
Towards lab-on-a-chip diagnostics for malaria elimination
N.
Kolluri
,
C. M.
Klapperich
and
M.
Cabodi
 *
*
Department of Biomedical Engineering, Boston University, Boston, MA, USA. E-mail: cabodi@bu.edu
First published on 3rd November 2017
Abstract
Malaria continues to be one of the most devastating diseases impacting global health. Although there have been significant reductions in global malaria incidence and mortality rates over the past 17 years, the disease remains endemic throughout the world, especially in low- and middle-income countries. The World Health Organization has put forth ambitious milestones moving toward a world free of malaria as part of the United Nations Millennium Goals. Mass screening and treatment of symptomatic and asymptomatic malaria infections in endemic regions is integral to these goals and requires diagnostics that are both sensitive and affordable. Lab-on-a-chip technologies provide a path toward sensitive, portable, and affordable diagnostic platforms. Here, we review and compare currently-available and emerging lab-on-a-chip diagnostic approaches in three categories: (1) protein-based tests, (2) nucleic acid tests, and (3) cell-based detection. For each category, we highlight the opportunities and challenges in diagnostics development for malaria elimination, and comment on their applicability to different phases of elimination strategies.
 Nikunja Kolluri | Nikunja Kolluri is a Ph.D. candidate in Biomedical Engineering at Boston University. She obtained her Masters in Biotechnology from the University of Pennsylvania in 2015 and received her B.S. with Honors in Chemical Engineering with a double major in Biomedical Engineering from Carnegie Mellon University in 2011. Nikunja previously worked as a process engineer in vaccine manufacturing and commercialization at Merck & Co, Inc. Her research interests include the development of novel isothermal assays and paper-fluidic devices for the diagnosis of malaria and other blood-borne pathogens at the point of care. She is broadly interested in translational research and development of medical devices for low-resource settings. |
 Catherine Klapperich | Dr. Catherine Klapperich is the Associate Dean for Research and the Founding Director of the Precision Diagnostics Center at Boston University. She is a Professor of Biomedical Engineering and the Dorf-Ebner Distinguished Faculty Fellow. Dr. Klapperich's research is focused on engineering diagnostic tests for use in low resource settings and at the point of care. Current projects are focused on disposable microfluidic and paper fluidic diagnostics that incorporate on-board sample preparation to enable molecular testing. Recently, Dr. Klapperich and colleagues founded a startup company to commercialize these technologies. |
 Mario Cabodi | Mario Cabodi received an MSci degree in Physics from Imperial College in London and a Ph.D. in Physics from Cornell University, where he was also a Postdoctoral Research Associate in Chemical Engineering. At Cornell, he used micro- and nano-fluidics for analytical separations of biomolecules, and later developed active biomaterials, based on embedded microfluidic structures. He joined the Boston University research faculty as Research Assistant Professor of Biomedical Engineering in 2008. His research interests at BU focus on microfluidic sample preparation techniques to enhance the detection of analytes in biological fluids, specifically integrating plastic and paper-fluidic technologies with nucleic acid-based tests for point-of-care applications. |
Introduction
Malaria affects billions of people around the world, especially in low- and middle-income countries. In the year 2000, the United Nations General Assembly identified malaria as an important global health challenge and pledged to halt and reduce malaria incidence as a Millennium Development Goal.1 Although there have been strides towards the elimination of malaria, the disease continues to be a major threat to world health. According to the World Health Organization (WHO), there were 212 million new cases and 429![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2015 and approximately 2.7 billion people remain at risk globally, with most of the disease burden falling on resource-constrained countries in sub-Saharan Africa or South-East Asia.2 The WHO has set ambitious milestones towards a malaria-free world, including worldwide reduction of 40% in global incidence and mortality, and achieving malaria elimination in 10 countries by 2020.3
000 deaths in 2015 and approximately 2.7 billion people remain at risk globally, with most of the disease burden falling on resource-constrained countries in sub-Saharan Africa or South-East Asia.2 The WHO has set ambitious milestones towards a malaria-free world, including worldwide reduction of 40% in global incidence and mortality, and achieving malaria elimination in 10 countries by 2020.3
Malaria is caused by the infection of red blood cells by five protozoan parasites of the Plasmodium species. Malaria infections begin with symptoms similar to a minor systemic viral illness, including fever, chills, abdominal discomfort, fatigue, and vomiting. When diagnosed and treated early, patients make a full recovery, but, if left untreated, malaria can progress rapidly to a severe, potentially lethal infection within hours to days.4 This is especially true for P. falciparum infections, which are almost always fatal without treatment,3 and account for 99% of malaria deaths.2 The recommended treatment for malaria is artemisinin-combination therapy (ACT), which is generally effective and well-tolerated by patients.4 However, ACT therapies can be over-prescribed due to lack of access to affordable diagnostics, leading to concerns about drug resistance in Plasmodium species. ACT therapy is often prescribed to all patients presenting with a fever, even without a definite malaria diagnosis,5 and, in the absence of an affordable diagnostic test, patients without any symptoms are often prescribed ACTs to prevent initial occurrence or relapse of malaria infection.4,6 ACTs are also used in mass drug administration programs where all healthy members of a population are treated with a therapeutic concentration of antimalarial drugs to cure possible asymptomatic infections.4 These programs may be enacted because sufficiently sensitive and affordable diagnostics are not yet available.
Challenges for malaria elimination
Elimination of malaria is defined as country-wide interruption of mosquito-borne transmission of a specific Plasmodium parasite, resulting in zero indigenous cases for 3 years or more.7 A significant challenge for elimination efforts is the large number of infected patients who are asymptomatic or remain undiagnosed. Asymptomatic infections do not result in a fever or the other acute symptoms described above. These infections can include: early-stage, low-parasite count infections which have not yet triggered an immune response; chronic infections with small numbers of parasites (low parasitemia) that will never trigger an immune response and become symptomatic; or infections with mild enough symptoms that lead patients not to seek treatment.8 Current malaria diagnostics lack the sensitivity to identify asymptomatic patients, thereby curbing malaria elimination efforts (Fig. 1).8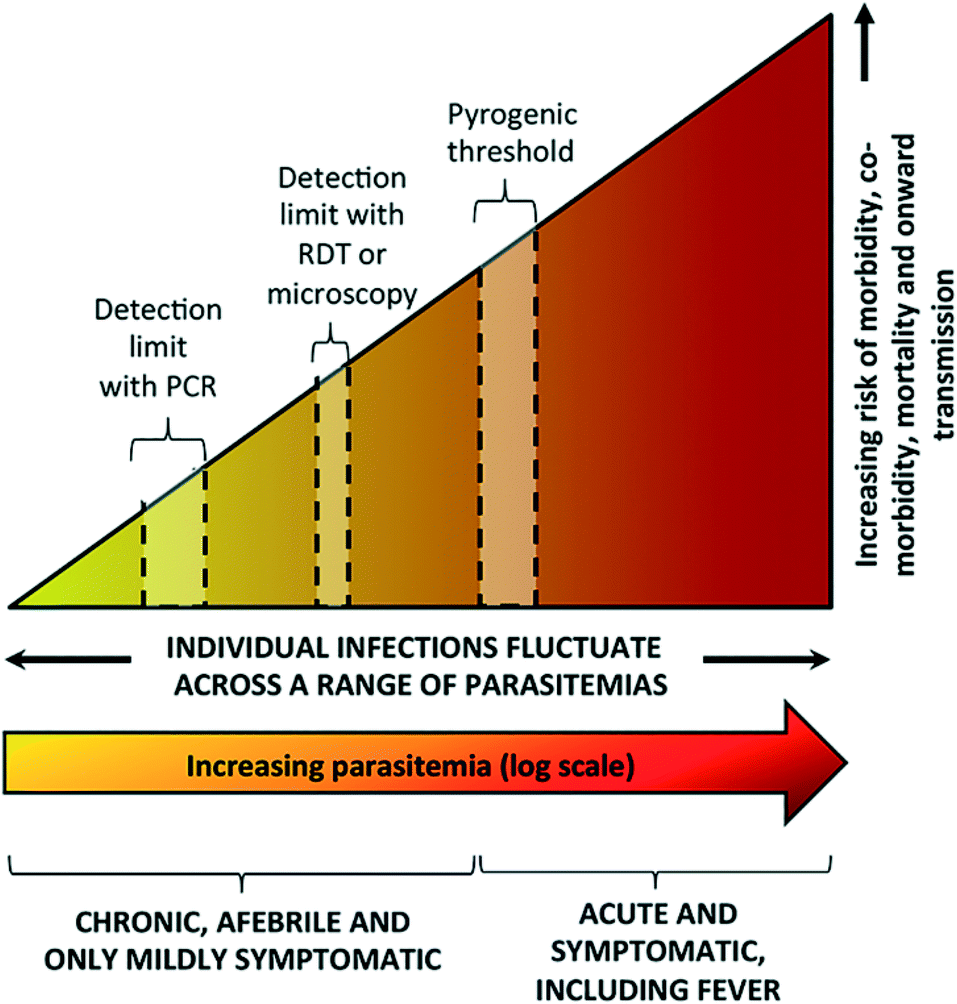 | ||
| Fig. 1 Spectrum of malaria infection. As parasite levels in blood increase, there is an increasing risk of morbidity, co-morbidity, mortality, and onward transmission of infection. Microscopy and rapid diagnostic tests (RDTs) are sensitive enough to detect symptomatic infections at or above the pyrogenic threshold, but are not sensitive enough to detect low parasite density infections. While polymerase chain reaction (PCR) is more sensitive, it is unable to detect infections with very low parasite density. Reproduced with permission from ref. 8 (2016), license CC BY 4.0. | ||
These asymptomatic patients constitute infectious reservoirs, where parasite transmission continues but is invisible to the health system. While major gains have been made in controlling the disease by timely diagnosis and treatment of symptomatic patients and mosquito vector control measures, asymptomatic patients who constitute infectious reservoirs must be identified and treated to achieve malaria elimination.3
An additional consideration in diagnostic development is the complex life cycle of the malaria parasite and its implications for disease transmission (Fig. 2). Sexual stage gametocytes are the only parasites that can transmit from the human host to the mosquito vector, and thus must necessarily be a consideration for any diagnostic tool aimed at elimination. Only a small fraction of parasites in the bloodstream are sexual stage gametocytes (<5% of the total biomass for P. falciparum infections), and late stage gametocytes circulate in the blood stream at extremely low concentrations that cannot be detected by current diagnostic techniques (0.1 parasites per μL – 100 parasites per μL).9 Identifying and treating these infections is an important part of malaria elimination efforts, and will further improve epidemiological understanding of malaria transmission.
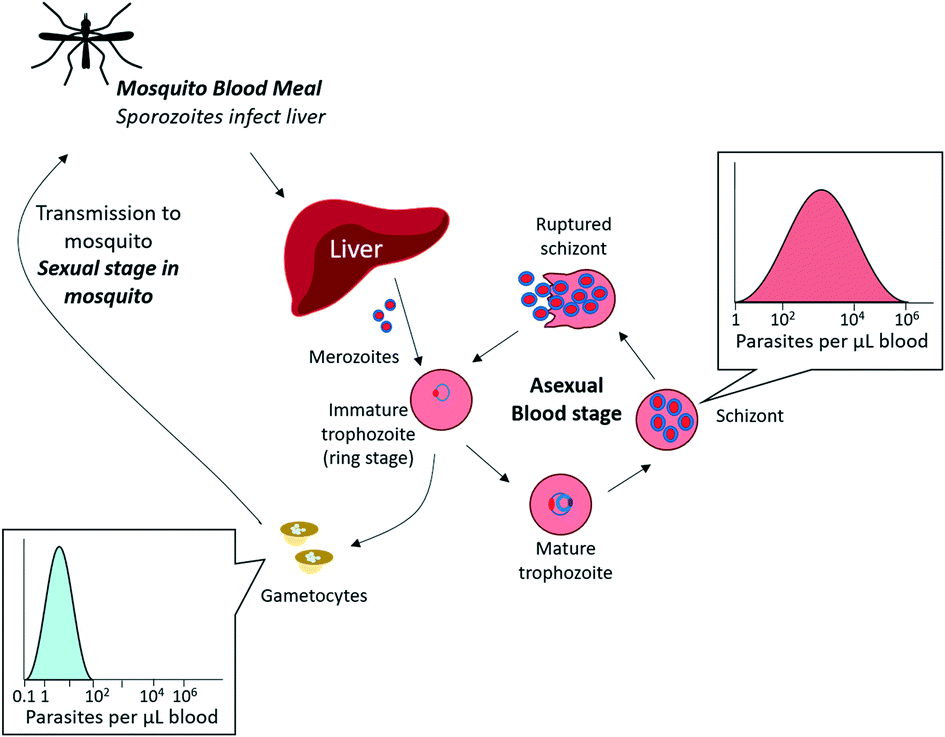 | ||
| Fig. 2 Malaria asexual multiplication cycle. The majority of circulating parasites in blood are in the asexual stage, found in varying concentrations depending on the extent of infection. A small fraction of asexual parasites can produce sexual gametocytes, which are transmissible to mosquitos. The sexual multiplication stage (not pictured) occurs in the mosquito. Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Microbiology, ref. 9, Copyright 2014. | ||
The technical challenges presented by asymptomatic infectious reservoirs and low-concentration gametocyte infection will need to be overcome to achieve the elimination targets set forth by the WHO.3 Countries in the malaria elimination phase employ a targeted strategy to identify, treat, and monitor new and recurring malaria infection. The elimination strategy involves (1) stratifying a geographic area by transmission intensity, (2) implementing vector control strategies to prevent further transmission, and (3) performing active surveillance and case management to prevent further infection transmission.7 Diagnostic devices for elimination-phase countries must therefore meet performance criteria for different purposes: case management of symptomatic infections (>100 parasites per μL in blood), parasite screening to monitor low-density (i.e., asymptomatic and circulating gametocyte) infections and study infection transmission (≤2 parasites per μL asexual stage parasites, ≤0.1 parasites per μL sexual stage gametocytes), and population surveillance to allow for active intervention to prevent transmission (≤2 parasites per μL). Bell et al. summarized the target product profiles to meet the demands for elimination-phase populations in a separate review.10 Current diagnostic methods are mainly focused on P. falciparum and P. vivax infections, as these constitute the vast majority of fatal infections in the elimination-phase countries.3 There is some advantage to distinguishing between the Plasmodium species for epidemiological studies to understand species prevalence and co-infection patterns, but it is more important to specifically diagnose P. falciparum, which causes the most severe infection.10
Limitations of current WHO-recommended malaria diagnostic methods
Since 2010, the WHO has recommended laboratory diagnosis of malaria using diagnostic light microscopy or rapid diagnostic tests (RDTs).4 Microscopic diagnosis relies on the collection of finger-prick blood, preparation of thin- or thick-blood smears on microscope slides, Giemsa-staining, and manual inspection of stained slides.11 The process is expensive, labor-intensive, and time-consuming, requiring trained operators who can accurately identify early-stage parasite infection in red blood cells. Notably, even for experienced technicians, the lower limit of detection (LLOD) of infection for this method is 50–100 parasites per μL.11 RDTs, the other WHO-recommended diagnostic method, rely on immunochromatographic detection of malaria-specific antigens using antigen-specific antibodies immobilized onto lateral flow test strips. Histidine-rich protein 2 (HRP-2) is the most common target for P. falciparum, while parasite lactate dehydrogenase (pLDH) antigen is commonly targeted for pan-Plasmodium detection. Although RDTs provide a clinical diagnosis within 15–20 minutes, they have a LLOD similar to that of microscopy (50–100 parasites per μL).9,11,12Microscopy and RDTs are able to diagnose fully-symptomatic patients, and are thus still the gold-standard for malaria diagnosis for case management of symptomatic infections in many settings. However, as countries move towards malaria elimination, these methods will no longer be sufficient for parasite screening or population surveillance activities, as parasite loads in these cases are much lower (0.1–2 parasites per μL). Notably, in areas with low transmission intensity—including countries moving towards elimination goals—sub-microscopic infections represent 70–80% of diagnosed malaria infections.9 The WHO strategy for these elimination-phase countries involves focused (within a specifically-defined area) or mass (within a broad geographical area) screening and treatment. Therefore, in addition to higher sensitivity, diagnostics must be inexpensive, portable, and available in large quantities to ensure the success of these mass screening and treatment programs.5
Newly developed diagnostics are needed to enable (1) case management of symptomatic infections in low-resourced settings, (2) screening for asymptomatic infections to aid elimination programs, (3) surveillance of continued transmission, and (4) monitoring of therapeutic efficacy.10 Here, we outline advances in the development of technologies appropriate for point-of-care (POC) settings, and evaluate these advances for suitability to achieve malaria elimination goals. Most of these technologies aim to achieve the target LLOD stated by the WHO for new malaria diagnostics, ≤2 parasites per μL. Because different target analytes are measured in different units, Table 1 provides conversions between reported concentrations for P. falciparum.
| Target | Reported concentration | Parasite concentration | Ref. |
|---|---|---|---|
| In this Critical Review, we focus on demonstrated technologies that will be directly applicable for malaria elimination programs. Emerging and upcoming technologies will certainly contribute towards this goal, but are not considered within the scope of this review. | |||
| HRP-2 antigen | 15.3 ± 17.0 ng mL−1 | 200 parasites per μL | 13 |
| pLDH antigen | 15.3 ± 11.3 ng mL−1 | 200 parasites per μL | 13 |
| Parasitemia | 1% | 50000 infected red blood cells per μL |
14 |
| Genomic DNA content | 0.0235 pg | 1 parasite (assuming 1 genome copy/parasite) | 15, 16 |
Improving gold standard techniques
An active area of research is the improvement of current WHO-recommended gold standard techniques for malaria diagnosis, RDTs and microscopy. RDTs are the most commonly used malaria diagnostic tests due to their relative ease-of-use and availability in public-sector health facilities.17 RDTs target malaria-specific antigens such as pLDH or HRP-2, but currently lack the necessary sensitivity to detect asymptomatic infections. Current best-in-class RDTs can detect as low as 0.8 ng mL−1 HRP-2, and 25 ng mL−1 pLDH for P. falciparum and pan-Plasmodium diagnosis, respectively, but most available RDTs do not reach this sensitivity.18 In addition, if the antigen concentration in the sample is too dilute, there is insufficient antigen to bind to the test line to enable visual readout, producing a false negative result. Recent efforts have focused on sample preparation and concentration strategies to improve existing RDT sensitivity, improving on the existing RDT sample workflow.19,20Several approaches to improve RDT signal by incorporating signal enhancement reagents and enzymes have been reviewed elsewhere.21 Sample concentration strategies have also been developed to improve malaria RDTs. For example, Davis et al. have developed several technologies to purify and concentrate target analytes from whole blood using nickel nitrilotriacetic acid (Ni-NTA), which selectively binds histidine-rich proteins in whole blood. Using Ni-NTA-functionalized agarose beads, the group developed a method increase the concentration of malaria-specific HRP-2 antigen in blood, and integrated the method with commercially available RDTs.22–25 Their most recent device, called mBEADS (magnetically-enabled biomarker extraction and delivery system), is a portable unit designed to concentrate the sample and deposit it onto commercially-available RDTs (Fig. 3(i)). The device was demonstrated with five commonly-used RDT brands, adding minimal processing time and cost per assay, and resulting in approximately 10-fold increase in sensitivity compared to conventional RDT results. Other RDT improvement strategies have focused on improving the visual readout from RDTs. Portable readers are being developed to enhance the signal from the RDT test line, allowing for an increase in sensitivity. For example, one technique exploits the thermal signature of irradiated gold nanoparticles bound to a test line using thermal contrast imaging,26 while other techniques involve the development of adapters for cell-phone cameras to better read RDT signal.27
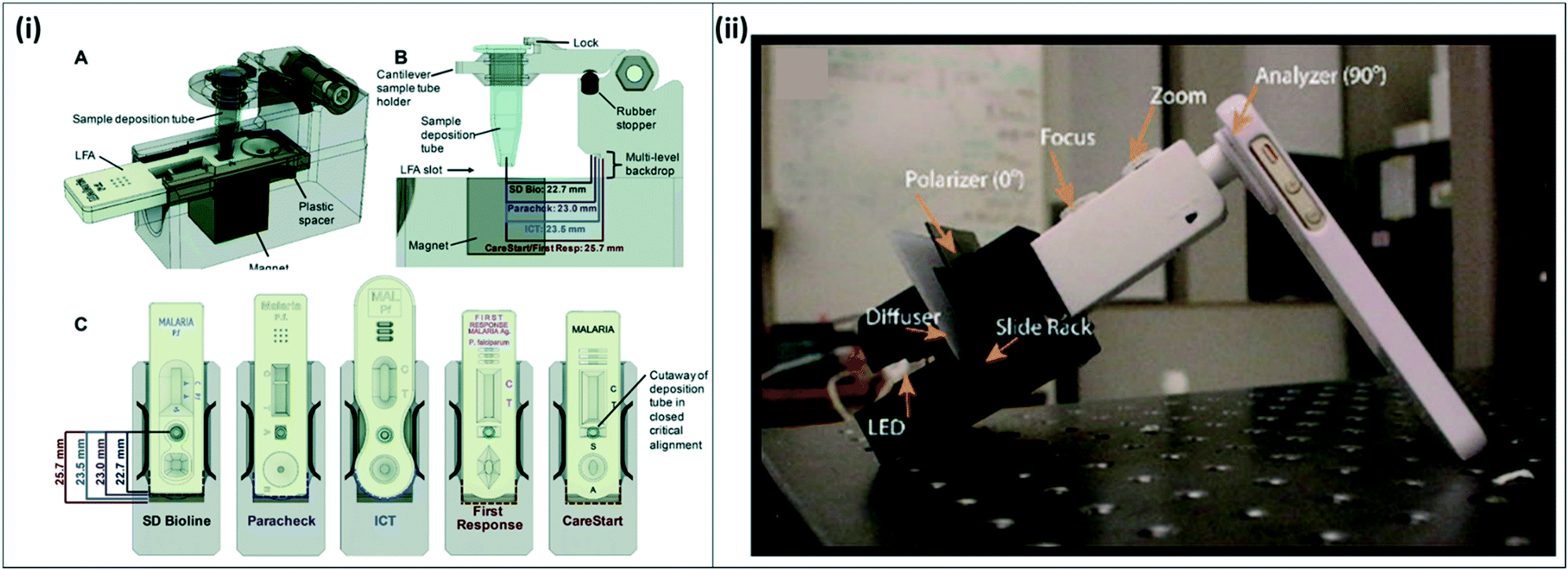 | ||
| Fig. 3 Improvements for existing gold standard techniques (i) mBEADS system for sample concentration of HRP-2 from lysed whole blood. A) Lysed whole blood is incubated with Ni-NTA beads in a tube, and the concentrated sample is dispensed onto commercial RDTs for more sensitive detection. B–C) The system is designed to accommodate 5 commonly-used RDT brands. Reproduced from ref. 24 with permission from the Royal Society of Chemistry. (ii) Cell-phone based transmission polarized light microscope. The MOPID system incorporates an iPhone 5S with polarizer sheets attached to a 3D-printed fitting holding a light source, diffuser, sample slide, and microscope attachment. Reproduced in part from ref. 29 (2015), license CC BY 4.0. | ||
Innovative approaches are also in development to improve microscopy, making it more portable for field use. A commercially-available handheld light microscope, the Newton Nm1, was demonstrated for malaria diagnosis from Giemsa-stained thick blood smears from 223 clinical samples from rural Côte d'Ivoire. The portable microscope showed 80.2% sensitivity and 100% specificity, indicating promising results for field deployment.28 In another study, a cell phone-based polarized microscope was developed to detect birefringence of the malaria-specific biomarker hemozoin in Giemsa-stained blood smears, making it an easy-to-deploy solution for standard malaria microscopy (Fig. 3(ii)).29 While not initially used for malaria diagnosis, the Foldscope, an origami-based, foldable paper microscope, and the Cellscope, a cell-phone based portable optical system,30 could be easily adapted to observe Giemsa-stained blood smears for malaria diagnosis in the field. These microscopy-based approaches can be deployed in the field to improve malaria case management; although they do not improve LLOD of microscopy, they obviate the need for expensive microscopes and clean lab environments, as they are much more inexpensive and portable.
Emerging lab-on-a-chip point of care diagnostics
Lab-on-a-chip strategies have the potential to fulfill the need for portable diagnostics with improved sensitivity. Over the past 15 years, there has been significant research on improving existing diagnostics and developing new microfluidic and paper-fluidic strategies to meet the needs outlined above. The solutions can be broadly divided into three categories based on the analyte of interest: protein-based tests, nucleic acid tests (NATs), and cell-based detection.Protein detection tests
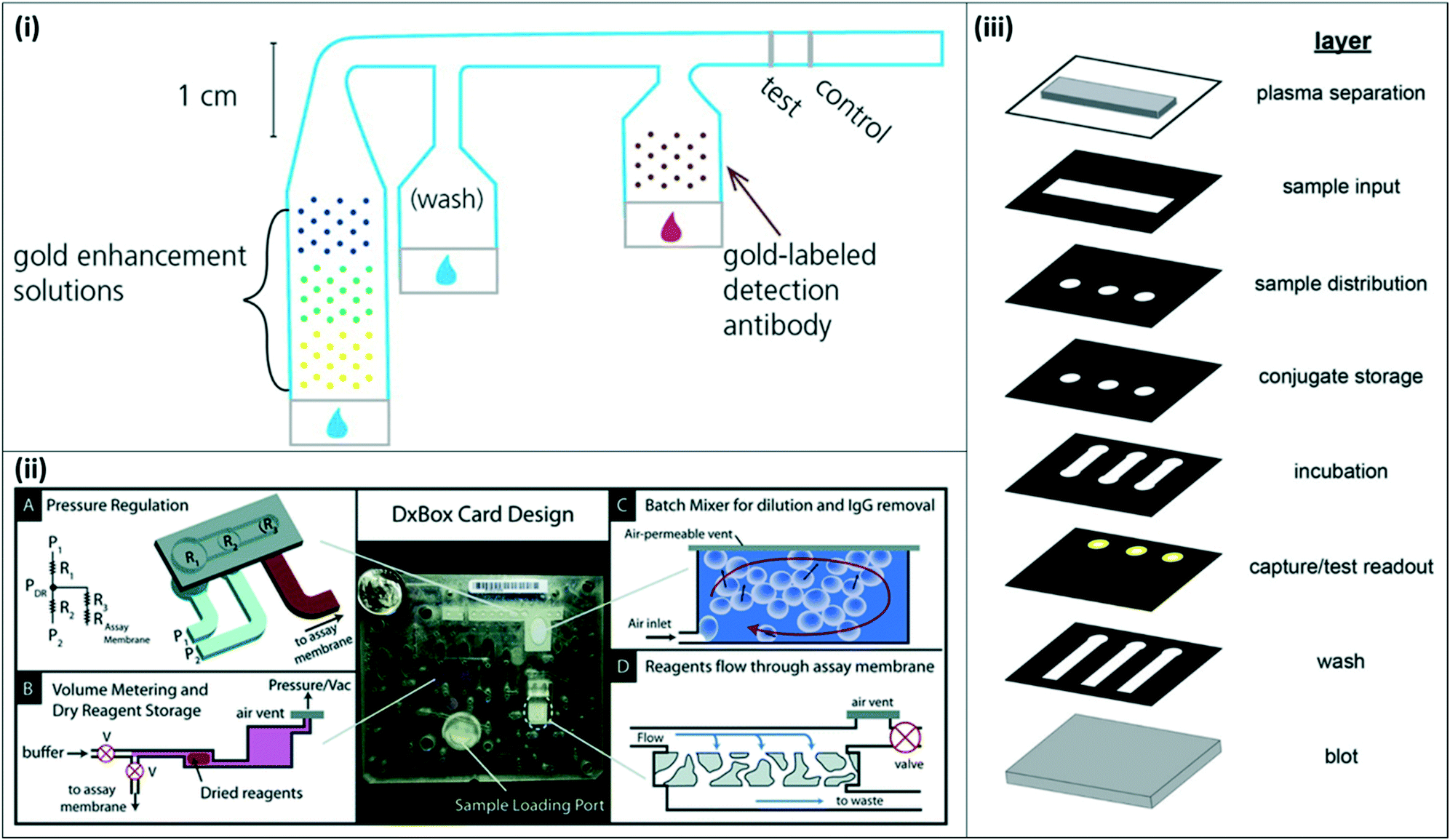 | ||
| Fig. 4 2-Dimensional and 3-dimensional paper-fluidic devices (i) 2-D paper matrix with dried and spotted gold enhancement reagents. The 2-D paper matrix allows for in situ mixing of the gold enhancement reagents upon sample addition and additional washing prior to incubation with detection antibody. Reproduced with permission from ref. 33. Copyright 2014, American Chemical Society. (ii) DxBox 3-D paper–plastic device for integrated sample processing and amplification. The 3-D setup incorporates pneumatic control for pressure regulation (A), allowing for controlled reagent storage and rehydration (B), batch mixing (C), and plasma separation (D). Reproduced from ref. 37 with permission from the Royal Society of Chemistry. (iii) Expanded view of 3-D paper device for integrated sample processing, distribution, and readout. The collapsed device enables sample distribution to three channels and multiple assay steps as the sample moves through the vertical layers. Reprinted with permission from ref. 38. Copyright 2016, American Chemical Society. | ||
Other groups have proposed novel 3-dimensional (3-D) paperfluidic immunoassays for integrated sample concentration and detection or for multiplexed detection of multiple targets. Pereira et al. developed a 3-D paper design to incorporate an aqueous two-phase system for simultaneous concentration and detection of pLDH.36 Lafleur et al. developed a combination paper–plastic system, called the DxBox, which relies on pneumatic control to regulate pressure and drive fluid flow through a custom immunoassay card (Fig. 4(ii)). The system incorporates plasma separation, batch mixing for sample dilution and purification, and controlled reagent flow for detection of multiple antigens.37 More recently, Deraney et al. demonstrated a completely paper-based 3-D microfluidic device to separate plasma and distribute sample to three different test zones, each of which contained reagents for a different target analyte, enabling the diagnosis of multiple co-presenting febrile illnesses from a single patient sample (Fig. 4(iii)).38 These devices showed successful detection of malaria antigens, along with other target antigens for other diseases.
The protein-based approaches described above demonstrate important progress toward more sensitive and inexpensive diagnostics for malaria, and are summarized in Table 2.
| Technique | Target parasite | Sample type | Target molecule | Limit of detection | Assay time/number of steps | Ref. | |
|---|---|---|---|---|---|---|---|
| a Original values were reported in parasites per μL; antigen levels were calculated based on previously-reported conversion factor.13 | |||||||
| Current RDTs | Current best-in-class RDTs | P. falciparum | Lysed whole blood | HRP-2 | 0.8 ng mL−1 | 15 minutes | 18 |
| Current best-in-class RDTs | Pan-Plasmodium | Lysed whole blood | pLDH | 25 ng mL−1 | 15 minutes | 18 | |
| RDT improvement | HRP-2 sample concentration for RDT | P. falciparum | Lysed whole blood | HRP-2 | 0.99–0.47 ng mL−1a (1.3–6.2 parasites per μL) | 3 minutes + standard RDT time | 22–25 |
| Thermal contrast amplification for RDT | P. falciparum, P. vivax | Lysed whole blood | HRP-2, pLDH | 0.125 ng mL−1 | 5 steps | 26 | |
| Novel microfluidic and paper-fluidic devices | Mobile phone-based ELISA | P. falciparum | Human serum | HRP-2 | 16 ng mL−1 | 15 minutes | 31 |
| 2-D paper network with gold enhancement reagents | P. falciparum | Fetal bovine serum | HRP-2 | 10 ng mL−1 | Not reported | 33, 34 | |
| 2-D paper network with biotin–streptavidin enhancement | P. falciparum | Fetal bovine serum | HRP-2 | 0.1 ng mL−1 | 90 minutes | 35 | |
| 3-D multiplexed paper-fluidic | P. falciparum, Salmonella typhii | Whole blood | HRP-2, IgM Ab to salmonella | 20 ng mL−1 HRP-2 | <30 minutes | 37 | |
| 3-D multiplexed paper-fluidic | P. falciparum, dengue fever | Lysed whole blood | HRP-2, pLDH, dengue S1 type2 | 46–100 ng mL−1a (600–1300 parasites per μL) | 20 minutes | 38 | |
| Novel detection strategies | Aptamer-based, colorimetric readout | P. falciparum, P. vivax | Lysed whole blood | pLDH | 0.06 ng mL−1 | 45 minutes | 39–42 |
| Magnetic beads, quantum dot florescent detection | P. falciparum | Urine, serum | HRP-2 | 0.5 ng mL−1 | Not reported | 43 | |
| Indicator displacement dye | P. falciparum | Serum | HRP-2 | 1110 ng mL−1 | Not reported | 44 | |
Nucleic acid tests
Molecular techniques that target parasite-specific nucleic acid sequences have emerged as alternatives to light microscopy or protein-based immunoassay diagnostics. These nucleic acid tests (NATs) are highly sensitive and specific for both pan-Plasmodium and species-specific targets, making them ideal for detecting low levels of parasitemia characteristic in elimination-phase populations. The 18S rRNA gene is the most commonly used target for amplification because it is present in multiple copies in the Plasmodium genome, is highly conserved, and has consensus regions across all five Plasmodium species as well as species-specific regions.45 Recently, genome-mining approaches have identified other targets, such as tandem repeat sequences dispersed throughout the genome, as targets for amplification, which significantly improve limits of detection of nucleic acid amplification assays for both P. falciparum and P. vivax malaria.46–48Several NATs, such as polymerase chain reaction (PCR), quantitative PCR (qPCR), and nested PCR (nPCR) assays, have been developed for malaria infections, and are widely considered the most sensitive diagnostic methods. However, PCR is generally thought to be too complex for field applications,4 requiring extensive sample preparation, sophisticated equipment for thermal cycling, expensive reagents, and skilled personnel. While PCR assays are currently not recommended by the WHO for case management and surveillance,7 microfluidic systems have the potential to bring these assays to the field to make them more widely adopted. Modifications to make PCR more appropriate for low-resource, point-of-care settings have been extensively reviewed elsewhere.49 Additionally, isothermal nucleic acid amplification assays—alternatives to PCR which operate at a single temperature, therefore eliminating the need for expensive thermal cycling equipment—have also been reviewed elsewhere.50 The approaches described below specifically rely on micro- and paper-fluidics to improve traditional NATs. An important consideration for NAT-based microfluidics is the potential for carry-over contamination, in which amplification products from one reaction may carry over to subsequent reactions, thereby increasing the likelihood of false positive results. Many microfluidic devices discussed here are proposed as closed systems that are disposable, minimizing the potential for contamination. In addition to creating closed systems, several techniques have been proposed to minimize cross-contamination, including pre- and post-amplification sterilization, enzymatic inactivation of carried over amplicons, and chemical inactivation of nucleic acids. These techniques have been extensively reviewed elsewhere.51
Over the last decade, portable, microfluidics-based PCR assays have become commercially available for field deployment. Although not all of these systems have been developed for malaria diagnosis, they have been successfully demonstrated for similar diseases at the point-of-care. The Palm PCR (Ahram Biosystems Inc.)53 and the TrueNat Uno (Molbio Diagnostics Pvt. Ltd.)54 systems are examples of portable systems with disposable microfluidic chips for PCR reactions, with off-chip DNA extraction and purification. The GeneXpert system (Cepheid, Inc.) is a fully-integrated system with heating/cooling plates, optical blocks, and an internal processor to drive fluid flow through a custom microfluidic cartridge using a syringe barrel (Fig. 5(i)). Unprocessed patient samples are placed into individual cartridges, and the system automatically moves the sample through specified chambers for lysis and nucleic acid extraction, purification, and amplification with a real-time fluorescence readout;55 the system has also been demonstrated for blood samples.56 The Accutas system (Aquila Diagnostics Systems, Inc.) was developed specifically for malaria diagnosis and uses a high-efficiency Omni KlenTaq enzyme to allow for amplification directly from blood without sample processing. In this system, a custom microfluidic chip with embedded PCR reagents is inserted into a benchtop unit for amplification and detection (Fig. 5(ii)).57,58 In a small-scale clinical study, the Accutas system showed good concordance (97%) with conventional real-time PCR results, and limit of detection of 2 parasites per μL. These commercially available techniques have been tested in the field with clinical samples; the reported clinical sensitivity and specificity for each commercial technique is detailed in Table 3.
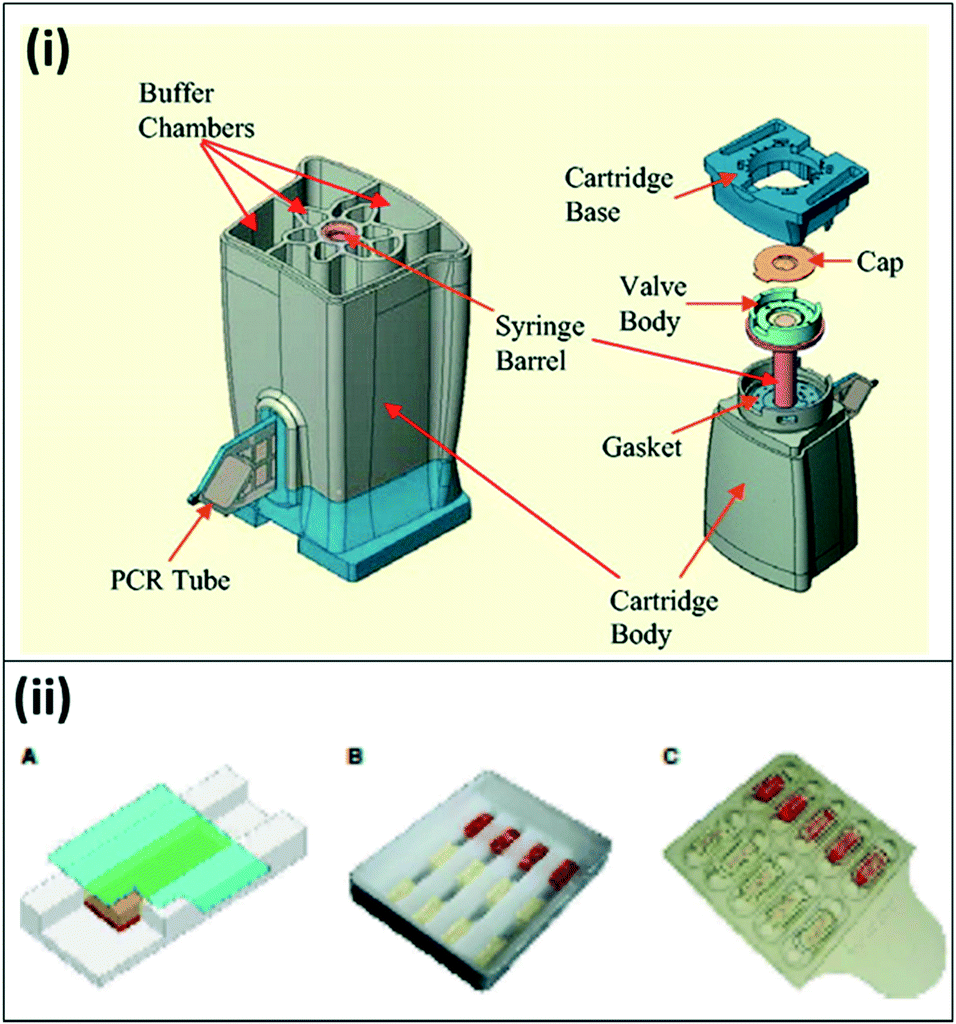 | ||
| Fig. 5 Commercially available lab-on-a-chip devices for PCR. (i) The Cepheid GeneXpert microfluidic PCR cartridge. The cartridge can be inserted into Cepheid GeneXpert system, which enables sample processing, thermal cycling for PCR amplification, and detection. Reproduced in part with permission from ref. 55. Copyright 2005, Clinical Chemistry. (ii) Microfluidic chip for Accutas system. A) Schematic of microfluidic chip for hydrogel PCR. A hydrogel embedded with PCR reagents adheres to the surface of the chip; blood flows in the channel underneath by capillary flow, and is absorbed by the hydrogel. B) Photo of a wax chip containing trenches overlaid with hydrogels bound to a glass coverslip. C) Photo of plastic chip made of two separate pieces; hydrogels are housed within wells on the top piece, with the second piece leaving space for capillary flow. Reproduced with permission from ref. 58, license CC BY 4.0. | ||
| Technique | Parasite | Sample | Sample processing | Assay target | Readout | Limit of detection | Assay time | Clinical Sensitivitya | Clinical Specificitya | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reference methods used for comparison are listed in parentheses. | |||||||||||
| Commercially available | Accutas® (PCR) | P. falciparum, P. vivax | Diluted blood | N/A | Not reported | Fluorescence (SYBR green) | 2 par μL−1 | Not reported | 97.4% (qPCR) | 93.8% (qPCR) | 57 |
| TrueNat Uno® (PCR) | P. falciparum, P. vivax | Whole blood | Qiagen extraction | Not reported | Fluorescence (probe) | 5 par μL−1 | 1 hour | 100% (nPCR), 95.5–99.9% (microscopy) | 100% (nPCR) | 54 | |
| Illumigene® LAMP | Pan-Plasmodium | Whole blood | DNA extraction, gravity-driven filtration | Mitochondrial DNA | Turbidity | 2 par μL−1 | Not reported | 92.6–99.1% (pet-PCR) | 76.6–94.2% (pet-PCR) | 60 | |
| LoopAmp® LAMP | Pan-Plasmodium, P. falciparum | Whole blood | Lysis + centrifugation | 18S rRNA gene | Turbidity | 5 par μL−1 | Not reported | 83.2–99.5%, (nPCR) | 71.4–92.9% (nPCR) | 61, 62 | |
| Microfluidic and paper-fluidic devices | Microfluidic RPA | P. falciparum | Whole blood | On chip dimethyl adipimidate/thin film extraction | Not reported | Interferometer, isothermal solid phase amplification | 1 par μL−1 | 1 hour | 63 | ||
| Rotational magnetic fluid flow, LAMP | P. falciparum | Whole blood | None | Not reported | Fluorescence | 0.6 par μL−1 | 40 minutes | 64 | |||
| Paper-fluidic RPA | pan-Plasmodium | Whole blood | Off-chip DNA extraction | 18S rRNA gene | Lateral flow strip | 5 copies per μL target DNA | 45 minutes | 82 | |||
| Paper-fluidic LAMP | P. falciparum, P. vivax, pan-Plasmodium | Whole blood | Off-chip heat lysis | Mitochondrial DNA | Fluorescence | 5 par μL−1 | 45 minutes | 83 | |||
| Droplet-based enzyme detection | P. falciparum | Whole blood | On-chip lysis by bead beating | pTopoisomeraseI | Fluorescence | 200 par μL−1 | Not reported | 94–97 | |||
| Benchtop POC techniques | LAMP | P. falciparum, P. vivax | Whole blood, dried blood spots | Saponin-chelex lysis/centrifugation, microwave irradiation | Not reported | Hydroxynaphthol blue (HNB) | 1–5 par μL−1 | Not reported | 85, 86, 91 | ||
| LAMP | P. falciparum, pan-Plasmodium | Whole blood, dried blood spots | Qiagen kit extraction, boil + centrifuge | Mitochondrial DNA | Malachite Green | 1–8 par μL−1 | 2 hours | 87 | |||
| Direct-blood LAMP | P. falciparum | Whole blood | None | Mitochondrial DNA | HNB + GelGreen | Not reported | 1 hour | 88 | |||
| Direct-blood LAMP | P. falciparum, P. vivax | Whole blood, saliva | Blood dilution + heat lysis, saliva Mobicol DNA isolation | Cox1 gene | Fluorescence | 1 par μL−1 | 30 minutes | 15 | |||
| Direct-blood HDA | P. falciparum, pan-Plasmodium | Whole blood | None | 18S rRNA gene | Lateral flow strip | 200 par μL−1 | ∼2.5 hours | 89 | |||
Non-commercial, research-grade microfluidic devices are being developed for NATs for both sample processing and amplification on-chip. For example, Liu et al. developed a two-stage system with sequential microfluidic devices for sample processing and amplification. The first stage utilizes dimethyl adipimidate/thin film sample processing to isolate DNA, while the second utilizes Mach–Zehnder interferometer–isothermal solid-phase DNA amplification and sensing. In this device, isothermal amplification is achieved using recombinase polymerase amplification (RPA). Combined, the device has a sensitive LLOD (1 parasite per μL) and requires approximately 1 hour of assay time.63
Benchtop centrifugal units have been developed to adapt NATs to field settings. These systems utilize centrifugal force to move fluids, obviating the need for a pump to drive fluid flow. However, centrifugal systems can have high power requirements, and fluid flow can be difficult to control precisely in these systems, making them unsuitable for POC applications. Choi et al. developed the AnyMDx system for malaria diagnosis, which relies on rotational magnetic interactions rather than centrifugal force. The AnyMDx system (Fig. 6) uses a custom-built disposable cartridge containing stationary reagent droplets in distinct chambers for nucleic acid extraction and purification, amplification, and detection. DNA-carrying magnetic beads move between chambers through a passive valve system, which retains the reagent droplets in the appropriate chambers while the beads move through. The system takes 10 μL of whole blood as an input, amplifies P. falciparum DNA with LAMP, and provides a quantitative output within 40 minutes with a LLOD of 0.6 parasites per μL. For a low-cost and simple user interface, the AnyMDx offers both sensitive and quantitative diagnosis, making it appropriate for applications in malaria elimination.64 Other diagnostic devices use centrifugal force to separate plasma from whole blood and isolate malaria parasites; this technique was demonstrated in a POC device in a paper-based centrifuge.65
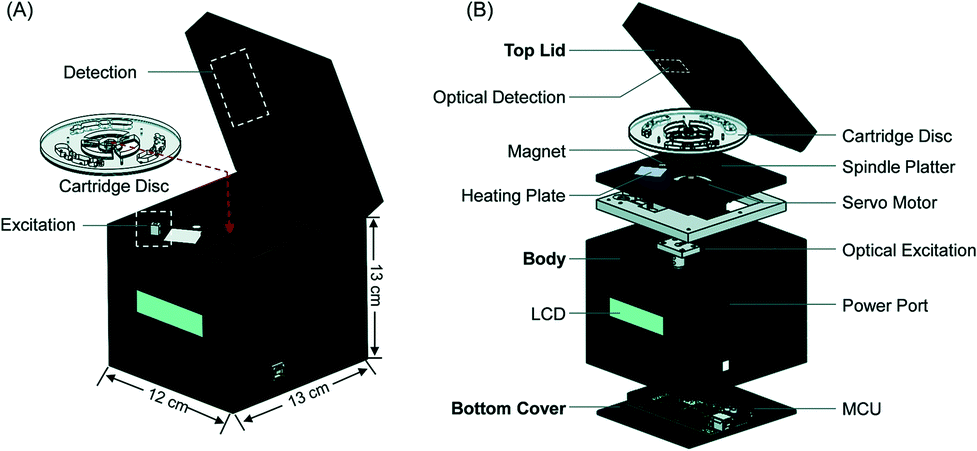 | ||
| Fig. 6 AnyMDx benchtop system for malaria LAMP. A) Each disposable cartridge disc can perform three independent tests. Each test region is designed to integrate sample processing and amplification B) the cartridges are placed into a custom unit for amplification. MCU, microcontroller unit; LCD, liquid crystal display. Reproduced from ref. 64 with permission of The Royal Society of Chemistry. | ||
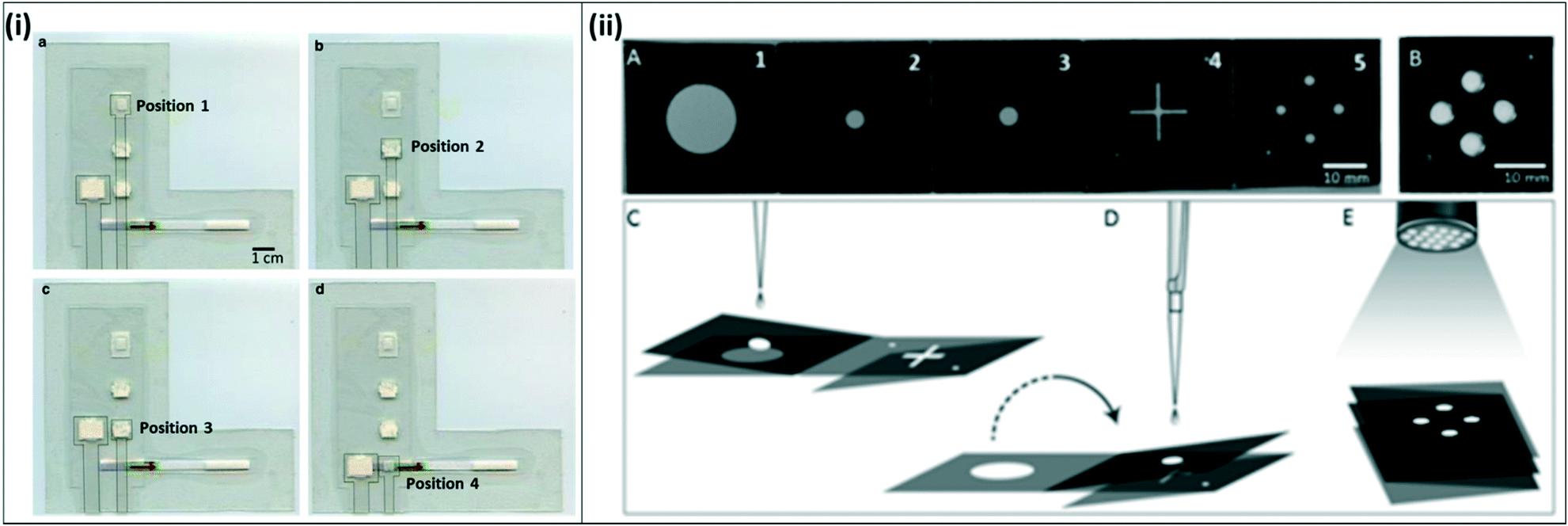 | ||
| Fig. 7 Integrated paper-fluidic devices for isothermal amplification (i) integrated paper-and-plastic device for amplification with RPA. a) The sample slider starts at position 1 for sample mixing with RPA reagents and amplification; b) sample slider is moved to position 2 to absorb RPA buffer; c) sample slider is moved to position 3 for dilution with buffer; d) sample slider is moved to position 4 for contact with lateral flow strip and visual readout. Reproduced with permission from ref. 82 (2015), license CC BY 4.0. (ii) Paper origami based multiplex LAMP assay. A) The device consists of filter paper with 5 distinct panels; hydrophobic wax was printed onto the paper to form channels; B) fully folded device, C) for extraction, panels 2 and 3 are folded together and rested on top of panel 1. The sample is dispensed onto panel 3 and extracted using capillary flows vertically from panel 3 to panel 1. D) The 2–3 panel is flipped over onto the 4–5 panel to transfer the sample to LAMP spots on panel 5 where the reaction is carried out. E) A UV flashlight at 365 nm is used to read out the signal. Reproduced with permission from ref. 83 (2016), license CC BY 4.0. | ||
There are several techniques in development for benchtop amplification assays that can be incorporated into paper-fluidic diagnostics. One potential avenue for improvement is the incorporation of colorimetric dyes onto paper-based tests to enable visual readout. Hydroxy naphtol blue (HNB), a metal ion indicator responsive to the concentration of Mg2+ ions in the reaction, is commonly used as a colorimetric indicator of the LAMP reaction84 and has been demonstrated for several LAMP-based malaria assays.72,75,85,86 Other colorimetric dyes such as the intercalating dye malachite green87 or custom-made dyes,88 have also been demonstrated in LAMP-based malaria assays. Colorimetric assays also provide a yes/no readout rather than a quantitative readout, which would be best suited for screening programs.
In addition to improving assay readout, paper-fluidic devices must minimize or eliminate sample pre-processing. It has been demonstrated that robust isothermal assays, such as helicase dependent amplification (HDA) or LAMP, are capable of amplifying DNA directly from a sample of whole blood with minimal assay inhibition.15,88,89 These assays still require some sample processing, such as pre-dilution of the whole blood sample, or sample denaturation by boiling. Still, these technologies obviate the need for more complex DNA isolation and purification from biological samples, and could therefore be adapted to paper-fluidic devices. A potential challenge for these direct amplification techniques might be their relatively small sample volumes (<5 μL blood). In cases where parasite concentration is relatively dilute, there is a need to process larger volumes (60–100 μL) of sample in order to minimize variability.90 For larger sample volumes with dilute parasite concentrations, benchtop techniques to concentrate and purify nucleic acids from biological samples may be adapted to paper-fluidic formats. These techniques include microwave irradiation from whole blood,91 nucleic acid isolation using magnetic beads,92 and filtration through blood separation paper.93
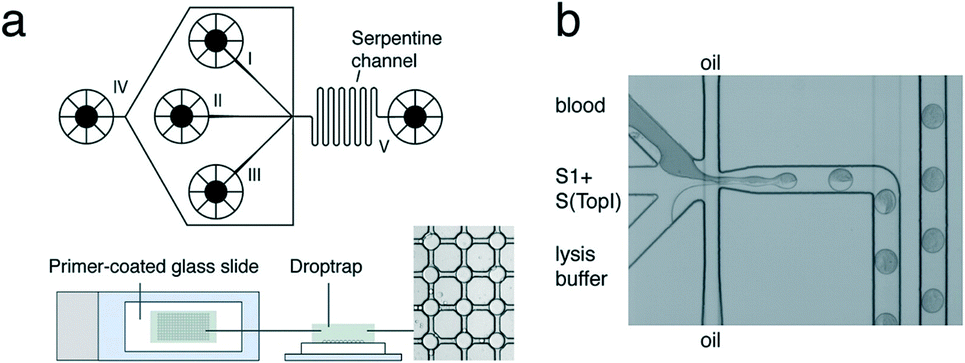 | ||
| Fig. 8 REEAD microfluidic system. (a) Top: Schematic of the microfluidic channel device. Sample is loaded into channel I, S1 and S(TopI) are loaded into channel II, and lysis buffer is loaded into channel III. Oil is fed in through channel IV to encapsulate the three components into droplets. The droplets move through a serpentine channel (V) to ensure mixing of droplet content and are deposited into the drop-trap device. Bottom: Drop-trap device confines droplets in cavities at the intersections in the drop-trap and exsiccates them onto a primer-coated glass slide for RCA. (b) Light-microscopic view of the microfluidic platform showing blood, substrates, and lysis buffer encapsulation in picoliter oil droplets. Reprinted with permission from ref. 94. Copyright 2012 American Chemical Society. | ||
NAT development is an active area of research for malaria diagnostic development; NATs reviewed here are summarized in Table 3.
Cell-based detection
Another active field of research for malaria diagnosis is the development of diagnostics based on the direct detection of parasites, or of cells infected with parasites. Malaria infection is known to alter red blood cell mechanical properties, including shape and stiffness, and their magnetic properties. In addition, malaria parasites produce hemozoin as a metabolic by-product inside the red blood cells; the biophysical properties of this molecule can be exploited to identify infected red blood cells. Several groups have developed techniques to distinguish infected red blood cells from uninfected red blood cells based on one or more of these cellular characteristics.Malaria-infected red blood cells can also be sorted based on their magnetic properties.107 Parasite digestion of hemoglobin results in aggregates of haem groups which form an insoluble pigment called haemozoin. Electron paramagnetic resonance and Moessbaur spectroscopy have shown that haemozoin has magnetic properties.102 This property of infected red blood cells has been utilized by several groups to develop novel microfluidic devices for malaria diagnosis. Several groups have exploited the magnetic properties of infected red blood cells to develop microfluidic cell sorters; many of these are described in detail elsewhere.102 In one example of a magnet-based microfluidic device, Nam et al. used a ferromagnetic nickel wire was embedded in the microfluidic device with a permanent magnet was affixed to the outside to induce an external magnetic field. The cells were first aligned by hydrodynamic focusing, and as the sample flowed through the device, infected red blood cells gathered near the nickel wire while the remaining cells stayed toward the top of the fluid stream. A separation channel at the end of the device allowed sorting and collection of the infected red blood cell population (Fig. 9). As in other cell-based microfluidic devices, the authors demonstrated their device using isolated red blood cells rather than whole blood, and propose using this device as a means to concentrate infected red blood cells prior to clinical microscopy.
 | ||
| Fig. 9 Magnetic sorting of malaria-infected red blood cells. a) Schematic of separation using paramagnetic characteristics of hemozoin in infected RBCs. b) Working principle of magnetophoretic separation with a ferromagnetic nickel wire in an external magnetic field. c) Image of the microfluidic device with affixed permanent magnet. Reprinted with permission from ref. 107. Copyright 2013, American Chemical Society. | ||
Other groups have investigated alternative approaches to malaria diagnostics based on other physical characteristics of malaria-infected cells. One such property is the increased stiffness of malaria-infected red blood cells. Microfluidic devices have been developed to take advantage of the naturally-occurring phenomenon of margination, where larger and stiffer leukocytes migrate toward the walls of blood vessels, while smaller and more deformable red blood cells migrate toward the center of the vessel. Hou et al. mimicked this phenomenon in a one-inlet, three-outlet microfluidic device, in which a high-hematocrit, infected blood sample was pumped through a long, straight channel (Fig. 10(i)).108 Due to the margination effect, infected red blood cells, which are stiffer than healthy red blood cells, migrated toward the walls of the channel and were successfully collected in outlets stemming from the walls. Uninfected red blood cells, which were more deformable and remained in the middle of the straight channel, were collected in an outlet stemming from the middle of the channel. The authors reported ∼75% recovery of early-stage infected red blood cells, and >90% recovery of late-stage infected red blood cells from a high-hematocrit sample. Additionally, they showed that the presence of leukocytes did not impact the margination effect of red blood cells; while the larger leukocytes were also collected into the side outlets, their presence did not alter the percent recovery of infected red blood cells.
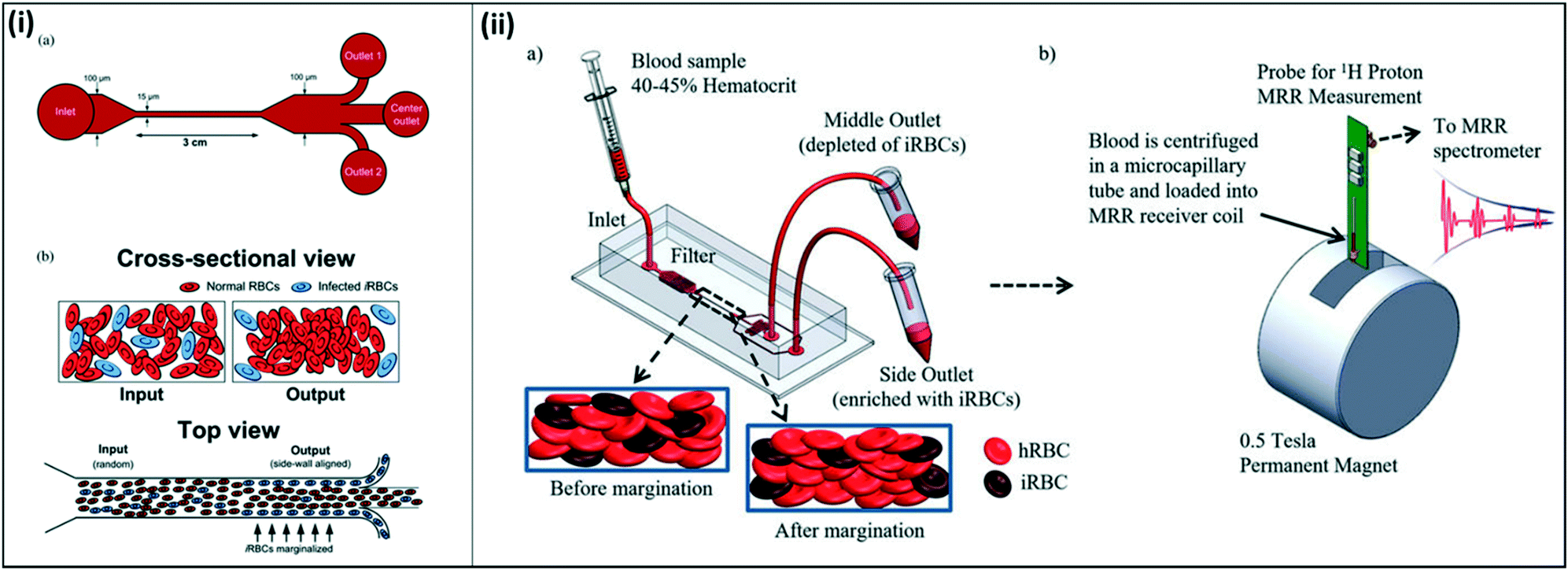 | ||
| Fig. 10 Microfluidic devices for margination of infected red blood cells. (i) Straight channel design for microfluidic margination. (a) Outlets 1 and 2 collect infected red blood cells, and the center outlet collects uninfected red blood cells. (b) Infected red blood cells align along the walls of the microfluidic channel due to the margination effect. Reproduced from ref. 108 with permission from The Royal Society of Chemistry. (ii) Margination combined with magnetic relaxometry. (a) Cells are sorted using a microfluidic device according to margination properties (b) both uninfected red blood cells from the middle outlet and infected red blood cells from the side outlets are analyzed using magnetic resonance relaxometry to determine a relative relaxation rate for each sample. Reproduced with permission from ref. 110 (2015), license CC BY 4.0. | ||
Kong et al. expanded on this concept to take advantage of both the increased stiffness and magnetic properties of infected red blood cells for diagnosis (Fig. 10(ii)). Here, the authors first utilized a microfluidic device to sort and separate infected red blood cells through margination, which allowed for increased concentration of the infected blood cells in the sample. Similar to the previously described device, this microfluidic device also required high-hematocrit samples to effectively enrich infected red blood cells from 1.9 to 32.1 times, depending on the stage of the parasite life cycle. The authors subsequently tested the concentrated sample using magnetic resonance relaxometry, in which a portable permanent magnet provides a strong polarizing magnetic field and a radiofrequency detection probe measures the relaxation rate of the emission signal from the sample.109 The increased magnetic susceptibility of infected red blood cells compared to healthy red blood cells resulted in a significant difference in relaxation rate, allowing the authors to distinguish between the two cell types with high sensitivity. Notably, the technique employed here eliminates the need to rely on an absolute relaxation rate, instead allowing to compare the change in relaxation rate between healthy and infected red blood cells within a single sample, since both are collected in the microfluidic device.110
Infected red blood cells also exhibit characteristic differences in optical properties, which can be used to distinguish them from healthy red blood cells. For instance, Banoth et al. developed a portable microfluidic device to detect the optical absorbance characteristics of red blood cells.111,112 In this system, a microfluidic chip with a narrow channel allowing for single-cell flow was incorporated into a custom-built portable optical system. The device allowed for real-time detection and analysis of optical absorbance of cells flowing through the single channel device. The device was fine-tuned to ensure that the optical absorbance signals of white blood cells and platelets in the sample would be distinct from those of red blood cells. Using the system, the authors showed that malaria-infected red blood cells displayed a unique absorbance signal, and could be distinguished from other cell types in whole blood.
Microfluidic optical devices have also been developed to analyze and identify infected red blood cells according to their compliance properties. Mauritz et al. developed a microfluidic optical stretcher to identify early-stage infected red blood cells in a high-throughput technique. An SU-8 pattern on a glass slide is used to support a capillary for sample flow and two optical fibers to deliver laser light. As cells flow through the capillary, they are trapped in the laser light and stretched by modulating the light intensity. The device is placed under a microscope-mounted camera, and analysis of cell elongation over time is used to calculate cell compliance. Infected red blood cells have reduced compliance compared to uninfected red blood cells; measuring compliance therefore enables single-cell diagnosis of infection.113 The study described here focused on samples containing only red blood cells; however, since the technique works at a single-cell level, it is reasonable to predict that leukocytes or other contaminating cells would not impact the results, as the mechanical signatures of other cell types would be distinct from red blood cells. Furthermore, although the device relies on a microscope mounted with a camera and a laser, there is potential for making the device more portable, allowing for field deployment, and enabling high-throughput analysis for parasite screening with high sensitivity.
Several studies have also used compliance and deformability characteristics to sort and isolate parasite-infected red blood cells. Kang et al. designed a device to sort malaria-infected cells based on their decreased deformability and increased cytoadherence. In this study, the authors designed a blood-on-a-chip device to monitor biophysical properties of blood using a microfluidic analog of a Wheatstone-bridge electric circuit (Fig. 11(i)).114 Here, the authors incorporate time-resolved microparticle image velocimetry to monitor the deformability and the viscosity of blood samples in a high-throughput and label-free manner. Guo et al. developed a similar system to sort red blood cells based on deformability.115 Here, the authors introduce two separate flows, one for filtration and a second for declogging as the sample moves through a matrix of constrictions. Uninfected red blood cells, which are more deformable, become lodged on the constrictions, while stiffer, infected red blood cells move through the device. Cells are varying deformabilities are collected at various stages throughout the device, with the final collection chamber collecting mainly pure infected red blood cells (Fig. 11(ii)). Bow, et al. also developed a deformability chamber. In this design, images are collected and analyzed as the sample moves through a microfluidic device with constrictions. Deformable healthy red blood cells are able to move through the constrictions with greater ease and progress through the chamber faster, while uninfected healthy red blood cells take a longer time to pass through.116 The proposed devices discussed here were tested with leukocyte-free samples. However, in all cases, it is possible to incorporate an additional chamber to remove contaminating leukocytes from a whole blood sample. Additionally, these microfluidic devices are proposed as enrichment chambers, to be used in conjunction with RDTs or other diagnostic methods, allowing for more efficient sample processing to achieve improved sensitivity. However, it is conceivable that they could be used as stand-alone diagnostic devices on their own.
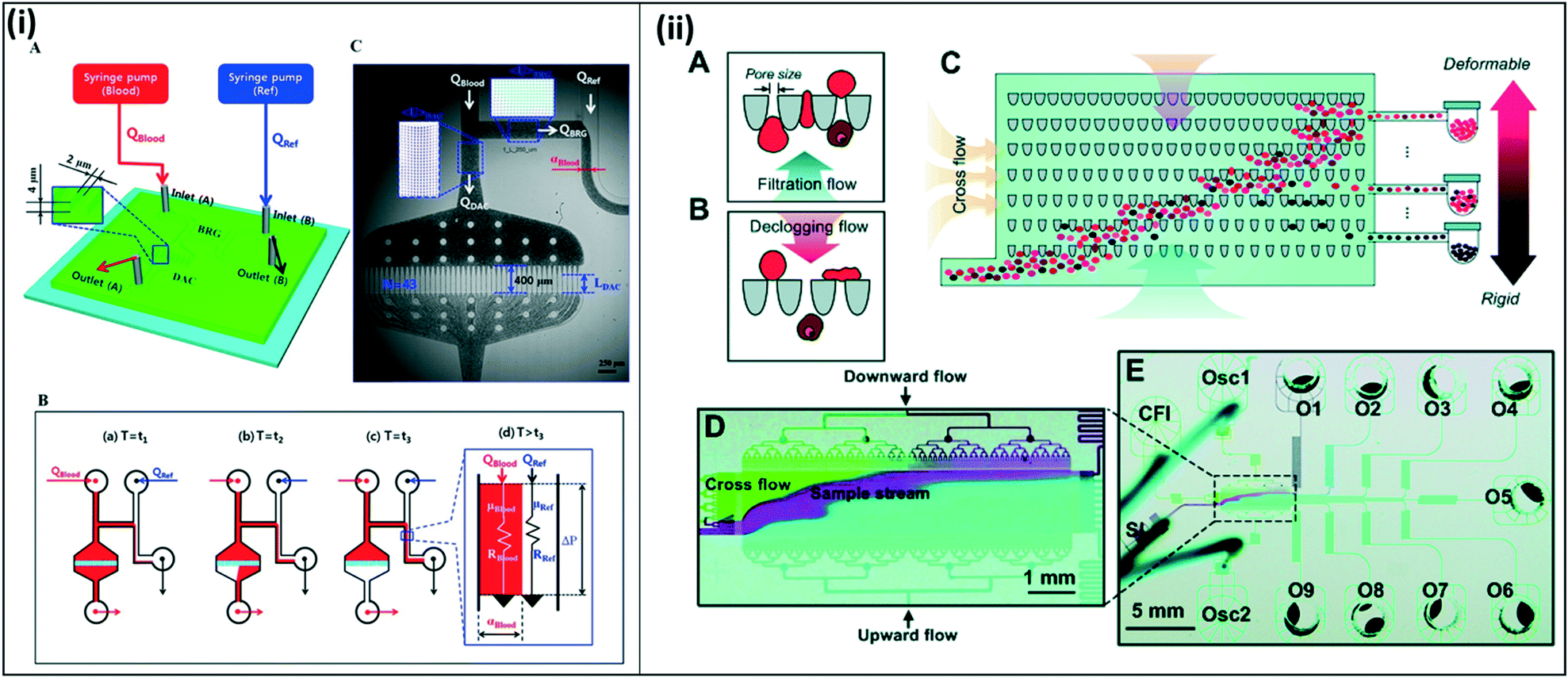 | ||
| Fig. 11 Blood on a chip devices to assess red blood cell deformability and viscosity. (i) Deformability chamber based on Wheatstone bridge circuit. (A) Two syringe pumps deliver blood sample and reference fluid into the microfluidic device. (B) All channels except the right upper side channels fill with blood. Fluid moves through the bridge channel (BRG), and the microfluidic filters in the deformability assessment chamber (DAC) become clogged due to RBC deformability. After t3, the blood and reference fluid flow into the lower right side channel. The blood filled width (αBlood) varies depending on viscosity and flow rate. C) Time-resolved images are used to calculate average velocities of blood flow in DAC and BRG channels. Reproduced in part from ref. 114. Copyright 2016, American Chemical Society. (ii) Deformability chamber using ratchet transport. (A and B) Each funnel constriction experiences oscillation excitation due to upward filtration flow and downward declogging flow, enabling unidirectional flow. (C–E) Sample is applied through the inlet (bottom left). The sample moves across the device in a diagonal trajectory due to the combined forces from the cross-flow inlet (CFI) and the oscillation excitation from the two oscillation inlets (Osc1 and Osc2). More deformable (uninfected) cells travel further up the chamber, while stiffer (infected) cells become blocked midway; samples are collected into outlets throughout the chamber (O1–O9). Reproduced from ref. 115 with permission from The Royal Society of Chemistry. | ||
Other microfluidic devices utilize the property of hydrodynamic lift forces on particles submerged and flowing within a liquid. Inertial hydrodynamic lift acts on all particles under intermediate Reynolds number flows, where shear forces of the moving fluid balance against lift forces from the wall of a chamber. Non-inertial lift forces, on the other hand, are important for compliant, deformable objects at low Reynolds number flows, where shear forces are sufficiently low such that inertial effects become negligible.119 Several microfluidic devices have been developed which exploit these hydrodynamic lift properties to sort infected red blood cells. For example, Warkiani et al. developed an inertial microfluidic device to sort and isolate parasite-infected red blood cells from lysed white blood cells for use in downstream PCR applications. The channels were designed with a high aspect ratio to obtain a high shear rate along the width of the channel, causing white blood cells to align along the channel walls, and infected red blood cells to remain in the center. The red blood cells were collected from the center channel for further downstream analysis.118 Conversely, Geislinger et al. developed a simple microfluidic device to sort cells based on non-inertial forces at low Reynolds number flows. Here, the stiffer, infected red blood cells would flow along the centerline of the flow path while more deformable, uninfected red blood cells would perturb the flow path and move away from the centerline.117
Cell-based detection strategies have the potential for single-cell level diagnosis and for infected red blood cell isolation and concentration using microfluidics. The strategies reviewed here are summarized in Table 4.
| Technique | Parasite | Sample type | Readout | Limit of detection | Assay time | Ref. | |
|---|---|---|---|---|---|---|---|
| In vivo monitoring, wearable monitors | Photoacoustic flow cytometry | P. yoelii in mice | In vivo | Photoacoustic signal, 2× amplification | Single cell level | 10 min–3 hours | 100, 101 |
| Confocal microscopy, proflavine-stained nucleii | P. falciparum, avian red blood cells | In vivo | Fluorescence | Not reported | Not reported | 98 | |
| Hemozoin detection by optical signatures | P. yoelii in mice | In vivo | Hemozoin absorbance and birefringence | 0.03% parasitemia | <1 min | 99 | |
| iRBC sorting, microfluidic | Dielectrophoretic cell sorting | P. falciparum | Red blood cells diluted in isotonic buffer, 55 mS m−1 conductivity | Post-sort flow cytometry | 1–10% parasitemia | ∼30 min | 104 |
| Dielectrophoresis for quantification of mechanical differences | P. falciparum | Red blood cells diluted in isotonic buffer, 55 mS m−1 conductivity | Red blood cell deformation | Not reported 1% hematocrit total | 700 RBCs mm−2 per second | 106 | |
| Magnetic separation of iRBCs | P. falciparum | Whole blood + NaNO2 | Post-sort Giemsa staining | 10% parasitemia | ∼40 min | 107 | |
| Margination | P. falciparum | Red blood cells, 40% hematocrit | Post-sort flow cytometry | 0.01–1% parasitemia | 20 million cells per min | 108 | |
| Margination, magnetic relaxometry | P. falciparum | Red blood cells, 40–50% hematocrit | Relaxation rate | 0.0005% parasitemia | Not reported | 110 | |
| Optofluidic absorption of RBCs | P. falciparum | Diluted whole blood | Optical absorbance | 1% parasitemia | <1 min | 111, 112 | |
| Optical stretching of RBCs | P. falciparum | Red blood cells | Red blood cell elongation | Single-cell level | Not reported | 113 | |
| Blood on a chip, RBC deformability and viscosity | P. falciparum | Diluted whole blood | Time-resolved microparticle image velocimetry | >2% parasitemia | <5 min | 114 | |
| Blood on a chip, RBC deformability | P. falciparum | Red blood cells | Post-sort fluorescence microscopy and RDT | <0.01% parasitemia | Not reported | 115 | |
| Blood on a chip, RBC deformability | P. falciparum | Diluted whole blood | Red blood cell deformation | Single cell-level up to 104 cells, 1–2% parasitemia | 1000 cells/10 min | 116 | |
| Non-inertial microfluidics | P. falciparum | Red blood cells, 2% hematocrit | Post-sort flow cytometry | 1.6–3.2% parasitemia | 12000 cells per hour |
117 | |
| Inertial microfluidics | P. falciparum | Saponin-lysed whole blood | Post-sort PCR | 2 par μL−1 | 15 min | 118 |
Opportunities and challenges for lab-on-a-chip strategies in malaria elimination efforts
Lab-on-a-chip strategies have made significant contributions to the development of portable and sensitive diagnostics for countries progressing towards malaria elimination. As stated previously, diagnostics for elimination-phase countries have different performance criteria depending on their use-case (case management of symptomatic infections, parasite screening for epidemiological studies, or active surveillance and case management to prevent further transmission). Table 5 summarizes some of the important performance criteria for different use cases, including cost, LLOD, number of tests, sample type and volume, assay time and throughput, and the target end user.| Cost per sample (USD) | Limit of detection (parasites per μL) | Tests/year (approx.) | Sample type/collection | Sample volume (whole blood) | Assay time/throughput | Target end user | |
|---|---|---|---|---|---|---|---|
| a Range of blood volumes was adapted and combined from multiple sources. All information presented was adapted from ref. 10 and 124 under license CC BY 4.0 and with permission from the Royal Society of Chemistry, respectively. | |||||||
| Case management of symptomatic infections | (O): ≤$1.00 | (O): ≤5 | 171 million | Finger prick blood | (O): 1–25 μL | 15–30 min | Patient, pharmacist |
| (M): ≤$1.00 | (M): 100–200 | (M): 1–50 μL | 10 tests per hour | ||||
| Low-density parasite screening for mass screening and treatment programs | (O): ≤$1.00 | (O): ≤2 | 33 million | (O): non-invasive (i.e. saliva, buccal swab, urine) | (O): 1–200 μLa | 15–30 min | Community health worker, trained lay person |
| (M): ≤$5.00 | (M): <20 | (M): 1–50 μL | |||||
| >10 tests per hour | |||||||
| (M): finger prick blood | |||||||
| Population surveillance to monitor for new infection and transmission | (O): ≤$1.00 | (O): ≤2 | 7000–10000 |
(O): non-invasive (i.e. saliva, buccal swab, urine) | (O): 1–200 μL | 15–30 min | Community health worker, trained lay person |
| (M): ≤$5.00 | (M): <20 | (M): 1–50 μL | >10 tests per hour | ||||
| (M): finger prick blood | |||||||
In some scenarios, ease-of-use and fast assay time take precedence over extremely low LLOD, while in others, high analytical sensitivity is more critical than device simplicity. In each of these cases, it is important to consider the end-user and the purpose of the test.
Diagnostics for case management of symptomatic infections will typically be purchased directly by patients showing febrile (fever-like) symptoms. Recent studies have shown that private sector sales—such as local pharmacies and shops—contribute to a large percentage of sales of both RDTs and antimalarial drugs for symptomatic patients. In order to be widely adopted in endemic regions, new diagnostics must cost less than ACT treatments readily available at pharmacies (∼$2),120 must be POC-applicable to minimize the patients' waiting times and number of clinic visits before treatment,121 and must be minimally instrumented, high-throughput, simple (≤3 user steps), and fast (≤60 minutes sample-to-answer time).122,123 Therefore, for case management of symptomatic patients, new diagnostic devices must be both simple and inexpensive relative to available antimalarial drugs, allowing them to be integrated into a patient's workflow with minimal extra effort. While cost and simplicity are the most important factors, the diagnostics must also have a short sample-to-answer time to incentivize patients to use the diagnostic rather than treating with available antimalarials without diagnosis. Notably, because case management typically occurs after symptoms occur, having an extremely low LLOD is not necessary for this case.
For parasite screening and population surveillance, the diagnostic tests are likely being administered by trained healthcare workers; therefore, diagnostics that require ≤4 hours of training with a minimal number of user steps are most appropriate. For these purposes, it is more important that the diagnostics are sensitive enough to detect low parasite densities characteristic of asymptomatic infections, or circulating gametocyte levels to enable effective intervention. Additionally, mass screening and treatment programs require rapid testing and treatment of large populations. Therefore, diagnostics must be appropriate for mass manufacturing and long-term storage to enable for easy administration to a large number of people. Further, the diagnostics must permit health workers to screen multiple patients in parallel in a short time to allow for simultaneous diagnosis and treatment; in other words, having high-throughput diagnostics in this case is key. It is also critical that these diagnostics have high accuracy, as the epidemiological data collected regarding parasite loads and transmission profiles will determine future strategy towards elimination. Finally, it is important that diagnostics for these purposes distinguish between Plasmodium species, especially P. falciparum and P. vivax, to enable better understanding of malaria transmission patterns and epidemiology.
Many of the LOC devices reviewed above meet the criteria for the use-cases described here. Protein-based approaches are the simplest point of entry for developing diagnostics for elimination settings, as protein-based RDTs are already considered a current gold standard for diagnosis. However, they also have some limitations. Although these methods are highly specific for HRP-2 or pLDH antigens, it has been shown that antigen levels may not directly correlate with parasite levels, making it difficult to determine parasitemia based on antigen detection alone.18 For example, HRP-2 can persist in the bloodstream even after parasite clearance, which may lead to false positive results. Conversely, deletion or mutation of the hrp-2 gene in the parasite may lead to the protein not being produced, resulting in false negatives.11 Therefore, protein-based LOC devices are suitable for case management of symptomatic infections due to their ease-of-use, but may not be appropriate for mass screening and treatment programs due to their potential limitations in determining parasite levels, as they may lead to incorrect representation of infection prevalence or transmission.
Wearable in vivo cell-based diagnostic approaches offer potential for powerful, highly sensitive detection of extremely low parasite levels. Similarly, cell-based techniques offer the advantage of single-cell precision diagnosis. However, many of these techniques currently require expensive equipment and are difficult to perform in parallel, leading to issues with scalability and throughput for mass screening and treatment programs. With more development, wearable diagnostics and cell-based microfluidic devices could offer promise in elimination settings.
NATs, while highly sensitive and specific, require multiple user steps for sample preparation. Therefore, in their current state, these tests may be more appropriate for parasite screening and surveillance programs, where skilled operators can process multiple tests in parallel, rather than for symptomatic case management settings. Fully-integrated paper-based devices offer a promising direction for NATs due to their ease of use, high accuracy, high throughput, and potential for inexpensive mass manufacturing and shipping. If these integrated devices could be further developed to distinguish not only parasite species, but also circulating gametocytes, they would be a powerful tool for surveillance in malaria elimination settings. Additionally, adapting these integrated devices to diagnose infection from non-invasive samples, such as saliva or urine, would allow for even simpler mass screening.
It is also important to note that for all cases, it is imperative that the diagnostic does not lose clinical sensitivity or specificity compared to gold standard techniques. False positive or false negative readings would impact patient treatment, and would result in inaccurate understanding of progress toward malaria elimination. Many of the techniques reviewed here are in the initial proof-of-concept phases, and do not yet report clinical sensitivity and specificity; in addition to the factors listed above, comparing these devices to current gold standard techniques will be a key step towards commercialization.
The WHO has set ambitious milestones for malaria elimination in several countries, and the development of sensitive and affordable diagnostics is an important component to achieve and maintain these goals. Factors including cost, sensitivity, field-deployability, and test throughput are important considerations for mass screening and treatment, or case management programs. Lab-on-a-chip platforms offer solutions to many of the challenges for malaria diagnostics for ‘elimination-phase’ settings. New lab-on-a-chip technologies, especially those that can integrate all processing steps and be easily incorporated into the existing screening and treatment workflow, have the potential to truly impact malaria elimination programs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the U.S. National Science Foundation, Division of Industrial Innovation and Partnerships, under Award 1650504. C. M. K. acknowledges funding from the U.S. National Institutes of Health, under award EB015403.References
- World Health Organization and UNICEF.
- World Health Organization, World Malaria Report 2016, 2016 Search PubMed.
- World Health Organization, Global technical strategy for malaria 2016-2030, 2015 Search PubMed.
- World Health Organization.
- World Health Organization.
- Centers for Disease Control, in CDC Health Information for International Travel 2016, Oxford University Press, New York, 2016 Search PubMed.
- World Health Organization.
- I. Chen, S. E. Clarke, R. Gosling, B. Hamainza, G. Killeen, A. Magill, W. O'Meara, R. N. Price and E. M. Riley, PLoS Med., 2016, 13, e1001942 Search PubMed.
- T. Bousema, L. Okell, I. Felger and C. Drakeley, Nat. Rev. Microbiol., 2014, 12, 833–840 CrossRef CAS PubMed.
- D. Bell, A. E. Fleurent, M. C. Hegg, J. D. Boomgard and C. C. McConnico, Malar. J., 2016, 15, 406 CrossRef PubMed.
- C. Wongsrichanalai, M. J. Barcus, S. Muth, A. Sutamihardja and W. H. Wernsdorfer, Am. J. Trop. Med. Hyg., 2007, 119–127 Search PubMed.
- D. Bell, C. Wongsrichanalai and J. W. Barnwell, Nat. Rev. Microbiol., 2006, 4, S7–S20 CrossRef PubMed.
- World Health Organization.
- P. A. Zimmerman and R. E. Howes, Curr. Opin. Infect. Dis., 2015, 28, 446–454 CrossRef CAS PubMed.
- S. S. Modak, C. A. Barber, E. Geva, W. R. Abrams, D. Malamud and Y. S. Y. Ongagna, Infect. Dis., 2016, 9, 1–9 Search PubMed.
- J. Mu, K. B. Seydel, A. Bates and X.-Z. Su, Curr. Genomics, 2010, 11, 279–286 CrossRef CAS PubMed.
- ACTwatch Group, K. Hanson and C. Goodman, Malar. J., 2017, 16, 205 CrossRef PubMed.
- A. Jimenez, R. R. Rees-Channer, R. Perera, D. Gamboa, P. L. Chiodini, I. J. Gonzalez, A. Mayor and X. C. Ding, Malar. J., 2017, 16, 128 CrossRef PubMed.
- World Health Organization, WHO | How malaria RDTs work, http://www.who.int/malaria/areas/diagnosis/rapid-diagnostic-tests/about-rdt/en/, (accessed 26 May 2017) Search PubMed.
- World Health Organization.
- S. Shan, W. Lai, Y. Xiong, H. Wei, H. Xu and J. Agric, Food Chem., 2015, 63, 745–753 CrossRef CAS PubMed.
- K. M. Davis, L. E. Gibson, F. R. Haselton and D. W. Wright, Analyst, 2014, 139, 3026–3031 RSC.
- K. M. Ricks, N. M. Adams, T. F. Scherr, F. R. Haselton and D. W. Wright, Malar. J., 2016, 15, 399 CrossRef PubMed.
- W. S. Bauer, D. W. Kimmel, N. M. Adams, L. E. Gibson, T. F. Scherr, K. A. Richardson, J. A. Conrad, H. K. Matakala, F. R. Haselton and D. W. Wright, Analyst, 2017, 142, 1569–1580 RSC.
- K. M. Davis, J. D. Swartz, F. R. Haselton and D. W. Wright, Anal. Chem., 2012, 84, 6136–6142 CrossRef CAS PubMed.
- Y. Wang, Z. Qin, D. R. Boulware, B. S. Pritt, L. M. Sloan, I. J. Gonzalez, D. Bell, R. R. Rees-Channer, P. Chiodini, W. C. W. Chan and J. C. Bischof, Anal. Chem., 2016, 88, 11774–11782 CrossRef CAS PubMed.
- D. J. You, T. S. Park and J.-Y. Yoon, Biosens. Bioelectron., 2013, 40, 180–185 CrossRef CAS PubMed.
- J. T. Coulibaly, M. Ouattara, J. Keiser, B. Bonfoh, E. K. N'goran, J. R. Andrews and I. I. Bogoch, Am. J. Trop. Med. Hyg., 2016, 95, 831–834 CrossRef PubMed.
- C. W. Pirnstill and G. L. Coté, Nature Scientific Reports, 2015, 5, 13368 CrossRef CAS PubMed.
- R. K. D. Ephraim, E. Duah, J. S. Cybulski, M. Prakash, M. V D'ambrosio, D. A. Fletcher, J. Keiser, J. R. Andrews and I. I. Bogoch, Am. J. Trop. Med. Hyg., 2015, 92, 1253–1256 CrossRef PubMed.
- P. B. Lillehoj, M.-C. Huang, N. Truong and C.-M. Ho, Lab Chip, 2013, 13, 2950 RSC.
- T. Laksanasopin, T. W. Guo, S. Nayak, A. A. Sridhara, S. Xie, O. O. Olowookere, P. Cadinu, F. Meng, N. H. Chee, J. Kim, C. D. Chin, E. Munyazesa, P. Mugwaneza, A. J. Rai, V. Mugisha, A. R. Castro, D. Steinmiller, V. Linder, J. E. Justman, S. Nsanzimana and S. K. Sia, Sci. Transl. Med., 2015, 7(273), 273re1 CrossRef PubMed.
- G. E. Fridley, H. Le and P. Yager, Anal. Chem., 2014, 86, 6447–6453 CrossRef CAS PubMed.
- E. Fu, T. Liang, P. Spicar-Mihalic, J. Houghtaling, S. Ramachandran and P. Yager, Anal. Chem., 2012, 84, 4574–4579 CrossRef CAS PubMed.
- B. D. Grant, C. A. Smith, K. Karvonen and R. Richards-Kortum, Anal. Chem., 2016, 88, 2553–2557 CrossRef CAS PubMed.
- D. Y. Pereira, R. Y. T. Chiu, S. C. L. Zhang, B. M. Wu and D. T. Kamei, Anal. Chim. Acta, 2015, 882, 83–89 CrossRef CAS PubMed.
- L. Lafleur, D. Stevens, K. McKenzie, S. Ramachandran, P. Spicar-Mihalic, M. Singhal, A. Arjyal, J. Osborn, P. Kauffman, P. Yager and B. Lutz, Lab Chip, 2012, 12, 1119 RSC.
- R. N. Deraney, C. R. Mace, J. P. Rolland and J. E. Schonhorn, Anal. Chem., 2016, 88, 6161–6165 CrossRef CAS PubMed.
- R. M. Dirkzwager, S. Liang and J. A. Tanner, ACS Sens., 2016, 1, 420–426 CrossRef CAS.
- Y.-W. Cheung, J. Kwok, A. W. L. Law, R. M. Watt, M. Kotaka and J. A. Tanner, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15967–15972 CrossRef CAS PubMed.
- S. Lee, K.-M. Song, W. Jeon, H. Jo, Y.-B. Shim and C. Ban, Biosens. Bioelectron., 2012, 35, 291–296 CrossRef CAS PubMed.
- W. Jeon, S. Lee, M. DH and C. Ban, Anal. Biochem., 2013, 439, 11–16 CrossRef CAS PubMed.
- P. C. Searson, Y. E. Castro-Sesquen, C. Kim, R. H. Gilman and D. J. Sullivan, Am. J. Trop. Med. Hyg., 2016, 95, 354–357 CrossRef PubMed.
- B. Chakma, P. Jain, N. K. Singh and P. Goswami, Anal. Chem., 2016, 88, 10316–10321 CrossRef CAS PubMed.
- E. Patsoula, G. Spanakos, D. Sofianatou, M. Parara and N. C. Vakalis, Ann. Trop. Med. Parasitol., 2003, 97, 15–21 CrossRef CAS PubMed.
- A. Demas, J. Oberstaller, J. Debarry, N. W. Lucchi, G. Srinivasamoorthy, D. Sumari, A. M. Kabanywanyi, L. Villegas, A. A. Escalante, S. P. Kachur, J. W. Barnwell, D. S. Peterson, V. Udhayakumar and J. C. Kissinger, J. Clin. Microbiol., 2011, 49, 2411–2418 CrossRef CAS PubMed.
- N. Hofmann, F. Mwingira, S. Shekalaghe, L. J. Robinson, I. Mueller and I. Felger, PLoS Med., 2015, 12, e1001788 Search PubMed.
- H. Gupta, S. Srivastava, S. Chaudhari, T. G. Vasudevan, M. H. Hande, S. C. D'souza, S. Umakanth and K. Satyamoorthy, Acta Trop., 2016, 160, 15–22 CrossRef CAS PubMed.
- M. S. Cordray and R. R. Richards-Kortum, Am. J. Trop. Med. Hyg., 2012, 87, 223–230 CrossRef CAS PubMed.
- E. C. Oriero, J. Jacobs, J.-P. Van Geertruyden, D. Nwakanma and U. D'alessandro, J. Antimicrob. Chemother., 2015, 70, 2–13 CrossRef CAS PubMed.
- J. Aslanzadeh, Ann. Clin. Lab. Sci., 2004, 34, 389–396 CAS.
- J. Wu, R. Kodzius, W. Cao and W. Wen, Microchim. Acta, 2014, 181, 1611–1631 CrossRef CAS.
- S. Lim, H. Nan, M.-J. Lee and S. H. Kang, J. Chromatogr., B, 2014, 963, 134–139 CrossRef CAS PubMed.
- C. B. Nair, J. Manjula, P. A. Subramani, P. B. Nagendrappa, M. N. Manoj, S. Malpani, P. K. Pullela, P. V. Subbarao, S. Ramamoorthy and S. K. Ghosh, PLoS One, 2016, 11, e0146961 Search PubMed.
- S. Raja, J. Ching, L. Xi, S. J. Hughes, R. Chang, W. Wong, W. Mcmillan, W. E. Gooding, K. S. Mccarty, M. Chestney, J. D. Luketich and T. E. Godfrey, Clin. Chem., 2005, 51(5), 882–890 CAS.
- P. P. Banada, R. Koshy and D. Alland, J. Clin. Microbiol., 2013, 51, 2317–2322 CrossRef PubMed.
- S. K. Yanow, Point of Care: The Journal of Near-Patient Testing, 2016, 15, 41–42 CrossRef.
- B. J. Taylor, A. Howell, K. A. Martin, D. Manage, W. Gordy, S. D. Campbell, S. Lam, A. Jin, S. D. Polley, R. A. Samuel, A. Atrazhev, A. J. Stickel, J. Birungi, A. K. Mbonye, L. M. Pilarski, J. P. Acker and S. K. Yanow, Malar. J., 2014, 13, 179 CrossRef PubMed.
- Y. Mori, K. Nagamine, N. Tomita and T. Notomi, Biochem. Biophys. Res. Commun., 2001, 289, 150–154 CrossRef CAS PubMed.
- N. W. Lucchi, M. Gaye, M. A. Diallo, I. F. Goldman, D. Ljolje, A. B. Deme, A. Badiane, Y. D. Ndiaye, J. W. Barnwell, V. Udhayakumar and D. Ndiaye, Sci. Rep., 2016, 6, 36808 CrossRef CAS PubMed.
- M. Sema, A. Alemu, A. G. Bayih, S. Getie, G. Getnet, D. Guelig, R. Burton, P. Labarre and D. R. Pillai, Malar. J., 2015, 14, 44 CrossRef PubMed.
- A. N. Mohon, L. D. Y. Lee, A. G. Bayih, A. Folefoc, D. Guelig, R. A. Burton, P. LaBarre, W. Chan, B. Meatherall and D. R. Pillai, Diagn. Microbiol. Infect. Dis., 2016, 85, 149–153 CrossRef PubMed.
- Q. Liu, J. Nam, S. Kim, C. T. Lim, M. K. Park and Y. Shin, Biosens. Bioelectron., 2016, 82, 1–8 CrossRef PubMed.
- G. Choi, D. Song, S. Shrestha, J. Miao, L. Cui and W. Guan, Lab Chip, 2016, 16, 4341–4349 RSC.
- M. S. Bhamla, B. Benson, C. Chai, G. Katsikis, A. Johri and M. Prakash, Nat. Biomed. Eng., 2017, 1, 9 CrossRef.
- World Health Organization, Guidelines for the Treatment of Malaria, Geneva, 3rd edn, 2015 Search PubMed.
- G. E. A. Strom, M. G. Tellevik, K. Hanevik, N. Langeland and B. Blomberg, Trans. R. Soc. Trop. Med. Hyg., 2014, 108, 488–494 CrossRef CAS PubMed.
- B. J. Taylor, K. A. Martin, E. Arango, O. M. Agudelo, A. Maestre, S. K. Yanow, P. Wilairatana, S. Looareesuwan, J. Rosenblatt, S. Kachur, J. Barnwell, D. Peterson, V. Udhayakumar and J. Kissinger, Malar. J., 2011, 10, 244 CrossRef CAS PubMed.
- P. Li, Z. Zhao, Y. Wang, H. Xing, D. M. Parker, Z. Yang, E. Baum, W. Li, J. Sattabongkot, J. Sirichaisinthop, S. Li, G. Yan, L. Cui and Q. Fan, Malar. J., 2014, 13, 175 CrossRef PubMed.
- S. Bereczky, A. Martensson, J. P. Gil and A. Farnert, Am. J. Trop. Med. Hyg., 2005, 72, 249–251 Search PubMed.
- C. Liu, E. Geva, M. Mauk, X. Qiu, W. R. Abrams, D. Malamud, K. Curtis, S. M. Owen and H. H. Bau, Analyst, 2011, 136, 2069–2076 RSC.
- P. Prompamorn, P. Sithigorngul, S. Rukpratanporn, S. Longyant, P. Sridulyakul and P. Chaivisuthangkura, Lett. Appl. Microbiol., 2011, 52, 344–351 CrossRef CAS PubMed.
- S. Yongkiettrakul, W. Jaroenram, N. Arunrut, W. Chareanchim, S. Pannengpetch, R. Suebsing, W. Kiatpathomchai, W. Pornthanakasem, Y. Yuthavong and D. Kongkasuriyachai, Parasitol. Int., 2014, 63, 777–784 CrossRef CAS PubMed.
- S. Kersting, V. Rausch, F. F. Bier and M. Von Nickisch-Rosenegk, Malar. J., 2014, 13, 99 CrossRef PubMed.
- J. R. Port, C. Nguetse, S. Adukpo, T. P. Velavan, A. Maestre, S. Yanow, B. Agaba, D. Kyabayinze, C. Sutherland and M. Perkins, Malar. J., 2014, 13, 454 CrossRef PubMed.
- S. Y. Ongagna-Yhombi, P. Corstjens, E. Geva, W. R. Abrams, C. A. Barber, D. Malamud and S. Mharakurwa, Malar. J., 2013, 12, 74 CrossRef PubMed.
- M. Jauset-Rubio, M. Svobodová, T. Mairal, C. Mcneil, N. Keegan, A. Saeed, M. N. Abbas, M. S. El-Shahawi, A. S. Bashammakh, A. O. Alyoubi and C. K. O'sullivan, Nature Scientific Reports, 2016, 6, 37732 CrossRef CAS PubMed.
- R. Tang, H. Yang, Y. Gong, M. You, Z. Liu, J. R. Choi, T. Wen, Z. Qu, Q. Mei, F. Xu, F. Xu, I. Bosch, D. M. Dudley, D. H. O'Connor, L. Gehrke and J. J. Collins, Lab Chip, 2017, 17, 1270–1279 RSC.
- M. Mahalanabis, J. Do, H. Almuayad, J. Y. Zhang and C. M. Klapperich, Biomed. Microdevices, 2010, 12, 353–359 CrossRef CAS PubMed.
- J. T. Connelly, J. P. Rolland and G. M. Whitesides, Anal. Chem., 2015, 87, 7595–7601 CrossRef CAS PubMed.
- N. M. Rodriguez, W. S. Wong, L. Liu, R. Dewar and C. M. Klapperich, Lab Chip, 2016, 16, 753–763 RSC.
- M. S. Cordray and R. R. Richards-Kortum, Malar. J., 2015, 14, 472 CrossRef PubMed.
- G. Xu, D. Nolder, J. Ulien Reboud, M. C. Oguike, D. A. Van Schalkwyk, C. J. Sutherland and J. M. Cooper, Angew. Chem., Int. Ed., 2016, 55, 15250–15253 CrossRef CAS PubMed.
- D. V. M. Motoki Goto, E. Honda, A. Ogura, A. Nomoto and K.-I. Hanaki, BioTechniques, 2009, 46, 167–172 CrossRef PubMed.
- S. Britton, Q. Cheng, C. J. Sutherland and J. S. McCarthy, Malar. J., 2015, 14, 335 CrossRef PubMed.
- S. Britton, Q. Cheng, M. J. Grigg, C. B. Poole, C. Pasay, T. William, K. Fornace, N. M. Anstey, C. J. Sutherland, C. Drakeley and J. S. McCarthy, PLoS Neglected Trop. Dis., 2016, 10, e0004443 Search PubMed.
- N. W. Lucchi, D. Ljolje, L. Silva-Flannery, V. Udhayakumar, L. Gomes and C. Fontes, PLoS One, 2016, 11, e0151437 Search PubMed.
- K. Hayashida, K. Kajino, H. Simukoko, M. Simuunza, J. Ndebe, A. Chota, B. Namangala and C. Sugimoto, Parasites Vectors, 2017, 10, 26 CrossRef PubMed.
- Y. Li, N. Kumar, A. Gopalakrishnan, C. Ginocchio, R. Manji, M. Bythrow, B. Lemieux and H. Kong, J. Mol. Diagn., 2013, 15, 634–641 CrossRef CAS PubMed.
- M. M. Bond and R. Richards-Kortum, Am. J. Clin. Pathol., 2015, 144, 885–894 CrossRef CAS PubMed.
- J. R. Port, C. Nguetse, S. Adukpo and T. P. Velavan, Malar. J., 2014, 13, 454 CrossRef PubMed.
- A. L. Bitting, H. Bordelon, M. L. Baglia, K. M. Davis, A. E. Creecy, P. A. Short, L. E. Albert, A. V. Karhade, D. W. Wright, F. R. Haselton and N. M. Adams, J. Lab. Autom., 2016, 21, 732–742 CrossRef CAS PubMed.
- S. M. McFall, R. L. Wagner, S. R. Jangam, D. H. Yamada, D. Hardie, D. M. Kelso and J. Virol, Methods, 2015, 214, 37–42 CAS.
- S. Juul, C. J. F. Nielsen, R. Labouriau, A. Roy, C. Tesauro, P. W. Jensen, C. Harmsen, E. L. Kristoffersen, Y.-L. Chiu, R. Frohlich, P. Fiorani, J. Cox-Singh, D. Tordrup, J. Koch, A.-L. Bienvenu, A. Desideri, S. Picot, E. Petersen, K. W. Leong, Y.-P. Ho, M. Stougaard and B. R. Knudsen, ACS Nano, 2012, 6, 10676–10683 CrossRef CAS PubMed.
- M. Hede, P. Okorie, S. Fruekilde, S. Fjelstrup, J. Thomsen, O. Franch, C. Tesauro, M. Bugge, M. Christiansen, S. Picot, F. Lötsch, G. Mombo-Ngoma, J. Mischlinger, A. Adegnika, F. Pedersen, Y.-P. Ho, E. Petersen, M. Stougaard, M. Ramharter and B. Knudsen, Micromachines, 2015, 6, 1505–1513 CrossRef.
- S. Juul, J. M. Obliosca, C. Liu, Y.-L. Liu, Y.-A. Chen, D. M. Imphean, B. R. Knudsen, Y.-P. Ho, K. W. Leong and H.-C. Yeh, Nanoscale, 2015, 7, 8332–8337 RSC.
- A. Givskov, E. L. Kristoffersen, K. Vandsø, Y.-P. Ho, M. Stougaard, B. R. Knudsen, E. Tamiya, Y. Murakami, T. Endo and D. Vestergaard, Sensors, 2016, 16, 1916 CrossRef PubMed.
- J. Burnett and R. Richards-Kortum, in Biomedical Optics and 3-D Imaging, OSA, Washington, D.C., 2012, p. BSu3A.26 Search PubMed.
- J. L. Burnett, J. L. Carns and R. Richards-Kortum, Biomed. Opt. Express, 2015, 6, 3462–3474 CAS.
- Y. A. Menyaev, K. A. Carey, D. A. Nedosekin, M. Sarimollaoglu, E. I. Galanzha, J. S. Stumhofer and V. P. Zharov, Biomed. Opt. Express, 2016, 7, 3643–3658 CrossRef PubMed.
- C. Cai, K. A. Carey, D. A. Nedosekin, Y. A. Menyaev, M. Sarimollaoglu, E. I. Galanzha, J. S. Stumhofer and V. P. Zharov, Cytometry, Part A, 2016, 89, 531–542 CrossRef CAS PubMed.
- S. Kasetsirikul, J. Buranapong, W. Srituravanich, M. Kaewthamasorn and A. Pimpin, Malar. J., 2016, 15, 358 CrossRef PubMed.
- P. Gascoyne, C. Mahidol, M. Ruchirawat, J. Satayavivad, P. Watcharasit and F. F. Becker, Lab Chip, 2002, 2, 70–75 RSC.
- P. Gascoyne, J. Satayavivad and M. Ruchirawat, Acta Trop., 2004, 89, 357–369 CrossRef PubMed.
- Y. Huang, X. B. Wang, F. F. Becker and P. R. Gascoyne, Biophys. J., 1997, 73, 1118–1129 CrossRef CAS PubMed.
- E. Du, M. Dao and S. Suresh, Extreme Mech. Lett., 2014, 1, 35–41 CrossRef CAS PubMed.
- J. Nam, H. Huang, H. Lim, C. Lim and S. Shin, Anal. Chem., 2013, 85, 7316–7323 CrossRef CAS PubMed.
- H. W. Hou, A. A. S. Bhagat, A. G. Lin Chong, P. Mao, K. S. Wei Tan, J. Han and C. T. Lim, Lab Chip, 2010, 10, 2605 RSC.
- W. K. Peng, T. F. Kong, C. S. Ng, L. Chen, Y. Huang, A. A. S. Bhagat, N.-T. Nguyen, P. R. Preiser and J. Han, Nat. Med., 2014, 20, 1069–1073 CrossRef CAS PubMed.
- T. Fook Kong, W. Ye, W. K. Peng, H. Wei Hou, P. R. Preiser Marcos, N.-T. Nguyen and J. Han, Sci. Rep., 2015, 5, 11425 CrossRef PubMed.
- E. Banoth, V. K. Kasula, V. K. Jagannadh and S. S. Gorthi, J. Biophotonics, 2016, 9, 610–618 CrossRef CAS PubMed.
- E. Banoth, V. K. Kasula and S. S. Gorthi, Appl. Opt., 2016, 55, 8637 CrossRef PubMed.
- J. M. A. Mauritz, T. Tiffert, R. Seear, F. Lautenschläger, A. Esposito, V. L. Lew, J. Guck and C. F. Kaminski, J. Biomed. Opt., 2010, 15, 30517 CrossRef PubMed.
- Y. J. Kang, Y.-R. Ha and S.-J. Lee, Anal. Chem., 2016, 88, 2912–2922 CrossRef CAS PubMed.
- Q. Guo, S. P. Duffy, K. Matthews, X. Deng, A. T. Santoso, E. Islamzada and H. Ma, Lab Chip, 2016, 16, 645–654 RSC.
- H. Bow, I. Pivkin, M. Diez-Silva, S. J. Goldfless, M. Dao, J. C. Niles, S. Suresh and J. Han, Lab Chip, 2011, 11, 1065–1073 RSC.
- T. M. Geislinger, S. Chan, K. Moll, A. Wixforth, M. Wahlgren and T. Franke, Malar. J., 2014, 13, 375 CrossRef PubMed.
- M. E. Warkiani, A. Kah Ping Tay, B. L. Khoo, X. Xiaofeng, J. Han and C. T. Lim, Lab Chip, 2015, 15, 1101–1109 RSC.
- T. M. Geislinger and T. Franke, Adv. Colloid Interface Sci., 2014, 208, 161–176 CrossRef CAS PubMed.
- K. S. Hansen, D. Pedrazzoli, A. Mbonye, S. Clarke, B. Cundill, P. Magnussen and S. Yeung, Health Policy Plan., 2013, 28, 185–196 CrossRef PubMed.
- S. Loubiere and J.-P. Moatti, Clin. Microbiol. Infect., 2010, 16, 1070–1076 CrossRef CAS PubMed.
- X. C. Ding, M. P. Ade, J. K. Baird, Q. Cheng, J. Cunningham, M. Dhorda, C. Drakeley, I. Felger, D. Gamboa, M. Harbers, S. Herrera, N. Lucchi, A. Mayor, I. Mueller, J. Sattabongkot, A. Ratsimbason, J. Richards, M. Tanner and I. J. González, PLoS Neglected Trop. Dis., 2017, 11(4), e0005516 Search PubMed.
- Program for Appropriate Technology in Health; PATH, 2014, p. 45 Search PubMed.
- P. A. Zimmerman and R. E. Howes, Curr. Opin. Infect. Dis., 2015, 28, 446–452 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |