
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFactors affecting the structure of lyotropic liquid crystals and the correlation between structure and drug diffusion
Yiming Huang a and
Shuangying Gui*abc
a and
Shuangying Gui*abc
aDepartment of Pharmacy, Anhui University of Chinese Medicine, No. 1, Qianjiang Road, Hefei, Anhui 230012, China. E-mail: guishy0520@126.com; Fax: +86-551-68129121; Tel: +86-551-68129121
bInstitute of Pharmaceutics, Anhui Academy of Chinese Medicine, Hefei, Anhui 230012, China
cKey Laboratory of Xin'an Medicine, Ministry of Education, Hefei, Anhui 230012, China
First published on 13th February 2018
Abstract
Lyotropic liquid crystals (LLCs) formed by the self-assembly of amphiphilic molecules in a solvent (usually water) have attracted increasingly greater attention in the last few decades, especially the lamellar phase (Lα), the reversed bicontinuous cubic phase (Q2) and the reversed hexagonal phase (H2). Such phases offer promising prospects for encapsulation of a wide range of target molecules with various sizes and polarities owing to the unique internal structures. Also, different structures of mesophases can give rise to different diffusion coefficients. The bicontinuous cubic phase and the hexagonal phase have been demonstrated to control and sustain the release of active molecules. Furthermore, the structures are susceptible to many factors such as water content, temperature, pH, the presence of additives etc. Many researchers have been studying these influencing factors in order to accurately fabricate the desired phase. In this paper, we give a review of the characteristics of different structures of liquid crystalline phases, the influencing factors on the phase transition of liquid crystals and the relationship between structures of LLC and drug diffusion. We hope our review will provide some insights into how to manipulate in a controlled manner the rate of incorporating and transferring molecules by altering the structure of lyotropic mesophases.
1. Introduction
Sustained-release preparations are able to maintain drug concentrations in the range of therapeutic concentrations for a prolonged time frame.1,2 This can reduce the frequency of administration and toxicity and minimize side effects compared with general preparations.LLCs, based on the self-assembly of amphiphilic molecules in excess water, have received considerable attention as a carrier for sustained release drugs.3–5 The internal structures of the LLCs are composed of a hydrophilic domain, lipophilic domain and lipid layer. Therefore, LLCs can encapsulate a wide range of active drugs (i.e. hydrophilic, hydrophobic, and amphiphilic) and protect them from hydrolysis and enzymolysis.6–9 Normally, hydrophilic drugs are located near the lipid polar head or in the water channels, while the lipophilic drugs are loaded in the lipid layer and the amphiphilic drugs are at the lipid layer interface.10
The most commonly studied types of LLCs are the lamellar, the bicontinuous cubic, the hexagonal mesophase and the micellar cubic phases.11,12 Moreover, the release rates of loaded drugs are distinguishable in different liquid crystal phases.13,14 It is the dimensions of the lyotropic liquid crystal structures that determine the release rate of the entrapped drug. The lamellar phase is a one-dimensional structure made up of lipid bilayers and layers of water.15 The bicontinuous cubic is a “honeycomb” structure in which the amphiphilic molecules are stacked in three-dimensional space.16 The two water channels are consecutive but not crossed and are separated by a lipid bilayer. Therefore, the cubic phase can sustain the release of active molecules.17 In addition, the hexagonal mesophase is arranged in hexagons by a closed cylindrical micelle.18 The release rate of drugs is slower than in the bicontinuous cubic phase.19,20 As for the reversed micellar cubic of Fd3m, it is a closed structure consisting of closely packed inverse micelles of two sizes.21 Fd3m has been of increasing interest to researchers because the release of hydrophilic substances from the reversed micellar cubic phase is slower than that from the reversed hexagonal phase.22
The structures of the LLCs are affected by many factors including temperature,19 pH,23–26 light,27–32 magnetic field,33,34 the type of amphiphilic,35 water content36,37 and additives.38,39 Therefore, by altering the preparation conditions, desired phase structures can be achieved and intelligent carriers can be fabricated. Fong et al.19 have shown the possibility of using temperature to trigger release. More specifically, they successfully switched the hexagonal mesophase to the bicontinuous cubic phase by changing the temperature above or below physiological temperature. The in vitro release studies showed that the release rate of hydrophilic drug glucose from the hexagonal phase was slower than that from the cubic phase. There are also many studies using pH to regulate the release rate of drug according to the pH changing within human body.40,41 For example, linoleic acid is susceptible to PH, so addition of linoleic acid to MLO/water or MO/water can control the formation of mesophases by controlling the pH, thereby affecting drug release.23,24
Besides, some researchers have also tried to change the internal structures of LLCs by encapsulating reconstitute membrane proteins within to control the release of the drug.26,42,43 Zabara et al.26 designed a novel perforated bicontinuous cubic phase (Pn3m), which was formed by adding outer membrane protein F (OmpF) to monolinolein (MLO)–water system. The results showed that the presence of OmpF did not alter the structure of Pn3m compared with the Pn3m formed by MLO–water. The OmpF proteins are located at the lipid bilayer of Pn3m cubic phase and therefore they can connect the two parallel sets of aqueous channels. Structural data showed that the OmpF contains a constriction site with two amino acid halfrings (one positively charged and the other negatively charged) which lead to sensitivity to PH. Therefore, OmpF acted as “pH-triggered release switch”. At pH = 7.4, the model drug glucose in OmpF-doped mesophase was released faster than in the blank (without OmpF). However, when the pH = 4.8, the release rates of glucose in two kinds of Pn3m were similar.
In this review, we initially briefly introduce the different structures of LLCs. We then offer an overview of the influencing factors on the phase structures, including prescription factors (i.e. amphiphilic molecules, water content, additives) and external factors (i.e. temperature, pressure, light, magnetic field). In the end, the relationship between structure and drug diffusion is discussed.
2. The structure characteristic of lyotropic liquid crystals
Generally, with the an increase amphiphilic concentrations, different well-defined structures are derived, such as the normal micelle (oil in water), the normal cubic phase, the normal hexagonal phase, the lamellar phase, the reversed cubic phase (water in oil) and the reversed hexagonal phase. Additionally, the reversed cubic phases can be divided into two categories, the reversed bicontinuous cubic phase and the reversed micellar cubic phase. Besides, a nomenclature for mesophases was proposed44,45 using various capital letter and subscripts. For example, L stands for the lamellar phase, Q for the cubic phase, V for the bicontinuous cubic phase, I for the micellar cubic phase and H for the hexagonal phase. Furthermore, subscripts I (1) and II (2) represent for normal and reversed phases respectively. Among all of the structures, the lamellar phase, the reversed cubic phase and the reversed hexagonal phase have received much attention for their highly ordered internal structures, which gives these phases great the potential as drug delivery systems. These common structures of LLCs diagram are presented in Fig. 1.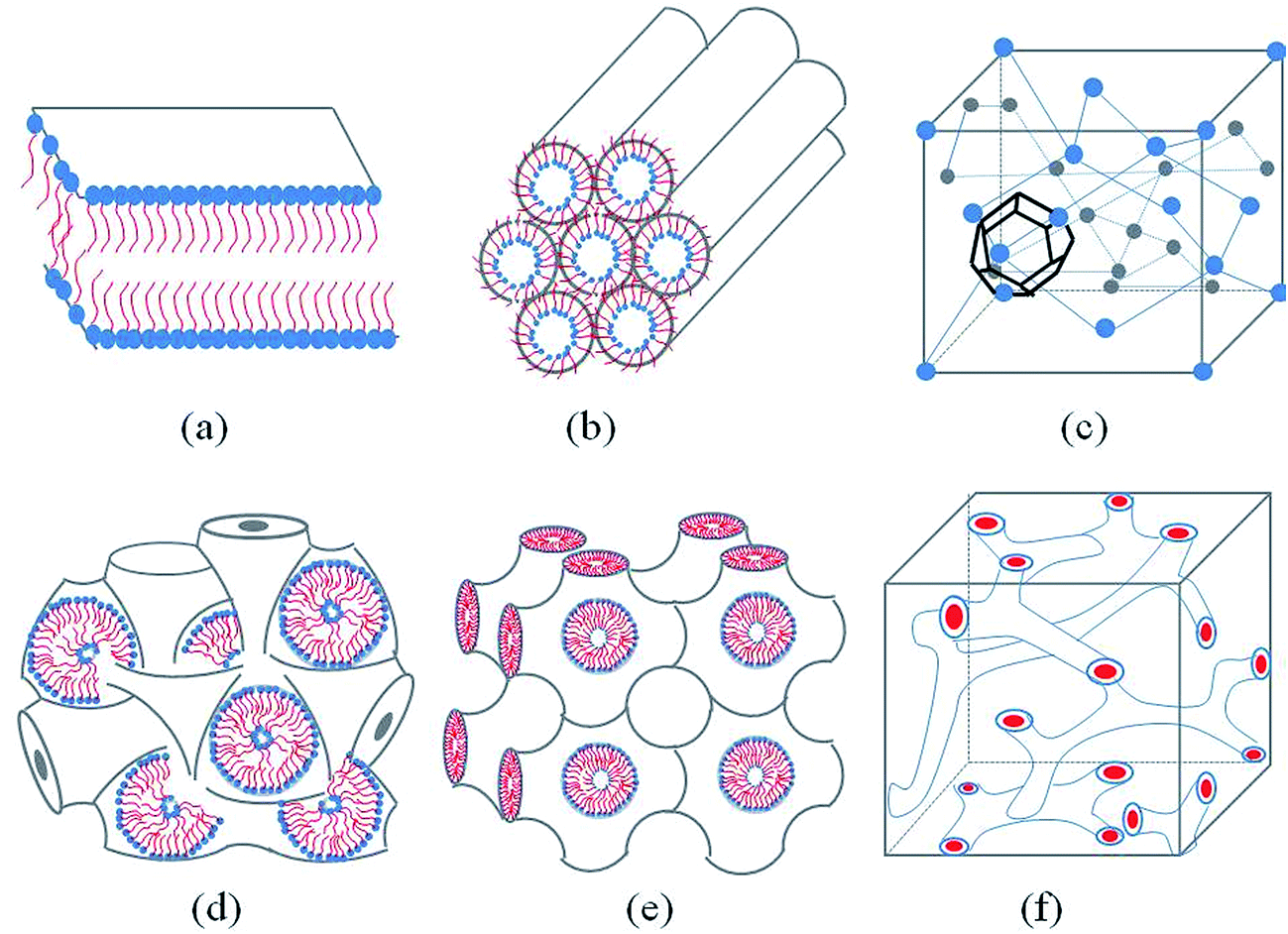 | ||
| Fig. 1 Schematic representation of the lyotropic liquid crystalline phases commonly found in neutral lipid/water systems. (a) Lamellar phase (b) reverse hexagonal phase (c) reversed micellar cubic of Fd3m (d) reversed bicontinuous cubic (Im3m) (e) reversed bicontinuous cubic (Pn3m) (f) reversed bicontinuous cubic (Ia3d). | ||
2.1 The lamellar phase
The lamellar phase extensively exists in organisms.46 For example, it is the basic building block of cell membranes. The lamellar phase is a planar structure, consisting of lipid bilayers separated by water, where the polar head groups of the amphiphilic molecules associate and are in contact with water directly, while the hydrophobic tails are away from water.47,48 The rheology gives the information that the lamellar structure is less viscous compared with the hexagonal structure. Furthermore, the lamellar phase can be characterized by Crossed Polarized Light Microscopy (CPLM), exhibiting “streaky” or “mosaic” like texture. In addition, Bragg peaks with relative positions at the ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:4 are obtained from the Small-Angle X-ray Scattering (SAXS) measurement.
:2:3:4 are obtained from the Small-Angle X-ray Scattering (SAXS) measurement.
2.2 The reversed bicontinuous cubic
The reversed bicontinuous cubic phase is complex and comprises two continuous but nonintersecting water channels separated by a curved bicontinuous lipid bilayer.49 Based on the theory of infinite periodic minimal surface (IPMS), the reversed bicontinuous cubic phase can be regarded as a three dimensional periodic lipidic bilayer with zero mean curvature at each point. It should be noted that the reversed bicontinuous cubic phase can be divided into three types based on X-ray crystallographic studies, namely, the primitive lattice (Im3m, Q229), the double-diamond lattice (Pn3m, Q224), and the gyroid lattice (Ia3d, Q230). Furthermore, the bulk phase is a transparent, viscous gel, which can be characterized by CPLM and SAXS.50,51 Under the CPLM, all types of the bulk phase do not display any optical textures. However, the SAXS can be used to distinguish different types. The ratios of Bragg peaks at different positions are as follow: Im3m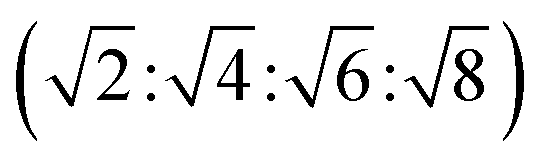 , Pn3m
, Pn3m 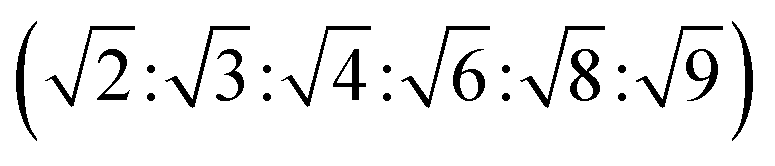 , Ia3d
, Ia3d 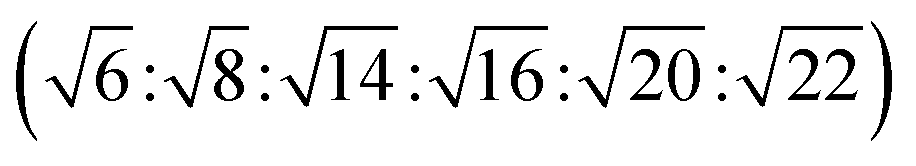 .
.
2.3 The reverse hexagonal phase
The reverse hexagonal phase is made up by a large quantity of densely packed water filled cylindrical micelles, following a long-range order two-dimensional lattice.52 Significantly, the diameter of the cylindrical micelle is about 1–2 nm, containing 30–60% water by weight. Furthermore, it is reported that the water inside the cylindrical micelles is not in contact with the water outside directly. The reverse hexagonal phase is less viscous than the reversed bicontinuous cubic. In other words, the reverse hexagonal phase is of intermediate viscosity compared with the lamellar phase and the reversed bicontinuous cubic phase. By CPLM, the reverse hexagonal phase appears “fan-like” textures and the ratios of Bragg peaks at different positions are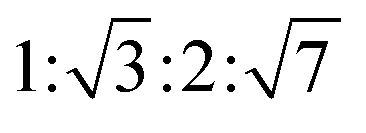 according to the SAXS measurement.53,54
according to the SAXS measurement.53,54
2.4 The reversed micellar cubic
The reversed micellar cubic is also referred as the discontinuous cubic phase which consists of micelles arranged in a cubic lattice. There are two main micellar cubic phases: Fd3m and Fm3m. The Fd3m micellar cubic phase is a closed structure composed of micelles of two different sizes organized in a double diamond network.21 The Bragg peaks are in the ratios of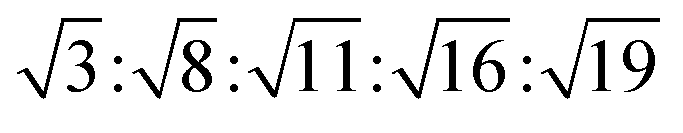 .55,56 The Fd3m phase is currently the most extensively studied structure for sustained drug release. The physical properties of Fd3m and the mesophases mentioned above are summarized in Table 1.
.55,56 The Fd3m phase is currently the most extensively studied structure for sustained drug release. The physical properties of Fd3m and the mesophases mentioned above are summarized in Table 1.
| Types of LLC phase | Optical properties | CPLM | SAXS |
|---|---|---|---|
| Lamellar phase | Anisotropic | Streaky-like | 1:2:3:4 |
| Im3m | Isotropic | Dark field | 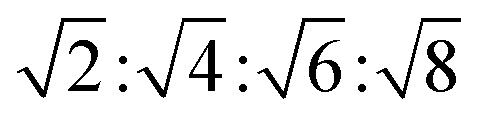 |
| Pn3m | Isotropic | Dark field | 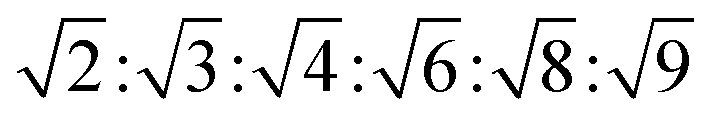 |
| Ia3d | Isotropic | Dark field | 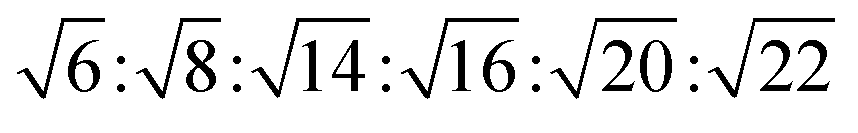 |
| Fd3m | Isotropic | Dark field | 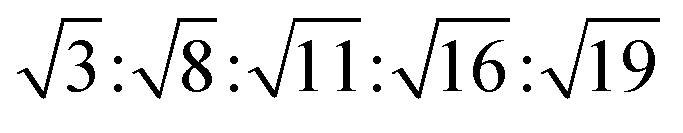 |
| Reverse hexagonal | Anisotropic | Fan-like | 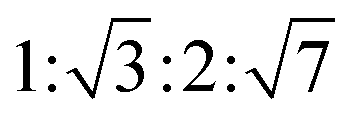 |
Reverse micellar cubic mesophase of symmetry Fm3m was reported by I. Martiel which consists of ternary mixtures of soy bean phosphatidylcholine (PC), water, and an organic solvent, including cyclohexane, (R)-(+)-limonene, and isooctane.57 It is composed of a compact packing of remarkably large and monodisperse reverse micelles in a face-centered cubic (fcc) lattice. Therefore, the drug loading may increase. In addition, Fm3m is stable in equilibrium with excess water, so in principle it can be dispersed into particles for use as a delivery system.
3. The influencing factors of phase transition
Studies have shown that phases can be altered by many factors. Therefore, in the recent years, scientific efforts have converged toward studying the influencing factors in order to control the preparation conditions of the desired phases. The influencing factors can be divided into external factors, such as temperature, pressure, light, and magnetic field and prescription factors, such as presence of amphiphilic molecules, water content and the third additives. The mechanism of the structural transitions affected by factors mentioned above can be explained by the critical packing parameter (CPP), which is based on the spatial stacking of amphiphilic molecules to predict the structure of the system may be formed.58,59 CPP is defined as CPP = V/al, where V represents the hydrophobic chain volume, a represents the cross-sectional area of the hydrophilic head group, and l represents the hydrophobic chain length in the molten state. According to the theory of CPP, different phases correspond to different CPP values. When CPP < 1, normal phases are formed which means that the cross-sectional area of the hydrophilic head group is larger than that of the hydrophobic tails, such as the normal micelle (L1), the normal discontinuous cubic (II) phase, the normal hexagonal (HI) phase and the normal bicontinuous cubic (QI) phase. Conversely, reversed phases such as the reversed bicontinuous cubic (QII) phase, the reversed hexagonal (HII) phase, the reversed discontinuous cubic (III) phase and the reversed micelle (L2) are developed with CPP > 1.60 When the polar head group area and the tail are almost equal, the lamellar phase is formed (Fig. 2).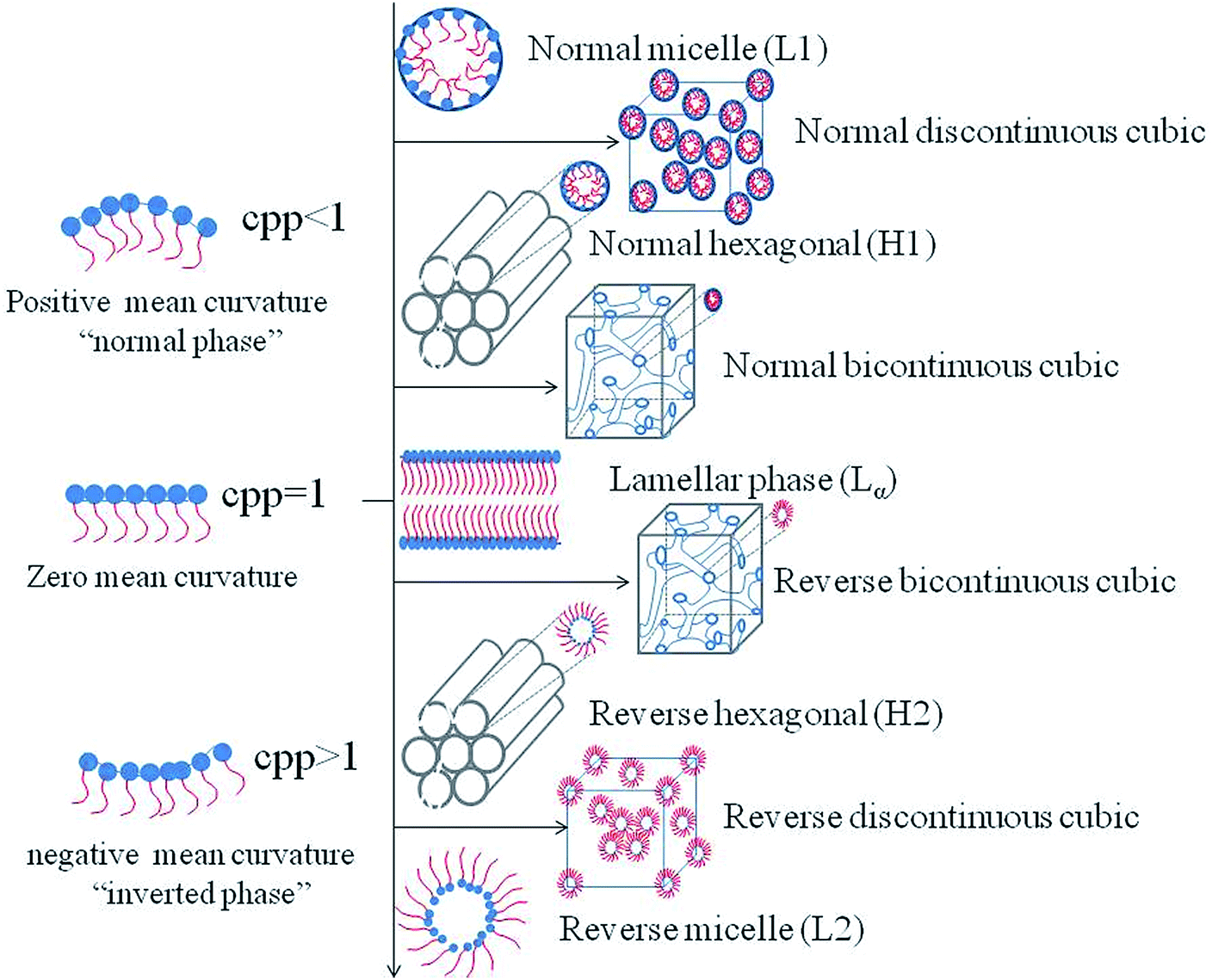 | ||
| Fig. 2 Schematic representations of common structures and their corresponding CPP. | ||
3.1 Prescription factors
Among the lipid materials mentioned above, GMO, MLO and PT have become the most widely used materials for encapsulation of a wide range of target molecules with various sizes and polarities.
Lyotropic liquid crystals are formed by adding the amphiphilic molecules to a polar solvent. Amphiphilic molecules contain a polar hydrophilic head and a non-polar hydrophobic tail. When getting in contact with water, the polar head gets close to water, while the hydrophobic tails keep away from water and are packed tightly for minimizing the free energy of the system. When the water content of the system is higher than the concentration of amphiphilic molecules, the amphiphilic molecules are tiled at the water interface. When the concentration of amphiphilic molecules exceeds the critical micelle concentration (CMC), micelles of different shapes will be formed. If the concentration continues to increase, the micelles will be further assembled into LLC with different structures.75,76
Shah and Paradkar38 explored the effect of different hydrophilic-lipophilic balance (HLB) on the system of GMO and water. Results indicated that additives with the HLB values of 1.5, 3, 4, and 5 formed the reversed hexagonal phase, while the lamellar phase was present with additives of HLB 7, 10, and 11 after 12 hours. The authors offered an explanation for these results: the lipophilic additives interacted with the tail of GMO, which in turn caused an increase in the CPP value and triggered the phase transition. It is not difficult to understand that the hydrophilic additives introduced the opposite phase transition. With the goal of obtaining a stable preparation method of the hexagonal phase at room temperature, Amar-Yuli and Garti39 studied the effects of TAGs with different chain lengths (C2–C18) on the effects of GMO/water system. It was found that TAG with very short chains like triacetin did not affect the phase behaviors, and the phases remained the Q2 phase and L2 phase. By contrast, TAG with middle chains like tricaprylin gave rise to the transformation of L2 phase to the H2 phase directly and it was the most stable and effective additive to trigger the transformation. This is because tricaprylin was incorporated between the GMO hydrophobic chains and filled the void volumes, and therefore increased the CPP value. Furthermore, in the other studies of Garti's group, tricaprylin was utilized to get the H2 phase. However, there was not much research on long-chain TAG since it would take at least one month to solvate the tails of GMO.
Hydrophobic additives such as vitamin E acetate (VitEA),78–83 tetradecane,81,84 hexadecane,22 triolein,81 limonene,21,65,81 cyclohexane65 and fatty acids25,85 have been reported and these additives are listed in Table 2. VitEA is currently the most widely used additive for forming the hexagonal phase at room temperature. The work of Dong et al.78 showed that the presence of hydrophobic VitEA in PT–water systems resulted in the formation of hexagonal phase at 25 °C. The bulk LLC could be dispersed into nanoparticles while keeping the unique internal structures. These nanoparticles are called liposomes, hexosomes, and cubosomes, respectively. The dispersed hexosomes were successfully prepared by Kim et al. when adding VitEA to PT–Pluronic F-127 (stabilizer)–water systems.79 The influence of hexadecane in PT and GMO was investigated respectively by Phan et al.22 SAXS data indicated that in GMO–water systems at 25 °C, Pn3m, H2, micellar cubic phase (Fd3m), and inversed micelles (L2) appeared when the concentration of hexadecane was 0–4% (w/w), 4–25%, 25–40% and >40% respectively. However, with an increase in the concentration of hexadecane in PT–water systems, the phase transitioned from Pn3m to H2 to L2 without forming the micellar cubic phase.
| Composition | Addition | Type of phase | Reference |
|---|---|---|---|
| GMO/water | Tricaprylin | H2 | 39 |
| PT/water | VitEA | H2 | 78 |
| PT/F-127/water | VitEA | Hexosomes | 79 |
| Phosphatidylcholine/sorbitan monooleate/water | Tocopherol acetate | Lα/H2 | 80 |
| MLO/water | Tetradecane | H2/L2/Fd3m | 84 |
| GMO/water | Hexadecane | H2/Q2/L2/Fd3m | 22 |
| MLO/water | Limonene | Fd3m/Lα/Pn3m | 21 |
| Phosphatidylcholine/water | Limonene | Fm3m/H2 | 65 |
| MLO/water | Linoleic acid | Im3m/H2 | 25 |
| GMO/water | Oleic acid | H2/L2/Fd3m | 85 |
| GMO/water | Octyl glucoside | Pn3m | 86 |
| MLO/water | Sucrose stearate | Pn3m/Im3m | 87 |
| MLO/F-127/water | Diglycerol monooleate | Hexosomes/cubosomes (Im3m) | 88 |
| PT/water | Ethanol | L2/Lα/Q2 | 61 |
| PT/water | Ethanol | L2/Lα/Q2 | 89 |
| PT/water | Propylene glycol | L2/Lα/Q2 | 90 |
| GMO/TAG/water | Transcutol | H2 (low viscosity) | 91 |
The addition of hydrophilic additives to the system has two main purposes. One is introduction of a hydration-enhancing agent such as octyl glucoside (OG),86 sucrose stearate87 and diglycerol monooleate (DGMO),88 which can interact with the polar head of amphiphilics to increase the water channel dimensions and enable the LLC to encapsulate large molecules, such as proteins and peptides. Angelov et al.86 developed a novel cubic phase (Pn3m) with large water channels using OG as hydration-enhancing agent. SAXS showed that the phase structure did not change but the diameter of channel was twice of that of the normal phase. Negrini and Mezzenga87 enlarged the diameter of water channels in the monolinolein/water system by adding sucrose stearate. Studies showed that the phase transitioned from Pn3m to Im3m when the concentration of sucrose stearate was 10% (w/w) and the size of water channels was increased from 3.85 to 12 nm with the concentration ranging from 0 to 25% (w/w). That is because the sucrose stearate interacted with the polar head, reducing the CPP value and inducing the phase change from Pn3m to Im3m.
Another purpose is the addition of solvents such as ethanol,61,89 propylene glycol,90 transcutol91 etc. to form a low-viscosity lamellar phase and a micelle phase for injection administration. Chen et al.89 developed injectable in situ liquid crystals for intraarticular administration by adding ethanol to the PT–water system. The results indicated that isotropic solutions with low viscosity were formed.
Pharmaceutical compounds of different polarities have been encapsulated in lyotropic liquid crystals such as the hydrophilic drugs rhodamine B,92 acyclovir,93 lidocaine hydrochloride94 and the lipophilic drugs lidocaine94 cinnarizine,95 vitamin K,96 indomethacin,97 diclofenac sodium98,99 and so on. Similar to the additives, the entrapment of the hydrophilic drug causes a phase change from the cubic phase to the lamellar phase, as opposed to the hydrophobic drugs.
3.2 External factors
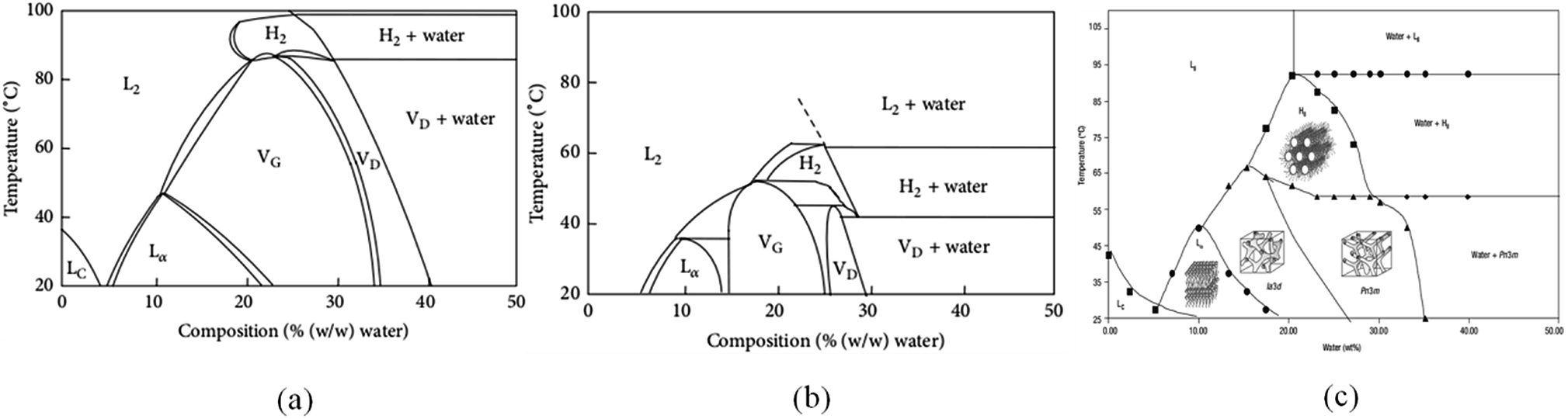
The structure of lyotropic liquid crystals is extremely susceptible to temperature. In practical applications, sometimes, the prepared temperature of lyotropic liquid crystals is below 20 °C. Qiu et al.62 prepared lyotropic liquid crystals at 0 °C. The samples were then stored at 4 °C and 20 °C respectively. The results showed that the lamellar crystal was formed when stored at 4 °C and transformed into cubic phase at 20 °C. The authors noted that the LC phase was formed in the low-temperature region of the GMO/water phase diagram. However, it is metastable. Therefore, the prepared samples need to be stored at 20–25 °C. For example, the samples can be placed in the incubator to maintain a constant temperature and avoid structural changes.
4. The correlation between structure and drug diffusion
LLC is capable of solubilizing drugs with different polarities (hydrophilic, hydrophobic, amphiphilic) due to the unique internal structures. The position of these drugs are different. Generally, hydrophilic drugs are located close to the polar head or in the water channels, while hydrophobic drugs are loaded in the lipid layer, and amphiphilic drugs at the interface. Furthermore, according to previous research, the release of drugs from LLC conformed to Higuchi diffusion kinetics, where the cumulative release of drugs was proportional to the square root of the time.102,103 The Higuchi diffusion equation is given as follow:where Mt and M∞ are the amount of drug released at time t and at infinite time respectively, and KH is the release rate constant.104 However, the release rate of the solubilized drugs were related to many factors, such as the polarities of drugs,65,87,105 the molecular weight of drugs,87,106 and the internal structures,22 etc. In fact, the drug diffusion kinetics depend in a complex way on the geometry, as demonstrated theoretically and experimentally by Antognini et al.107 In the case of the lamellar phase, the flat water layer is separated by hydrophobic bilayers. As for the hexagonal phase, there exist one-dimensional parallel water cylinders, and the water channels of the cubic phase are continuous but nonintersecting. Moreover, hydrophilic drugs are located in the polar core of the inverse micellar cube. Consequently, the release rates of loaded drugs are in the order as follows: Dlamellar phase > Dcubic phase > Dhexagonal phase > Dmicellar cubic.108 Many researchers have studied the release of drugs with different properties from different liquid crystal structures and we will discuss this in the following sections.
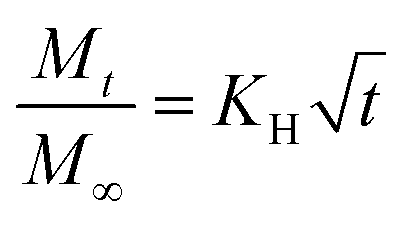
4.1 Release of hydrophilic drugs
Phan et al.22 investigated the release rate of model hydrophilic drug glucose in four GMO-based mesophases which were the reversed bicontinuous cubic phase (V2), the inverse hexagonal phase (H2), the micellar cubic phase (I2), and the inverse miceller phase (L2) respectively. The results indicated that the drug release in four mesophases followed first-order diffusion kinetics. The study demonstrated that LLC structures could be used to control the rate of release. The release rates of glucose were in the order as follow: V2 > L2 > H2 > I2. Lee et al.108 explored the relationship between hydrophilic drugs of different molecular weight (14C-glucose, Allura Red and FITC-dextrans) and the cubic phase formed by GMO (QGMO), the cubic phase formed by PT (QPT) and the hexagonal phase formed by PT and VE (HPT). It was found that the release rate of drugs was the greatest in QGMO, followed by QPT and HPT. In addition, molecular weight also affected the release of drugs and the release of those with a small molecular weight (14C-glucose) was the fastest. Furthermore, there have been many studies on the in vitro release of macromolecules such as protein drugs and release processes that follow Higuchi diffusion kinetics.4.2 Release of hydrophobic drugs
Hydrophobic drugs irinotecan,109 paclitaxel,87 caffeine,65 proflavine,65 diazepam,110 propofol,111 rifampicin112 were used to study the release behavior. In vitro release experiments showed that the release of encapsulated hydrophobic drugs followed Higuchi diffusion kinetics too. Boyd et al.109 focused their research on the release of irinotecan in hexosome dispersions. The results showed that the release of irinotecan was controlled by diffusion, despite being essentially instantaneous.5. Conclusion and outlook
LLC systems have received considerable attention and research as drug carriers. In recent years, more and more researchers are focusing on the structure of LLCs and the release mechanism of drugs, especially R. Mezzenga's research team.This article mainly reviews the influencing factors on LLC structures and the relationship between drugs diffusion and structures. Fully understanding the factors that affect the structure of lyotropic liquid crystals is conducive to controlling the preparation conditions and obtaining a stable liquid crystal phase. Furthermore, we can utilize these factors to design intelligent LLC carriers. Additionally, studying the relationship between drug release and structures can provide a reference for encapsulating different types of drugs.
Certainly, there are many difficulties to overcome for the clinical application of lyotropic liquid crystals, among which are the biocompatibility and safety of lipid materials. It is reported that GMO cause hemolysis, especially for intravenous (IV) administration.113 PEGylation (polyethylene glycol, PEG) can be used to improve the hemocompatibility of MO-based systems.114,115 Furthermore, some new materials such as dioleoyl phosphatidylethanolamine (DOPE),116 glycerol dioleate (GDO) and soy phosphatidylcholine (SPC)117 can reduce hemolysis. Nevertheless, LLC is still considered a good candidate for drug delivery, and further research is expected for deeper exploration of applications of LLC as drug carriers.
Conflicts of interest
The authors declare that there is no conflict of interests regarding the publication of this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81274099).References
- S. B. Rizwan, B. J. Boyd, T. Rades and S. Hook, Expert Opin. Drug Delivery, 2010, 7(10), 1133 CrossRef CAS PubMed.
- J. Clogston and M. Caffrey, J. Controlled Release, 2005, 107(1), 97 CrossRef CAS PubMed.
- T. Kaasgaard and C. J. Drummond, Phys. Chem. Chem. Phys., 2006, 8(43), 4957–4975 RSC.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Adv. Drug Delivery Rev., 2001, 47(2), 229–250 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Acc. Chem. Res., 2011, 44(2), 147–156 CrossRef CAS PubMed.
- L. Sagalowicz, R. Mezzenga and M. E. Leser, Curr. Opin. Colloid Interface Sci., 2006, 11(4), 224–229 CrossRef CAS.
- D. Libster, A. Aserin, E. Wachtel, G. Shoham and N. Garti, J. Colloid Interface Sci., 2007, 308(2), 514 CrossRef CAS PubMed.
- V. Luzzati, Curr. Opin. Struct. Biol., 1997, 7(5), 661–668 CrossRef CAS PubMed.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4(6), 449–456 CrossRef CAS.
- L. Sagalowicz, M. E. Leser, H. J. Watzke and M. Michel, Trends Food Sci. Technol., 2006, 17(5), 204–214 CrossRef CAS.
- S. Z. Mohammady, M. Pouzot and R. Mezzenga, Biophys. J., 2009, 96(4), 1537–1546 CrossRef CAS PubMed.
- P. S. Pershan, Phys. Today, 1982, 35(5), 34–39 CrossRef CAS.
- J. Barauskas, A. M. Johnsson and F. Tiberg, Nano Lett., 2005, 5(8), 1615–1619 CrossRef CAS PubMed.
- T. H. Nguyen, T. Hanley, C. J. Porter and B. J. Boyd, J. Controlled Release, 2011, 153(2), 180 CrossRef CAS PubMed.
- K. Holmberg, Pigm. Resin Technol., 2002, 10(5), 383–402 Search PubMed.
- K. Larsson, J. Phys. Chem., 1989, 93(21), 512–513 CrossRef.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2000, 5(1), 64–69 CrossRef CAS.
- R. Laughlin, J. Am. Chem. Soc., 1995, 117(42), 10603 CrossRef.
- W. K. Fong, T. Hanley and B. J. Boyd, J. Controlled Release, 2009, 135(3), 218 CrossRef CAS PubMed.
- H. Chung, J. Kim, J. Y. Um, I. C. Kwon and S. Y. Jeong, Diabetologia, 2002, 45(3), 448–451 CrossRef CAS PubMed.
- M. Pouzot, R. Mezzenga, M. Leser, L. Sagalowicz, S. Guillot and O. Glatter, Langmuir, 2007, 23(19), 9618–9628 CrossRef CAS PubMed.
- S. Phan, W. K. Fong, N. Kirby, T. Hanley and B. J. Boyd, Int. J. Pharm., 2011, 421(1), 176–182 CrossRef CAS PubMed.
- C. M. Chang and R. Bodmeier, J. Controlled Release, 1997, 46(3), 215–222 CrossRef CAS.
- T. K. Kwon and J. C. Kim, Drug Dev. Ind. Pharm., 2011, 37(1), 56 CrossRef CAS PubMed.
- R. Negrini and R. Mezzenga, Langmuir, 2011, 27(9), 5296 CrossRef CAS PubMed.
- A. Zabara, R. Negrini, O. Onaca-Fischer and R. Mezzenga, Small, 2013, 9(21), 3602–3609 CrossRef CAS PubMed.
- W. K. Fong, T. L. Hanley, B. Thierry, N. Kirby and B. J. Boyd, Langmuir, 2010, 26(26), 6136–6139 CrossRef CAS PubMed.
- W. K. Fong, T. L. Hanley, B. Thierry, N. Kirby, L. J. Waddington and B. J. Boyd, Langmuir, 2012, 28(40), 14450 CrossRef CAS PubMed.
- A. Yaghmur, L. Paasonen, M. Yliperttula, A. Urtti and M. Rappolt, J. Phys. Chem. Lett., 2010, 1(6), 962–966 CrossRef CAS.
- W. K. Fong, T. L. Hanley, B. Thierry, A. Hawley, B. J. Boyd and C. B. Landersdorfer, J. Controlled Release, 2016, 228, 67 CrossRef CAS PubMed.
- K. J. Tangso, W. K. Fong, T. Darwish, N. Kirby, B. J. Boyd and T. L. Hanley, J. Phys. Chem. B, 2013, 117(35), 10203–10210 CrossRef CAS PubMed.
- W. K. Fong, N. Malic, R. A. Evans, A. Hawley, B. J. Boyd and T. L. Hanley, Biointerphases, 2012, 7(1–4), 1–5 Search PubMed.
- J. J. Vallooran, S. Bolisetty and R. Mezzenga, Adv. Mater., 2011, 23(34), 3932 CrossRef CAS PubMed.
- J. J. Vallooran, R. Negrini and R. Mezzenga, Langmuir, 2013, 29(4), 999–1004 CrossRef CAS PubMed.
- B. J. Boyd, D. V. Whittaker, S. M. Khoo and G. Davey, Int. J. Pharm., 2006, 309(1–2), 218 CrossRef CAS PubMed.
- P. Wadsten-Hindrichsen, J. Bender, J. Unga and S. Engström, J. Colloid Interface Sci., 2007, 315(2), 701–713 CrossRef CAS PubMed.
- J. Barauskas and T. Landh, Langmuir, 2003, 19(23), 9562–9565 CrossRef CAS.
- M. Shah and A. Paradkar, Eur. J. Pharm. Biopharm., 2007, 67(1), 166–174 CrossRef CAS PubMed.
- I. Amar-Yuli and N. Garti, Colloids Surf., B, 2005, 43(2), 72 CrossRef CAS PubMed.
- A. Asokan and M. J. Cho, J. Pharm. Sci., 2002, 91(4), 903–913 CrossRef CAS PubMed.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56(6), 1194–1198 CAS.
- C. Speziale, M. L. Salvati, C. Manatschal, E. M. Landau and R. Mezzenga, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(27), 7491 CrossRef CAS PubMed.
- C. Speziale, A. F. Zabara, C. J. Drummond and R. Mezzenga, ACS Nano, 2017, 11(11), 11687–11693 CrossRef CAS PubMed.
- S. T. Hyde, Curr. Opin. Solid State Mater. Sci., 1996, 1, 653 CrossRef CAS.
- P. J. Collings, M. Hird and C. C. Huang, Am. J. Phys., 1997, 66(6), 551 CrossRef.
- N. Garti, D. Libster and A. Aserin, Food Funct., 2012, 3(7), 700 CAS.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2009, 14(1), 16–20 CrossRef CAS.
- V. Luzzati and F. Husson, J. Cell Biol., 1962, 12(2), 207 CrossRef CAS PubMed.
- B. D. Kruijff, Nature, 1997, 386, 129–130 CrossRef PubMed.
- D. C. Turner, Z. G. Wang, S. M. Gruner, D. A. Mannock and R. N. Mcelhaney, J. Phys. B: At., Mol. Opt. Phys., 1992, 2(11), 2039–2063 CAS.
- J. Briggs, H. Chung and M. Caffrey, J. Phys. B: At., Mol. Opt. Phys., 1996, 6(5), 723–751 CAS.
- J. M. Seddon, Biochim. Biophys. Acta, 1990, 1031(1), 1 CrossRef CAS.
- U. S. Schwarz and G. Gompper, Langmuir, 2001, 17(7), 2084–2096 CrossRef CAS.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21(8), 3322–3333 CrossRef CAS PubMed.
- J. M. Seddon, Biochemistry, 1990, 29(34), 7997–8002 CrossRef CAS PubMed.
- J. M. Seddon, J. Robins, T. G. Krzywicki and H. Delacroix, Phys. Chem. Chem. Phys., 2000, 2(2), 4485–4493 RSC.
- I. Martiel, L. Sagalowicz and R. Mezzenga, Langmuir, 2013, 29(51), 15805–15812 CrossRef CAS PubMed.
- W. B. Lee, R. Mezzenga and G. H. Fredrickson, Phys. Rev. Lett., 2007, 99(18), 187801 CrossRef PubMed.
- J. Israelachvili, Colloids Surf., A, 1994, 91, 1–8 CrossRef CAS.
- C. Fong, T. Le and C. J. Drummond, Chem. Soc. Rev., 2012, 41(3), 1297–1322 RSC.
- K. Han, X. Pan, M. W. Chen, R. Wang, Y. H. Xu and M. Feng, et al., Eur. J. Pharm. Sci., 2010, 41(5), 692 CrossRef CAS PubMed.
- H. Qiu and M. Caffrey, Biomaterials, 2000, 21(3), 223–234 CrossRef CAS PubMed.
- J. C. Li, N. Zhu, J. X. Zhu, W. J. Zhang, H. M. Zhang and Q. Q. Wang, et al., Med. Sci. Monit., 2015, 21, 3298–3310 CrossRef PubMed.
- I. Martiel, L. Sagalowicz, S. Handschin and R. Mezzenga, Langmuir, 2014, 30(48), 14452–14459 CrossRef CAS PubMed.
- I. Martiel, N. Baumann, J. J. Vallooran, J. Bergfreund, L. Sagalowicz and R. Mezzenga, J. Controlled Release, 2015, 204, 78 CrossRef CAS PubMed.
- B. J. Boyd, S. M. Khoo, D. V. Whittaker, G. Davey and C. J. Porter, Int. J. Pharm., 2007, 340(1–2), 52 CrossRef CAS PubMed.
- B. M. Bertelsen, K. S. Korsholm, F. Rose, P. Nordly, H. Franzyk and P. Andersen, et al., RSC Adv., 2013, 3(43), 20673–20683 RSC.
- M. Sjölund, G. Lindblom, L. Rilfors and G. Arvidson, Biophys. J., 1987, 52(2), 145–153 CrossRef.
- P. O. Eriksson and G. Lindblom, Biophys. J., 1993, 64(1), 129–136 CrossRef CAS PubMed.
- L. T. Boni and S. W. Hui, Biochim. Biophys. Acta, 1983, 731(2), 177–185 CrossRef CAS.
- B. Tenchov, M. Rappolt, R. Koynova and G. Rapp, Biochim. Biophys. Acta, 1996, 1285(1), 109–122 CrossRef.
- B. Tenchov, R. Koynova and G. Rapp, Biophys. J., 1998, 75(2), 853–866 CrossRef CAS PubMed.
- M. H. And and H. Minamikawa, Langmuir, 1996, 12(6), 1658–1665 CrossRef.
- C. Fong, D. Wells, I. Krodkiewska, J. Booth and P. G. Hartley, J. Phys. Chem. B, 2007, 111(6), 1384 CrossRef CAS PubMed.
- K. Didehban and H. Namazib, Eur. Polym. J., 2010, 46(9), 1923–1931 CrossRef CAS.
- S. Milak and A. Zimmer, Int. J. Pharm., 2015, 478(2), 569–587 CrossRef CAS PubMed.
- R. Mezzenga, P. Schurtenberger, A. Burbidge and M. Michel, Nat. Mater., 2005, 4(10), 729–740 CrossRef CAS PubMed.
- Y. D. Dong, I. Larson, T. Hanley and B. J. Boyd, Langmuir, 2006, 22(23), 9512–9518 CrossRef CAS PubMed.
- D. H. Kim, S. Lim, J. Shim, J. E. Song, J. S. Chang and K. S. Jin, et al., ACS Appl. Mater. Interfaces, 2015, 7(36), 20438–20446 CAS.
- Y. M. Báez-Santos, A. Otte, E. A. Mun, B. K. Soh, C. G. Song and Y. N. Lee, et al., Int. J. Pharm., 2016, 514(1), 314–321 CrossRef PubMed.
- L. Sagalowicz, S. Guillot, S. Acquistapace, B. Schmitt, M. Maurer and A. Yaghmur, et al., Langmuir, 2013, 29(26), 8222–8232 CrossRef CAS PubMed.
- L. Bitancherbakovsky, I. Yuliamar, A. Aserin and N. Garti, Langmuir, 2010, 26(5), 3648–3653 CrossRef CAS PubMed.
- L. Bitancherbakovsky, I. Yuliamar, A. Aserin and N. Garti, Langmuir, 2009, 25(22), 13106–13113 CrossRef CAS PubMed.
- A. Yaghmur, C. L. De, S. Salentinig, L. Sagalowicz, M. E. Leser and O. Glatter, Langmuir, 2006, 22(2), 517–521 CrossRef CAS PubMed.
- J. Borné, A. T. Nylander and A. Khan, Langmuir, 2001, 17(25), 7742–7751 CrossRef.
- B. Angelov, A. Angelova, V. M. Garamus, G. Lebas, S. Lesieur and M. Ollivon, et al., J. Am. Chem. Soc., 2007, 129(44), 13474 CrossRef CAS PubMed.
- R. Negrini and R. Mezzenga, Langmuir, 2012, 28(47), 16455–16462 CrossRef CAS PubMed.
- A. Yaghmur, L. D. Campo, L. Sagalowicz, M. E. Leser and O. Glatter, Langmuir, 2006, 22(24), 9919–9927 CrossRef CAS PubMed.
- Y. Chen, X. Liang, P. Ma, Y. Tao, X. Wu and X. Chu, et al., AAPS PharmSciTech, 2015, 16(4), 846–854 CrossRef CAS PubMed.
- X. Jiang, X. Liang, S. Wang, J. Guo, Y. Tao and S. Gui, J. Pharm. Sci., 2017, 72(5), 249–256 CAS.
- I. A. Yuli, E. Wachtel, D. E. Shalev, A. Aserin and N. Garti, J. Phys. Chem. B, 2008, 112(13), 3971–3982 CrossRef PubMed.
- J. Bender, C. Simonsson, M. Smedh, S. Engström and M. B. Ericson, J. Controlled Release, 2008, 129(3), 163–169 CrossRef CAS PubMed.
- L. S. Helledi and L. Schubert, Drug Dev. Ind. Pharm., 2001, 27(10), 1073 CrossRef CAS PubMed.
- S. Engström and L. Engström, Int. J. Pharm., 1992, 79(2–3), 113–122 CrossRef.
- B. J. Boyd, S. M. Khoo, D. V. Whittaker, G. Davey and C. J. Porter, Int. J. Pharm., 2007, 340(1–2), 52–60 CrossRef CAS PubMed.
- L. B. Lopes, F. F. Speretta and M. V. Bentley, Eur. J. Pharm. Sci., 2007, 32(3), 209 CrossRef CAS PubMed.
- E. Esposito, R. Cortesi, M. Drechsler, L. Paccamiccio, P. Mariani, C. Contado, E. Stellin, E. Menegatti, F. Bonina and C. Puglia, Pharm. Res., 2005, 22(12), 2163–2173 CrossRef CAS.
- D. Yariv, R. Efrat, D. Libster, A. Aserin and N. Garti, Colloids Surf., B, 2010, 78(2), 185–192 CrossRef CAS PubMed.
- M. Cohenavrahami, A. Aserin and N. Garti, Colloids Surf., B, 2010, 77(2), 131–138 CrossRef CAS PubMed.
- J. M. Seddon, A. M. Squires, C. E. Conn, O. Ces, A. J. Heron and X. Mulet, et al., Philos. Trans. R. Soc., A, 2006, 364(1847), 2635–2655 CrossRef CAS PubMed.
- A. Yaghmur, M. Kriechbaum, H. Amenitsch, M. Steinhart, P. Laggner and M. Rappolt, Langmuir, 2010, 26(2), 1177–1185 CrossRef CAS PubMed.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm., 2010, 67(3), 217 CAS.
- P. Costa and J. M. S. Lobo, Eur. J. Pharm. Sci., 2001, 13(2), 123 CrossRef CAS PubMed.
- P. Costa, D. C. Ferreira and J. M. S. Lobo, Rev. Port. Farm., 1996, 46(1), 2–8 Search PubMed.
- E. Rosenbaum, S. Tavelin and L. B. Johansson, Int. J. Pharm., 2010, 388(1), 52–57 CrossRef CAS PubMed.
- A. R. Ahmed, A. Dashevsky and R. Bodmeier, Eur. J. Pharm. Biopharm., 2008, 70(3), 765–769 CrossRef CAS PubMed.
- L. M. Antognini, S. Assenza, C. Speziale and R. Mezzengaa, J. Chem. Phys., 2016, 145(8), 084903 CrossRef PubMed.
- K. W. Lee, T. H. Nguyen, T. Hanley and B. J. Boyd, Int. J. Pharm., 2009, 365(1–2), 190–199 CrossRef CAS PubMed.
- B. J. Boyd, D. V. Whittaker, S. M. Khoo and G. Davey, Int. J. Pharm., 2006, 318(1–2), 154–162 CrossRef CAS PubMed.
- T. H. Nguyen, T. Hanley, C. J. Porter, I. Larson and B. J. Boyd, J. Pharm. Pharmacol., 2010, 62(7), 844–855 CrossRef CAS PubMed.
- M. Johnsson, J. Barauskas, A. Norlin and F. Tiberg, J. Nanosci. Nanotechnol., 2006, 6(9–10), 3017–3024 CrossRef CAS PubMed.
- R. Parmar, R. Misra and S. Mohanty, Colloids Surf., B, 2015, 129, 198–205 CrossRef CAS PubMed.
- J. C. Bode, J. Kuntsche, S. S. Funari and H. Bunjes, Int. J. Pharm., 2013, 448(1), 87–95 CrossRef CAS PubMed.
- M. S. Freag, Y. S. R. Elnaggar, D. A. Abdelmonsif and O. Y. Abdallah, Int. J. Nanomed., 2016, 11, 4799–4818 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, M. Drechsler, V. M. Garamus and S. Lesieur, Int. J. Pharm., 2013, 454(2), 625–632 CrossRef CAS PubMed.
- J. Barauskas, M. Johnsson and F. Tiberg, Nano Lett., 2005, 5(8), 1615–1619 CrossRef CAS PubMed.
- J. Barauskas, C. Cervin, M. Jankunec, M. Špandyreva, K. Ribokaite, F. Tiberg and M. Johnsson, Int. J. Pharm., 2010, 391(1–2), 284–291 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |