
Palladium-catalyzed olefination of aryl/alkyl halides with trimethylsilyldiazomethane via carbene migratory insertion†
Qiu-Chao
Mu
abc,
Xing-Ben
Wang
a,
Fei
Ye
a,
Yu-Li
Sun
a,
Xing-Feng
Bai
ab,
Jing
Chen
*b,
Chun-Gu
Xia
b and
Li-Wen
Xu
 *ab
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn; Fax: +86 2886 7756; Tel: +86 2886 7756
bState Key Laboratory for Oxo Synthesis and Selective Oxidation Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, and University of the Chinese Academy of Sciences, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing, P. R. China
First published on 25th October 2018
Abstract
The direct olefination of aryl/alkyl halides with trimethylsilyldiazomethane (TMSD) as a C1- or C2-unit was achieved successfully via a metal carbene migratory insertion process, which offered a new access to afford (E)-vinyl silanes and (E)-silyl-substituted α,β-unsaturated amides in good yields and high chemoselectivity.
Transition-metal-catalyzed cross-coupling reactions via carbene migratory insertion (CMI) have received much attention and have been extensively established as reliable and versatile methods for carbon–carbon bond-forming reactions in the past decades.1 Accordingly, the key step of carbene coupling reactions is the migratory insertions of an aryl or alkyl group into the metal–carbene intermediate, which makes metal carbene migratory insertion as a powerful strategy for carbon–carbon bond-forming transformations.2 In particular, since Van Vranken and co-workers3 reported the first example of palladium carbene migratory insertion in 2001, palladium-catalyzed carbene migratory insertion of aryl halides has been well demonstrated for the synthesis of versatile molecules through a cross-coupling process, and the corresponding intermediate or mechanistic process has been revealed by several groups (Scheme 1, eqn (1) and (2)).1,2 Thus, in principle these diazo compounds or other carbene precursors can be used in the construction of alkenes by carbon–carbon bond-forming carbene migratory insertion and subsequent β-hydrogen elimination. Among various alkenes, vinyl silanes occupy an important position in organic synthesis,4 which have wide applications due to the characteristic properties of silicon and privileged functions as versatile precursors and potent nucleophiles. Vinyl silanes would be used to construct many characteristic molecules by organic transformation involving the Hiyama cross-coupling reactions5 and crotylation reactions,6 even the Hosomi–Sakurai-type allylation7 and so on. For these reasons, numerous efforts have been directed to the preparation of vinyl silanes, which utilize carbonyl compounds,8 alkynes,9 vinyl halides,10 terminal olefins,11 and aryl iodies12 as starting materials. However, limitations still exist with many of the current methods, which often make the preparation of vinyl silanes challenging, especially in the control of chemoselectivity. Therefore, the development of novel and efficient olefination reactions with new catalytic systems is highly desirable in synthetic chemistry. As reported, little attention has been paid to a simple olefination process to provide alkenes containing a silyl group by carbene migratory insertion.13 Meanwhile, chemical transformations involving two molecules of diazo compounds in the carbene migratory insertion have not been well realized in the past.14 Inspired by previous works on the metal carbene migratory insertion process,1–3 attempts to obtain vinyl silanes with TMSD as a C1- or C2-unit have been made in this work (Scheme 1).
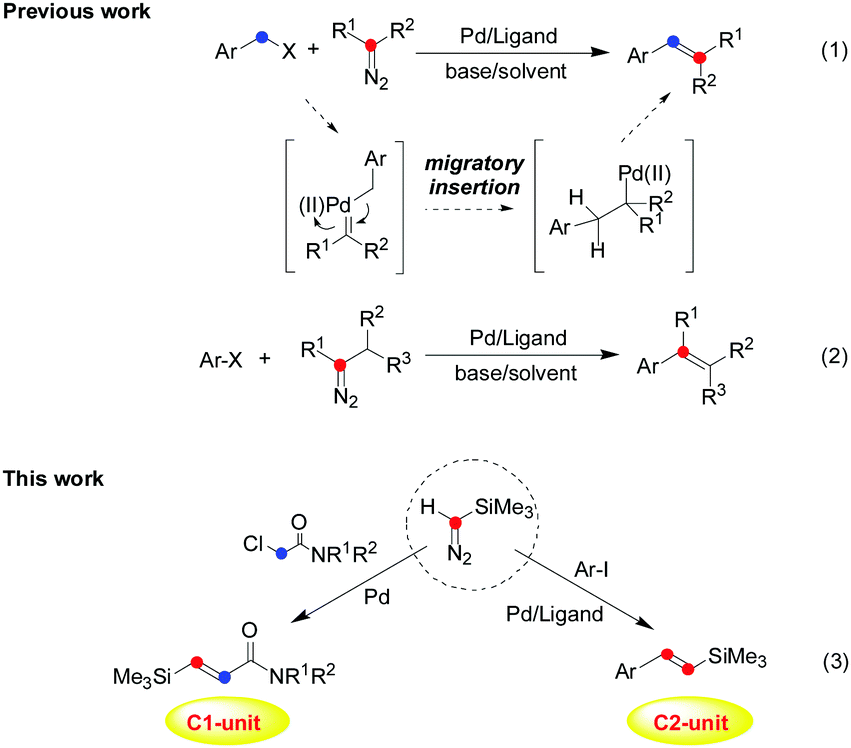 | ||
| Scheme 1 Palladium-catalyzed cross-coupling reaction through metal carbene migratory insertion (CMI): from the classic method (previous work) to controllable carbene migratory insertion (this work). | ||
Our effort began by selecting aryl iodide 2a as a model substrate to examine the carbene insertion reaction of TMSD. By optimizing various reaction parameters, we found that the combination of [Pd(π-cinnamyl)Cl2]2, di(1-ad)-n-butylphosphine, t-BuOLi, KOAc in dry 1,4-dioxane at 100 °C gave the best result after 24 hours (Table 1). As described in Table 1, replacing [Pd(π-cinnamyl)Cl2]2 with PdCl2, Pd(MeCN)2Cl2 or others diminished the reaction yield and stereoselectivity (entries 2–5). Several bases were investigated (entries 6–9), and t-BuOLi was found to be the best choice (entry 1). Additionally, the effect of the solvent on the selectivity and catalytic efficiency was evaluated, and 1,4-dioxane proved to be effective (entry 1 and entries 10–13). When various phosphine ligands (PPh3, DavePhos, PCy3, Xphos, and XantPhos) were tested, 3a could also be detected but in relatively lower yields and stereoselectivities (entries 14–18). Increasing the catalyst loading did not influence the conversation significantly (entry 19). When KOAc was removed, only 20% yield of (E)-3a was detected (entry 20).
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yieldb (%) |
| 2a/(E)-3a/(Z)-3a | ||
| a 2a (0.1 mmol), TMSD (3.0 equiv.), [Pd(π-cinnamyl)Cl2]2 (5 mol%), di(1-ad)-n-butylphosphine (20 mol%), t-BuOLi (1.0 equiv.), KOAc (1.0 equiv.), dry dioxane (1 mL), 100 °C, 24 hours. b The yield was determined by GC-MS. c DIEA = N,N-diisopropylethylamine. | ||
| 1 | None | 0/99/<1 |
| 2 | 10 mol% PdCl2 | 64/30/6 |
| 3 | 10 mol% Pd(MeCN)2Cl2 | 45/50/5 |
| 4 | 10 mol% Pd(PhCN)2Cl2 | 38/57/5 |
| 5 | 10 mol% Pd(PPh3)4 | 65/23/12 |
| 6 | K2CO3 as the base | 70/30/0 |
| 7c | DIEA as the base | 70/30/0 |
| 8 | Ag2CO3 as the base | 0/38/62 |
| 9 | Li2CO3 as the base | 81/19/0 |
| 10 | THF instead of dioxane | 24/50/26 |
| 11 | Toluene instead of dioxane | 64/36/0 |
| 12 | DCE instead of dioxane | 75/25/0 |
| 13 | DMSO instead of dioxane | 0/73/27 |
| 14 | PPh3 as the ligand | 53/14/33 |
| 15 | DavePhos as the ligand | 62/15/23 |
| 16 | PCy3 as the ligand | 60/20/20 |
| 17 | Xphos as the ligand | 67/15/18 |
| 18 | XantPhos as the ligand | 80/14/6 |
| 19 | Using 10 mol% [Pd(π-cinnamyl)Cl2]2 | 0/97/3 |
| 20 | Without KOAc | 40/20/40 |

With the optimized reaction conditions in hand, we explored the substrate scope of aryl iodides to yield the corresponding (E)-vinyl silanes. As shown in Table 2, good yields and stereoselectivity were obtained for the ortho-substituted aryl iodide with electron-rich substituents (2a, 2c, and 2d), whereas lower E/Z selectivity was obtained for the ortho-substituted aryl iodides with electron-deficient substituents (2b and 2e–2h), suggesting that the electronic effect would be crucial for E/Z stereocontrol. Substrates (2i–2v) having m- or p-substituents were also suitable for the olefination reaction, giving moderate to good yields. In particular, the reactions of 2k, 2n, 2r, and 2v proceeded smoothly to afford the corresponding (E)-vinyl silanes with excellent E/Z selectivity (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The alkene 3w containing a sterically hindered t-Bu group was isolated in 36% yield. Notably, the carboxylic ester moiety was also tolerated in this reaction (2z), even though a strong base was required. Surprisingly, some aryl bromides could also be used for the olefination reaction (Scheme 2). Both 3a and 3x afforded moderate yields and excellent E/Z selectivity.
:1). The alkene 3w containing a sterically hindered t-Bu group was isolated in 36% yield. Notably, the carboxylic ester moiety was also tolerated in this reaction (2z), even though a strong base was required. Surprisingly, some aryl bromides could also be used for the olefination reaction (Scheme 2). Both 3a and 3x afforded moderate yields and excellent E/Z selectivity.
|
|
||||
|---|---|---|---|---|
| Entry | Ar-I | Product | Yieldb (%) | Ratioc (E/Z) |
| a 2 (0.75 mmol), TMSD (3.0 equiv.), [Pd(π-cinnamyl)Cl2]2 (5 mol%), di(1-ad)-n-butylphosphine (20 mol%), t-BuOLi (1.0 equiv.), KOAc (1.0 equiv.), dry dioxane (1 mL), 100 °C, 24 hours. b Isolated yields. c Determined by NMR. | ||||
| 1 | 2-MeC6H4 (2a) | 3a | 65 | 99:1 |
| 2 | 2-FC6H4 (2b) | 3b | 30 | 82:18 |
| 3 | 2-ClC6H4 (2c) | 3c | 69 | 99:1 |
| 4 | 2-MeOC6H4 (2d) | 3d | 45 | 99:1 |
| 5 | 2-(CF3)C6H4 (2e) | 3e | 38 | 88:12 |
| 6 | 2-NO2C6H4 (2f) | 3f | 32 | 88:12 |
| 7 | 2-(CF3O)C6H4 (2g) | 3g | 36 | 74:26 |
| 8 | 2-CNC6H4 (2h) | 3h | 56 | 88:12 |
| 9 | 3-MeC6H4 (2i) | 3i | 53 | 91:9 |
| 10 | 3-ClC6H4 (2j) | 3j | 42 | 83:17 |
| 11 | 3-MeOC6H4 (2k) | 3k | 49 | 99:1 |
| 12 | 3-(CF3)C6H4 (2l) | 3l | 44 | 87:13 |
| 13 | 3-NO2C6H4 (2m) | 3m | 36 | 88:12 |
| 14 | 3-CNC6H4 (2n) | 3n | 60 | 99:1 |
| 15 | 4-MeC6H4 (2o) | 3o | 59 | 95:5 |
| 16 | 4-FC6H4 (2p) | 3p | 31 | 81:19 |
| 17 | 4-ClC6H4 (2q) | 3q | 54 | 85:15 |
| 18 | 4-MeOC6H4 (2r) | 3r | 51 | 99:1 |
| 19 | 4-(CF3)C6H4 (2s) | 3s | 49 | 92:8 |
| 20 | 4-(CF3O)C6H4 (2t) | 3t | 41 | 85:15 |
| 21 | 4-NO2C6H4 (2u) | 3u | 31 | 86:14 |
| 22 | 4-CNC6H4 (2v) | 3v | 57 | 99:1 |
| 23 | 4-tBuC6H4 (2w) | 3w | 36 | 99:1 |
| 24 | C6H5 (2x) | 3x | 50 | 85:15 |
| 25 | 3,5-(Me)2C6H3 (2y) | 3y | 41 | 99:1 |
| 26 | 2-CO2MeC6H4 (2z) | 3z | 45 | 99:1 |
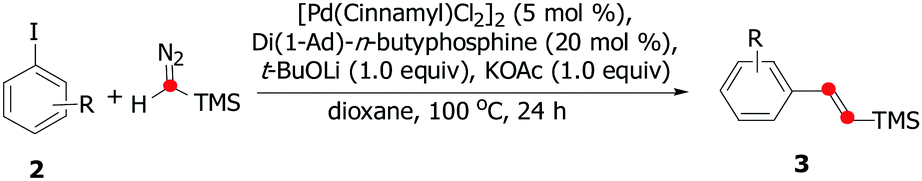
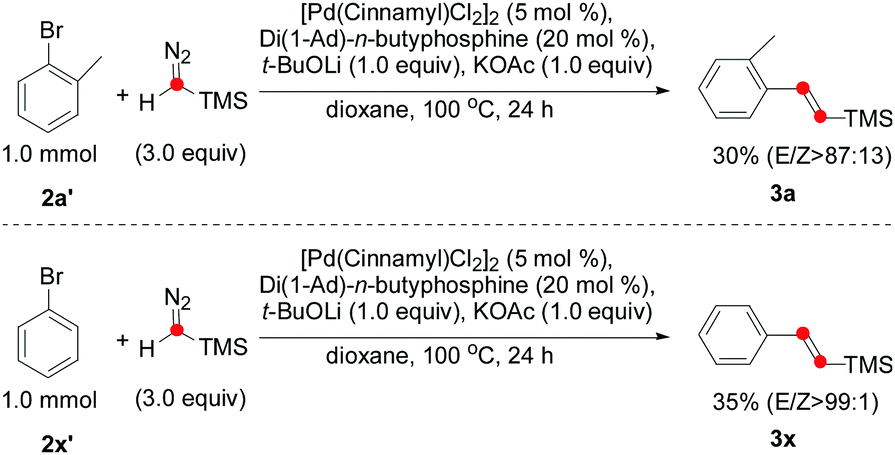 | ||
| Scheme 2 The palladium-catalyzed olefination of aryl bromides with trimethylsilyldiazomethane. | ||
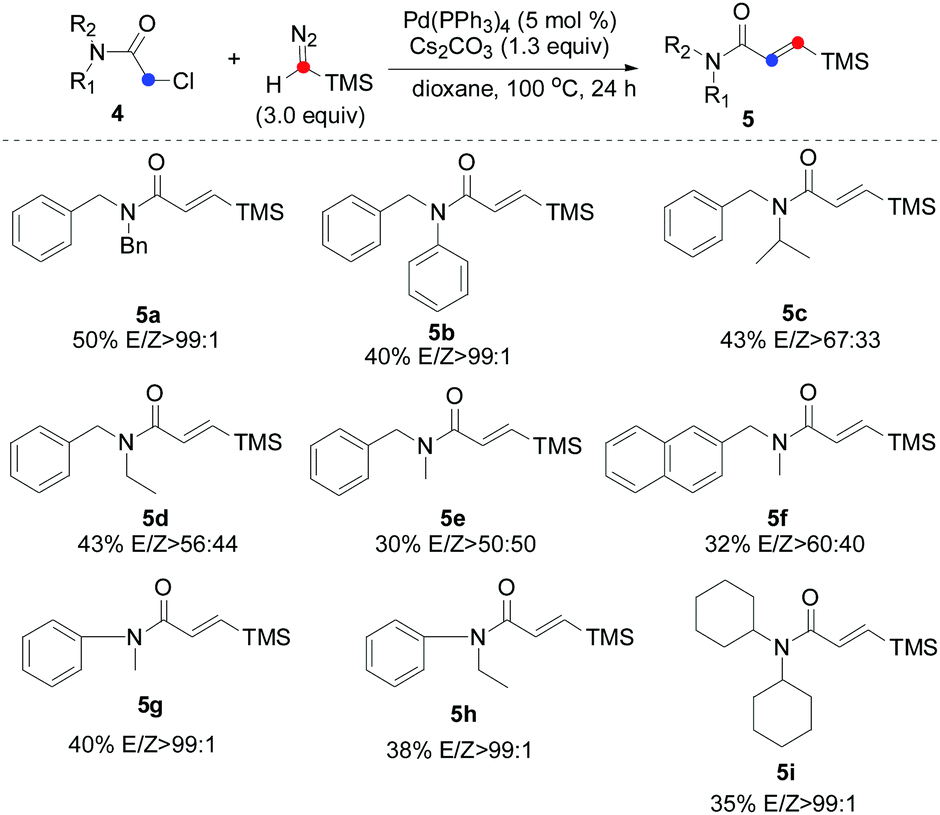
Encouraged by these initial findings, we then further explored the efficiency and practicality of this method. Considering the importance of the β-silyl-α,β-unsaturated amide scaffolds in the organic syntheses which can be employed into diverse types of chemical transformation such as Michael addition,15 total syntheses of (+)-lactacystin,16 tandem Stille reaction17 and so on,18 we thus examined the carbene insertion reaction of chloride-substituted acetamide 4 with TMSD. Unfortunately, β-silyl-α,β-unsaturated amide was not detected under the aforementioned optimized reaction conditions using 4a as a model substrate. To our delight, when KOAc was removed, a small amount of 5a was detected. After screening several parameters such as different Pd sources, bases and solvents (for details, see the ESI†), we found that a protocol based on Pd(PPh3)4 and Cs2CO3 in dry dioxane at 100 °C for 24 hours provided the desired product in 50% yield and excellent E/Z selectivity (99:1).
We next evaluated the scope and limitations on the α-chloroacetamide partners using the optimized conditions. As described in Table 3, compounds 4a and 4b smoothly participated in the reaction to afford 5a and 5b in 50% and 40% yields with excellent E/Z selectivity (99:1), whereas the reaction of 4c, 4d, 4e, and 4f resulted in a lower stereoselectivity (5c, 5d, 5e, and 5f), suggesting that the steric-hindrance effect would be crucial for the E/Z stereocontrol. The results show that variation of the amide by replacing the benzyl moiety with a phenyl group had significant influence on the course of the reaction (5g and 5h). Interesting, bulkier dicyclohexyl-substituted acetamide 4i was also a suitable substrate, giving the corresponding product 5i in satisfactory yield and excellent E/Z selectivity. To show the synthetic potential of our developed catalytic system, a palladium-catalyzed carbene coupling reaction between 4a (α-chloroacetamides) and TMSD was carried out on a gram scale, furnishing the corresponding product 5a (1.23 g, 38%) (Scheme 3).
|
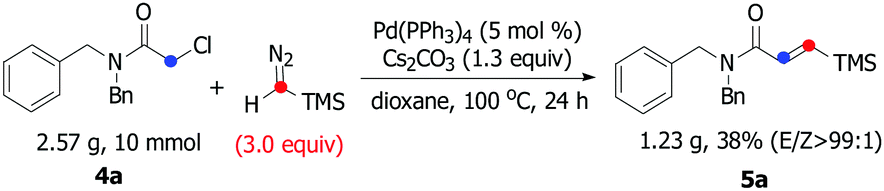 | ||
| Scheme 3 Gram-scale synthesis of 5a. | ||
On the basis of our experimental observations and previous works on related reactions and the DFT calculations on the conformer energetics of F,13,14 the mechanism was proposed as shown in Scheme 4. Initially, oxidative addition of 2a/4a generates the arylpalladium/alkylpalladium species B, which reacts with one molecule of TMSD to form the palladium carbene complex C. Then intermediate D was formed by the migratory insertion of C. 5a would be obtained via β-H elimination of intermediate D. From intermediate D, the carbene complex E was afforded through insertion of a second molecule of TMSD, followed by migratory insertion to give intermediate F. Due to the steric reasons, the anti-relationship between two TMS groups will be more favorable in this insertion process. Finally, the product 3a was obtained through β-silyl-elimination of F with high stereoselectivity.
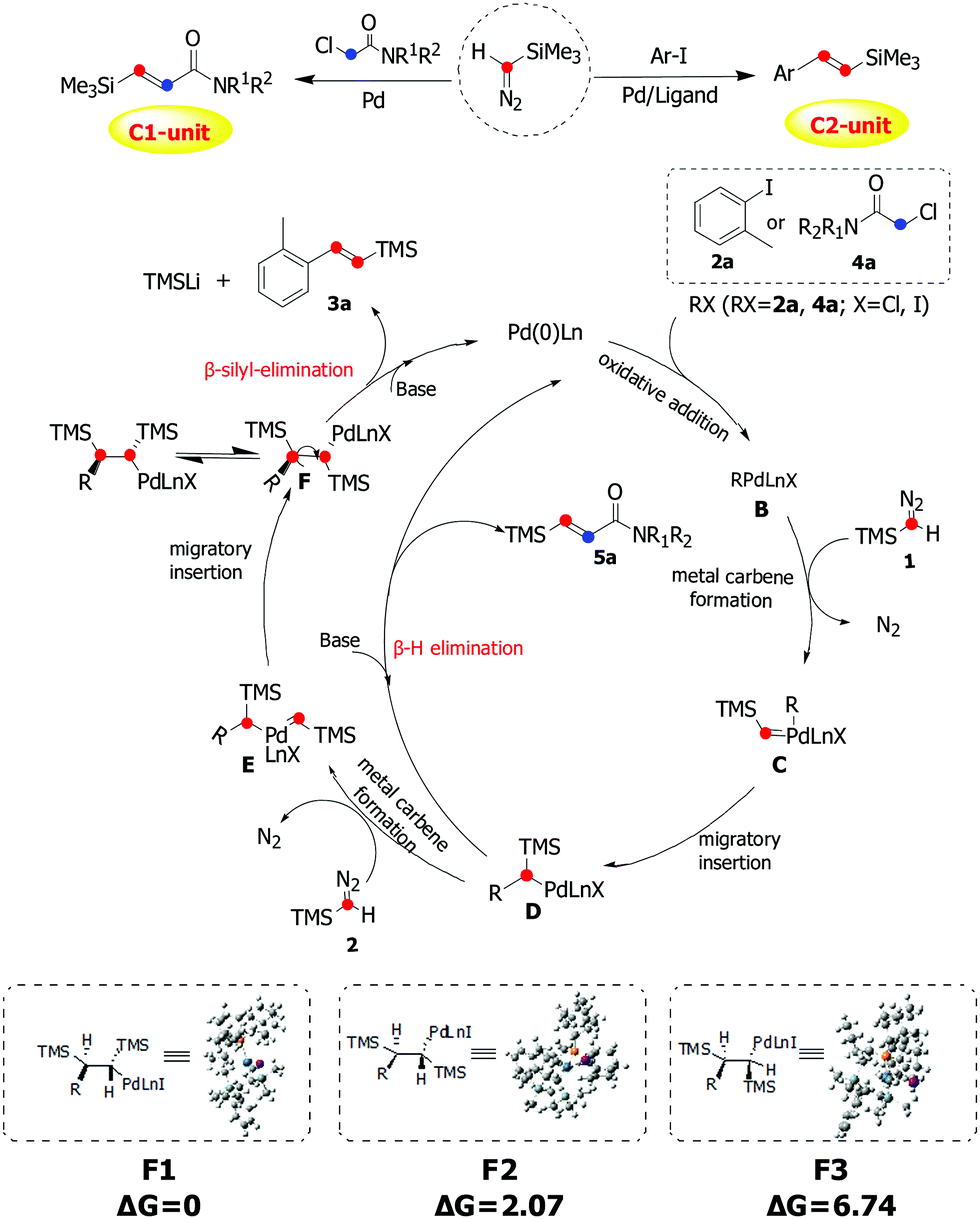 | ||
| Scheme 4 Plausible mechanistic pathway via carbene migratory insertion for palladium-catalyzed olefination of aryl/alkyl halides with trimethylsilyldiazomethane. | ||
In conclusion, we have developed novel and stereospecific protocols for obtaining (E)-vinyl silanes and (E)-silyl-substituted α,β-unsaturated amides in good yields with a good level of E/Z stereoselectivities, which would be a worthwhile valuable complement to the existing methods. These methods turned out to be convenient, with easily available and inexpensive aryl/alkyl halides as starting materials. Notably, it is the first example which shows that two molecules of diazo compounds could be used as a C2-unit in the palladium-catalyzed olefination of aryl halides, in which the trimethylsilyldiazomethane (TMSD)-initiated double carbene migratory insertion was for the first time realized in this work. In addition, it is possible that the corresponding vinyl silanes were produced with a possible pathway involving palladium carbene formation, migratory insertion and finally β-syn-elimination or reductive elimination. Further investigation into the application of this protocol is currently underway in our lab.
We thank the National Natural Science Foundation of China (NSFC, No. 21472031, 21703051, 21702211, and 21773051), the Natural Science Foundation of Jiangsu Province (BK20170421), and the Zhejiang Provincial Natural Science Foundation of China (ZJNSFC, No. LZ18B020001, LY16E030009, LY17E030003, and LY17B030005) for financial support of this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews, see: (a) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (b) Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306 RSC; (c) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS; (d) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (e) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 1002 Search PubMed; (f) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC; (g) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS PubMed; (h) Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015 CrossRef.
- For recent examples, see: (a) X. Hu, X. Chen, Y. Shao, H. Xie, Y. Deng, Z. Ke, H. Jiang and W. Zeng, ACS Catal., 2018, 8, 1308 CrossRef CAS; (b) Y. Gao, G. Wu, Q. Zhou and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 2716 CrossRef CAS PubMed; (c) K. Wang, Y. Pang, T. Chang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 13140 CrossRef CAS PubMed; (d) Q. Zhou, S. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 16013 CrossRef CAS PubMed; (e) Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W. D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970 CrossRef CAS PubMed; (f) H. Zhang, G. Wu, H. Yi, T. Sun, B. Wang, Y. Zhang, G. Dong and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 3945 CrossRef CAS PubMed , and references cited therein.
- K. L. Greenman, D. S. Carter and D. L. Van Vranken, Tetrahedron, 2001, 57, 5219 CrossRef CAS.
- (a) T. A. Blumenkopf and L. E. Overman, Chem. Rev., 1986, 86, 857 CrossRef CAS; (b) I. Fleming, I. A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed; (c) T. Hiyama and E. Shirakawa, Top. Curr. Chem., 2002, 219, 61 CrossRef CAS.
- (a) S. E. Denmark and J. H.-C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978 CrossRef CAS PubMed; (b) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (c) C. Thiot, C. Mioskowski and A. Wagner, Eur. J. Org. Chem., 2009, 3219 CrossRef CAS.
- (a) C. E. Masse and J. S. Panek, Chem. Rev., 1995, 95, 1293 CrossRef CAS; (b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed.
- A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1976, 941 CrossRef CAS.
- (a) D. R. Williams, A. I. Morales-Ramos and C. M. Williams, Org. Lett., 2006, 8, 4393 CrossRef CAS PubMed; (b) J. McNulty and P. Das, Chem. Commun., 2008, 1244 RSC.
- (a) O. Buisine, G. Berthon-Gelloz, J.-F. Briere, S. Sterin, G. Mignani, P. Branlard, B. Tinant, J.-P. Declercq and I. E. Marko, Chem. Commun., 2005, 3856 RSC; (b) T. Konno, K. Taku, S. Yamada, K. Moriyasu and T. Ishihara, Org. Biomol. Chem., 2009, 7, 1167 RSC; (c) C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419 RSC.
- (a) K. Fugami, K. Oshima, K. Utimoto and H. Nozaki, Tetrahedron Lett., 1986, 27, 2161 CrossRef CAS; (b) M. Murata, S. Watanabe and Y. Masuda, Tetrahedron Lett., 1999, 40, 9255 CrossRef CAS; (c) A. Krasovskiy and B. H. Lipshutz, Org. Lett., 2011, 13, 3818 CrossRef CAS PubMed.
- (a) B. Marciniec, E. Walczuk-Gusciora and C. Pietraszuk, Organometallics, 2001, 20, 3423 CrossRef CAS; (b) Y. Jiang, O. Blacque and H. Berke, Dalton Trans., 2011, 40, 2578 RSC; (c) J. R. McAtee, S. E. S. Martin, D. T. Ahneman, K. A. Johnson and D. A. Watson, Angew. Chem., Int. Ed., 2012, 51, 3663 CrossRef CAS PubMed; (d) J. R. McAtee, S. E. S. Martin, A. P. Cinderella, W. B. Reid, K. A. Johnson and D. A. Watson, Tetrahedron, 2014, 70, 4250 CrossRef CAS PubMed; (e) J. R. McAtee, S. B. Krause and D. A. Watson, Adv. Synth. Catal., 2015, 357, 2317 CrossRef CAS PubMed; (f) H. Yamashita, T. Hayashi, T. Kobayashi, M. Tanaka and M. Goto, J. Am. Chem. Soc., 1988, 110, 4417 CrossRef CAS; (g) H. Yamashita, M. Tanaka and M. Goto, Organometallics, 1997, 16, 4696 CrossRef CAS; (h) F. Stchr, D. Sturmayr, G. Kickelbick and U. Schubert, Eur. J. Inorg. Chem., 2002, 2305 Search PubMed; (i) S. Gatard, C. H. Chen, B. Foxman and O. Ozerov, Organometallics, 2008, 27, 6257 CrossRef CAS; (j) J. Gu and C. Cai, Chem. Commun., 2016, 52, 10779 RSC; (k) P. Pawluc, J. Szudkowska, G. Hreczycho and B. Marciniec, J. Org. Chem., 2011, 76, 6438 CrossRef CAS PubMed.
- (a) K. Karabelas and A. Hallberg, Tetrahedron Lett., 1985, 26, 3131 CrossRef CAS; (b) K. Karabelas and A. Hallberg, J. Org. Chem., 1986, 51, 5286 CrossRef CAS; (c) T. Jeffery, Tetrahedron Lett., 1999, 40, 1673 CrossRef CAS.
- (a) T. Aoyama, S. Toyama, N. Tamaki and T. Shioiri, Chem. Pharm. Bull., 1983, 31, 2957 CrossRef CAS; (b) N. Hashimoto, T. Aoyama and T. Shioiri, Chem. Pharm. Bull., 1982, 30, 119 CrossRef CAS; (c) T. Aoyama and T. Shioiri, Chem. Pharm. Bull., 1981, 29, 3249 CrossRef CAS; (d) N. Hashimoto, T. Aoyama and T. Shioiri, Heterocycles, 1981, 15, 975 CrossRef CAS; (e) N. Hashimoto, T. Aoyama and T. Shioiri, Chem. Pharm. Bull., 1981, 29, 1475 CrossRef CAS; (f) T. Aoyama and T. Shioiri, Tetrahedron Lett., 1980, 21, 4461 CrossRef CAS; (g) S. Xu, Y. Gao, R. Chen, K. Wang, Y. Zhang and J. Wang, Chem. Commun., 2016, 52, 4478 RSC; (h) S. Xu, R. Chen, Z. Fu, Q. Zhou, Y. Zhang and J. Wang, ACS Catal., 2017, 7, 1993 CrossRef CAS.
- (a) D. Sole, L. Vallverdu, X. Solans, M. Font-Bardia and J. Bonjoch, Organometallics, 2004, 23, 1438 CrossRef CAS; (b) R. Kudirka, L. David and V. Vranken, J. Org. Chem., 2008, 73, 3585 CrossRef CAS PubMed.
- (a) H. Tomoko, T. Nana, K. Satoko, K. Tiyako, T. Nahoko, C. Nao, S. Chikako, N. Aya and A. Morio, Chem. Lett., 2007, 36, 54 CrossRef; (b) G. W. Klumpp, A. J. C. Mierop, J. J. Vrielink, A. Brugman and M. Schakel, J. Am. Chem. Soc., 1985, 107, 6742 CrossRef; (c) K. Mikiko, N. Satomi, C. Nao, T. Nahoko and A. Morio, Chem. Lett., 2007, 36, 736 CrossRef; (d) M. Ihara, Y. Ishida, Y. Tokunaga, C. Kabuto and K. Fukumoto, J. Chem. Soc., Chem. Commun., 1995, 2085 RSC; (e) I. Fleming and N. D. Kindon, J. Chem. Soc., Chem. Commun., 1987, 1177 RSC.
- (a) E. P. Balskus and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 6810 CrossRef CAS PubMed; (b) J. Clayden, D. W. Watson, M. Helliwell and M. Chambers, Chem. Commun., 2003, 2582 RSC.
- (a) K. Cherry, A. Duchene, J. Thibonnet, J. L. Parrain and M. Abarbri, Synthesis, 2005, 2349 CAS; (b) K. Cherry, M. Abarbri, J. L. Parrain and A. Duchene, Tetrahedron Lett., 2003, 44, 5791 CrossRef CAS.
- (a) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2004, 126, 13942 CrossRef CAS PubMed; (b) S. R. Wilson and M. J. Di Grandi, J. Org. Chem., 1991, 56, 4766 CrossRef CAS; (c) Y. Karibe, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2012, 51, 6214 CrossRef CAS PubMed; (d) X. Wang, N. Masaki, S. Eloisa and M. Ruben, J. Am. Chem. Soc., 2016, 138, 15531 CrossRef CAS PubMed; (e) J. L. Pan, C. Chen, Z. G. Ma, J. Zhou, L. R. Wang and S. Y. Zhang, Org. Lett., 2017, 19, 5216 CrossRef CAS PubMed; (f) P. K. Kundu and S. K. Ghosh, Org. Biomol. Chem., 2009, 7, 4611 RSC; (g) M. J. C. Buckle, I. Fleming, S. Gil and K. L. C. Pang, Org. Biomol. Chem., 2004, 2, 749 RSC; (h) W. Oppolzer, R. J. Mills, W. Pachinger and T. Stevenson, Helv. Chim. Acta, 1986, 69, 1542 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07664b |
| This journal is © The Royal Society of Chemistry 2018 |