
Ultrafine molybdenum phosphide nanocrystals on a highly porous N,P-codoped carbon matrix as an efficient catalyst for the hydrogen evolution reaction†
Jin-Tao
Ren
ab,
Lei
Chen
ab,
Chen-Chen
Weng
ab and
Zhong-Yong
Yuan
 *ab
*ab
aNational Institute for Advanced Materials, School of Materials Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: zyyuan@nankai.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China
First published on 25th June 2018
Abstract
High-performance and affordable electrocatalysts from earth-enriched materials are desirable for many sustainable energy conversion and storage systems. Highly crystalline MoP nanocrystals with ultrasmall sizes embedded in an N,P-codoped carbon matrix (MoP@NPCS) were designed and fabricated for hydrogen evolution. Due to the highly active catalytic nature of the MoP nanocrystals, the ultrasmall MoP nanoparticles on the conductive carbon matrix, the high surface area of the carbon matrix with abundant N and P dopants, and the strong coupling effect between the MoP nanoparticles and the carbon matrix, the designed MoP@NPCS catalysts show excellent hydrogen evolution behaviour, with an onset potential of −47 mV and an overpotential of 107 mV, to achieve a current density of 10 mA cm−2 in 1.0 M KOH, along with robust operational durability. Specially, the fabricated N,P-codoped carbon matrix (NPCS) exhibits high OER catalytic activity with an overpotential of 390 mV to afford 10 mA cm−2 and excellent stability. Finally, a two-electrode electrolyzer using MoP@NPCS and NPCS as the cathode and anode electrodes, respectively, requires a low voltage of 1.70 V to achieve 10 mA cm−2 over long-term operation. Due to this impressive performance, MoP@NPCS is a promising electrocatalyst for practical hydrogen production from electrocatalytic water splitting.
1. Introduction
Water splitting through electrochemistry is particularly essential for the production of renewable hydrogen energy to replace traditional fossil fuels. As the half-reaction of the electrochemical overall water-splitting, the hydrogen evolution reaction (HER) plays a significant role in the efficiency of the reaction.1,2 Currently, Pt-based catalysts and their alloys are the state-of-the-art catalysts for HER; however, their scarcity and high cost impede their widespread practical applications.3,4 Developing efficient catalysts using earth-abundant elements is necessary from the viewpoint of actual development.5,6 To date, various affordable alternatives have been fabricated for HER, including transition-metal chalcogenides, carbides, and phosphides as well as other metal compounds.7–10 Recently, molybdenum-based materials, including molybdenum carbides, molybdenum phosphides, molybdenum sulfides and molybdenum nitrides, have exhibited high HER electrocatalytic activity due to their Pt-like electronic structures and high hydrogen adsorption capacities.11–14 For example, Chen et al. used phosphomolybdic acid to initiate the polymerization of pyrrole and obtained N,P-codoped Mo2C@C nanospheres which exhibited impressive water reduction activity.12 Gao's group reported Mo2C nanocrystals supported on carbon sheets which demonstrated superior HER and OER performance.15 MoS2 nanosheets also exhibit high hydrogen evolution activity; their catalytic active sites are mainly located on the edges of the nanosheets.16–18 However, molybdenum phosphide materials have been rarely reported to be used for the hydrogen evolution reaction, in contrast to other nonprecious transition metal phosphides which exhibit high HER catalytic activity. Recently, Li's group used a MOFs-assisted strategy to prepare MoP nanoparticles that were well dispersed in porous carbon, exhibiting remarkable electrocatalytic capability for HER in acidic electrolyte.19 Thus, there is much room to improve the electrocatalytic efficiency of molybdenum phosphides.Usually, to obtain the phase formation of molybdenum oxides to phosphides, high-temperature treatment is necessary.3,19,20 Although the high temperature facilitates the acquisition of well-crystallized nanocrystals, they are inevitably accompanied by aggregation and coalescence.20,21 Thus, it still a challenge to simultaneously obtain molybdenum phosphides with ultrafine nanostructuring and sufficient highly active sites through a cost-effective and controllable strategy. Recently, the structure design of hybridized carbonaceous materials has emerged as an efficient strategy to improve the catalytic performance of molybdenum-based electrocatalysts. For example, N,P-codoped Mo2C@C nanospheres,12 molybdenum carbide and phosphide hybrids on an N-doped carbon matrix,22 and porous molybdenum phosphide–carbon nano-octahedrons19 have been synthesized and exhibit excellent HER electrocatalytic activities. The inclusion of carbonaceous materials effectively alleviates the aggregation of molybdenum-based nanoparticles, improving their electric conductivity and affording synergistic effects between them.23–26
Herein, we rationally designed and prepared an electrocatalyst consisting of ultrafine MoP nanocrystals supported on an N,P-codoped carbon matrix (MoP@NPCS) through a facile polymerization strategy of aniline and ammonium molybdenum in the presence of organophosphonic acid, wherein graphitic carbon nitride (g-C3N4) was used as a template for the formation of sponge-like structures. The presence of molybdate (Mo7O246−) at the atomic scale in the polymer monolith ensures the ultrasmall size of the molybdenum-based nanoparticles. The phosphorus source of organophosphonic acid cross-linked in the polyaniline polymer network allows in situ phosphorization and sufficient chemical doping into the final carbonaceous materials. The carbon matrix derived from carbonization of a polyaniline polymer efficiently alleviates sintering and agglomeration of the molybdenum-based nanoparticles. Especially, the thermal decomposition of polymeric g-C3N4 creates a hollow sponge-like structure, providing a larger specific surface area and exposing abundant active centers. Most importantly, the structure and composition of the prepared catalysts can be easily tuned by adjusting the ratio of the reactants to obtain optimal catalytic activity. Due to the high electronic conductivity of the heteroatom-doped carbon monoliths, the abundant active centers of the ultrafine MoP nanoparticles, and the efficient mass transportation of the hollow sponge-like structures, the prepared MoP@NPCS exhibits high catalytic capability and renders an onset potential at −47 mV and a current density of 10 mA cm−2 at an overpotential of 107 mV for hydrogen evolution without evident activity degradation over long-term operation in alkaline medium. Especially, the prepared N,P-codoped carbon matrix (NPCS) exhibits high OER catalytic behavior and requires an overpotential of 390 mV to afford a current density of 10 mA cm−2. Moreover, an alkaline water electrolyzer equipped with MoP@NPCS and NPCS as the cathode and anode, respectively, only requires a voltage of about 1.70 V to obtain 10 mA cm−2, with slight activity decay over 20 h. This work successfully develops a molybdenum phosphide–carbon hybrid material as an efficient and inexpensive catalyst for electrochemical hydrogen reduction; especially, it also provides a new strategy for the fabrication of heteroatom-doped carbon materials with well-designed porous morphologies.
2. Experimental section
2.1. Material preparation
Ammonium molybdate [(NH4)6Mo7O24], melamine, aniline, ammonium persulfate (APS) and molybdenum phosphide (MoP) were obtained from Tianjin Chemical Reagent Co.; all chemicals were of analytical grade and were used as received without further purification. Commercial Pt/C (20 wt%), IrO2, and Nafion (5 wt%) were purchased from Sigma–Aldrich. Hydroxyethylidene diphosphonic acid (HEDP) was received from Henan Qingyuan Chemical Co., Ltd.2.2. Physicochemical characterization
Scanning electron microscopy (SEM) images were recorded on a JEOL JSM-7500L microscope with an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) images were obtained on a JEOL JSM-2800 microscope at 200 kV. X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Focus diffractometer. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Scientific ESCALAB 250Xi spectrometer using a monochromatic Al-Kα X-ray source (1486.6 eV). Thermogravimetry analysis (TGA) was performed on a TA SDT Q600 instrument. The specific surface areas and pore structures of the samples were analyzed on a Quantachrome Autosorb-1 sorption analyzer at liquid nitrogen temperature (77 K). The surface areas were calculated by the multi-point Brunauer–Emmett–Teller (BET) method. The pore-size distributions were obtained from the adsorption branches according to the Barrett–Joyner–Halenda (BJH) model. Also, the total pore volumes were estimated from the volumes adsorbed at a relative pressure (P/P0) of 0.99. Raman spectra were obtained on a Thermo-Fisher Scientific DXR spectrometer with 532 nm wavelength incident laser light.2.3. Electrochemical measurements
The catalyst suspension was prepared by dispersing the catalyst (5 mg) in a mixed solution containing ethanol (950 μL) and 5 wt% Nafion (50 μL) under sonication. Then, the obtained catalyst suspension (10 μL) was dropped onto a glassy carbon electrode (GCE, 4 mm in diameter), followed by evaporation in air to obtain a thin catalyst layer on the GCE; the resulting electrode was used as the working electrode.All electrochemical measurements were tested on a WaveDriver 20 Bipotentiostat/Galvanostat (Pine Research Instrumentation, USA) electrochemical workstation in a three-electrode configuration, wherein Ag/AgCl and graphitic carbon rod were used as the reference and counter electrodes, respectively. The LSV polarization curves were obtained in 1.0 M KOH with a scan rate of 2 mV s−1 at a rotating rate of 1600 rpm during the tests. The current densities were normalized with the geometric surface areas of the GCE electrode, and the measured potentials vs. Ag/AgCl were referenced to the reversible hydrogen electrode (RHE) by adding the potential of (0.205 + 0.059pH) V. The electrolyte was purged with N2 or O2 before the HER or OER tests, respectively. All polarization curves were corrected for the iR compensation within the cell. The electrode durability was measured by both cyclic voltammogram (CV) and chronopotentiometric response. The Tafel slope was calculated according to the Tafel equation as below:
η = a + b × log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) J J |
The electrical double layer capacitances (Cdl) of the as-prepared electrodes were obtained using cyclic voltammograms (CVs) in a non-faradaic potential range. Before recording the electrochemical data, the working electrodes were scanned for several cycles until the current was stabilized. The slope of the plot of the current density at a certain potential against the scan rate is the double layer capacitance (Cdl). Electrochemical impedance spectroscopy (EIS) measurements were performed in potentiostatic mode from 0.1 to 100 kHz with an AC voltage of 5 mV and were recorded on a Zahner IM6eX (Zahner, Germany) workstation.
For the overall-water-splitting tests, the fabricated catalysts coated on carbon cloth (CFC) with mass loadings of 1 mg cm−2 were directly used as electrodes for the measurements, wherein MoP@NPCS and NPCS were used as the cathode and anode, respectively. The polarization curves were obtained from LSV tests in 1.0 M KOH with a sweep rate of 2 mV s−1. The electrolyte was purged with N2 for 30 min before the tests. For the solar-driven water splitting, the alkaline electrolyzer was directly driven by a commercial silicon photovoltaic device (open circuit voltage of 5 V).
3. Results and discussion
3.1. Material synthesis and characterization
The fabrication of ultrafine highly crystalline MoP nanocrystals strongly coupled to a porous N,P-codoped carbon matrix (MoP@NPCS) began with self-polymerization between Mo7O246− and the aniline monomer on the basis of oxidation to initiate the formation of polyaniline, wherein hydrogen bonding interactions between HEDP (hydroxyethylidene diphosphonic acid) and the polyaniline chains efficiently coordinated the abundant phosphonic acid groups (Scheme 1). Especially, the residual aniline monomer was polymerized in the presence of excess oxidizing agent (ammonium persulfate, APS). The subsequent shape-preserving pyrolysis at different temperatures carbonized the polyaniline to the carbon matrix and simultaneously converted Mo7O246− to MoP nanoparticles through a carbothermal phosphorization process in nitrogen atmosphere, wherein the polyaniline and organophosphonic moieties induced the doping of N and P heteroatoms into the carbonaceous backbones and the phosphorization process. Furthermore, the Mo7O246− present in the polyaniline polymer with organophosphonic molecules ensured that the in situ formed MoP nanoparticles were homogeneously distributed in the carbon matrix without aggregation. Also, the high-temperature thermal decomposition of the g-C3N4 monolith template allowed the formation of a porous morphology (see the Experimental section for details). Thermogravimetric analysis (TGA) (Fig. S1, ESI†) confirmed that the thermal decomposition of g-C3N4 monolith and the carbonization of polyaniline occurred simultaneously.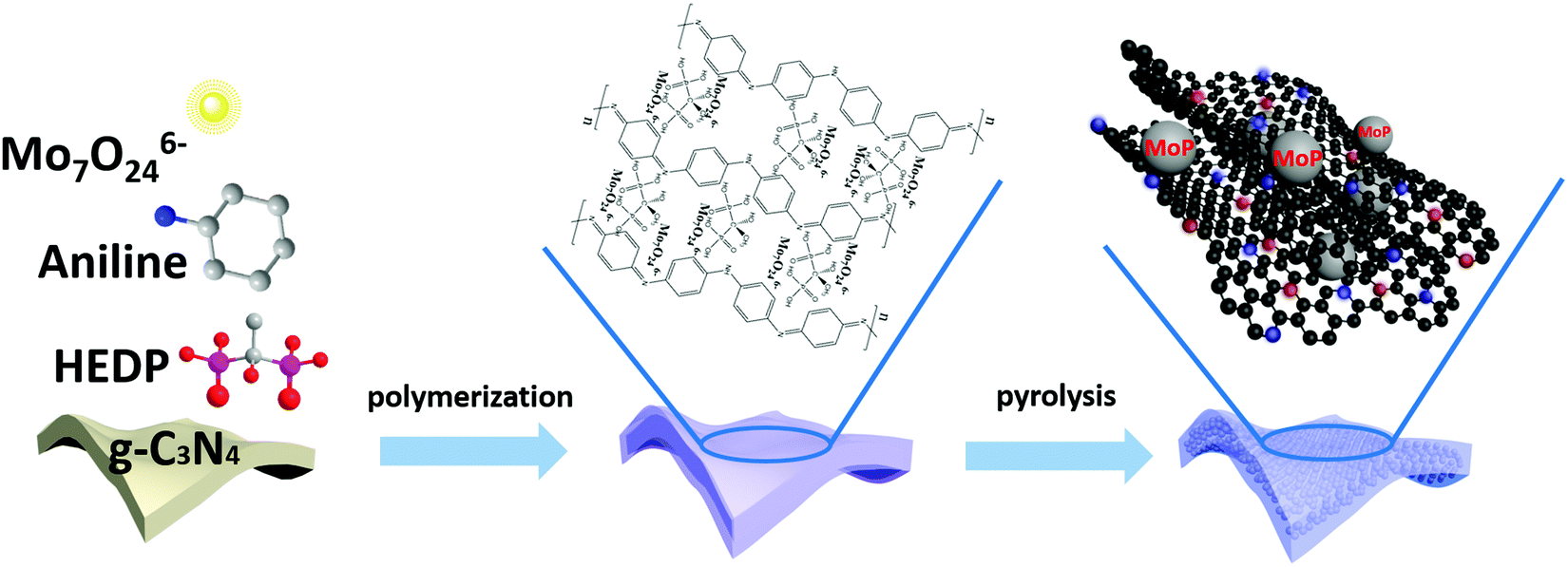 | ||
| Scheme 1 Schematic of the fabrication of ultrafine MoP nanocrystals on the N,P-doped carbon matrix. | ||
Fig. 1a shows the X-ray diffraction (XRD) patterns of NPCS and MoP@NPCS. For the polymeric precursor of MoP@NPCS, there were no obvious peaks except for the diffraction peak at 28° (2θ), which is attributed to the (100) plane of graphitic carbon nitride. After pyrolysis at 900 °C, the broad diffraction peak observed at 24° (2θ) for NPCS can be ascribed to the typical (002) plane of graphitic carbon materials with low graphitization degrees. Several diffraction peaks of MoP@NPCS can be indexed to hexagonal MoP phase (JCPDS No. 65-6487). Also, the depressed peak intensity indicates that the formed MoP nanoparticles have small particle sizes and are highly dispersed in the carbon matrix. The structural defects of the prepared carbonaceous materials were detected by Raman spectroscopy. MoP@NPCS and NPCS both exhibit two components at 1359 and 1588 cm−1, corresponding to typical D and G bands (Fig. 1b). Also, the prominent Raman peaks of MoP@NPCS in the range of 300 to 1000 cm−1 may be related to the MoP species.13,19,27 The intensity ratio of ID/IG represents the defects in carbonaceous materials. The ID/IG values are 1.14 for MoP@NPCS and 1.25 for NPCS; this clearly indicates that the heteroatom doping can cause structural defects or microstructural rearrangement of carbon atoms, which is beneficial to electrocatalytic processes. Usually, more structural defects contribute to remarkably enhanced electrocatalytic capability.28,29
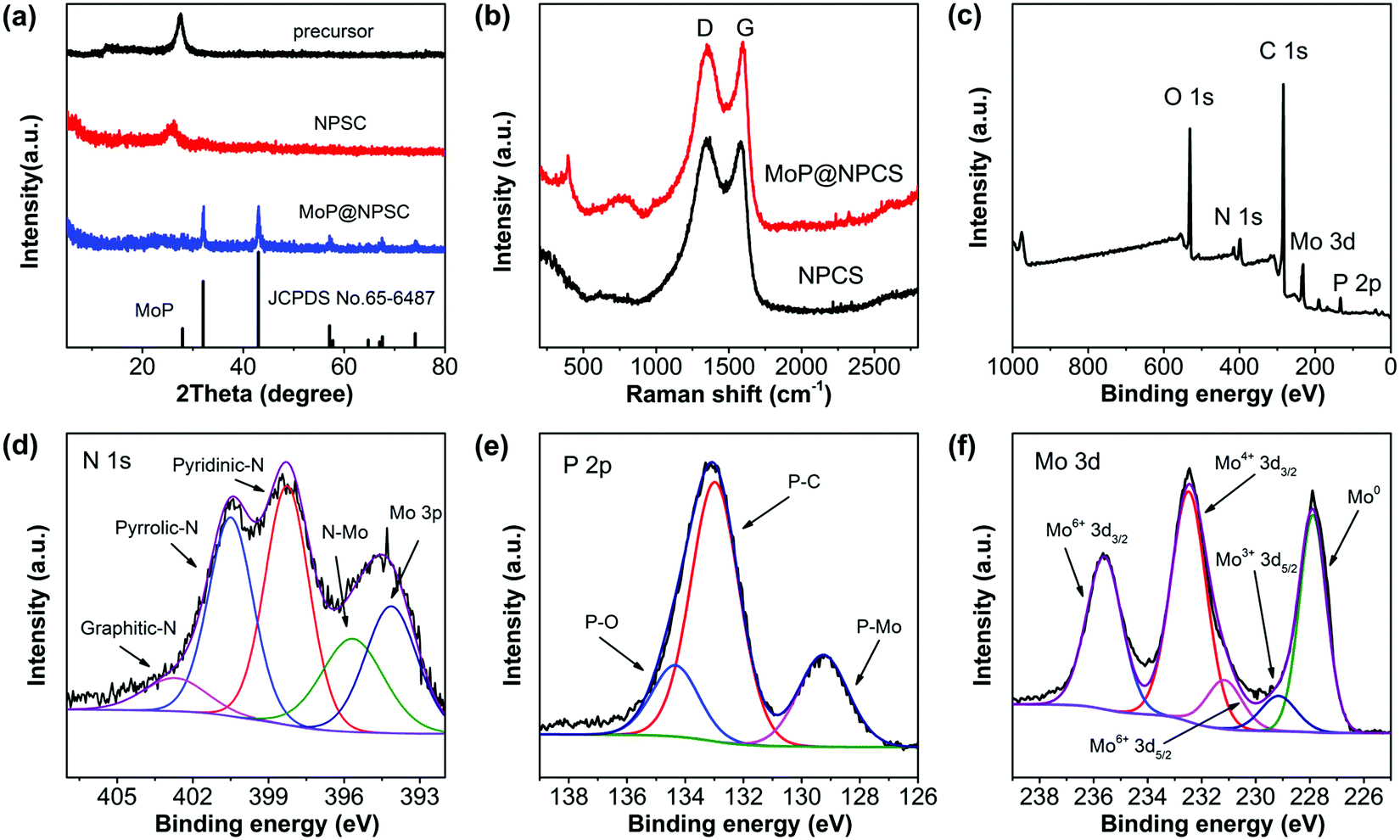 | ||
| Fig. 1 (a) Wide-angle XRD patterns of the polymeric precursors MoP@NPCS, NPCS and MoP@NPCS. (b) Raman spectra of MoP@NPCS and NPCS. (c) XPS survey spectrum of MoP@NPCS; high-resolution (d) N 1s, (e) P 2p and (f) Mo 3d XPS spectra of MoP@NPCS. | ||
The composition and surface structural information of the synthesized carbon-based materials were illustrated by X-ray photoelectron spectroscopy (XPS). The XPS survey scans (Fig. 1c) exhibit the signals of the elements C, N, O, P, and Mo, suggesting the doping of N and P functional groups in MoP@NPCS. To further understand the bonding states of the prepared molybdenum–carbon hybrid materials, the XPS spectrum of NPCS was also recorded as a reference (Fig. S2a, ESI†), revealing N and P contents of 7.78 and 5.67 at%, respectively. The molecular interactions between the polymeric moieties containing sufficient organic functionalities, such as amino and phosphonic groups, in their bridging components induced abundant heteroatom doping under high-temperature carbonization.12,30,31 For the C 1s spectra (Fig. S2b and S3, ESI†), the fitted peak at about 286 eV can be ascribed to the C–N and C–P in the carbon backbone, which are attributed to the nitrogen and phosphonic functional groups in the formed polymeric analogues.19,22,32 Peak deconvolution of N 1s of NPCS (Fig. S2c, ESI†) suggests three contributions located at 398.8 eV (pyridinic-N), 400.7 eV (pyrrolic-N) and 402.8 eV (graphitic-N). Meanwhile, for the N 1s spectra of MoP@NPCS (Fig. 1d), in addition to pyridinic-N, pyrrolic-N and graphitic-N at the binding energies of 398.2, 400.5 and 402.3 eV, respectively, the low fitted peak at 401.1 eV corresponds to the Mo–N bonds of molybdenum nitrides; this peak is commonly observed in N-doped molybdenum–carbon materials.12,15,22 In the high-resolution P 2p spectra of MoP@NPCS (Fig. 1e) and NPCS (Fig. S2d, ESI†), the signals at 133.0 and 133.9 eV reveal the presence of P–C and P–O, indicating that phosphate-like structures may have resulted from the oxidation of P species exposed to air.33,34 Meanwhile, the peak at 129.1 eV of MoP@NPCS is ascribed to P–Mo bonding in MoP@NPCS.35,36 The high-resolution Mo 3d XPS spectrum reveals five components (Fig. 1f) at 227.9 eV (Mo0), 229.2 eV (Mo3+), 231.2 eV (Mo6+), 232.5 eV (Mo4+) and 235.6 eV (Mo6+).9,20,22,25,35 Mo4+ and Mo6+ correspond to oxidized molybdenum species (MoO2 and MoO3) due to surface oxidation in air. The Mo3+ is related to molybdenum phosphide (MoP) and nitrides (MoN) originating from the phosphorization and nitridation processes. Additionally, Mo0 is assigned to molybdenum carbide (Mo2C), which forms the active sites for HER. Therefore, the XPS analysis indicates the successful doping of N and P dopants and the formation of MoP nanoparticles for the MoP@NPCS hybrid materials. It should be mentioned that the binding energies of N 1s and P 2p for MoP@NPCS are both positively shifted in comparison to the carbonaceous material of NPSC, indicating the presence of strong interactions between the MoP nanoparticles and porous N,P-codoped carbon.
Scanning electron microscopy (SEM) and transition electron microscopy (TEM) images of the fabricated hybrid materials are shown in Fig. 2. After pyrolysis at 900 °C, the fabricated NPCS exhibits a sponge-like structure with wide meso-/macropores (Fig. 2a), which can provide sufficient pathways for the fast transportation of electrolytes and products.15,37 Meanwhile, MoP@NPCS (Fig. 2b) inherited the well-designed porous character of NPCS. Also, the rough surface indicates the homogeneous dispersion of MoP nanoparticles on the carbon substrates. However, MoP@NPCF prepared without the use of g-C3N4 monolith exhibits a featureless structure (Fig. S5, ESI†) which is evidently different from the morphology of MoP@NPCS; this indicates that the presence of g-C3N4 monolith is critical to obtain the sponge-like structure. Also, the NPCF fabricated in the absence of g-C3N4 monolith exhibits a nanofiber morphology (Fig. S6, ESI†). Random open pores can be clearly observed in the TEM images of NPCS (Fig. 2c and d). The high-magnification TEM image (Fig. 2e) indicates the amorphous structure of NPCS, which is consistent with the selected area electron diffraction (SAED) pattern (Fig. 2e inset). The homogeneous dispersion of N and P elements is also revealed by the typical element mapping with energy-dispersive X-ray spectroscopy (EDX) under the scanning transition electron microscopy (STEM) model of NPCS (Fig. 2f), confirming the strong interaction between the polyaniline oligomers and organophosphonic linkages and demonstrating the formation of the chemically modified carbonaceous material. The low-magnification TEM images in Fig. 2g and h exhibit that nanoparticles are uniformly spread on the carbon monolith. Also, the high-resolution TEM (HR-TEM) images (Fig. 2i and j) indicate the crystalline nature of the nanoparticles; the apparent lattice finger distance of 0.28 nm is ascribed to the (100) plane of hexagonal MoP. The SAED pattern (Fig. 2j inset) displays three observed diffraction rings that are assigned to the (001), (101) and (002) planes of hexagonal MoP. The evident bright-dark contrast further demonstrates the uniform dispersion of the nanoparticles in the carbon matrix, as observed from the high angle bright field (HAABF) image (Fig. 2k). The homogeneous distribution of C, N, P, and Mo elements in MoP@NPCS was verified by STEM-EDX element mapping images over a widespread region (Fig. 2m). The above results indicate that the Mo7O246−-induced polyaniline polymeric network with sufficient nitrogen- and phosphate-containing functional groups can be employed as a reversible precursor for the fabrication of chemically modified carbon with well-developed porosity. The multifunctional amino groups of polyaniline ensure efficient hydrogen bonding interactions with the phosphonic acid groups in HEDP.38–40 Therefore, many organic functional groups are distributed uniformly in the obtained polymeric precursor, and the heteroatom-doped carbon matrix is finally formed upon high-temperature carbonization.41–43 In addition, high-temperature pyrolysis can result in sublimation of elemental phosphorus due to the carbothermal reduction of organophosphonic acid, which can further react with the Mo7O246− moieties through a facile phosphorization process to form uniformly-distributed MoP nanoparticles decorating the porous carbon monolith.43,44 As such, the MoP nanoparticles with particle sizes below 5 nm are homogeneously dispersed throughout MoP@NPCS. In comparison, the MoP particles in MoP@NPCF are larger (Fig. S7, ESI†), clearly demonstrating that the g-C3N4 monolith has an evident effect on the formation of the porous morphology with ultrafine MoP particles.
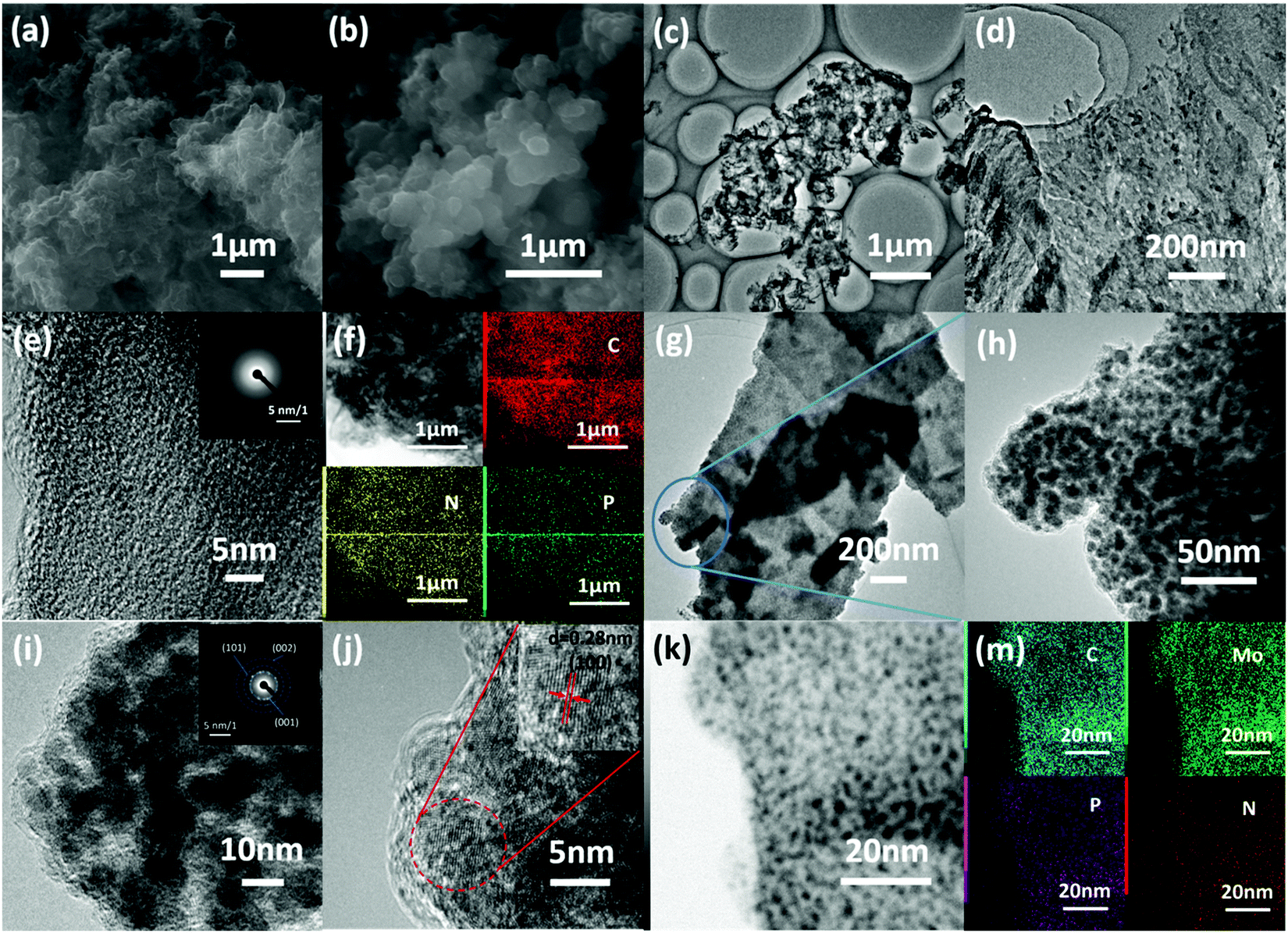 | ||
| Fig. 2 SEM images of (a) NPCS and (b) MoP@NPCS. (c–e) TEM images and (inset e) the SEAD pattern of NPCS. (f) STEM-EDX mapping images of C, N, P and the corresponding HAADF-STEM image of NPCS. (g–j) TEM images of MoP@NPCS and (inset i) the SEAD pattern. (k) HAABF-STEM image of MoP@NPCS and (m) STEM-EDX mapping images of C, Mo, P and N of MoP@NPCS. | ||
The textural properties of the prepared materials were investigated by N2 adsorption–desorption measurements. The N2 sorption isotherms (Fig. S8a, ESI†) are type IV with distinct H3-type hysteresis loops, indicating the formation of large amounts of mesopores stacked from turbostratic carbon.45,46 The enhanced volumes of absorbed nitrogen at relatively high pressures are attributed to the large pores arising from the secondary porosity, consistent with the pore size distribution curves determined by the Barrett−Joyner−Halenda (BJH) model from the adsorption branches (Fig. S8b, ESI†). The thermal decomposition and gas escape at high temperatures result in abundant pores. The Brunauer−Emmett−Teller (BET) specific surface areas were calculated to be 867 and 298 m2 g−1 for NPCS and MoP@NPCS, respectively. The decreased surface area of MoP@NPCS may result from the destruction of the slit-like carbonaceous network after introducing the MoP nanoparticles. This well-developed morphology with abundant pores provides MoP@NPCS with sufficient and accessible active centers to contact the electrolyte and enable diffusion of the electrolyte/products, which is helpful to enhance the electrocatalytic activity.41,44,47 In contrast, the NPCF and MoP@NPCF have low specific surface areas of 142 and 76 m2 g−1, respectively, indicating the effects of the g-C3N4 monolith template on the morphology and textural properties of MoP@NPCS. In addition, the Mo contents of MoP@NPCS, MoP@NPCF and MoP/NPCS were determined to be approximately 45.55 wt%, 44.63 wt%, and 43.37 wt%, respectively (Fig. S9, ESI†). These results indicate that the present strategy is facile and efficient to prepare an N,P-codoped porous carbon matrix with uniformly dispersed MoP nanocrystals which is designed to contain highly active sites towards HER.
3.2. Electrocatalytic performance
The electrocatalytic activities of the synthesized materials toward hydrogen evolution were evaluated with the use of a classical three-electrode apparatus in alkaline medium (1.0 M KOH), wherein a glassy carbon electrode (GCE) was coated with the catalysts as the working electrodes. For the purpose of comparison, pristine GCE and commercial Pt/C (20 wt%) were also investigated. From the linear sweep voltammogram (LSV) polarization curves (Fig. 3a), the Pt/C electrode exhibits outstanding activity toward HER and presents a small onset potential (Eonset) of nearly zero, while the pristine GCE electrode shows no evident measured current. The dual-doped NPCS manifests a high onset potential of over −226 mV; this performance is superior to those of previously reported N,P-doped porous carbon,43 P-doped graphene,48 and other carbon-based catalysts (Table S1, ESI†), indicating the enhanced effects of heteroatom doping on the proton adsorption and reduction abilities and, thereby, enhancement of the HER performance.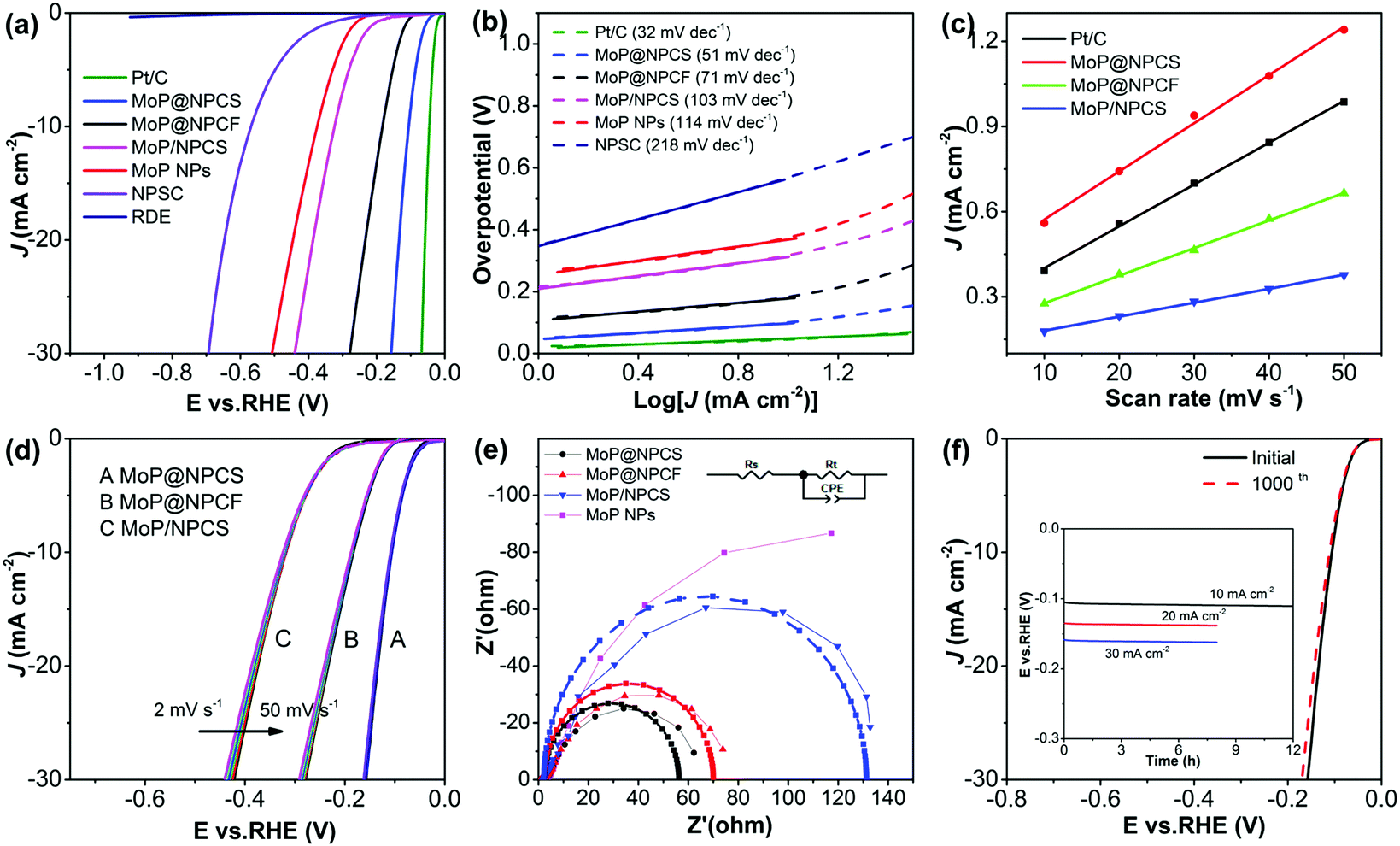 | ||
| Fig. 3 (a) LSV polarization curves and (b) Tafel plots of NPCS, MoP@NPCS, MoP@NPCF, MoP/NPCS and MoP NPs together with Pt/C and bare GCE as a control (1.0 M KOH; scan rate: 2 mV s−1). (c) Plot of the current density at 0.18 V vs. the scan rate of MoP@NPCS, MoP/NPCS, MoP@NPCF and Pt/C. (d) LSV profiles of MoP@NPCS, MoP/NPCS and MoP@NPCF at different scan rates from 2 to 50 mV s−1. (e) EIS of the catalysts; the measured data on the electrochemical workstation were fitted by ZView software. Inset in (e): The corresponding equivalent circuit diagram. (f) Polarization curves of MoP@NPCS before and after 1000 potential cycles. Inset in (f): Chronoamperometry curve of MoP@NPCS during electrolysis at stable potentials. | ||
In sharp contrast, MoP@NPCS presents an onset potential of −47 mV with evidently increased cathodic current at high biases. The obtained onset potential is very close to that of the Pt/C benchmark, indicating the promising potential of MoP@NPCS as a candidate cathode for water reduction to produce hydrogen. In addition, cathodic current densities of 10 mA cm−2 (J10) and 20 mA cm−2 (J20) are afforded at the overpotentials of 107 and 133 mV, respectively, for MoP@NPCS. It should be noted that these results for MoP@NPCS are comparable or even superior to the performance of previously reported molybdenum-based electrocatalysts in alkaline medium (Table S1, ESI†). For example, MoC–Mo2C nanoparticles dispersed on nitrogen-rich carbon nanowires maintain 10 mA cm−2 at overpotential of 120 mV,49 and molybdenum phosphide nanocrystals embedded in carbon spheres afford 10 mA cm−2 at an overpotential of 169 mV.50 An overpotential of 194 mV is required to afford 10 mA cm−2 for semimetallic MoP2 nanoparticle films on a metal Mo plate.51 However, the sample prepared by post-loading MoP nanoparticles on NPCS via direct phosphorization (denoted as MoP/NPSC) exhibits inferior performance, with a large onset potential of −123 mV, compared with MoP@NPCS; this is probably due to aggregation of the MoP nanoparticles (Fig. S10, ESI†) and inadequate interaction of MoP and NPCS. Also, MoP@NPCF exhibits inferior HER catalytic activity (Eonset at −104 mV and J10 at 179 mV), which is mainly related to its monotonous porosity.
The reaction kinetics was identified by employing the Tafel slope obtained from the LSV data based on the Tafel equation: η = a + blogJ, where η indicates the overpotential, b represents the Tafel slope, and J corresponds to the current density. Usually, a typical two-electron reaction model for HER involves two steps. A discharge step, namely, H2O + e− → Hads + OH− (Volmer reaction), is followed by a desorption step of H2O + Hads + e− → H2 + OH− (Heyrovsky reaction) or a recombination step of Hads + Hads → H2 (Tafel reaction), where Hads corresponds to H adsorbed on the active site of the electrocatalyst.12,52 Additionally, the rate-controlling steps of the Volmer, Heyrovsky, and Tafel reactions toward the HER process in alkaline medium are associated with Tafel slopes of 120, 40, and 30 mV dec−1, respectively. The fitted Tafel slope for MoP@NPCS is 51 mV dec−1 (Fig. 3b), implying that the water evolution behavior occurring on the active centers follows a Volmer–Heyrovsky mechanism. The significantly improved electrocatalytic performance with low Tafel slopes of MoP@NPCS indicates a low activation energy for the formation of hydrogen, which can be ascribed to the initiated contact of MoP and NPCS and the well-designed architectures for the exposure of active sites.22,28
To further investigate their intrinsic electrochemical reaction capacities, the exchange current densities (J0) of the catalysts were determined. Calculated from the expansion of the Tafel plot, the exchange current density of MoP@NPCS is 0.127 mA cm−2, which is close to that of commercial Pt/C (0.314 mA cm−2); this is evidently superior to the other controllable samples, including NPCS (0.025 mA cm−2), MoP@NPCF (0.031 mA cm−2), MoP/NPSC (0.009 mA cm−2) and MoP NPs (0.006 mA cm−2). Commonly, the exchange current density of an electrocatalyst is considered to be associated with its electrochemically active surface area (ECSA). Especially, the electrochemically active surface area is deemed to be proportional to the electrochemical double layer capacitance (Cdl) at the solid–liquid interface between the catalyst and electrolyte.12,53 Also, Cdl can be easily measured by employing cyclic voltammetry (Fig. S11, ESI†). The Cdl value for MoP@NPCS, calculated from the slope of the current density against the scan rate, is 15.9 mF cm−2 (Fig. 3c); this exceeds the values of MoP/NPCS (4.9 mF cm−2) and MoP@NPCF (9.7 mF cm−2) and is comparable with the capacitance of commercial Pt/C (14.7 mF cm−2). Ultrafine MoP nanoparticles with natural activity are directly anchored on the heteroatom-modified carbon matrix with well-developed pores; this provides a large catalytic surface area and facilitates the transfer of electrolyte and products, thereby affording increased Cdl and improved water reduction capability in comparison with MoP/NPCS and MoP@NPCF. In accordance with their Cdl values, MoP@NPCS reveals a large value of ECSA (422.4 cm2), which is even superior to that of Pt/C (367.5 cm2) (see ESI†); this indicates the presence of sufficient active sites for the electrochemical reaction. Furthermore, the LSV polarization curves of the fabricated electrocatalysts were normalized to ECSA (Fig. S12a, ESI†). MoP@NPCS shows impressive electrocatalytic activity for HER which is much better than that of the control samples. The Tafel slopes and corresponding J0,ECSA values on the basis of ECSA were also measured (Fig. S12b, ESI†) and are shown in Table 1. These results are consistent with the performance obtained from the corresponding LSV polarization curves, revealing the impressive intrinsic electrocatalytic activity of MoP@NPCS.
| Electrocatalyst | η 10 (mA cm−2) | Tafel slope (mV dec−1) | J 0 (mA cm−2) | C dl (mF cm−2) | ECSA (cm2) | Tafel slope ECSA (mV dec−1) | J 0,ECSA (μA cm−2) |
|---|---|---|---|---|---|---|---|
| Pt/C | 45 | 32 | 0.314 | 14.7 | 367.5 | 29 | 0.498 |
| MoP@NPCS | 107 | 51 | 0.127 | 16.9 | 422.4 | 43 | 0.1 |
| MoP@NPCF | 183 | 71 | 0.031 | 9.7 | 242.5 | 49 | 0.01 |
| MoP/NPSC | 315 | 103 | 0.009 | 4.9 | 122.5 | 87 | 0.027 |
| MoP NPs | 371 | 114 | 0.006 | — | — | — | — |
| NPCS | 570 | 218 | 0.025 | — | — | — | — |
The enhanced HER activity of MoP@NPCS can be rationally ascribed to its well-defined porous morphology, its conductive carbonaceous backbone, the direct grafting of MoP nanoparticles with ultrasmall size, and the strong synergistic effect between the MoP nanoparticles and the porous N,P-codoped carbon matrix. First, the electric conductivity of the N,P-doped carbonaceous skeleton can efficiently facilitate charge transfer from the carbon backbone to the MoP nanoparticles, and the unique porous architecture supplies open spaces to improve the mass transfer.13,43,54 It was noted that the current density of MoP@NPCS is not sensitive to the scan rate even at 50 mV s−1 (Fig. 3d); this is because the well-defined porosity facilitates the infiltration of electrolytes, the efficient transfer of reactants (e.g., OH−), and the fast diffusion of gas products (e.g., H2). Meanwhile, the polarization curves of MoP@NPCF and MoP/NPCS show negative shifts with increasing scan rate.
Second, the ultrafine MoP nanoparticles allocated on the carbon matrix can be beneficial for the exposure of active sites. As observed from the XPS spectra, the shifts of the binding energies of N 1s and P 2p of MoP@NPCS in comparison with pristine NPCS, indicate the strong interaction of MoP and NPCS. On one hand, sufficient N and P functional groups in the carbon skeleton can not only alter the electron state of adjacent carbonaceous backbones, but also strongly interact with MoP nanoparticles. On the other hand, the strong coupling effect between MoP and the carbonaceous materials can change the electronic structure of MoP, thereby causing synergistic effects of the MoP@NPCS electrocatalyst systems for improving their electrocatalytic hydrogen evolution capacity.15,22,44 It should be mentioned that the coupling effect between the heteroatom-doped carbonaceous skeleton and transition metal species has been proved for several electrocatalyst systems, including Mo2C/N,P-doped carbon nanospheres,12 and N-doped porous molybdenum carbide and phosphide hybrids,22 to enhance the electrocatalytic hydrogen evolution capability. In addition, the inferior cathodic current measured on the control samples of MoP NPs and MoP/NPCS (Fig. 3a) further demonstrates that the synergistic effect can efficiently improve the electrocatalytic activity.
Finally, the in situ formed MoP nanoparticles on the porous carbon matrix enable intimate electrical connection between them, resulting in favorable reaction kinetics.43,44,55 Electrochemical impedance spectroscopy (EIS) was measured to investigate the HER kinetics. The arc diameter of MoP@NPCS is smaller than that of other control samples (Fig. 3e), including MoP@NPCF, MoP/NPCS and MoP NPs, indicating small contact impedance and fast charge transport between the two sides and thereby improving the HER kinetics. Consequently, the conductive carbonaceous backbone and the direct grafting of MoP nanoparticles both contribute to the highly favorable reaction kinetics of MoP@NPCS for water reduction.
In addition to impressive activity, long-term durability is a critical indicator to evaluate the electrocatalytic capacity of electrocatalysts. For MoP@NPCS, to evaluate the operation stability in strongly alkaline electrolyte, accelerated degradation measurements were performed using continuous cyclic voltammograms at an enlarged sweep rate of 100 mV s−1 (Fig. 3f). The polarization curve recorded after 1000 cycling tests shows a slight positive shift, and only 2.3% degradation of the cathodic current density was observed in comparison with the initial density at −0.10 V; this is due to the alternate accumulation of hydrogen bubbles on the active centers and the slight peeling-off of active species resulting from the extended operation, highlighting the superior stability of this hybrid material to resist accelerated degradation. Furthermore, arising from the practical operation, the chronoamperometric response was also measured within an extended period (Fig. 3f inset). The cathodic current of MoP@NPCS collected at a fixed overpotential of 106 mV exhibits a steady value of about 10 mA cm−2 over 12 h without observable change, signifying the high stability of the fabricated catalyst to resist accelerated degradation.
Moreover, the coupling of N and P dopants with abundant defects on the porous carbon matrix (NPCS) produces sufficient active sites, contributing to the electrocatalytic activity toward OER.56,57Fig. 4a shows the LSV curves of the fabricated electrocatalysts and the state-of-the-art OER catalyst IrO2 for the purpose of comparison. IrO2 renders a sharp onset potential and requires an overpotential of 310 mV to provide 10 mA cm−2. Interestingly, NPCS exhibits high OER catalytic activity, only requiring overpotentials of 390 and 450 mV to supply 10 and 20 mA cm−2, respectively, along with a low Tafel slope of 78 mV dec−1 (Fig. 4b); these results are evidently better than those of other control samples and are comparable to those of reported carbonaceous electrocatalysts, as summarized in Table S2 (ESI†). The large difference between the specific surface areas of NPCS and NPCF (Fig. S8, ESI†) may be the dominant reason for the inferior catalytic performance of NPCF toward OER.16,41 Additionally, the higher content of heteroatom-containing functional groups of NPCS compared with that of NCS, based on XPS analysis (Fig. S15, ESI†), corresponds to the significant OER electrocatalytic activity. On one hand, the sufficient N and P dopants in the carbonaceous backbones can change the electronic structure and redistribute the spin density of adjacent carbon, consequently promoting the adsorption of water oxidation intermediates (i.e., OH− and OOH−) and thereby enhancing the oxygen evolution process.58,59 On the other hand, the N,P-doped carbon with well-developed porous character provides a large electrocatalytic surface area and facilitates the fast transfer of electrolyte and products, as shown by the fact that NPCS is not sensitive to scan rate (Fig. 4c); thus, its OER activity is enhanced.41,44 Furthermore, the stability of NPCS was tested by continuously cycling the catalyst for 1000 cycles ranging from 1.0 to 1.8 V with an accelerated scan rate of 50 mV s−1. After the cycling process, NPCS afforded a similar LSV curve to the initial curve, with negligible current loss (Fig. 4d inset). The long-term durability was also measured by electrolysis under an anodic current of 10 mA cm−2 (Fig. 4d). NPCS exhibits a negligible anodic current attenuation of 6.6%, evidently lower than that of IrO2 (21.7%). These results clearly indicate that NPCS is a suitable electrocatalyst for OER, with high catalytic efficiency and excellent durability.
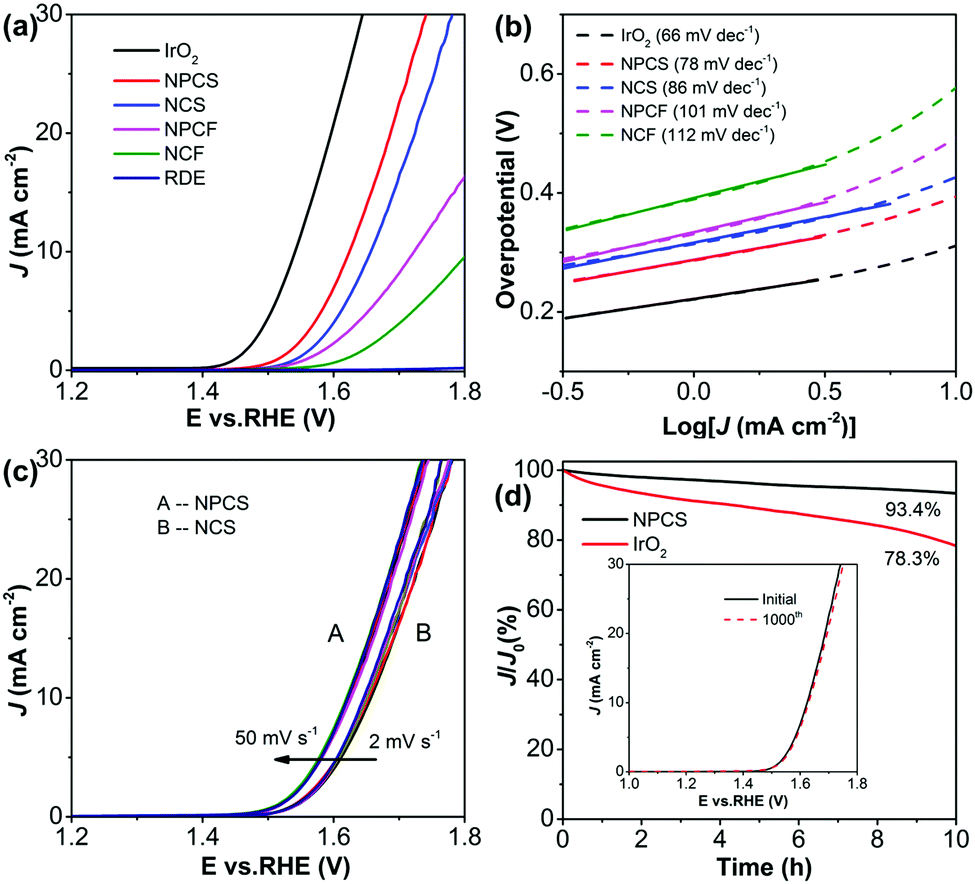 | ||
| Fig. 4 (a) LSV polarization curves and (b) Tafel plots of NPCS, NCS, NPCF, and NCF, together with IrO2 and bare GCE as a control (1.0 M KOH; scan rate: 2 mV s−1). (c) LSV profiles of NPCS and NCS at different scan rates from 2 to 50 mV s−1. (d) Chronoamperometry curves of NPCS and IrO2 during electrolysis over 10 h. Inset in (d): polarization curves of NPCS before and after 1000 potential cycles. | ||
Finally, the porous carbon material NPCS was used as an anode to pair with an MoP@NPCS cathode in a two-electrode electrolyzer to achieve overall water splitting in 1.0 M KOH (Fig. 5a). A current density of 10 mA cm−2 was obtained at a cell voltage of 1.70 V, which is comparable to that of reported OER/HER bifunctional catalysts (Table S3, ESI†). The long-term stability of this fabricated electrolyzer was also assessed. The electrolyzer showed a stable current output even under a larger current density of 40 mA cm−2. This reveals that MoP@NPCS and NPCS have significant potential as stable and efficient cathodic and anodic electrocatalysts for overall water splitting. Most importantly, this fabricated two-electrode electrolyzer can be driven by the energy input of commercial silicon photovoltaic devices stimulated by solar light; as illustrated in Fig. 5c, large numbers of hydrogen (right) and oxygen (left) bubbles accumulate on the electrode surface.
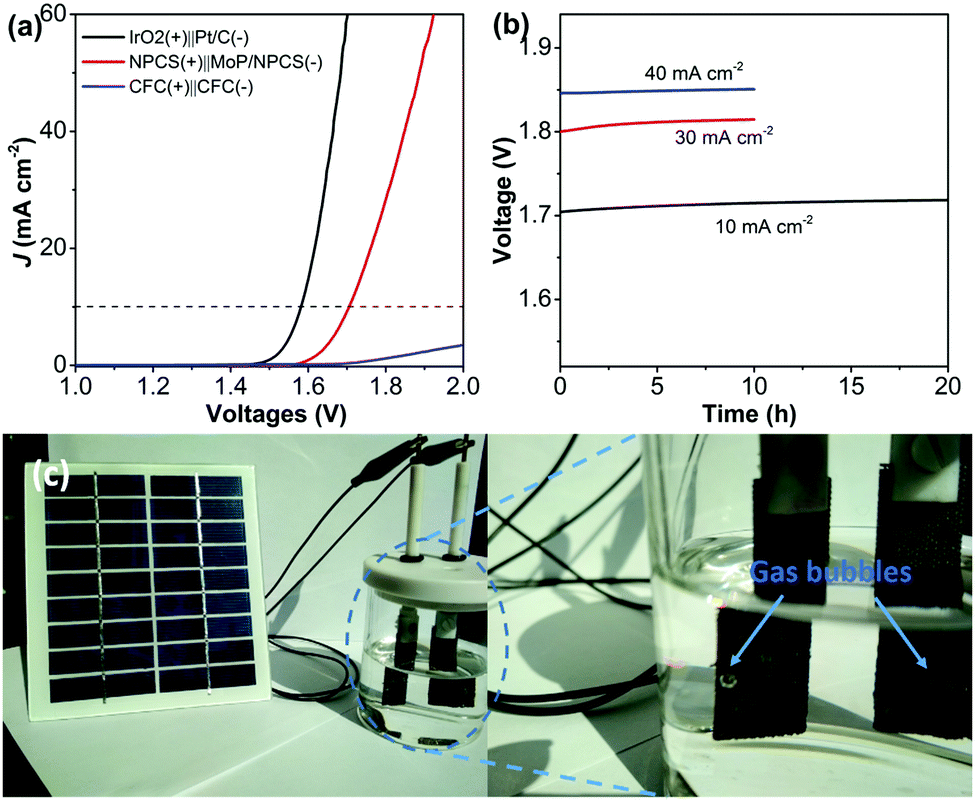 | ||
| Fig. 5 (a) Polarization curves of a two-electrode alkaline electrolyzer with different electrocatalyst couples (1.0 M KOH; scan rate: 2 mV s−1). (b) Galvanostatic water electrolysis at a stationary potential for the fabricated electrolyzer NPCS(+)||MoP/NPCS(−). (c) Digital photograph of the electrolyzer, revealing H2 (left) and O2 (right) production towards overall water splitting with energy input by sunlight. | ||
4. Conclusions
A well-developed porous N,P-doped carbon matrix functionalized with ultrafine MoP nanoparticles was directly fabricated by pyrolyzing a polyaniline-organophosphonic polymer with molybdate in the presence of g-C3N4 monolith, wherein the g-C3N4 can efficiently tailor the micro-structure of the obtained materials. The synthesized materials exhibit high electrocatalytic hydrogen evolution activity and excellent stability, which can be ascribed to the sufficient electrocatalytic sites, enhanced chemically active surface area, unique structural characteristics and synergistic effect between the MoP nanoparticles and heteroatom-modified carbonaceous skeleton. Moreover, this method is versatile and can be applied to prepare N,P-codoped carbon materials that exhibit high OER electrocatalytic activities with impressive durability in alkaline medium. Remarkably, the fabricated MoP@NPCS and NPCS can be used as the cathode and anode, respectively, in a two-electrode alkaline electrolyzer, affording an overall water splitting current density of 10 mA cm−2 at a voltage of 1.70 V and robust durability. This work provides a facile and effective strategy to develop hierarchical structures decorated with nanocrystals; it has significant potential for applications in sustainable energy-related systems.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21421001, 21573115), the 111 project (B12015), and the Fundamental Research Funds for the Central Universities (63185015).Notes and references
- Z. Xing, Q. Liu, A. M. Asiri and X. Sun, Adv. Mater., 2014, 26, 5702–5707 CrossRef PubMed.
- W. Zhang, S. Zhu, R. Luque, S. Han, L. Hu and G. Xu, Chem. Soc. Rev., 2015, 45, 715–752 RSC.
- M. A. R. Anjum and J. S. Lee, ACS Catal., 2017, 7, 3030–3038 CrossRef.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef PubMed.
- J. Jiang, C. Zhang and L. Ai, Electrochim. Acta, 2016, 208, 17–24 CrossRef.
- M. Wang, J. Jiang and L. Ai, ACS Sustainable Chem. Eng., 2018, 6, 6117–6125 CrossRef.
- A. Sun, Y. Shen, Z. Wu and D. Wang, Int. J. Hydrogen Energy, 2017, 42, 14566–14571 CrossRef.
- K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef PubMed.
- Z. Wu, J. Wang, K. Xia, W. Lei, X. Liu and D. Wang, J. Mater. Chem. A, 2018, 6, 616–622 RSC.
- J. T. Ren, G. G. Yuan, C. C. Weng, L. Chen and Z. Y. Yuan, Nanoscale, 2018, 10, 10620–10628 RSC.
- Y. Zhu, G. Chen, X. Xu, G. Yang, M. Liu and Z. Shao, ACS Catal., 2017, 7, 3540–3547 CrossRef.
- Y. Y. Chen, Y. Zhang, W. J. Jiang, X. Zhang, Z. Dai, L. J. Wan and J. S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef PubMed.
- L. Chen, H. Jiang, H. Jiang, H. Zhang, S. Guo, Y. Hu and C. Li, Adv. Energy Mater., 2017, 7, 1602782 CrossRef.
- J. Jiang, Q. Liu, C. Zeng and L. Ai, J. Mater. Chem. A, 2017, 5, 16929–16935 RSC.
- H. Wang, Y. Cao, C. Sun, G. Zou, J. Huang, X. Kuai, J. Zhao and L. Gao, ChemSusChem, 2017, 10, 3540–3546 CrossRef PubMed.
- L. R. L. Ting, Y. Deng, L. Ma, Y.-J. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 861–867 CrossRef.
- Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693–1705 CrossRef.
- Z. Ye, J. Yang, B. Li, L. Shi, H. Ji, L. Song and H. Xu, Small, 2017, 13, 1700111 CrossRef PubMed.
- J. Yang, F. Zhang, X. Wang, D. He, G. Wu, Q. Yang, X. Hong, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2016, 128, 13046–13050 CrossRef.
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J.-Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC.
- H. Yan, Y. Jiao, A. Wu, C. Tian, X. Zhang, L. Wang, Z. Ren and H. Fu, Chem. Commun., 2016, 52, 9530–9533 RSC.
- Y. Huang, J. Ge, J. Hu, J. Zhang, J. Hao and Y. Wei, Adv. Energy Mater., 2018, 8, 1701601 CrossRef.
- C. Lu, D. Tranca, J. Zhang, F. R. Hernandez, Y. Su, X. Zhuang, F. Zhang, G. Seifert and X. Feng, ACS Nano, 2017, 11, 3933–3942 CrossRef PubMed.
- W. Gao, Y. Shi, Y. Zhang, L. Zuo, H. Lu, Y. Huang, W. Fan and T. Liu, ACS Sustainable Chem. Eng., 2016, 4, 6313–6321 CrossRef.
- Y. Mu, Y. Zhang, L. Fang, L. Liu, H. Zhang and Y. Wang, Electrochim. Acta, 2016, 215, 357–365 CrossRef.
- Y. Li, X.-K. Tian, J. Wang, L.-l. Ma, C. Dai, c. Yang and C. Zhou, Nanoscale, 2016, 8, 1676–1683 RSC.
- L. N. Zhang, S.-H. Li, H.-Q. Tan, S. U. Khan, Y.-Y. Ma, H.-Y. Zang, Y.-H. Wang and Y.-G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 16270–16279 CrossRef PubMed.
- Y.-Y. Ma, C.-X. Wu, X.-J. Feng, H.-Q. Tan, L.-K. Yan, Y. Liu, Z.-H. Kang, E.-B. Wang and Y.-G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC.
- J. Yang, F. Zhang, X. Wang, D. He, G. Wu, Q. Yang, X. Hong, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2016, 128, 13046–13050 CrossRef.
- H. Wei, Q. Xi, X. a. Chen, D. Guo, F. Ding, Z. Yang, S. Wang, J. Li and S. Huang, Adv. Sci., 2018, 5, 1700733 CrossRef PubMed.
- L. Peng, Y. Nie, L. Zhang, R. Xiang, J. Wang, H. Chen, K. Chen and Z. Wei, ChemCatChem, 2017, 9, 1588–1593 CrossRef.
- W. Tian, H. Zhang, H. Sun, A. Suvorova, M. Saunders, M. Tade and S. Wang, Adv. Funct. Mater., 2016, 26, 8651–8661 CrossRef.
- C. Xiao, Y. Li, X. Lu and C. Zhao, Adv. Funct. Mater., 2016, 26, 3515–3523 CrossRef.
- C. Wang, J. Jiang, T. Ding, G. Chen, W. Xu and Q. Yang, Adv. Mater. Interfaces, 2015, 3, 1500454 CrossRef.
- X. Sun, L. Lu, Q. Zhu, C. Wu, D. Yang, C. Chen and B. Han, Angew. Chem., Int. Ed., 2018, 57, 2427–2431 CrossRef PubMed.
- D. Wang, X. Zhang, D. Zhang, Y. Shen and Z. Wu, Appl. Catal., A, 2016, 511, 11–15 CrossRef.
- X.-D. Wang, Y.-F. Xu, H.-S. Rao, W.-J. Xu, H.-Y. Chen, W.-X. Zhang, D.-B. Kuang and C.-Y. Su, Energy Environ. Sci., 2016, 9, 1468–1475 RSC.
- T. Yang, R. Yang, H. Chen, F. Nan, T. Ge and K. Jiao, ACS Appl. Mater. Interfaces, 2015, 7, 2867–2872 CrossRef PubMed.
- D. Xu, C. Chen, J. Xie, B. Zhang, L. Miao, J. Cai, Y. Huang and L. Zhang, Adv. Energy Mater., 2016, 6, 1501929 CrossRef.
- C. Long, D. Qi, T. Wei, J. Yan, L. Jiang and Z. Fan, Adv. Funct. Mater., 2014, 24, 3953–3961 CrossRef.
- Y. P. Zhu, Y. L. Liu, Y. P. Liu, T. Z. Ren, G. H. Du, T. H. Chen and Z. Y. Yuan, J. Mater. Chem. A, 2015, 3, 11725–11729 RSC.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 16850–16856 CrossRef PubMed.
- J. T. Ren, G. G. Yuan, C. C. Weng and Z. Y. Yuan, Electrochim. Acta, 2018, 261, 454–463 CrossRef.
- Y. P. Zhu, X. Xu, H. Su, Y. P. Liu, T. Chen and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 28369–28376 CrossRef PubMed.
- J. Liu, Y. Xie, Y. Nan, G. Gou, X. Li, Y. Fang, X. Wang, Y. Tang, H. Yang and J. Ma, Electrochim. Acta, 2017, 257, 233–242 CrossRef.
- L. Ai, T. Tian and J. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 4771–4777 CrossRef.
- T. Tian, L. Huang, L. Ai and J. Jiang, J. Mater. Chem. A, 2017, 5, 20985–20992 RSC.
- B. Zhang, H.-H. Wang, H. Su, L.-B. Lv, T.-J. Zhao, J.-M. Ge, X. Wei, K.-X. Wang, X.-H. Li and J.-S. Chen, Nano Res., 2016, 9, 2606–2615 CrossRef.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, Chem. Sci., 2016, 7, 3399–3405 RSC.
- Z. Wu, J. Wang, R. Liu, K. Xia, C. Xuan, J. Guo, W. Lei and D. Wang, Nano Energy, 2017, 32, 511–519 CrossRef.
- Z. Pu, I. Saana Amiinu, M. Wang, Y. Yang and S. Mu, Nanoscale, 2016, 8, 8500–8504 RSC.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- T. Zhou, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, A. Borgna and R. Xu, ACS Catal., 2017, 7, 6000–6007 CrossRef.
- C. Qiu, L. Ai and J. Jiang, ACS Sustainable Chem. Eng., 2018, 6, 4492–4498 CrossRef.
- J. T. Ren, G. G. Yuan, C. C. Weng and Z. Y. Yuan, ACS Sustainable Chem. Eng., 2018, 6, 707–718 CrossRef.
- J. C. Li, P. X. Hou, S. Y. Zhao, C. Liu, D. M. Tang, M. Cheng, F. Zhang and H. M. Cheng, Energy Environ. Sci., 2016, 9, 3079–3084 RSC.
- G. L. Tian, M. Q. Zhao, D. Yu, X. Y. Kong, J. Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef PubMed.
- R. Ma, R. Xing, G. Lin, Y. Zhou, Q. Liu, M. Yang, C. Hu, K. Yan and J. Wang, Mater. Chem. Front., 2018 10.1039/c8qm00160j.
- C. Wu, Y. Zhang, D. Dong, H. Xie and J. Li, Nanoscale, 2017, 9, 12432–12440 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00226f |
| This journal is © the Partner Organisations 2018 |