
Transition-metal-free radical cleavage of a hydrazonyl N–S bond: tosyl radical-initiated cascade C(sp3)–OAr cleavage, sulfonyl rearrangement and atropisomeric cyclopropanation†
Yao-Zhong
Chen
a,
Teng
Liu
a,
Jie
Zhu
 a,
Hui
Zhang
b and
Lei
Wu
*ac
a,
Hui
Zhang
b and
Lei
Wu
*ac
aJiangsu Key Laboratory of Pesticide Science and Department of Chemistry, College of Sciences, Nanjing Agricultural University, Nanjing 210095, China. E-mail: rickywu@njau.edu.cn
bNanjing Forestry University, Nanjing 210037, China
cBeijing National Laboratory for Molecular Sciences and Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 2nd November 2018
Abstract
For the first time, the combination of catalytic 1,10-phenanthroline and potassium carbonate enables a radical cleavage of a hydrazonyl N–S bond to deliver a tosyl radical and a diazo compound, along with releasing molecular hydrogen spontaneously. The tosyl radical and hydrogen evolution are witnessed by radical trapping, EPR experiment and gas chromatography. An innovative application of this strategy into metal-free coupling of N-tosylhydrazone and phosphinyl allene provides a one-step synthesis of novel atropisomeric 3-tosyl-1-enyl-cyclopropyl-diphenylphosphine oxide derivatives with excellent diastereoselectivity and E-selectivity. Moreover, these atropisomeric products, verified by 1H-NMR, DFT calculations and X-ray crystallography, are controllably formed. Mechanistic studies indicate that the multistep cascade reaction occurs through radical hydrazonyl N–S bond cleavage, radical C(sp3)–OAr bond cleavage, sulfonyl rearrangement and atropisomeric cyclopropanation.
Introduction
In the past decade, the green and sustainable strategy for free-radical generation from a diamine/alkali metal salt system has contributed greatly to cross-coupling reactions, which received significant attention from scientific and industrial communities.1 Contributions by Hayashi, Shi, Lei, Murphy and others have witnessed the blooming of this chemistry,2 lately classified as base-promoted homolytic aromatic substitution (BHAS),3 in various inter/intramolecular C–C couplings between aryl halides and arenes or olefins (Scheme 1a). Although potassium t-butoxide alone was reported to enable the couplings,4 a wide range of organic additives including 1,10-phenanthroline derivatives, aliphatic 1,2-diamines, imines, amino acids, hydrazine derivatives, N-heterocyclic carbenes, and diphosphines have been used in initiating the radical chain process more efficiently.2a–g,3b,5 From both experimental facts and theoretical calculation considerations,2i a single electron transfer process has been verified, for example, 1,10-phenanthroline/t-BuOK initiated radical cleavage of the aryl–I bond will deliver an aryl radical and an iodide anion. We believe that this strategy should be ideally suitable for developing novel radical chain reactions other than C–C couplings; however, to the best of our knowledge, this field remains untouched so far.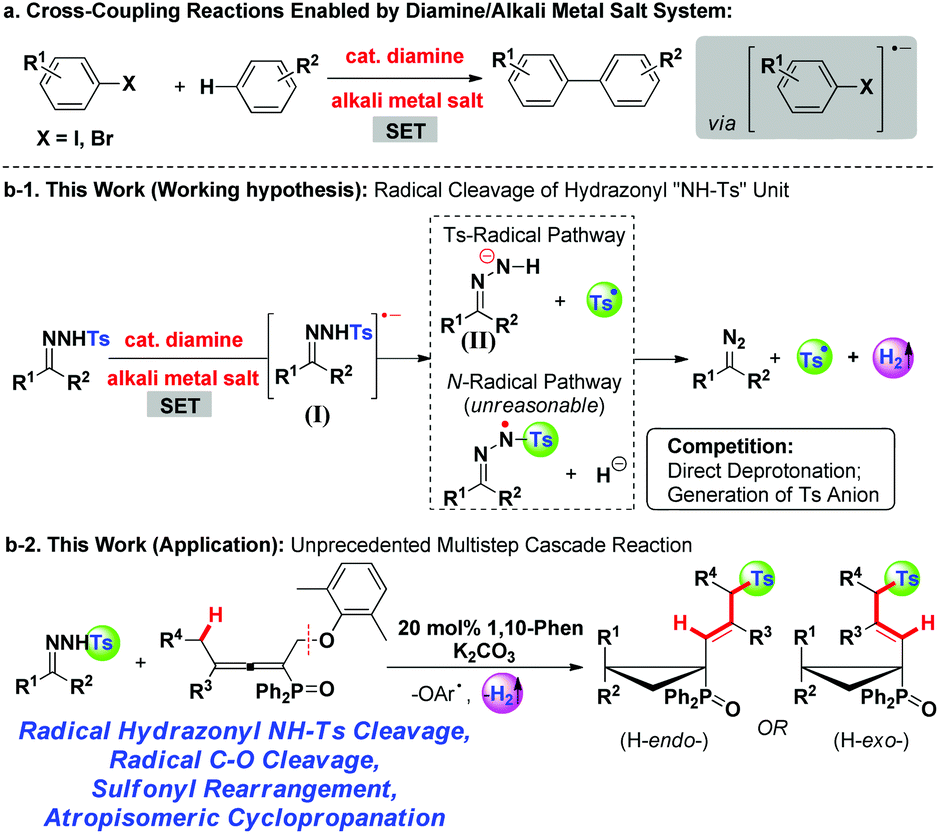 | ||
| Scheme 1 Previous work with diamine/alkali metal salt combination (a) and this work (b-1, b-2). | ||
The pursuit of N-tosylhydrazone chemistry dates back to the mid-20th century, as well-known as the Bamford–Stevens reaction and Shapiro reaction.6 Since the first report of palladium-catalyzed coupling of N-tosylhydrazones with arylbromides in 2007, this chemistry has been attracting extensive interest in providing diazo precursors in building versatile functionalities for synthetic compounds, led by the groups of Barluenga and Valdés,7 Wang8 and others.9 Despite these great achievements, only a handful of methods have been established regarding free-radical generation from N-tosylhydrazone, albeit confined to a transition metal-catalysis system.10 For example, Xiao's group has established a photocatalytic hydroamination of N-tosylhydrazones with intramolecular alkenes, in which N-radical formation from single-electron oxidation of deprotonated N-tosylhydrazones was involved.10a,b Very recently, cobaltIII–carbene radicals, generated through metalloradical activation of N-tosylhydrazones by cobaltII complexes, were developed in the reactions of N-tosylhydrazones and olefins, alkynes or alkanes.10d–f,10h–i Inspired by the aforementioned studies and our previous studies,11 we hypothesized that, a diamine/alkali metal salt excited hydrazonyl radical anion might undergo a probable Ts-radical pathway (N–S bond cleavage)12 to furnish a diazo compound, tosyl radical and molecular hydrogen (Scheme 1b-1), and this unprecedented strategy should offer a powerful and important platform for discovering new radical chemistry of N-tosylhydrazones with allenes toward complex molecules.
Results and discussion
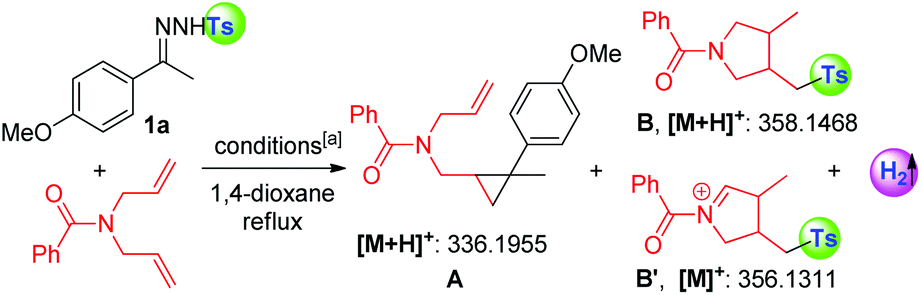
With an aim to rationalize our hypothesis, an EPR experiment for the reaction of 1-(1-(4-methoxyphenyl)ethylidene)-2-tosylhydrazine (1a), 20 mol% of 1,10-phen and 1.5 equivalents of t-BuOK was conducted to monitor the formation of radical species, which indicated the formation of the tosyl radical (see more details in the ESI†).13 The tosyl radical could also be captured by TEMPO as monitored by HR-MS (in ESI†). As shown in Table 1, N,N-diallylbenzamide was subsequently applied to trap the fragments cleaved from N-tosylhydrazone (1a). In preliminary results, 20 mol% of 1,10-phen and 1.5 equivalents of t-BuOK or K2CO3 successfully enabled radical cleavage of the “NHTs” unit (entries 1 and 2), which was strongly supported by the detection of adducts A, B and B′, deriving from the diazo precursor and the tosyl radical, respectively. Interestingly, a catalytic amount of potassium carbonate afforded the same result of fragment abundance in the HRMS spectrum, even for the case with high purity reagents (entries 3 and 4). A parallel experiment without 1,10-phen gave only the cyclopropanation product (A, entry 5). The observations in entries 3 and 4 deserve to be highlighted since the tosyl anion could be generated in the presence of excess base, and a single-electron oxidation of the tosyl anion by metal catalysts has been reported to afford the tosyl radical;14 however, the negative result of sodium 4-methylbenzene-sulfinate and N,N-diallyl-benzamide entirely ruled out the possibility of forming the tosyl radical from the tosyl anion under the current conditions (entry 6). Although TBAI/TBHP evidenced a positive result in producing the trapping products (B and B′) from the tosyl radical as monitored by HR-MS, it was found that the diazo precursor decomposed rapidly (entry 7). The results in entries 8 and 9 showed that a palladium catalyst would not inhibit the radical process but did not deliver the tosyl radical without the participation of 1,10-phen. Intriguingly, molecular hydrogen evolution was detected by gas chromatography in entries 1–4 and 8, indicating that the hydrogen radical or hydrogen anion might be produced in the system (in ESI†).|
|
|||
|---|---|---|---|
| Entry | Radical initiator | HR-MS result | H2 evolution |
| a N-Tosylhydrazone (1a, 0.1 mmol), N,N-diallylbenzamide (0.12 mmol), catalyst or oxidant in 3 mL refluxing 1,4-dioxane for 2 hours. b High purity reagents: 1,10-phen (anhydrous, purity > 99 + %), K2CO3 (purity: 99.997%). c TsNa was used instead of 1a. | |||
| 1 | 20 mol% 1,10-phen/1.5 equiv. t-BuOK | A + B + B′ | Yes |
| 2 | 20 mol% 1,10-phen/1.5 equiv. K2CO3 | A + B + B′ | Yes |
| 3 | 20 mol% 1,10-phen/15 mol% K2CO3 | A + B + B′ | Yes |
| 4b | 20 mol% 1,10-phen/15 mol% K2CO3 | A + B + B′ | Yes |
| 5 | 1.5 equiv. K2CO3 | Only A | No |
| 6c | 20 mol% 1,10-phen/15 mol% K2CO3 | — | No |
| 7 | 0.2 equiv. TBAI/2 equiv. TBHP | B + B′ | No |
| 8 | 5 mol% Pd(OAc)2/20 mol% 1,10-phen/15 mol% K2CO3 | A + B + B′ | Yes |
| 9 | 5 mol% Pd(PPh3)2Cl2/15 mol% K2CO3 | Only A | No |
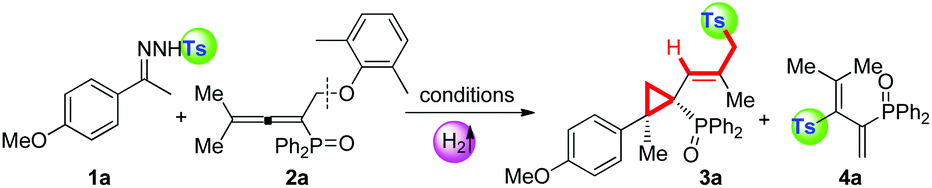
Subsequently, we applied this strategy in the coupling of N-tosylhydrazones and phosphinyl allenes (Scheme 1b-2). This choice was based on our previous report of a transition-metal-catalyzed coupling of N-tosylhydrazones with phosphinyl allenes to form phosphinyl [3]dendralenes,11a which we believe would be a model reaction suitable for providing an expeditious platform for investigating the reactivity of the tosyl radical and diazo compound in situ generated from N-tosylhydrazones and differentiating the 1,10-phen/K2CO3 system from metal contamination.1a,15 The study was initiated by examining the coupling of 1a with phosphinyl allene (2a). As shown in Table 2, for a preliminary experiment with a catalytic amount of 1,10-phen and t-BuOK, 6% yield of (E)-(2-(4-methoxy phenyl)-2-methyl-1-(2-methyl-3-tosylprop-1-en-1-yl)cyclopropyl)diphenyl-phosphine oxide (3a) was obtained with high diastereoselectivity, along with 50% yield of tosylated 1,3-diene (4a) (entry 2). The ratios of 3a/4a changed in proportion to the amount of base used (entries 3–5), in which 2.5 equivalents of K2CO3 improved the isolated yield of 3a up to 74% (Note: this observation does not conclude that stoichiometric base is necessary to generate the tosyl radical, instead, excess base plays a vital role in converting 4a into 3a in a sulfonyl rearrangement step). A series of bases, including inorganic and organic bases, were then screened. Weaker inorganic bases, such as Na2CO3, NaHCO3 and CsF, enabled the cascade reactions with yields ranging from 26 to 56%, whereas organic bases entirely inhibited the reaction (see the ESI†). Other catalysts of pyridine or phenanthroline derivatives were tested, which exhibited considerable activities as well (entries 10–13). The cascade reaction was also susceptible to solvents and catalyst loadings (in ESI†). Control experiments under transition-metal catalysis were conducted as a comparison. Transition metals, including palladium, cobalt, nickel, rhodium, copper, zinc, manganese, iron and iridium, were investigated; however, all of the results were not comparable (entries 14–17, also in the ESI†). Most importantly, a high-impurity of 1,10-phenanthroline and potassium carbonate delivered the final product 3a in a slightly better yield of 75% (entry 18), excluding the possibility of a metal-catalytic pathway and keeping in line with the HR-MS evaluations given in Table 1. The above observations indicated that the tosyl radical produced by 1,10-phenanthroline and an alkali metal cation plays a key role in cleavage of the C(sp3)–OAr bond, probably via a radical pathway, achieving a multistep cascade reaction with high efficiency.
|
|
|||
|---|---|---|---|
| Entry | Catalyst (20 mol%) | Base | Yield (%) (3a/4a)b |
a Reaction conditions: N-Tosylhydrazone (1a, 0.9 mmol), phosphinyl allene (2a, 0.3 mmol), catalyst (20 mol%) and base in 3 mL refluxing 1,4-dioxane for 8 hours.
b Isolated yield by column chromatography (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, only E-isomer for all cases).
c 5 mol% palladium catalyst.
d The major product was phosphinyl[3]dendralenes, see our previous work in ref. 11a.
e High purity reagents: 1,10-phen (anhydrous, purity > 99 + %), K2CO3 (purity: 99.997%). :1, only E-isomer for all cases).
c 5 mol% palladium catalyst.
d The major product was phosphinyl[3]dendralenes, see our previous work in ref. 11a.
e High purity reagents: 1,10-phen (anhydrous, purity > 99 + %), K2CO3 (purity: 99.997%).
|
|||
| 1 | 1,10-Phen | t-BuOK (15 mol%) | 0/14 |
| 2 | 1,10-Phen | t-BuOK (40 mol%) | 6/50 |
| 3 | 1,10-Phen | K2CO3 (15 mol%) | <5/85 |
| 4 | 1,10-Phen | K2CO3 (1.5 equiv.) | 35/30 |
| 5 | 1,10-Phen | K 2 CO 3 (2.5 equiv.) | 74/0 |
| 6 | 1,10-Phen | NaHCO3 (2.5 equiv.) | 56/0 |
| 7 | 1,10-Phen | CsF (2.5 equiv.) | 53/0 |
| 8 | 1,10-Phen | Cs2CO3 (2.5 equiv.) | 17/0 |
| 9 | 1,10-Phen | DBU (2.5 equiv.) | 0/0 |
| 10 | 2,2′-Bipyridine | K2CO3 (2.5 equiv.) | 38/13 |
| 11 | 2,9-Dimethyl-1,10-phenanthroline | K2CO3 (2.5 equiv.) | 36/11 |
| 12 | 2,2′:6′,2′′-Terpyridine | K2CO3 (2.5 equiv.) | 42/9 |
| 13 | 4,7-Dimethoxy-1,10-phenanthroline | K2CO3 (2.5 equiv.) | 50/0 |
| 14c,d | Pd(PPh3)2Cl2 | K2CO3 (2.5 equiv.) | 0/0 |
| 15c | Pd(OAc)2/1,10-phen | K2CO3 (2.5 equiv.) | 40/0 |
| 16c | Cu(OAc)2/1,10-phen | K2CO3 (2.5 equiv.) | 0/0 |
| 17c | CoCl2/1,10-phen | K2CO3 (2.5 equiv.) | 21/0 |
| 18e | 1,10-Phen | K2CO3 (2.5 equiv.) | 75/0 |
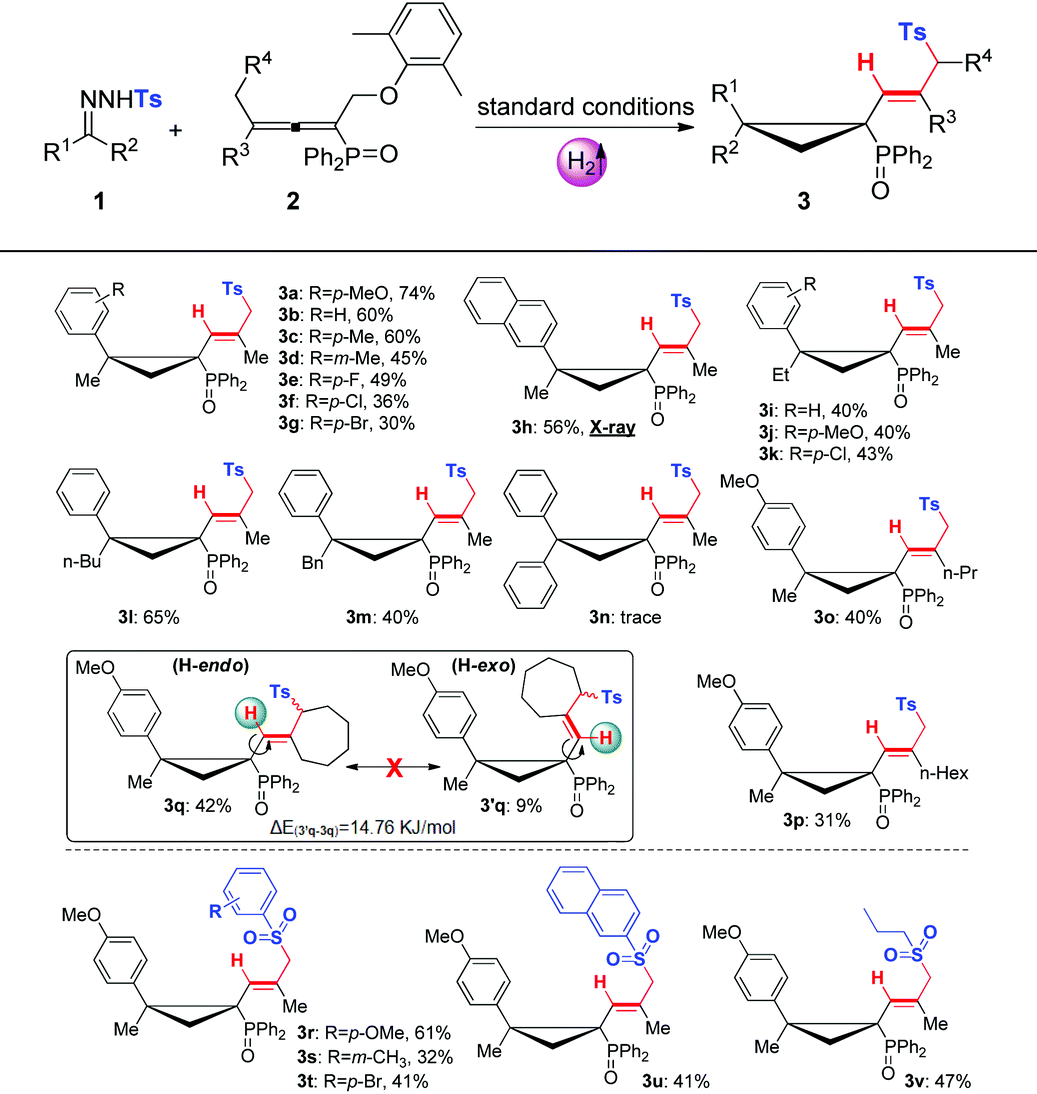
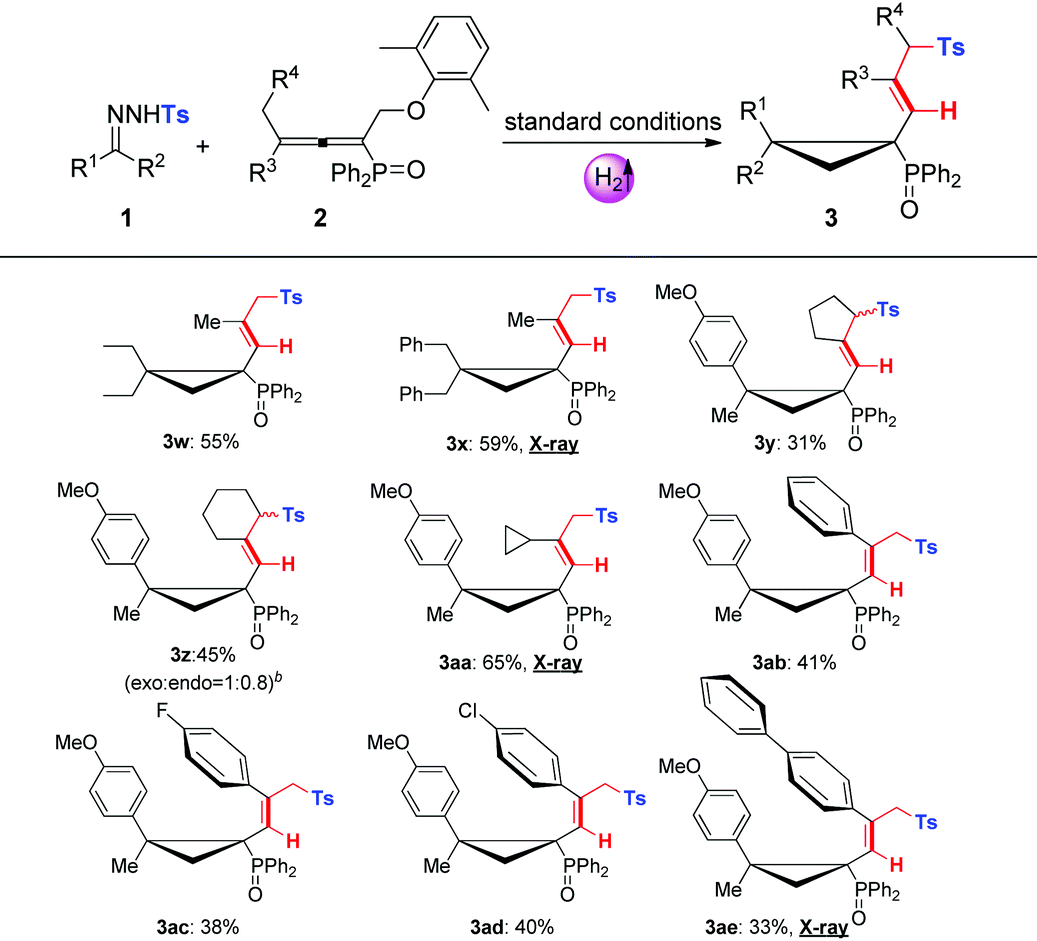
With the optimal conditions in hand, we then evaluated the substrate scopes of various N-tosylhydrazones (1) with phosphinyl allenes (2). As summarized in Table 3, the cascade reactions gave adducts (3) in yields ranging from acceptable to medium with excellent diastereoselectivities and E-selectivities, relying on the structure and electronic properties of substituents. For instance, electron-neutral N-tosylhydrazones and para-electron-rich ones afforded the products 3a–3c with medium yields. Otherwise, electron-deficient substituents, including p-F, p-Cl, and p-Br, impaired the efficiency to some extent, with yields dropping to below 50%. N-Tosylhydrazones bearing strong electron-deficient substituents of p-CF3 and p-NO2 performed very sluggishly without formation of the corresponding products. 56% yield of 3h was obtained for β-naphthyl substituted substrates. Notably, the X-ray crystal structure of 3h unambiguously displayed the location of the tosyl group and the dimensional orientations of anchored moieties,16 with methyl and diphenylphosphine oxide groups at syn-positions. Ethyl, n-butyl, and benzyl derived N-tosylhydrazones were also applicable to the system with acceptable to medium yields (3i–3m). Nevertheless, diphenyl derived N-tosyl-hydrazone performed sluggishly under current conditions. The phosphinyl allenes could be expanded to other aliphatic substitutions. For instance, the allenes with distal n-propyl, n-hexyl substituents furnished the target products (3o, 3p) in yields of 40% and 31%, respectively. Other N-sulfonylhydrazone derivatives, including p-methoxyphenyl, m-methylphenyl, p-bromophenyl, polyaromatic and aliphatic ones, maintained certain reactivities in the radical cleavage and sulfonyl rearrangement step, giving exclusively E-isomers (3r–3v) with slightly lower yields. Moreover, it is worth mentioning that most of the proton chemical shifts of products (3a–3p, 3r–3v) locate in a range of 5.20–5.46 ppm, with alkenyl-H pointing to the top side of the three-membered ring (denoted as the H-endo-isomer). An exceptional case was found in the reaction of allenes bearing cycloheptyl substitution with 1a. Interestingly, 42% yield of H-endo-isomer 3q and 9% yield of H-exo-isomer 3′q were both isolated, and 1H-NMR spectra of 3′q showed a significant downfield shift to 6.04 ppm. No reciprocal transformation was detected for these two isomers under thermal conditions, indicating that the cyclopropanation step should be decisive to obtain either atropisomer. Besides, this spatial configuration could be attributed to the congestion of moieties on a three-membered ring, in which aryl and diphenylphosphine oxide substituents strongly restricted the single bond rotation of the alkenyl moiety. DFT calculations showed that 3′q possesses higher energy than 3q with an energy gap of 20 kJ mol−1, and the torsion energy associated with the dihedral angle between 3q and 3′q is as high as 59 kJ mol−1 (see details in the ESI†).
|
a Isolated yield by column chromatography, dr > 20:1, only E-isomer.
|
|---|
|
To our delight, dominant H-exo-isomers appeared while altering the substitutions on N-tosylhydrazones and phosphinyl allenes (Table 4). In the case of diethyl and dibenzyl derived N-tosyl-hydrazones, products 3w and 3x were characterized as H-exo-configurations, evidenced by distinct alkene-H chemical shifts (5.62, 5.86 ppm) and an X-ray crystal structure of 3x.16 The ORTEP drawing of 3x unambiguously manifested the spatial proximity of the alkene-H atom to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety, thus the deshielding effect resulted in an apparent downfield chemical shift. The scope of phosphinyl allenes was subsequently investigated. In general, the allenes bearing distal cyclopropyl, cyclopentyl and various aryl substituents furnished the corresponding H-exo-isomers exclusively in yields of 31–65% (3y–3ae), except for the cyclohexyl one (3z, exo:endo = 1:0.8). Meanwhile, the DFT calculation results were also well consistent with the ratio of product configurations (in the ESI†). X-ray crystal structures of 3aa and 3ae supported the formation of single atropisomers as well. Distinct from the substituents on hydrazones, electron effects were not apparent for allenes with terminal aromatics. Electron-deficient groups of para-fluoro and para-chloro gave indistinctive results compared to that of the electron-neutral substrate. Note that the development of atropisomers is currently a central task and hot research topic in organic chemistry.17 However, as far as we know, atropisomeric cyclopropanation has never been reported and this cascade reaction provides highly efficient construction of multi-substituted atropisomers in one pot.
O moiety, thus the deshielding effect resulted in an apparent downfield chemical shift. The scope of phosphinyl allenes was subsequently investigated. In general, the allenes bearing distal cyclopropyl, cyclopentyl and various aryl substituents furnished the corresponding H-exo-isomers exclusively in yields of 31–65% (3y–3ae), except for the cyclohexyl one (3z, exo:endo = 1:0.8). Meanwhile, the DFT calculation results were also well consistent with the ratio of product configurations (in the ESI†). X-ray crystal structures of 3aa and 3ae supported the formation of single atropisomers as well. Distinct from the substituents on hydrazones, electron effects were not apparent for allenes with terminal aromatics. Electron-deficient groups of para-fluoro and para-chloro gave indistinctive results compared to that of the electron-neutral substrate. Note that the development of atropisomers is currently a central task and hot research topic in organic chemistry.17 However, as far as we know, atropisomeric cyclopropanation has never been reported and this cascade reaction provides highly efficient construction of multi-substituted atropisomers in one pot.
Next, in order to understand the reaction mechanism, control experiments were performed. Delightfully, while shortening the reaction time to 1 hour, we isolated the key intermediate, tosylated 1,3-dienes (4a), in a good yield of 85% (eqn (a), Scheme 2). Kinetic studies on this step verified that it proceeds through a radical pathway, and the aromatic ether moiety left in the radical form is trapped by BHT (in ESI†). The parallel reaction of tosyl sodium and allene 2a left all substrates intact (eqn (b)), implying that the tosyl anion cannot cleave the C–O bond to initiate the cascade reaction. Eqn (c) presented that the intermediate 4a could be converted into a sulfonyl rearrangement product 5a,18 but stoichiometric K2CO3 turned out to be necessary, otherwise, this step severely retarded the cascade process. A crossover experiment with sodium 4-methoxybenzenesulfinate in eqn (d) indicated that the sulfonyl rearrangement step occurs inter-molecularly. After that, 5a underwent atropisomeric cyclopropanation rapidly to afford final product 3a in 60% yield. Besides, deuterium labelling experiments were conducted to investigate the kinetic isotope effects (KIE) (eqn (e)). The reaction of 1a and 2a-D6 afforded deuterated product 4a-D6 in 84% yield, which transformed into 5a-D in 60% yield with deuterium atoms distributed over all of the alkenyl positions. The KIE value from two parallel reactions between 4a and 4a-D6 was found to be kH/kD = 1.5, suggesting that the rate-determining step probably involves in the sulfonyl rearrangement step, but does not contain a C(sp3)–H bond cleavage. Therefore, the mechanism studies and results in Table 1 collectively pointed out that the cascade reaction consists of four steps: 1,10-phen/K2CO3 catalysed cleavage of the NH-Ts bond to generate the tosyl radical, radical cleavage of the C(sp3)–OAr bond, base-promoted sulfonyl rearrangement, and atropisomeric cyclopropanation.
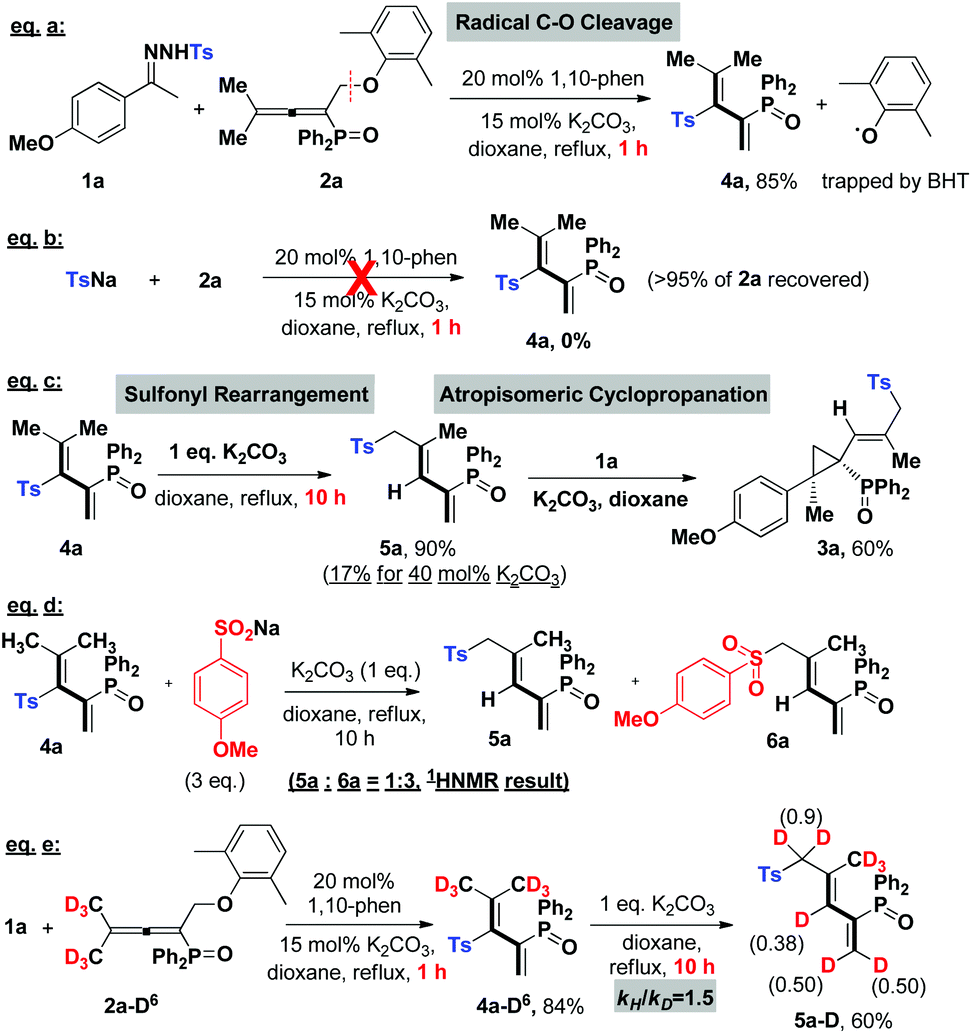 | ||
| Scheme 2 Mechanism studies. | ||
Based on our experimental facts and previous reports,2a,10g,19 a proposed mechanism is outlined in Scheme 3. Initially, single electron transfer (SET) from the 1,10-phen/K2CO3 complex to N-tosylhydrazone (1) gives a radical anion (I), which undergoes N–S bond cleavage to result in a tosyl radical and an N-anion intermediate (II) ((II) could be trapped by the CF3 cation source and detected by HR-MS, see details in the ESI†). The N-anion intermediate (II) will leave the hydrogen anion rapidly to give a diazo compound [C]. Molecular hydrogen evolution comes from the deprotonation of 1 by the in situ generated hydrogen anion. Meanwhile, the accompanying tosyl anion helps to quench the associated radical cation (IV) to end the catalytic cycle and releases another portion of the tosyl radical. This proposed catalytic cycle keeps in line with the result of catalytic amounts of 1,10-phen and K2CO3 in eqn (a), Scheme 2. Afterwards, the tosyl radical attacks allene 2 to yield tosylated 1,3-butadienes (4). Base-promoted sulfonyl rearrangement of 4 leads to the formation of thermally reversible (E)-S-trans or (E)-S-cis isomers (5/5′), depending on the nature of substituents on the olefin double bond. Eventually, these two isomers undertake 1,3-dipolar cycloaddition with diazo intermediate [C] to give atropisomers 3 in the H-exo or H-endo configuration, along with the extrusion of nitrogen. Above all, the equilibrium ratios of 5 and 5′ are associated directly with the dominant stereo-configuration of atropisomers. For instance, when the R3 group is a bulky phenyl group, an (E)-S-cis isomer is favourably formed and rapidly transforms into an H-exo-isomer (3ab).
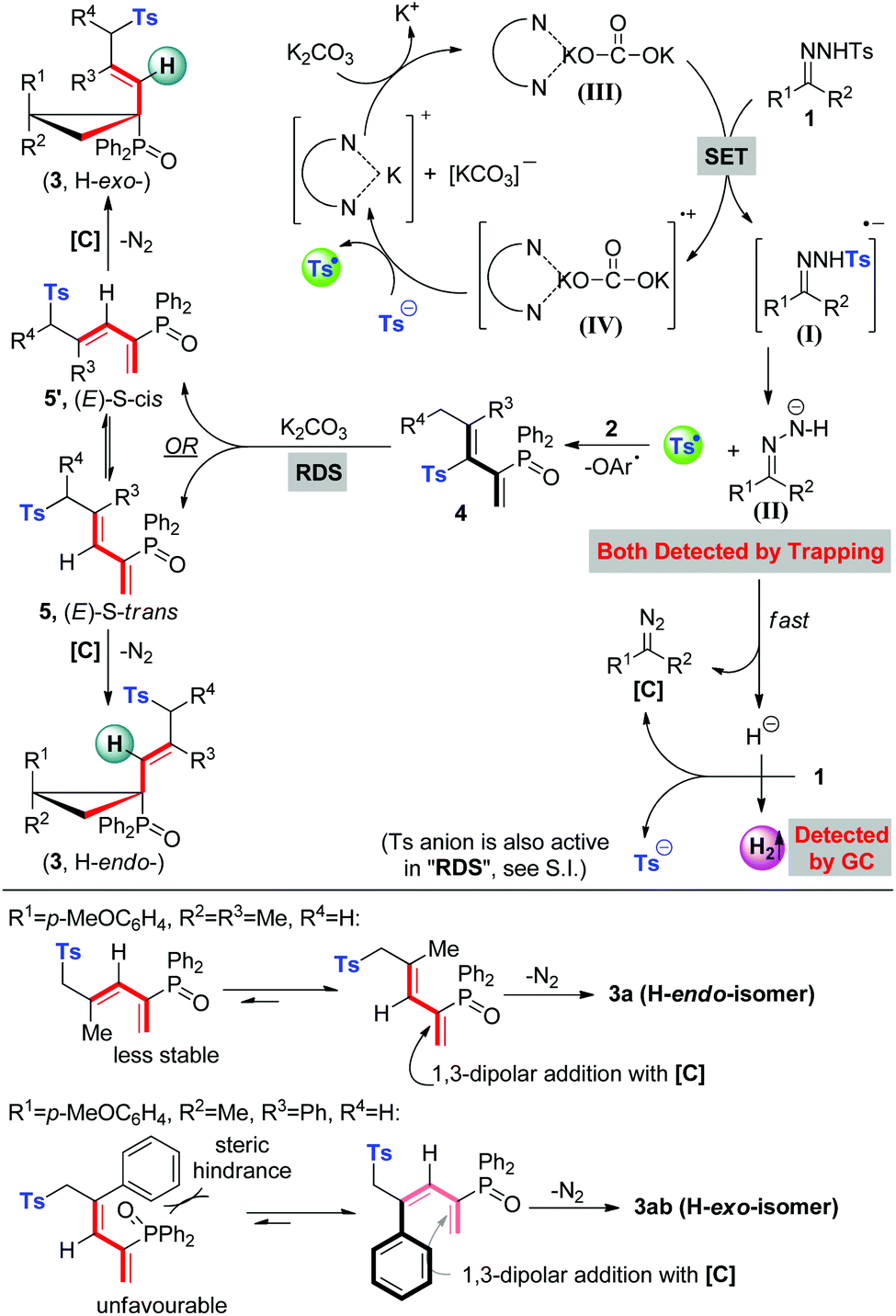 | ||
| Scheme 3 Proposed mechanism and rationalization of atropisomer formation. | ||
Experimental
General procedure
To a 15 mL sealed reaction tube were added N-tosylhydrazones (1a, 286 mg, 0.9 mmol), phosphinyl allenes (2a, 120 mg, 0.3 mmol), 1,10-phen (12 mg, 20 mol%), K2CO3 (103.5 mg, 0.75 mmol), and 3 mL degassed 1,4-dioxane. The reaction mixture was then heated to reflux for 8 hours until the complete consumption of 2a monitored by TLC. After all of the volatiles were removed under vacuum, the crude product was purified by flash chromatography (eluent: 1:2:1 (v/v) of ethyl acetate/petroleum ether) to afford product 3a as a white solid.
Conclusions
In conclusion, we disclosed, for the first time, a transition-metal-free radical cleavage of a hydrazonyl “NH-Ts” unit with a catalytic amount of 1,10-phenanthroline and potassium carbonate. The cleavage of N-tosylhydrazone through a radical anion pathway toward a tosyl radical, diazo compound and molecular hydrogen was rationalized by DFT calculations and verified by radical trapping, EPR experiment and gas chromatography. Further application of this novel strategy into metal-free coupling of N-tosylhydrazones and phosphinyl allenes provides expeditious synthesis of unprecedented atropisomeric 3-tosyl-1-enyl-cyclopropyldiphenyl-phosphine oxide derivatives with excellent diastereoselectivity and E-selectivity. A variety of atropisomeric products, confirmed by 1H NMR, DFT calculations, and X-ray crystallography, were controllably formed depending on the substitutions of N-tosyl-hydrazone and phosphinyl allenes. Mechanistic studies indicate that the cascade reaction proceeds through a tosyl radical initiated successive C(sp3)–OAr cleavage, sulfonyl rearrangement and atropisomeric cyclopropanation. We expect that this strategy will enrich the blooming radical chemistry and offer new scaffolds for the development of potential chiral molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Fundamental Research Funds for the Central Universities (Grant No. KYDZ201904 and KYTZ201604) and the “333 High Level Talent Project” of JiangSu Province. Special thanks go to Dr Peng-Cheng Wang (Nanjing University of Science & Technology) for DFT calculations.Notes and references
- For selected reviews: (a) N. E. Leadbeater, Nat. Chem., 2010, 2, 1007 CrossRef CAS PubMed; (b) A. Stude and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef PubMed; (c) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed.
- (a) E. Shirakawa, K. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537 CrossRef CAS PubMed; (b) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS PubMed; (c) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS PubMed; (d) M. Rueping, M. Leiendecker, A. Das, T. Poisson and L. Bui, Chem. Commun., 2011, 47, 10629 RSC; (e) C.-L. Sun, Y.-F. Gu, W.-P. Huang and Z.-J. Shi, Chem. Commun., 2011, 47, 9813 RSC; (f) H. Zhang, R. Shi, A. Ding, L. Lu, B. Chen and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 12542 CrossRef CAS PubMed; (g) S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818 CrossRef CAS PubMed; (h) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (i) L. Zhang, H. Yang and L. Jiao, J. Am. Chem. Soc., 2016, 138, 7151 CrossRef CAS PubMed; (j) J. Ahmed, P. Sreejyothi, G. Vijaykumar, A. Jose, M. Raj and S. K. Mandal, Chem. Sci., 2017, 8, 7798 RSC.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed; (b) A. Dewanji, S. Murarka, D. P. Curran and A. Studer, Org. Lett., 2013, 15, 6102 CrossRef CAS PubMed.
- (a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673 CrossRef CAS PubMed; (b) M. P. Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Angew. Chem., Int. Ed., 2015, 54, 10587 CrossRef PubMed; (c) Q. Shi, S. Zhang, J. Zhang, V. F. Oswald, A. Amassian, S. R. Marder and S. B. Blakey, J. Am. Chem. Soc., 2016, 138, 3946 CrossRef CAS PubMed; (d) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402 CrossRef CAS PubMed; (e) J. Chen and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 3951 CrossRef CAS PubMed.
- (a) W.-C. Chen, Y.-C. Hsu, W.-C. Shih, C.-Y. Lee, W.-H. Chuang, Y.-F. Tsai, P. P.-Y. Chen and T.-G. Ong, Chem. Commun., 2012, 48, 6702 RSC; (b) S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476 RSC; (c) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., Int. Ed., 2016, 55, 4492 CrossRef CAS PubMed; (d) M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139, 16327 CrossRef CAS PubMed; (e) Z. Guo, M. Li, X.-Q. Mou, G. He, X.-S. Xue and G. Chen, Org. Lett., 2018, 20, 1684 CrossRef CAS PubMed.
- (a) W. R. Bamford and T. S. Stevens, J. Chem. Soc., 1952, 4735 RSC; (b) R. H. Shapiro, J. H. Duncan and J. C. Clopton, J. Am. Chem. Soc., 1967, 89, 471 CrossRef CAS; (c) R. H. Shapiro and M. J. Heath, J. Am. Chem. Soc., 1967, 89, 5734 CrossRef CAS; (d) R. H. Shapiro, Org. React., 1976, 23, 405 CAS; (e) R. M. Adlington and A. G. M. Barrett, Acc. Chem. Res., 1983, 16, 55 CrossRef CAS. For selected recent reviews, see: (f) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 8, 7486 CrossRef PubMed; (g) Y. Zhang and J. Wang, Top. Curr. Chem., 2012, 1, 1 Search PubMed; (h) Z.-H. Shao and H.-B. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC; (i) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS; (j) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134 CrossRef CAS PubMed; (k) Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306 RSC; (l) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed.
- (a) J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS PubMed; (b) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494 CrossRef CAS PubMed; (c) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 122, 5113 CrossRef; (d) J. Barluenga, M. Escribano, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 6856 CrossRef CAS PubMed; (e) J. Barluenga, N. Quiñones, M.-P. Cabal, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 2350 CrossRef CAS PubMed; (f) J. Barluenga, M. Tomás-Gamasa and C. Valdés, Angew. Chem., Int. Ed., 2012, 51, 5950 CrossRef CAS PubMed; (g) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2013, 52, 7219 CrossRef PubMed; (h) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2015, 54, 13729 CrossRef PubMed; (i) M. Plaza and C. Valdés, J. Am. Chem. Soc., 2016, 138, 12061 CrossRef CAS PubMed; (j) M. Paraja and C. Valdés, Org. Lett., 2017, 19, 2034 CrossRef CAS PubMed.
- (a) Q. Xiao, J. Ma, Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2009, 11, 4732 CrossRef CAS PubMed; (b) Z.-H. Zhang, Y.-Y. Liu, M.-X. Gong, X.-K. Zhao, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1139 CrossRef CAS PubMed; (c) Q. Xiao, Y. Xia, H. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2011, 50, 1114 CrossRef CAS PubMed; (d) X. Zhao, G.-J. Wu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 3296 CrossRef CAS PubMed; (e) F. Ye, X.-S. Ma, Q. Xiao, H. Li, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2012, 134, 5742 CrossRef CAS PubMed; (f) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (g) Q. Xiao, B. Wang, L. Tian, Y. Yang, J. Ma, Y. Zhang, S. Chen and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 9305 CrossRef CAS PubMed; (h) F. Hu, Y. Xia, F. Ye, Z. Liu, C. Ma, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 1364 CrossRef CAS PubMed; (i) Z.-X. Liu, H.-C. Tan, L. Wang, T.-R. Fu, Y. Xia, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 3056 CrossRef CAS PubMed; (j) S. Xu, G.-J. Wu, F. Ye, X. Wang, H. Li, X. Zhao, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 4669 CrossRef CAS PubMed; (k) Z. Liu, H. Tan, T. Fu, Y. Xia, D. Qiu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2015, 137, 12800 CrossRef CAS PubMed; (l) W.-D. Chu, L. Zhang, Z.-K. Zhang, Q. Zhou, F.-Y. Mo, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2016, 138, 14558 CrossRef CAS PubMed; (m) Q. Zhou, S. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 16013 CrossRef CAS PubMed.
- (a) T. Yao, K. Hirano, T. Satoh and M. Mira, Angew. Chem., Int. Ed., 2012, 51, 775 CrossRef CAS PubMed; (b) van S. S. Berkel, S. Brauch, L. Gabriel, M. Henze, S. Stark, D. Vasilev, L. A. Wessjohann, M. Abbas and B. Westermann, Angew. Chem., Int. Ed., 2012, 51, 5343 CrossRef PubMed; (c) X. Li, X. Liu, H. Chen, W. Wu, C. Qi and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 14485 CrossRef CAS PubMed; (d) W. Xiong, C. Qi, H. He, L. Ouyang, M. Zhang and H. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3084 CrossRef CAS PubMed; (e) N. Su, J. A. Theorell, D. J. Wink and T. G. Driver, Angew. Chem., Int. Ed., 2015, 54, 12942 CrossRef CAS PubMed; (f) A. R. Reddy, F. Hao, K. Wu, C.-Y. Zhou and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 1810 CrossRef CAS PubMed; (g) S. Mai and Q. Song, Angew. Chem., Int. Ed., 2017, 56, 7952 CrossRef CAS PubMed; (h) T. Ishikawa, M. Kimura, T. Kumoi and H. Lida, ACS Catal., 2017, 7, 4986 CrossRef CAS.
- (a) X.-Q. Hu, J.-R. Chen, Q. Wei, F.-L. Liu, Q.-H. Deng, A. B. Beauchemin and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 12163 CrossRef CAS; (b) X.-Q. Hu, X. Qi, J.-R. Chen, Q.-Q. Zhao, Q. Wei, Y. Lan and W.-J. Xiao, Nat. Commun., 2016, 7, 11188 CrossRef PubMed; (c) S. Mao, Y.-R. Gao, X.-Q. Zhu, D.-D. Guo and Y.-Q. Wang, Org. Lett., 2015, 17, 1692 CrossRef CAS PubMed; (d) N. D. Paul, S. Mandal, M. Otte, X. Cui, X. P. Zhang and de B. Bruin, J. Am. Chem. Soc., 2014, 136, 1090 CrossRef CAS PubMed; (e) B. G. Das, A. Chirila, M. Tromp, J. N. H. Reek and de B. Bruin, J. Am. Chem. Soc., 2016, 138, 8968 CrossRef CAS PubMed; (f) Y. Wang, X. Wen, X. Cui, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2017, 139, 1049 CrossRef CAS PubMed; (g) F. Wang, P. Xu, S.-Y. Wang and S.-J. Ji, Org. Lett., 2018, 20, 2204 CrossRef CAS PubMed; (h) te C. Grotenhuis, van den N. Heuvel, van der J. I. Vlugt and de B. Bruin, Angew. Chem., Int. Ed., 2018, 57, 140 CrossRef PubMed; (i) Y. Wang, X. Wen, X. Cui and X. P. Zhang, J. Am. Chem. Soc., 2018, 140, 4792 CrossRef CAS PubMed.
- (a) M. Mao, L. Zhang, Y.-Z. Chen, J. Zhu and L. Wu, ACS Catal., 2017, 7, 181 CrossRef CAS; (b) L. Zhang, J. Zhu, J. Ma, L. Wu and W.-H. Zhang, Org. Lett., 2017, 19, 6308 CrossRef CAS PubMed; (c) J. Zhu, M. Mao, H.-J. Ji, J.-Y. Xu and L. Wu, Org. Lett., 2017, 19, 1946 CrossRef CAS PubMed; (d) T. Liu, X. Sun and L. Wu, Adv. Synth. Catal., 2018, 360, 2005–2012 CrossRef CAS; (e) X.-D. Wang, J.-J. Wu, X. Sun, W.-C. Yang, J. Zhu and L. Wu, Adv. Synth. Catal., 2018, 360, 3518 CrossRef CAS.
- Taking into account the single-electron reduction properties of a diamine/alkali metal salt system and thermodynamic stabilities, other pathways either through producing a (Ts)N-radical and hydrogen anion or direct single-electron oxidation of deprotonated N-tosylhydrazone (ref. 10a and b) are unlikely here.
- For the EPR study on a t-BuOK/1,10-phen system, see: H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545 RSC.
- For the selected tosyl radical generation from the tosyl anion: (a) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205 CrossRef CAS PubMed; (b) H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371 CrossRef CAS PubMed.
- I. Thomé, A. Nijs and C. Bolm, Chem. Soc. Rev., 2012, 41, 979 RSC.
- CCDC 1573911 (3h), 1573912 (3x), 1573906 (3aa) and 1573907 (3ae) contain the supplementary crystallographic data for this paper.†.
- For selected studies on synthesizing atropisomers: (a) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239 CrossRef CAS PubMed; (b) E. Kumarasamy, A. J.-L. Ayitou, N. Vallavoju, R. Raghunathan, A. Lyer, A. Clay, S. K. Kandappa and J. Sivaguru, Acc. Chem. Res., 2016, 49, 2713 CrossRef CAS PubMed; (c) S.-C. Zheng, S. Wu, Q. Zhou, L. W. Chung, L. Ye and B. Tan, Nat. Commun., 2017, 8, 15238 CrossRef PubMed; (d) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534 CrossRef CAS PubMed.
- Although the mechanism of this sulfonyl rearrangement needs to be further studied, preliminary results show that a non-radical pathway is involved, and an extramolecular sulfonyl source will render competitive rearrangement products; please see the details and proposed mechanism in the ESI.†.
- (a) H. Qiu, H. D. Srinivas, P. Y. Zavalij and M. P. Doyle, J. Am. Chem. Soc., 2016, 138, 1808 CrossRef CAS PubMed; (b) D. F. Taber and P. Guo, J. Org. Chem., 2008, 73, 9479 CrossRef CAS PubMed; (c) C. Zhu, J. Li, P. Chen, W. Wu, Y. Ren and H. Jiang, Org. Lett., 2016, 18, 1470 CrossRef CAS PubMed; (d) W. Zhang, Y. Yue, D. Yu, L. Song, Y.-Y. Xu, Y.-J. Tian and Y.-J. Guo, Adv. Synth. Catal., 2012, 354, 2283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1573906, 1573907, 1573911 and 1573912. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00873f |
| This journal is © the Partner Organisations 2018 |