
Rhodium-catalyzed highly diastereoselective intramolecular [4 + 2] cycloaddition of 1,3-disubstituted allene-1,3-dienes†
Yulin
Han
ab and
Shengming
Ma
 *ac
*ac
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cResearch Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, P. R. China
First published on 31st July 2018
Abstract
RhCl(PPh3)3-catalyzed [4 + 2] intramolecular cycloaddition of allene-1,3-dienes afforded cis-fused [3.4.0]-bicyclic products with three chiral centers in good yields with excellent chemo- and diastereoselectivity. The configuration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the 1,3-diene unit controls the relative configurations of the non-bridging tertiary carbon atom in the six-membered ring. Based on the experimental results, a mechanism involving cyclometalation has been proposed.
C bonds in the 1,3-diene unit controls the relative configurations of the non-bridging tertiary carbon atom in the six-membered ring. Based on the experimental results, a mechanism involving cyclometalation has been proposed.
Cycloaddition reactions are a practical and efficient method to construct cyclic products from simple acyclic starting materials by generating at least two bonds and one ring in only one step.1 For example, [4 + 2] cycloaddition between 1,3-dienes and alkenes or alkynes is one of the most powerful synthetic tools to assemble six-membered cyclic structures.2–4 In 1995, Wender and coworkers reported the intramolecular [4 + 2] cycloaddition reaction of disubstituted terminal allenes with 1,3-dienes to afford cis-6,6-fused rings or cis-6,5-fused rings with a nickel or rhodium complex as catalyst (Scheme 1, a).5 The chemoselectivity mainly depends on the length of the tether between the diene and terminal allene. In 2009, the groups of Toste and Mascareñas independently demonstrated the gold-catalyzed intramolecular [4 + 2] cycloaddition reaction of symmetric tetrasubstituted allene with 1,3-diene to give trans-6,5-fused rings (Scheme 1, b).6 So far, there have been very limited reports on applying the 1,3-disubstituted allene unit as the dienophile.7 The challenge is the Z/E selectivity of the remaining C
C bond as well as the diastereoselectivity of the three chiral centers generated during the reaction (Scheme 1, c).8 Recently, there have been many advances in the development of new synthetic approaches to prepare 1,3-disubstituted allenes.9,10 Herein, we present the RhCl(PPh3)3-catalyzed intramolecular [4 + 2] cycloaddition of 1,3-disubstituted allene–dienes, during which the internal CC bond of the allene moiety reacted with the 1,3-diene unit to produce cis-6,5-fused bicyclic products with an excellent Z/E selectivity for the remaining CC bond and an excellent diastereoselectivity referring to the three chiral centers (Scheme 1, c).
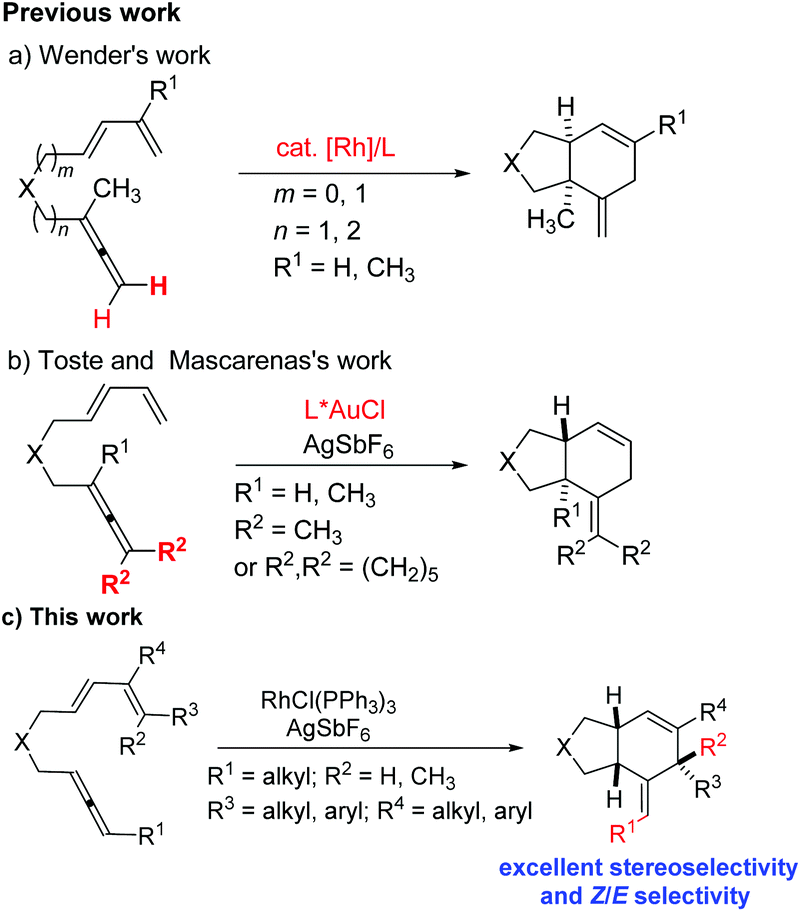 | ||
| Scheme 1 Metal-catalyzed intramolecular [4 + 2] cycloaddition of allene-1,3-dienes. | ||
We started our study on the reaction with 1,3-disubstituted allene-1,3-diene 1a as the model substrate. The desired product was not formed under the catalysis of Ni(COD)2 or [Rh(COD)2Cl]2 (entries 1 and 2, Table 1). The cationic rhodium catalyst [Rh(COD)2]BF4 also couldn't catalyze the reaction either (entry 3, Table 1). Interestingly, when [RhCl(CO)2]2 was used, the reaction processed smoothly to afford the expected bicyclic product 1a in 81% yield with a Z/E ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (entry 4, Table 1). The structure and relative configurations of Z-2a and E-2a were established via analogy with the X-ray single crystal diffraction study of 2c (Fig. 1)11 and the NOESY spectra (see the ESI†). The catalyst [(C10H8)Rh(COD)]+SbF6− used in Trost's report7a can facilitate the reaction to afford the cycloaddition product Z-2a with an incomplete conversion (entry 5, Table 1). To our delight, the yield was improved to 90% when RhCl(CO)(PPh3)2 was used with the exclusive formation of Z-2a (entry 6, Table 1). With RhCl(PPh3)3 as the catalyst, the yield was improved to 93% (entry 7, Table 1). The desired product could also be obtained in 92% and 91% yield at a lower temperature of 40 °C (entries 8 and 9, Table 1). The yield was 95% when running the reaction at a concentration of 0.2 M (entry 10, Table 1). The yield dropped to 85% with 1 mol% RhCl(PPh3)3 (entry 11, Table 1). Thus, the reaction conducted with 1a and RhCl(PPh3)3 (2 mol%) in toluene at 40 °C was chosen as the standard conditions for further study.
:50 (entry 4, Table 1). The structure and relative configurations of Z-2a and E-2a were established via analogy with the X-ray single crystal diffraction study of 2c (Fig. 1)11 and the NOESY spectra (see the ESI†). The catalyst [(C10H8)Rh(COD)]+SbF6− used in Trost's report7a can facilitate the reaction to afford the cycloaddition product Z-2a with an incomplete conversion (entry 5, Table 1). To our delight, the yield was improved to 90% when RhCl(CO)(PPh3)2 was used with the exclusive formation of Z-2a (entry 6, Table 1). With RhCl(PPh3)3 as the catalyst, the yield was improved to 93% (entry 7, Table 1). The desired product could also be obtained in 92% and 91% yield at a lower temperature of 40 °C (entries 8 and 9, Table 1). The yield was 95% when running the reaction at a concentration of 0.2 M (entry 10, Table 1). The yield dropped to 85% with 1 mol% RhCl(PPh3)3 (entry 11, Table 1). Thus, the reaction conducted with 1a and RhCl(PPh3)3 (2 mol%) in toluene at 40 °C was chosen as the standard conditions for further study.
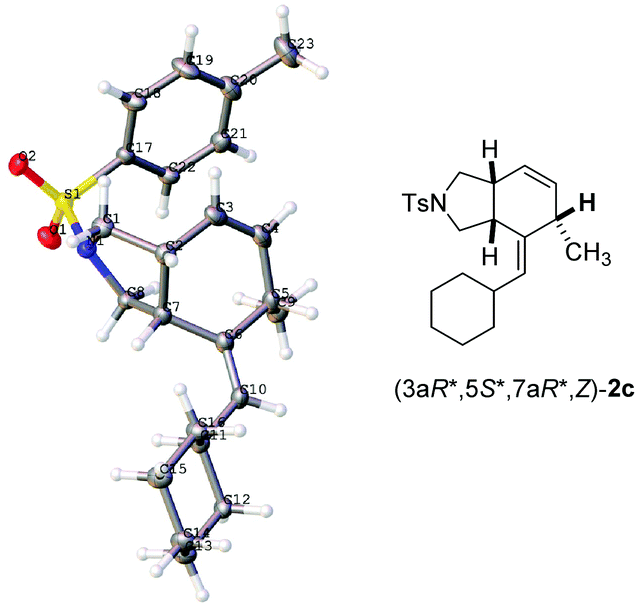 | ||
| Fig. 1 The ORTEP representations of (3aR*,5S*,7aR*,Z)-2c. | ||
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Rh] | T (°C) | t (h) | Yield of 2ab |
Z/E | Recovery of 1ab |
| a The reaction was conducted with 1a (0.2 mmol) and catalyst (2 mol%) in 2 mL of toluene. b Determined using 1H NMR analysis with mesitylene as the internal standard and N.D. = not detected. c The reaction was conducted with 1a (0.1 mmol) and catalyst (3 mol%) in 2 mL of 1,2-dichloroethane. d 1 mL of toluene was used. e 1 mol% RhCl(PPh3)3 and 1 mL of toluene were used. | ||||||
| 1 | Ni(COD)2 | 80 | 12 | N.D. | — | 68 |
| 2 | [Rh(COD)Cl]2 | 80 | 12 | N.D. | — | 90 |
| 3 | [Rh(COD)2]BF4 | 80 | 12 | N.D. | — | 77 |
| 4 | [RhCl(CO)2]2 | 80 | 12 | 81 | 50:50 |
N.D. |
| 5c | [(C10H8)Rh(COD)]+SbF6− | rt | 12 | 59 | 100:0 |
12 |
| 6 | RhCl(CO)(PPh3)2 | 80 | 12 | 90 | 100:0 |
N.D. |
| 7 | RhCl(PPh3)3 | 80 | 12 | 93 | 100:0 |
N.D. |
| 8 | RhCl(PPh3)3 | 60 | 5 | 92 | 100:0 |
N.D. |
| 9 | RhCl(PPh3)3 | 40 | 3 | 91 | 100:0 |
N.D. |
| 10d | RhCl(PPh3)3 | 40 | 3 | 95 | 100:0 |
N.D. |
| 11e | RhCl(PPh3)3 | 40 | 10 | 85 | 100:0 |
N.D. |

With the optimized conditions in hand, the scope of substrates was investigated. The substrates with NTs as the linker were investigated firstly. The reaction afforded the corresponding products Z-2a–2c in decent yields when R1 was methyl, n-butyl, and cyclohexyl, respectively (entries 1–3, Table 2). The relative configurations in 2c were established by the X-ray single crystal diffraction study11 (Fig. 1) – all three H atoms are cis oriented with a Z-CC bond. R2 may be H, methyl, or n-propyl (entries 1–4, and 10, Table 2). The substrates with R2 being aryl groups in the terminal of the diene moiety could afford the target products under the standard conditions with lower yields. The yields were improved by conducting the reactions at 80 °C (entries 5–7, Table 2). With a substituent such as o-Br, the reactions only afforded 2h in 15% yield with 36% recovery of 1h even at 80 °C for 36 h (entry 8, Table 2). To our delight, 66% yield of 2h was obtained when the reaction was conducted with the extra addition of AgSbF6 (3 mol%) (entry 9, Table 2). R3 may be H or alkyl. It is worth mentioning that the reactions produced products 2a–2h as the only diastereoisomers while in entry 10 of Table 2 a minor amount of other diastereoisomers was formed. Malonate could also be used as the tether of the substrates. However, the reaction of 1j became sluggish as compared to the substrates with NTs as the tether and the corresponding product 2j was afforded with the help of AgSbF6 (entry 11, Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | X/R1/R2/R3 (1) | T (°C) | t (h) | 2 |
| Isolated yield (%) | ||||
|
a Conditions A: The reaction was conducted with 1 (1.0 mmol) and RhCl(PPh3)3 (2 mol%) in 5 mL of toluene.
b The reaction was conducted on a 0.5 mmol scale.
c 36% yield of 1h was recovered and the yield of 2h was determined with the NMR spectrum of the crude reaction mixture.
d Conditions B: The reaction was conducted with 1 (0.5 mmol), RhCl(PPh3)3 (2 mol%), and AgSbF6 (3 mol%) in 2.5 mL of toluene.
e The ratios of 2i/3i in the isolated product and the crude product were 27:1 and 15:1, which were determined with the 1H NMR analysis of the isolated product and the crude product.
|
||||
| 1 | NTs/nBu/CH3/H ((2E,4E)-1a) | 40 | 3 | 81 ((3aR*,5S*,7aR*,Z)-2a) |
| 2 | NTs/CH3/CH3/H ((2E,4E)-1b) | 40 | 8 | 82 ((3aR*,5S*,7aR*,Z)-2b) |
| 3 | NTs/Cy/CH3/H ((2E,4E)-1c) | 40 | 3 | 73 ((3aR*,5S*,7aR*,Z)-2c) |
| 4 | NTs/nBu/n-C3H7/H ((2E,4E)-1d) | 40 | 4 | 78 ((3aR*,5S*,7aR*,Z)-2d) |
| 5b | NTs/CH3/Ph/H ((2E,4E)-1e) | 80 | 24 | 39 ((3aR*,5R*,7aR*,Z)-2e) |
| 6b | NTs/CH3/p-FC6H4/H ((2E,4E)-1f) | 80 | 21.5 | 71 ((3aR*,5R*,7aR*,Z)-2f) |
| 7b | NTs/CH3/p-ClC6H4/H ((2E,4E)-1g) | 80 | 15 | 63 ((3aR*,5R*,7aR*,Z)-2g) |
| 8b | NTs/nBu/o-BrC6H4/H ((2E,4E)-1h) | 80 | 36 | 15c ((3aR*,5S*,7aR*,Z)-2h) |
| 9d | NTs/nBu/o-BrC6H4/H ((2E,4E)-1h) | 80 | 8 | 66 ((3aR*,5S*,7aR*,Z)-2h) |
| 10 | NTs/Cy/H/CH3 ((E)-1i) | 40 | 4 | 87e ((3aR*,7aR*,Z)-2i) |
| 11d | C(CO2Me)2/nBu/Ph/H ((2E,4E)-1j) | 40 | 24 | 81 ((3aR*,5R*,7aR*,E)-2j) |

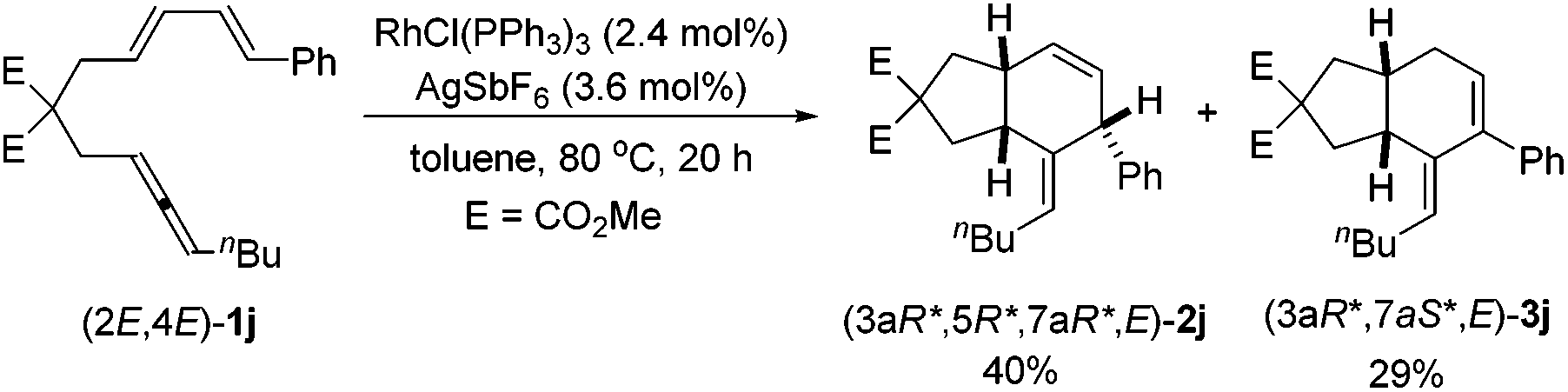
When the phenyl-substituted 1,3-diene-incorporated substrate (2E,4E)-1j was reacted with RhCl(PPh3)3 and AgSbF6 at 80 °C for 20 h, in addition to 40% yield of (3aR*,5R*,7aR*,E)-2j, 29% yield of (3aR*,7aS*,E)-3j was obtained (eqn (1)). The relative configuration of (3aR*,7aS*,E)-3j was also established using X-ray single crystal diffraction12 (Fig. 2) – the CC bond in the six-membered ring was conjugated with the exocyclic double bond.
 | (1) |
 | ||
| Fig. 2 The ORTEP representations of (3aR*,7aS*,E)-3j. | ||
In addition, when we applied the CC bond stereoisomer (2E,4Z)-1b and 1k, the reaction at 80 °C afforded the diastereoisomers (3aR*,5R*,7aR*,Z)-2b and (3aR*,5R*,7aR*,Z)-2k (Scheme 2). The relative configuration of (3aR*,5R*,7aR*,Z)-2b was also established by the X-ray single crystal diffraction studies (Fig. 3)13 – the H atom of C5 in the six-membered ring is trans to the two H atoms of the bridged carbon atoms C3a and C7a. Thus, the configuration of the C5 was controlled by the E/Z configuration of the distal CC bond in the diene moiety of the substrates (eqn (2), (3) vs. entry 2, Table 2).
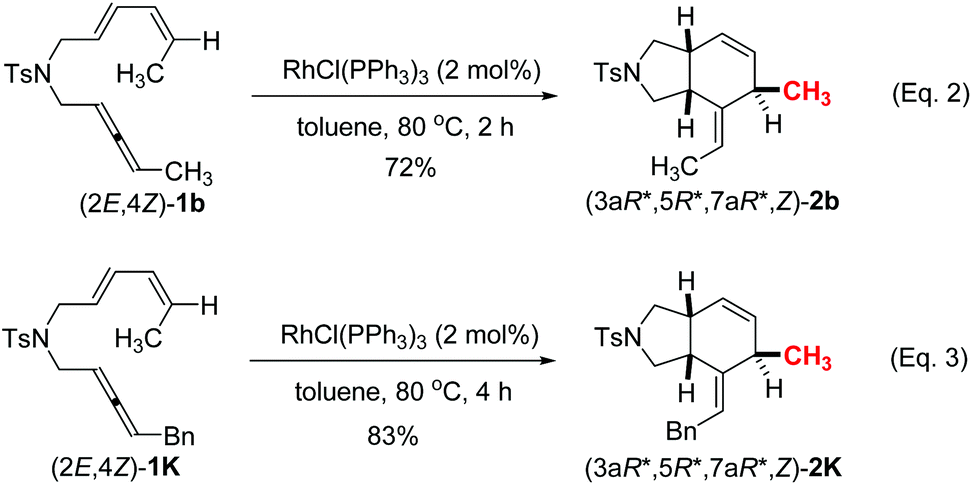 | ||
| Scheme 2 Rhodium-catalyzed intramolecular [4 + 2] cycloaddition of (2E,4Z)-1b and (2E,4Z)-1k. | ||
 | ||
| Fig. 3 The ORTEP representation of (3aR*,5R*,7aR*,Z)-2b. | ||
Based on the X-ray diffraction study of (3aR*,5S*,7aR*,Z)-2c, (3aR*,5R*,7aR*,Z)-2b and the observed diastereoselectivity, a mechanism is proposed (Scheme 3). Firstly, RhCl(PPh3)3 reacts with AgSbF6 to generate the cationic catalyst [Rh(PPh3)3]+SbF6−. The reaction of the R-isomer (Ra,2E,4E)-1 with the Rh catalyst would generate intermediate A, in which the “inner” CC bonds in allene and the s-cis-1,3-diene coordinated with the rhodium atom in such a way that the two H atoms in intermediate A are cis oriented in order to make the R1 group pointing away from the 1,3-diene unit. Subsequent cyclometalation leads to the formation of intermediate B. The allylic rearrangement leads to the formation of rhodiabicycloheptene C, in which the three tertiary hydrogen atoms are cis orientated. Subsequent reductive elimination and ligand exchange with (Ra,2E,4E)-1 provide the product (3aR,5S,7aR,Z)-2 and regenerate the catalytically active species A to finish the catalytic cycle. For the S-enantiomer (Sa,2E,4E)-1, intermediate A′ other than intermediate A″ (which has the same coordination mode as intermediate A) would be formed in order to keep the R1 group far away from the catalyst. Subsequent cyclometalation, η3–η1 rearrangement, reductive elimination, and ligand exchange with (Sa,2E,4E)-1 would afford the enantiomer (3aS,5R,7aS,Z)-2.
 | ||
| Scheme 3 A proposed mechanism for the rhodium-catalyzed intramolecular [4 + 2] cycloaddition of allene-dienes (2E,4E)-1 (R3 ≠ aryl). | ||
When the optically active allene-1,3-diene (Ra,2E,4E)-1a was reacted under the standard conditions, only one diastereomer (3aR,5S,7aR,Z)-2a was obtained in 76% yield; unfortunately, the efficiency of chirality transformation was low and the ee of (3aR,5S,7aR,Z)-2a was only 75% (eqn (4)). We are still working on this issue.
 | (4) |
In conclusion, we have developed the RhCl(PPh3)3-catalyzed intramolecular [4 + 2] cycloaddition of 1,3-disubstituted allene-1,3-dienes, giving cis-6,5-fused bicyclic products with a very high diastereoselectivity. In addition, the relative configurations in the C3a, C5, and C7a of the products are controlled by the configuration of the CC bonds in the 1,3-diene unit in the starting materials. The three H atoms of the tertiary carbon in the product are cis to each other when (2E,4E)-1,3-disubstituted allene–dienes were used; the two cis-H atoms of the bridged carbon and the H atom of the other tertiary carbon in the six-membered ring are trans to each other if the (2E,4Z)-1,3-disubstituted allene–dienes were applied. A concerted cyclometalation, allylic rearrangement, and reductive elimination has been proposed to account for the observed diastereoselectivity. Further studies of the chirality transfer of optically active allene-1,3-dienes are being conducted in our laboratory.
Conflicts of interest
There are no conflict of interest to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21690063) is greatly appreciated. We thank Mr Chaofan Huang in this group for reproducing the results of (3aR*,5R*,7aR*,Z)-2f and (3aR*,7aR*,Z)-2i in Table 2 and (3aR*,5R*,7aR*,Z)-2b in Scheme 2 presented in this study.Notes and references
- For selected reviews of cycloaddition, see: (a) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef PubMed; (b) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef PubMed; (c) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef PubMed; (d) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef PubMed; (e) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (f) W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon, Oxford, 1990, pp. 1–208 Search PubMed.
- For selected reviews of the Diels–Alder reaction, see: (a) W. Oppolzer, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 5, pp 315–399 Search PubMed. For selected reviews on applications of Diels–Alder reactions in total synthesis, see: (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef; (c) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779 CrossRef PubMed.
- For representative examples with 1,3-dienes and alkenes, see: (a) R. S. Jolly, G. Luedtke, D. Sheehan and T. Livinghouse, J. Am. Chem. Soc., 1990, 112, 4965 CrossRef; (b) L. McKinstry and T. Livinghouse, Tetrahedron, 1994, 50, 6145 CrossRef; (c) D. F. Harvey and E. M. Grenzer, J. Org. Chem., 1996, 61, 159 CrossRef; (d) D. J. R. O'Mahony, D. B. Belanger and T. Livinghouse, Synlett, 1998, 443 CrossRef; (e) H. Heath, B. Wolfe, T. Livinghouse and S. K. Bae, Synthesis, 2001, 2341 CrossRef; (f) S. R. Gilbertson and G. S. Hoge, Tetrahedron Lett., 1998, 39, 2075 CrossRef; (g) S. R. Gilbertson, G. S. Hoge and D. G. Genov, J. Org. Chem., 1998, 63, 10077 CrossRef; (h) B. Wang, P. Cao and X. Zhang, Tetrahedron Lett., 2000, 41, 8041 CrossRef; (i) Y. Sato, Y. Oonishi and M. Mori, Organometallics, 2003, 22, 30 CrossRef; (j) D. J. R. O'Mahony, D. B. Belanger and T. Livinghouse, Org. Biomol. Chem., 2003, 1, 2038 RSC.
- For representative intramolecular examples with 1,3-dienes and alkynes, see: (a) P. A. Wender and T. E. Jenkins, J. Am. Chem. Soc., 1989, 111, 6432 CrossRef; (b) R. S. Jolly, G. Luedtke, D. Sheehan and T. Livinghouse, J. Am. Chem. Soc., 1990, 112, 4965 CrossRef; (c) P. A. Wender and T. E. Smith, J. Org. Chem., 1996, 61, 824 CrossRef; (d) K. Kumar and R. S. Jolly, Tetrahedron Lett., 1998, 3047 CrossRef; (e) D. Motoda, H. Kinoshita, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2004, 43, 1860 CrossRef PubMed; (f) A. Fürstner and C. C. Stimson, Angew. Chem., Int. Ed., 2007, 46, 8845 CrossRef PubMed; (g) H. Kusama, Y. Karibe, Y. Onizawa and N. Iwasawa, Angew. Chem., Int. Ed., 2010, 49, 4269 CrossRef PubMed; (h) S. M. Kim, J. H. Park and Y. K. Chung, Chem. Commun., 2011, 47, 6719 RSC; (i) K. Aikawa, S. Akutagawa and K. Mikami, J. Am. Chem. Soc., 2006, 128, 12648 CrossRef PubMed; (j) W.-J. Yoo, A. Allen, K. Villeneuve and W. Tam, Org. Lett., 2005, 7, 5853 CrossRef PubMed. For intermolecular examples with 1,3-dienes and alkynes, see: (k) A. Carbonaro, A. Greco and G. Dall'Asta, J. Org. Chem., 1968, 33, 3948 CrossRef; (l) H. tom Dieck and R. Diercks, Angew. Chem., Int. Ed. Engl., 1983, 22, 778 CrossRef; (m) S.-J. Paik, S. U. Son and Y. K. Chung, Org. Lett., 1999, 1, 2045 CrossRef; (n) G. Hilt, W. Hess and K. Harms, Org. Lett., 2006, 8, 3287 CrossRef PubMed. For a related cycloaddition between unactivated vinylallenes and alkynes, see: (o) M. Murakami, M. Ubukata, K. Itami and Y. Ito, Angew. Chem., Int. Ed., 1998, 37, 2248 CrossRef. For representative examples of hetero-Diels–Alder reactions, respectively, see: (p) B. M. Trost, R. E. Brown and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 5877 CrossRef; (q) I. Koyama, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2009, 131, 1350 CrossRef PubMed.
- (a) P. A. Wender, T. E. Jenkins and S. Suzuki, J. Am. Chem. Soc., 1995, 117, 1843 CrossRef; (b) P. A. Wender, M. P. Croatt and N. M. Deschamps, Angew. Chem., Int. Ed., 2006, 45, 2459 CrossRef PubMed; (c) B. C. Lainhart and E. J. Alexanian, Org. Lett., 2015, 17, 1284 CrossRef PubMed.
- (a) P. Mauleón, R. M. Zeldin, A. Z. González and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6348 CrossRef PubMed; (b) I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020 CrossRef PubMed; (c) A. Z. González and F. D. Toste, Org. Lett., 2010, 12, 200 CrossRef PubMed.
- (a) B. M. Trost, D. R. Fandrick and D. C. Dinh, J. Am. Chem. Soc., 2005, 127, 14186 CrossRef PubMed; (b) J. K. Lam, Y. Schmidt and C. D. Vanderwal, Org. Lett., 2012, 14, 5566 CrossRef PubMed.
- F. López and J. L. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904 RSC.
- For selected reviews on the synthesis of allenes, see: (a) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, vol. 1 and 2 Search PubMed; (b) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC; (c) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795 CrossRef; (d) R. K. Neff and D. E. Frantz, ACS Catal., 2014, 4, 519 CrossRef; (e) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210 RSC; (f) M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259 CrossRef.
- For selected reports on the synthesis of 1,3-disubstituted allenes, see: (a) J. Kuang and S. Ma, J. Am. Chem. Soc., 2010, 132, 1786 CrossRef PubMed; (b) J. Kuang, H. Luo and S. Ma, Adv. Synth. Catal., 2012, 354, 933 CrossRef.
- Crystal data for compound (3aR*,5S*,7aR*,Z)-2c: C23H31NO2S, MW = 385.55, triclinic, space group P−1, final R indices [I > 2σ(I)], R1 = 0.0550, wR2 = 0.1388, R indices (all data) R1 = 0.0764, wR2 = 0.1388, a = 6.182(5) Å, b = 11.371(8) Å, c = 13.441(9) Å, α = 80.287(13)°, β = 84.840(13)°, γ = 77.986(12)°, V = 1002.2(12) Å3, T = 130 K, Z = 2, reflections collected/unique 14550/4638 (Rint = 0.0470), number of observations [>2σ(I)] 4638, parameters: 246. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1528081.†.
- Crystal data for compound (3aR*,7aS*,E)-3j: C24H30O4, MW = 382.48, monoclinic, space group P1(21)/c(1), final R indices [I > 2σ(I)], R1 = 0.0453, wR2 = 0.1060, R indices (all data) R1 = 0.0781, wR2 = 0.1227, a = 8.9632(10) Å, b = 26.894(3) Å, c = 9.2793(10) Å, α = 90°, β = 110.151(2)°, γ = 90°, V = 2099.9(4) Å3, T = 130 K, Z = 4, reflections collected/unique 21185/6552 (Rint = 0.0373), number of observations [>2σ(I)] 6552, parameters: 256. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1555723.†.
- Crystal data for compound (3aR*,5R*,7aR*,Z)-2b: C18H23NO2S, MW = 317.43, monoclinic, space group P21/c, final R indices [I > 2σ(I)], R1 = 0.0486, wR2 = 0.1232, R indices (all data) R1 = 0.0611, wR2 = 0.1316, a = 10.6868(14) Å, b = 20.200(3) Å, c = 8.0112(10) Å, α = 90°, β = 101.747(3)°, γ = 90°, V = 1693.2(4) Å3, T = 293(2) K, Z = 4, reflections collected/unique 9268/2981 (Rint = 0.0374), number of observations [>2σ(I)] 2981, parameters: 202. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1555722.†.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and characterisation data as well as 1H and 13C NMR spectra of all compounds. CCDC 1528081, 1555723 and 1555722. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00650d |
| This journal is © the Partner Organisations 2018 |