Indarto, J.-W. Choi, H. Lee and H. K. Song, Chin. Sci. Bull., 2008, 53, 2783–2792 Search PubMed.
- T. Tsuchiya and S. Iizuka, J. Environ. Eng. Technol., 2013, 2, 35–39 Search PubMed.
- K. Okazaki, T. Kishida, K. Ogawa and T. Nozaki, Energy Convers. Manage., 2002, 43, 1459–1468 CrossRef.
- L. S. Frost and A. V. Phelps, Phys. Rev., 1964, 136, A1538–A1545 CrossRef.
- K. Takechi and M. A. Lieberman, J. Appl. Phys., 2001, 90, 3205–3211 CrossRef.
- V. Léveillé and S. Coulombe, Plasma Processes Polym., 2006, 3, 587–596 CrossRef.
- B. Zhu, X. Li, J. Liu, X. Zhu and A. Zhu, Chem. Eng. J., 2015, 264, 445–452 CrossRef.
- A. Wu, J. Yan, H. Zhang, M. Zhang, C. Du and X. Li, Int. J. Hydrogen Energy, 2014, 39, 17656–17670 CrossRef.
- Y. P. Zhang, Y. Li, Y. Wang, C. J. Liu and B. Eliasson, Fuel Process. Technol., 2003, 83, 101–109 CrossRef.
- E. Cleiren, S. Heijkers, M. Ramakers and A. Bogaerts, ChemSusChem, 2017, 10, 4025–4036 CrossRef PubMed.
- L. M. Martini, G. Dilecce, G. Guella, A. Maranzana, G. Tonachini and P. Tosi, Chem. Phys. Lett., 2014, 593, 55–60 CrossRef.
- G. Scarduelli, G. Guella, D. Ascenzi and P. Tosi, Plasma Processes Polym., 2011, 8, 25–31 CrossRef.
- L. Jin, Y. Li, Y. Feng, H. Hu and A. Zhu, J. Anal. Appl. Pyrolysis, 2017, 126, 194–200 CrossRef.
- Y. Li, C. J. Liu, B. Eliasson and Y. Wang, Energy Fuels, 2002, 16, 864–870 CrossRef.
- N. Hayashi, T. Yamakawa and S. Baba, Vacuum, 2006, 80, 1299–1304 CrossRef.
- J. F. de la Fuente, S. H. Moreno, A. I. Stankiewicz and G. D. Stefanidis, Int. J. Hydrogen Energy, 2016, 41, 21067–21077 CrossRef.
- L. Guo, X. Ma, Y. Xia, X. Xiang and X. Wu, Fuel, 2015, 158, 843–847 CrossRef.
- I. Ihara, T. Ouro, T. Ochiai, M. Kiboku and Y. Iriyama, Bull. Chem. Soc. Jpn., 1996, 69, 241–244 CrossRef.
- T. Ihara, M. Kiboku and Y. Iriyama, Bull. Chem. Soc. Jpn., 1994, 67, 312–314 CrossRef.
- M. A. Lieberman and A. J. Lichtenberg, Principles of Plasma Discharges and Materials Processing: Second Edition, John Wiley & Sons, Inc., Hoboken, New Jersey, 2nd edn, 2005 Search PubMed.
- B. Hammer and J. K. Norskov, Adv. Catal., 2000, 45, 71–129 Search PubMed.
- P. A. Alaba, A. Abbas and W. M. W. Daud, J. Cleaner Prod., 2017, 140, 1298–1312 CrossRef.
- T. Nozaki, N. Muto, S. Kadio and K. Okazaki, Catal. Today, 2004, 89, 67–74 CrossRef.
- M. Li, C. Li, H. Zhan, J. Xu and X. Wang, Appl. Phys. Lett., 2008, 92, 031503-1–031503-3 Search PubMed.
- H. Puliyalil, G. Filipič and U. Cvelbar, Plasma Processes Polym., 2016, 13, 737–743 CrossRef.
- K. R. Ryan and I. C. Plumb, Plasma Chem. Plasma Process., 1984, 4, 271–283 CrossRef.
- D. Li, X. Li, M. Bai, X. Tao, S. Shang, X. Dai and Y. Yin, Int. J. Hydrogen Energy, 2009, 34, 308–313 CrossRef.
- Y. C. Kim and M. Boudart, Langmuir, 1991, 7, 2999–3005 CrossRef.
- S. Gomez, P. G. Steen and W. G. Graham, Appl. Phys. Lett., 2002, 81, 19–21 CrossRef.
- M. Cacciatore and M. Rutigliano, Plasma Processes Polym., 2009, 18, 23002 Search PubMed.
- M. Cacciatore, M. Rutigliano and G. D. Billing, J. Thermophys. Heat Transfer, 1999, 13, 195–203 CrossRef.
- E. C. Neyts, Plasma Chem. Plasma Process., 2016, 36, 185–212 CrossRef.
- H. L. Chen, H. M. Lee, S. H. Chen, Y. Chao and M. B. Chang, Appl. Catal., B, 2008, 85, 1–9 CrossRef.
- L. Baraton, Z. He, C. S. Lee, J.-L. Maurice, C. S. Cojocaru, A.-F. Gourgues-Lorenzon, Y. H. Lee and D. Pribat, Nanotechnology, 2011, 22, 85601 CrossRef PubMed.
- J. R. Conrad, J. L. Radtke, R. A. Dodd, F. J. Worzala and N. C. Tran, J. Appl. Phys., 1987, 62, 4591–4596 CrossRef.
- M. Meyyappan, L. Delzeit, A. Cassell and D. Hash, Plasma Processes Polym., 2003, 12, 205–216 Search PubMed.
- Q. Zhang, J. Q. Huang, M. Q. Zhao, W. Z. Qian and F. Wei, ChemSusChem, 2011, 4, 864–889 CrossRef PubMed.
- S. H. Lee, D. H. Lee, W. J. Lee and S. O. Kim, Adv. Funct. Mater., 2011, 21, 1338–1354 CrossRef.
- A. Obradović, B. Likozar and J. Levec, Int. J. Hydrogen Energy, 2013, 38, 1419–1429 CrossRef.
- U. P. M. Ashik and W. M. a. Wan Daud, RSC Adv., 2015, 5, 46735–46748 RSC.
- D. Mei, B. Ashford, Y.-L. He and X. Tu, Plasma Processes Polym., 2017, 1–13 Search PubMed.
- X. Zhu, P. Huo, Y. Zhang, D. Cheng and C. Liu, Appl. Catal., B, 2008, 81, 132–140 CrossRef.
- D. L. Trimm, Catal. Today, 1999, 49, 3–10 CrossRef.
- M. Młotek, J. Sentek, K. Krawczyk and K. Schmidt-Szałowski, Appl. Catal., A, 2009, 366, 232–241 CrossRef.
- K. Schmidt-Szałowski, K. Krawczyk and M. Młotek, Plasma Processes Polym., 2007, 4, 728–736 CrossRef.
- H. Puliyalil, G. Filipic, J. Kovac, M. Mozetic, S. Thomas and U. Cvelbar, RSC Adv., 2016, 6, 95120–95128 RSC.
- T. Witvrouwen, S. Paulussen and B. Sels, Plasma Processes Polym., 2012, 9, 750–760 CrossRef.
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef PubMed.
- Y. Zhang, J. Li, G. An and X. He, Sens. Actuators, B, 2010, 144, 43–48 CrossRef.
- T. Witvrouwen, S. Paulussen and B. Sels, Plasma Processes Polym., 2012, 9, 750–760 CrossRef.
- Y. Zhu, Z. Li, Y. Zhou, J. Lv and H. Wang, React. Kinet. Catal. Lett., 2005, 87, 33–41 CrossRef.
- X. Tang, K. Li, H. Yi, P. Ning, Y. Xiang, J. Wang and C. Wang, J. Phys. Chem. C, 2012, 116, 10017–10028 CrossRef.
- F. Guo, J.-Q. Xu and W. Chu, Catal. Today, 2015, 256, 124–129 CrossRef.
- J.-g. Wang, C.-j. Liu, Y.-p. Zhang, K.-l. Yu, X.-l. Zhu and F. He, Catal. Today, 2004, 89, 183–191 CrossRef.
- H. Peng, Y. Ma, W. Liu, X. Xu, X. Fang, J. Lian, X. Wang, C. Li, W. Zhou and P. Yuan, J. Energy Chem., 2015, 24, 416–424 CrossRef.
- X. Yu, F. Zhang, N. Wang, S. Hao and W. Chu, Catal. Lett., 2014, 144, 293–300 CrossRef.
- S. Kawi, Y. Kathiraser, J. Ni, U. Oemar, Z. Li and E. T. Saw, ChemSusChem, 2015, 8, 3556–3575 CrossRef PubMed.
- H. Taghvaei, M. Heravi and M. R. Rahimpour, Plasma Processes Polym., 2017, 1600204 CrossRef.
- A. Wang, M. Qin, J. Guan, L. Wang, H. Guo, X. Li, Y. Wang, R. Prins and Y. Hu, Angew. Chem., 2008, 120, 6141–6143 CrossRef.
- Y. Li, Z. Wei and Y. Wang, Front. Chem. Sci. Eng., 2014, 8, 133–140 CrossRef.
- X. Fang, J. Lian, K. Nie, X. Zhang, Y. Dai, X. Xu, X. Wang, W. Liu, C. Li and W. Zhou, J. Energy Chem., 2016, 25, 825–831 CrossRef.
- R. Benrabbah, C. Cavaniol, H. Liu, S. Ognier, S. Cavadias, M. E. Gálvez and P. Da Costa, Catal. Commun., 2017, 89, 73–76 CrossRef.
- Y. Zhao, Y. xiang Pan, Y. Xie and C. jun Liu, Catal. Commun., 2008, 1558–1562 CrossRef.
- X. Tang, F. Gao, Y. Xiang, H. Yi and S. Zhao, Catal. Commun., 2015, 64, 12–17 CrossRef.
- D. Ding, Z.-L. Song, Z.-Q. Cheng, W.-N. Liu, X.-K. Nie, X. Bian, Z. Chen and W. Tan, J. Mater. Chem. A, 2014, 2, 472–477 RSC.
- S. Z. Islam, A. Reed, D. Y. Kim and S. E. Rankin, Microporous Mesoporous Mater., 2016, 220, 120–128 CrossRef.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef PubMed.
- D. H. Kim, G. S. Han, W. M. Seong, J.-W. Lee, B. J. Kim, N.-G. Park, K. S. Hong, S. Lee and H. S. Jung, ChemSusChem, 2015, 8, 2392–2398 CrossRef PubMed.
- S. Y. Huang, S. Xu, J. W. Chai, Q. J. Cheng, J. D. Long and K. Ostrikov, Mater. Lett., 2009, 63, 972–974 CrossRef.
- A. P. Bhirud, S. D. Sathaye, R. P. Waichal, L. K. Nikam and B. B. Kale, Green Chem., 2012, 14, 2790–2798 RSC.
- X. Hu, Z. Zhou, Q. Lin, Y. Wu and Z. Zhang, Chem. Phys. Lett., 2011, 503, 287–291 CrossRef.
- Q. Wang, X. Wang, Z. Chai and W. Hu, Chem. Soc. Rev., 2013, 42, 8821–8834 RSC.
- K. (Ken) K. Ostrikov, U. Cvelbar and A. B. Murphy, J. Phys. D: Appl. Phys., 2011, 44, 174001 CrossRef.
- K. Bazaka, M. V. Jacob and K. Ostrikov, Chem. Rev., 2016, 116, 163–214 CrossRef PubMed.
- J. C. Whitehead, J. Phys. D: Appl. Phys., 2016, 49, 243001 CrossRef.
- H. H. Kim, Y. Teramoto, A. Ogata, H. Takagi and T. Nanba, Plasma Chem. Plasma Process., 2016, 36, 45–72 CrossRef.
- J. Lee, D. C. Sorescu and X. Deng, J. Am. Chem. Soc., 2011, 133, 10066–10069 CrossRef PubMed.
- R. Aerts, W. Somers and A. Bogaerts, ChemSusChem, 2015, 8, 702–716 CrossRef PubMed.
- G. Chen, V. Georgieva, T. Godfroid, R. Snyders and M. P. Delplancke-Ogletree, Appl. Catal., B, 2016, 190, 115–124 CrossRef.
- T. Nozaki, H. Tsukijihara, W. Fukui and K. Okazaki, Energy Fuels, 2007, 21, 2525–2530 CrossRef.
- J. Kim, M. S. Abbott, D. B. Go and J. C. Hicks, ACS Energy Lett., 2016, 1, 94–99 CrossRef.
- D. Park, J. Kim and T. Kim, Catal. Today, 2018, 299, 86–92 CrossRef.
- X. Tu, H. J. Gallon, M. V Twigg, P. A. Gorry and J. C. Whitehead, J. Phys. D: Appl. Phys., 2011, 44, 274007 CrossRef.
- J. Sentek, K. Krawczyk, M. Młotek, M. Kalczewska, T. Kroker, T. Kolb, A. Schenk, K. H. Gericke and K. Schmidt-Szałowski, Appl. Catal., B, 2010, 94, 19–26 CrossRef.
- Z. Zakaria and S. K. Kamarudin, Renewable Sustainable Energy Rev., 2016, 65, 250–261 CrossRef.
- E. M. Alayon, M. Nachtegaal, M. Ranocchiari and J. a. van Bokhoven, Chem. Commun., 2012, 48, 404 RSC.
- B. Eliasson, U. Kogelschatz, B. Xue and L.-M. Zhou, Ind. Eng. Chem. Res., 1998, 37, 3350–3357 CrossRef.
- L. Huang, X. Zhang, L. Chen and L. Lei, Plasma Chem. Plasma Process., 2011, 31, 67–77 CrossRef.
- H. H. Kim, S. Tsubota, M. Daté, A. Ogata and S. Futamura, Appl. Catal., A, 2007, 329, 93–98 CrossRef.
- M. Magureanu, N. B. Mandache, P. Eloy, E. M. Gaigneaux and V. I. Parvulescu, Appl. Catal., B, 2005, 61, 12–20 CrossRef.
- J. Van Durme, J. Dewulf, W. Sysmans, C. Leys and H. Van Langenhove, Appl. Catal., B, 2007, 74, 161–169 CrossRef.
- H. H. Kim, Y. Teramoto, N. Negishi and A. Ogata, Catal. Today, 2014, 256, 13–22 CrossRef.
- F. Holzer, Appl. Catal., B, 2002, 38, 163–181 CrossRef.
- F. Holzer, F. D. Kopinke and U. Roland, Plasma Chem. Plasma Process., 2005, 25, 595–611 CrossRef.
- C. Hibert, I. Gaurand, O. Motret and J. M. Pouvesle, J. Appl. Phys., 1999, 85, 7070 CrossRef.
- R. Ono, Y. Yamashita, K. Takezawa and T. Oda, J. Phys. D: Appl. Phys., 2005, 38, 2812–2816 CrossRef.
- W. S. Kang, J. M. Park, Y. Kim and S. H. Hong, IEEE Trans. Plasma Sci., 2003, 31, 504–510 CrossRef.
- E. J. H. Lee, K. Balasubramanian, R. T. Weitz, M. Burghard and K. Kern, Nat. Nanotechnol., 2008, 3, 486–490 CrossRef PubMed.
- K. Takaki, J. S. Chang and K. G. Kostov, IEEE Trans. Dielectr. Electr. Insul., 2004, 11, 481–490 CrossRef.
- A. Mizuno and H. Ito, J. Electrost., 1990, 25, 97–107 CrossRef.
- X. Tu and J. C. Whitehead, Appl. Catal., B, 2012, 125, 439–448 CrossRef.
- S. Jo, D. H. Lee, W. S. Kang and Y. H. Song, Phys. Plasmas, 2013, 20, 123507 CrossRef.
- Y. R. Zhang, K. Van Laer, E. C. Neyts and A. Bogaerts, Appl. Catal., B, 2016, 185, 56–67 CrossRef.
- Y.-R. Zhang, E. C. Neyts and A. Bogaerts, J. Phys. Chem. C, 2016, 120, 25923–25934 CrossRef.
- S. Jo, T. Kim, D. H. Lee, W. S. Kang and Y. H. Song, Plasma Chem. Plasma Process., 2014, 34, 175–186 CrossRef.
- H. H. Kim, J. H. Kim and A. Ogata, J. Phys. D: Appl. Phys., 2009, 42, 135210 CrossRef.
- H.-H. Kim and A. Ogata, Eur. Phys. J.: Appl. Phys., 2011, 55, 1–11 CrossRef.
- W. Wang, H. H. Kim, K. Van Laer and A. Bogaerts, Chem. Eng. J., 2018, 334, 2467–2479 CrossRef.
- L. Chen, X. Zhang, L. Huang and L. Lei, Chem. Eng. Technol., 2010, 33, 2073–2081 CrossRef.
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962–998 CrossRef PubMed.
- Y. Zhao, C. Cui, J. Han, H. Wang, X. Zhu and Q. Ge, J. Am. Chem. Soc., 2016, 138, 10191–10198 CrossRef PubMed.
- A. Gómez-Ramírez, V. J. Rico, J. Cotrino, A. R. González-Elipe and R. M. Lambert, ACS Catal., 2014, 4, 402–408 CrossRef.
- G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, pp. 1–34 Search PubMed.
- B. Zhao, Y. Liu, Z. Zhu, H. Guo and X. Ma, J. CO2 Util., 2018, 24, 34–39 CrossRef.
- N. Amin, Fuel, 2006, 85, 577–592 CrossRef.
- C. J. Liu, B. Xue, B. Eliasson, F. He, Y. Li and G. H. Xu, Plasma Chem. Plasma Process., 2001, 21, 301–310 CrossRef.
- Y. Li, G. Xu, C. Liu, B. Eliasson and B. Xue, Energy Fuels, 2001, 15, 299–302 CrossRef.
- F. J. Spiess, S. L. Suib, K. Irie, Y. Hayashi and H. Matsumoto, Catal. Today, 2004, 89, 35–45 CrossRef.
- M. Scapinello, L. M. Martini and P. Tosi, Plasma Processes Polym., 2014, 11, 624–628 CrossRef.
- K. Zhang, B. Eliasson and U. Kogelschatz, Ind. Eng. Chem. Res., 2002, 41, 1462–1468 CrossRef.
- X. Zhang, B. Dai, A. Zhu, W. Gong and C. Liu, Catal. Today, 2002, 72, 223–227 CrossRef.
- M. H. Pham, V. Goujard, J. M. Tatibouët and C. Batiot-Dupeyrat, Catal. Today, 2011, 171, 67–71 CrossRef.
- C. Sun, D. Mao, L. Han and J. Yu, RSC Adv., 2016, 6, 55233–55239 RSC.
- L. Chen, X. Zhang, L. Huang and L. Lei, J. Nat. Gas Chem., 2010, 19, 628–637 CrossRef.
- T. Pham Huu, S. Gil, P. Da Costa, A. Giroir-Fendler and A. Khacef, Catal. Today, 2015, 257, 86–92 CrossRef.
- A. Boulamanti and J. A. Moya, Renewable Sustainable Energy Rev., 2017, 68, 1205–1212 CrossRef.
- U. Kogelschatz, Plasma Chem. Plasma Process., 2003, 23, 1–46 CrossRef.
- S. Jo, D. Hoon Lee and Y.-H. Song, Chem. Eng. Sci., 2015, 130, 101–108 CrossRef.
- C. Xu and X. Tu, J. Energy Chem., 2013, 22, 420–425 CrossRef.
- B. Eliasson, U. Kogelschatz, B. Xue and L.-M. Zhou, Ind. Eng. Chem. Res., 1998, 37, 3350–3357 CrossRef.
- A. Gómez-Ramírez, V. J. Rico, J. J. Cotrino, A. R. González-Elipe, R. M. Lambert, R. Gonza and R. M. Lambert, ACS Catal., 2014, 4, 402–408 CrossRef.
- L. Chen, X. W. Zhang, L. Huang and L. C. Lei, Chem. Eng. Process., 2009, 48, 1333–1340 CrossRef.
- L. Huang, X. Zhang, L. Chen and L. Lei, Plasma Chem. Plasma Process., 2010, 31, 67–77 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |