
Asymmetric synthesis of polysubstituted methylenecyclobutanes via catalytic [2+2] cycloaddition reactions of N-allenamides†
Xia
Zhong
 ,
Qiong
Tang
,
Pengfei
Zhou
,
Ziwei
Zhong
,
Shunxi
Dong
*,
Xiaohua
Liu
and
Xiaoming
Feng
*
,
Qiong
Tang
,
Pengfei
Zhou
,
Ziwei
Zhong
,
Shunxi
Dong
*,
Xiaohua
Liu
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: xmfeng@scu.edu.cn; dongs@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
First published on 23rd August 2018
Abstract
A highly enantioselective [2+2] cycloaddition reaction of alkylidene malonates with the internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of N-allenamides was developed with a MgII/N,N′-dioxide complex as a catalyst. Various polysubstituted methylenecyclobutanes were afforded in good yields (up to 99%) and excellent enantioselectivities (up to 96% ee) under mild conditions. The utility of the donor–acceptor cyclobutane product was demonstrated as a masked 1,4-dipole in the formal [4+2] annulation reaction with a silyl enol ether.
C bond of N-allenamides was developed with a MgII/N,N′-dioxide complex as a catalyst. Various polysubstituted methylenecyclobutanes were afforded in good yields (up to 99%) and excellent enantioselectivities (up to 96% ee) under mild conditions. The utility of the donor–acceptor cyclobutane product was demonstrated as a masked 1,4-dipole in the formal [4+2] annulation reaction with a silyl enol ether.
Cyclobutanes are not only important structural motifs in numerous biologically significant molecules, but also used as key intermediates for the synthesis of many bioactive compounds and drugs.1 Among them, heteroatom-substituted cyclobutanes, and in particular the N-substituted ones, have received much attention. The introduction of aminocyclobutanes into peptides resulted in foldamers with interesting properties ranging from cell-penetrating agents to low-molecular-weight gelators.2 On the other hand, heteroatom-substituted cyclobutanes bearing electron-donating and withdrawing groups could serve as masked 1,4-dipoles in cycloaddition reactions.3 Regarding the methods for the construction of heteroatom-substituted cyclobutanes, the most efficient one is the [2+2] cycloaddition reaction (Scheme 1a). In the past several decades, the synthesis of heteroatom-substituted cyclobutanes via [2+2] cycloaddition were achieved successfully using photocatalysts, Lewis acids, amine-catalysts or transition metal catalysts.4 However, compared with the well-established chiral alkoxy5 and alkyl sulphanyl6 substituted cyclobutanes, the synthetic strategies of enantioenriched amino-cyclobutanes were mainly focused on photocatalysis,7 and other routes are rare.8
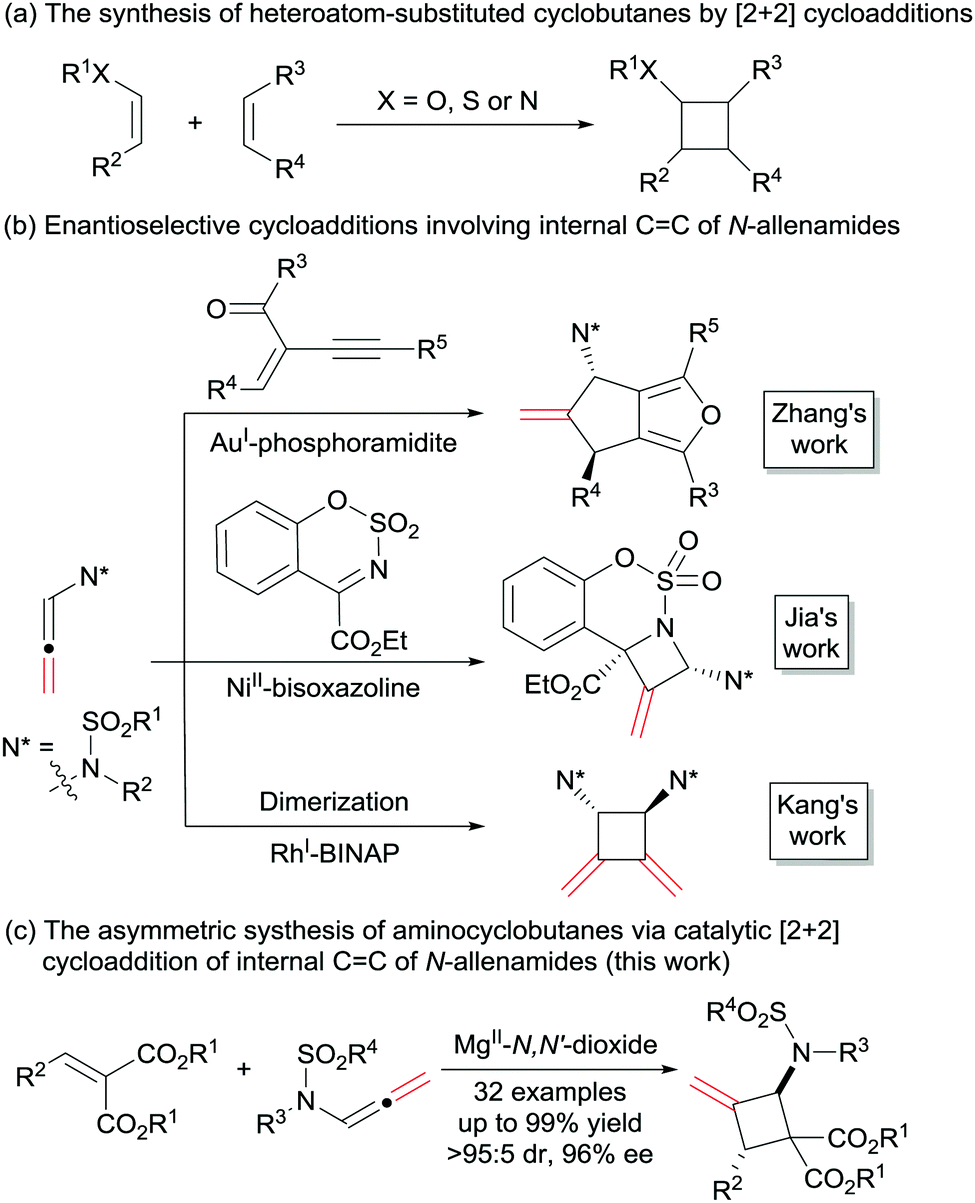 | ||
| Scheme 1 The synthesis of heteroatom-substituted cyclobutanes and cycloaddition reactions of internal CC of N-allenamides. | ||
Due to the presence of the electron-withdrawing group on nitrogen, N-allenamides show higher stability than the corresponding allenamines. In the past two decades, N-allenamides have been employed as versatile reagents in organic synthesis.9 In N-allenamides, the delocalization of the nitrogen lone pair toward the allenic moiety creates an electronic bias, leading to consecutive addition of electrophiles and nucleophiles in a highly regioselective manner. The impressive examples of such elegant transformations are cycloaddition reactions, in which either the terminal CC or internal CC bond could participate, leading to a diverse array of carbo- and heterocyclic structures. Highly enantioselective cycloaddition reactions involving the terminal CC bond were well studied.10 In contrast, only a few of the reactions occurring at the internal CC bond have been established in the asymmetric version to date. In 2015, Zhang and co-workers described the asymmetric formal [3+2] cycloaddition reaction of 2-(1-alkynyl)-2-alken-1-ones with the internal CC bond of N-allenamides using AuI-chiral phosphoramidite as the catalyst.11a Last year, an asymmetric [2+2] reaction of ketimines with the internal CC bond of N-allenamides was realized by Jia et al. in the presence of NiII-chiral bisoxazoline (Scheme 1b).11b Recently, the group of Kang developed the internal CC bond involved asymmetric dimerization of N-allenamides in the presence of RhI-BINAP.11c In addition, an elegant intramolecular [2+2] cycloaddition reaction of alkenes with the internal CC bond of N-allenamide was disclosed by Arisawa in 2016.12 Herein, we disclose a highly enantioselective intermolecular [2+2] cycloaddition reaction, providing a direct access to amino-substituted methylenecyclobutanes13 with a Mg(OTf)2/N,N′-dioxide complex as the catalyst.14 Diverse ranges of functionalized aminocyclobutanes were obtained in moderate to good yields with excellent diastereo- and enantioselectivities (Scheme 1c).
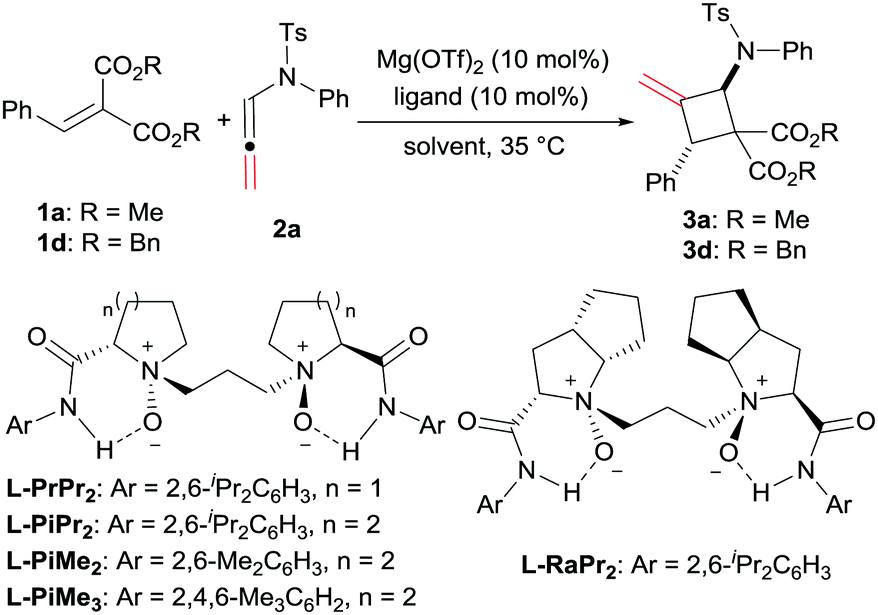
The optimization of the reaction conditions was carried out by taking the reaction of alkylidene malonate (1a) and N-allenamide (2a) as the model reaction. Initially, identification of the metal salts indicated that Mg(OTf)2 exhibited higher activity than other metal salts in the presence of chiral N,N′-dioxide L-PrPr2 (for details, see the ESI†). The desired product 3a was obtained in 18% yield with a moderate ee value (46% ee). Subsequently, various chiral N,N′-dioxide ligands complexing with Mg(OTf)2 were evaluated, suggesting that L-PiPr2 was superior to L-PrPr2 and L-RaPr2 in terms of enantioselectivities (Table 1, entries 1–3). Changing the 2,6-diisopropylaniline moiety to 2,4,6-trimethylaniline provided better results (65% yield, 75% ee; Table 1, entry 4). After careful screening of the solvents, CH2ClCH2Cl was proved to be the best choice, and the cycloaddition product can be obtained 63% yield with 77% ee (Table 1, entry 5). To our delight, the reactivity and enantioselectivity of the reaction increased with the addition of NaBArF4 as an additive, which was proposed to be used for exchanging the counterion (80% yield and 79% ee; Table 1, entry 6).15 Lowering the temperature to 20 °C and switching the alkylidene malonate 1a to 1d gained further improvement (86% yield and 90% ee; Table 1, entries 7 and 8). Finally, 99% yield was afforded when 2.0 equivalents of 2a were used (Table 1, entry 9).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | eec (%) |
a Unless otherwise noted, all reactions were performed with ligand (10 mol%), Mg(OTf)2 (10 mol%), 1a (0.10 mmol) and 2a (0.10 mmol) in solvent (1.0 mL) at 35 °C under N2 for 48 h. The dr values (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) were determined via1H NMR of the crude mixture.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d NaBArF4 {NaB[3,5-(F3C)2C6H3]4} (20 mol%) was added as an additive.
e At 20 °C for 48 h.
f
1d (0.10 mmol) was used instead of 1a.
g 0.20 mmol of 2a was used. :5) were determined via1H NMR of the crude mixture.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d NaBArF4 {NaB[3,5-(F3C)2C6H3]4} (20 mol%) was added as an additive.
e At 20 °C for 48 h.
f
1d (0.10 mmol) was used instead of 1a.
g 0.20 mmol of 2a was used.
|
||||
| 1 | L-PrPr2 | CH2Cl2 | 18 | 46 |
| 2 | L-RaPr2 | CH2Cl2 | 53 | 23 |
| 3 | L-PiPr2 | CH2Cl2 | 29 | 47 |
| 4 | L-PiMe3 | CH2Cl2 | 65 | 75 |
| 5 | L-PiMe3 | CH2ClCH2Cl | 63 | 77 |
| 6d | L-PiMe3 | CH2ClCH2Cl | 80 | 79 |
| 7de | L-PiMe3 | CH2ClCH2Cl | 80 | 85 |
| 8def | L-PiMe3 | CH2ClCH2Cl | 86 | 90 |
| 9defg | L-PiMe3 | CH2ClCH2Cl | 99 | 90 |
With the optimized reaction conditions in hand (Table 1, entry 9), the scope of alkylidene malonates 1 was investigated by reacting with N-allenamide 2a (Table 2). Both reactivities and enantioselectivities gradually reduced with the increase of steric hindrance of the ester group (Table 2, entries 1–3). The positions of substituents on the phenyl ring in alkylidene malonates 1 showed a significant influence on the results (Table 2, entries 4–15). Generally, alkylidene malonates 1e−j bearing para-substituents regardless of electron-donating and electron-withdrawing groups are all tolerated well, providing the desired products in high yields and ee values (92–99% yield, 92–95% ee; Table 2, entries 4–9). The reaction with 1g was performed on a gram scale and obtained comparable results (1.02 g, 92% yield and 95% ee, Table 2, entry 6). The meta-substituted ones displayed similar reactivities but the ee values of the products 3k–3m decreased slightly (70–99% yield, 85–89% ee; Table 2, entries 10–12), and the 3,4-disubstituted alkylidene malonate afforded comparable results (Table 2, entry 13). In contrast, a sharp decrease in both reactivities and enantioselectivities was observed for ortho-substituted substrates (Table 2, entries 14 and 15). Pleasingly, the reaction of fused-ring-substituted alkylidene malonate proceeded well, and the adduct 3q was isolated in 99% yield with 88% ee (Table 2, entry 16). Moreover, the heteroaromatic cycle derived ones also performed the reaction with moderate ee values (58–98% yield, 51–84% ee, Table 2, entries 17–20). It should be noted that only one of the diastereomers was detected in all of these cases. To our delight, the catalyst system was also effective for aliphatic substrates, affording excellent enantioselectivities (93–96% ee) and high diastereoselectivities (85:15–92:8 dr; Table 2, entries 21–24). Interestingly, cinnamyl-substituted alkylidene malonate was also suitable with high regioselectivity (Table 2, entry 25).
|
|
||||
|---|---|---|---|---|
| Entry | R1/R2 | Yield (%) | dr | ee (%) |
| a Reaction conditions are identical to those in entry 9 of Table 1. b The value in parentheses was obtained when conducted on a gram scale (1.5 mmol of 1g). c Carried out with L-PiEt2Me (10 mol%) in CHCl2CHCl2 (1.0 mL) for 48 h. | ||||
| 1 | Me/Ph (3a) | 99 | >95:5 |
87 |
| 2 | Et/Ph (3b) | 95 | >95:5 |
87 |
| 3 | iPr/Ph (3c) | 31 | >95:5 |
67 |
| 4 | Bn/4-FC6H4 (3e) | 94 | >95:5 |
93 |
| 5 | Bn/4-ClC6H4 (3f) | 99 | >95:5 |
95 |
| 6b | Bn/4-BrC6H4 (3g) | 99 (92) | >95:5 |
95 (95) |
| 7 | Bn/4-MeC6H4 (3h) | 93 | >95:5 |
93 |
| 8 | Bn/4-iPrC6H4 (3i) | 92 | >95:5 |
92 |
| 9 | Bn/4-PhC6H4 (3j) | 98 | >95:5 |
92 |
| 10 | Bn/3-ClC6H4 (3k) | 99 | >95:5 |
87 |
| 11 | Bn/3-BrC6H4 (3l) | 99 | >95:5 |
89 |
| 12 | Bn/3-MeC6H4 (3m) | 70 | >95:5 |
85 |
| 13 | Bn/3,4-Me2C6H3 (3n) | 91 | >95:5 |
84 |
| 14 | Bn/2-ClC6H4 (3o) | 52 | >95:5 |
39 |
| 15 | Bn/2-MeC6H4 (3p) | 32 | >95:5 |
48 |
| 16 | Bn/2-naphthyl (3q) | 99 | >95:5 |
88 |
| 17 | Bn/2-thienyl (3r) | 86 | >95:5 |
74 |
| 18 | Bn/2-benzothienyl (3s) | 98 | >95:5 |
84 |
| 19 | Bn/2-furanyl (3t) | 58 | >95:5 |
51 |
| 20 | Bn/2-benzofuranyl (3u) | 92 | >95:5 |
67 |
| 21c | Bn/iPr (3v) | 95 | 89:11 |
96 |
| 22c | Bn/cyclohexyl (3w) | 83 | 85:15 |
96 |
| 23c | Bn/iBu (3x) | 86 | 92:8 |
92 |
| 24c | Bn/nBu (3y) | 57 | 92:8 |
93 |
| 25c |
|
49 | 91:9 |
88 |

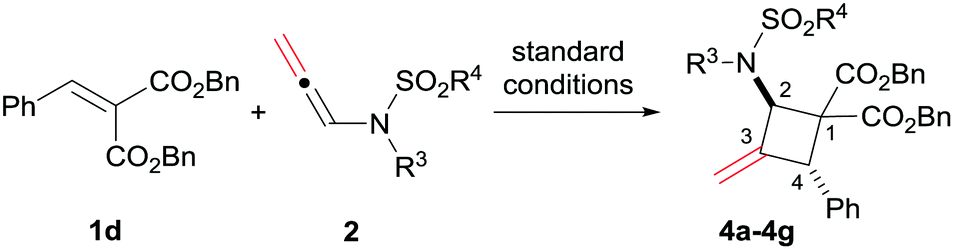
Then, various substituted N-allenamides were examined. As shown in Table 3, N-allenamides with different substituents were applicable, giving the corresponding products in 85–99% yield and 88–91% ee (Table 3, entries 1–6). The absolute configuration of compound 4d was determined to be (2R, 4R) by X-ray single crystallographic analysis.16
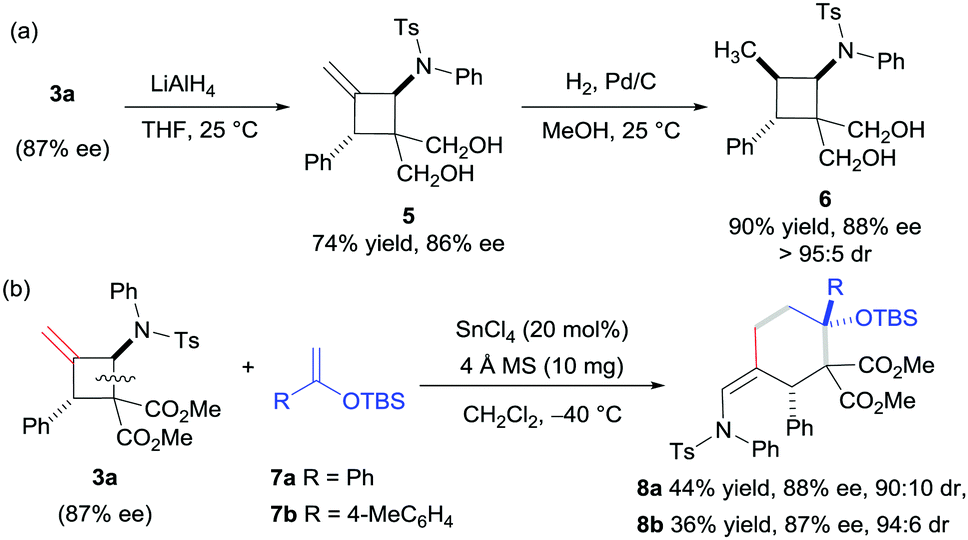
To evaluate the practicality of this catalytic system, the reduction of 3a with LiAlH4 generated the 1,3-diol derivative 5, which can be reduced continuously to the compound 6 in excellent yield with high diastereoselectivity (Scheme 2a). In addition, the product 3a was successfully used as a masked 1,4-dipole in the [4+2] annulation reaction with silyl enol ether 7 in the presence of SnCl4 (Scheme 2b), and the products 8a and 8b were obtained in moderate yields and high diastereoselectivities along with a maintained ee value (for the proposed mechanism, see the ESI†). The relative configuration of compound 8a was assigned via X-ray single crystallographic analysis.16
 | ||
| Scheme 2 Transformation of the products. | ||
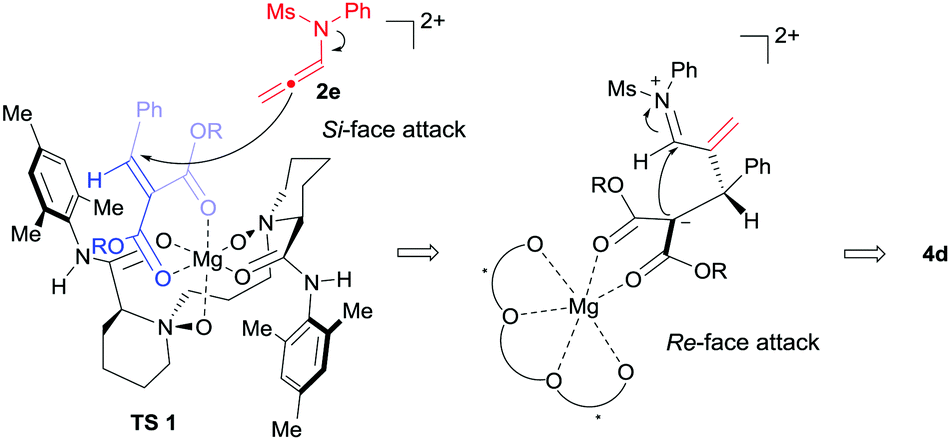
Based on the previous work14 and the absolute configuration of the product 4d, a possible transition state model was proposed to elucidate the origin of chiral induction in the [2+2] cycloaddition reaction. As shown in Fig. 1, chiral N,N′-dioxide and alkylidene malonate 1d coordinated to MgII in tetradentate and bidentate fashions respectively to form a slightly distorted hexahedral complex. The Re-face of the substrate 1d was shielded by the substituted aniline group on the ligand. Consequently, N-allenamide 2e approached from the Si-face of the substrate to form the zwitterionic intermediate, which subsequently underwent cyclization from the Re-face of the imine moiety to afford 4d.
 | ||
| Fig. 1 Proposed transition state model. | ||
In summary, we have developed a chiral MgII/N,N′-dioxide catalyst system to realize the asymmetric [2+2] cycloaddition of alkylidene malonates with the proximal CC bond of N-allenamides. A wide range of aminocyclobutanes was obtained in moderate to excellent yields (up to 99%) with excellent ee values (up to 96% ee). The utility of the product was demonstrated as a masked 1,4-dipole in highly diastereoselective cycloaddition with silyl enol ether 7. Furthermore, a possible transition state mode was proposed to explain the origin of the chiral induction.
We appreciate the National Natural Science Foundation of China (No. 21432006 and 21772127) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews, see: (a) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449 CrossRef PubMed; (b) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559 CrossRef PubMed; (c) J.-P. Deprés, P. Delair, J.-F. Poisson, A. Kanazawa and A. E. Greene, Acc. Chem. Res., 2016, 49, 252 CrossRef PubMed . For select examples, see: ; (d) K. Tanino, M. Takahashi, Y. Tomata, H. Tokura, T. Uehara, T. Narabu and M. Miyashita, Nat. Chem., 2011, 3, 484 CrossRef PubMed; (e) X.-K. Kuang, J. Zhu, L. Zhou, L. Wang, S. R. Wang and Y. Tang, ACS Catal., 2018, 8, 4991 CrossRef.
- For select examples on aminocyclobutanes as amino acids, see: (a) G. Jiménez-Osés, F. Corzana, J. H. Busto, M. Pérez-Fernández, J. M. Peregrina and A. Avenoza, J. Org. Chem., 2006, 71, 1869 CrossRef PubMed; (b) A. Fetnández-Tejada, F. Corzana, J. H. Busto, G. Jiménez-Osés, J. M. Peregrina and A. Avenoza, Chem. – Eur. J., 2008, 14, 7042 CrossRef PubMed; (c) E. Gorrea, P. Nolis, E. Torres, E. D. Silva, D. B. Amabilino, V. Branchadell and R. M. Ortuño, Chem. – Eur. J., 2011, 17, 4588 CrossRef PubMed; (d) A. Sorrenti, O. Illa, R. Pons and R. M. Ortuño, Langmuir, 2015, 31, 9608 CrossRef PubMed; (e) M. Alauddin, E. Gloaguen, V. Brenner, B. Tardivel, M. Mons, A. Zehnacker-Rentien, V. Declerck and D. J. Aitken, Chem. – Eur. J., 2015, 21, 16479 CrossRef PubMed; (f) A. Sorrenti, O. Illa, R. M. Ortuño and R. Pons, Langmuir, 2016, 32, 6977 CrossRef PubMed.
- For selected reviews on cyclobutanes as 1,4-dipoles, see: (a) F. de Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912 RSC; (b) H.-U. Ressig and R. Zimmer, Angew. Chem., Int. Ed., 2015, 54, 5009 CrossRef PubMed; (c) N. Vemula and B. L. Pagenkopf, Org. Chem. Front., 2016, 3, 1205 RSC . For select examples, see: ; (d) A. Levens, A. Ametovski and D. W. Lupton, Angew. Chem., Int. Ed., 2016, 55, 16136 CrossRef PubMed; (e) L. K. B. Garve, A. Kreft, P. G. Jones and D. B. Werz, J. Org. Chem., 2017, 82, 9235 CrossRef PubMed; (f) L.-W. Feng, H. Ren, H. Xiong, P. Wang, L. Wang and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 3055 CrossRef PubMed; (g) J.-L. Hu, L. Zhou, L. Wang, Z. Xie and Y. Tang, Chin. J. Chem., 2018, 36, 47 CrossRef.
- For reviews, see: (a) F. Secci, A. Frongia and P. P. Piras, Molecules, 2013, 18, 15541 CrossRef PubMed; (b) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918 CrossRef PubMed; (c) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef PubMed . For examples, see: ; (d) B. Brown and L. S. Hegedus, J. Org. Chem., 1998, 63, 8012 CrossRef; (e) A. Avenoza, J. H. Busto, N. Canal, J. M. Peregrina and M. Pérez-Fernández, Org. Lett., 2005, 7, 3597 CrossRef PubMed; (f) A. Avenoza, J. H. Busto, L. Mata, J. M. Peregrina and M. Pérez-Fernández, Synthesis, 2008, 743 Search PubMed; (g) A. Avenoza, J. H. Busto, N. Canal, F. Corzana, J. M. Peregrina, M. Pérez-Fernández and F. Rodríguez, J. Org. Chem., 2010, 75, 545 CrossRef PubMed; (h) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 9009 CrossRef PubMed; (i) Z. Dobi, T. Holczbauer and T. Soós, Eur. J. Org. Chem., 2017, 1391 CrossRef.
- (a) E. Canales and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 12686 CrossRef PubMed; (b) R. Brimioulle and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 12921 CrossRef PubMed; (c) F. Yagishita, N. Takagishi, H. Ishikawa, Y. Kasashima, T. Mino and M. Sakamoto, Eur. J. Org. Chem., 2014, 6366 CrossRef; (d) S. C. Coote, A. Pöthig and T. Bach, Chem. – Eur. J., 2015, 21, 6906 CrossRef PubMed; (e) K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186 CrossRef PubMed.
- (a) Y. Hayashi and K. Narasaka, Chem. Lett., 1989, 793 CrossRef; (b) K. Narasaka, Y. Hayashi, H. Shimadzu and S. Niihata, J. Am. Chem. Soc., 1992, 114, 8869 CrossRef; (c) K. Narasaka, K. Hayashi and Y. Hayashi, Tetrahedron, 1994, 50, 4529 CrossRef.
- (a) S. C. Coote and T. Bach, J. Am. Chem. Soc., 2013, 135, 14948 CrossRef PubMed; (b) R. Brimioulle and T. Bach, Science, 2013, 342, 840 CrossRef PubMed; (c) R. Brimioulle, A. Bauer and T. Bach, J. Am. Chem. Soc., 2015, 137, 5170 CrossRef PubMed; (d) H. Wang, X. Cao, X. Chen, W. Fang and M. Dolg, Angew. Chem., Int. Ed., 2015, 54, 14295 CrossRef PubMed; (e) A. Tröster, R. Alonso, A. Bauer and T. Bach, J. Am. Chem. Soc., 2016, 138, 7808 CrossRef PubMed; (f) C. Wang and Z. Lu, Org. Lett., 2017, 19, 5888 CrossRef PubMed.
- For selected examples on the asymmetric synthesis by other methods, see: (a) L. Ghosez, F. Mahuteau-Betzer, C. Genicot, A. Vallribera and J.-F. Cordier, Chem. – Eur. J., 2002, 8, 3411 CrossRef PubMed; (b) F. Mahuteau-Betzer and L. Ghosez, Tetrahedron, 2002, 58, 6991 CrossRef; (c) D. J. Aitken, P. Caboni, H. Eijsberg, A. Frogia, R. Guillot, J. Ollivier, P. P. Piras and F. Secci, Adv. Synth. Catal., 2014, 356, 941 CrossRef; (d) N. Melis, L. Ghisu, R. Guillot, P. Caboni, F. Secci, D. J. Aitken and A. Frongia, Eur. J. Org. Chem., 2015, 4358 CrossRef.
- (a) L.-L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773 CrossRef PubMed; (b) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 CrossRef PubMed; (c) M. R. Fructos and A. Prieto, Tetrahedron, 2016, 72, 355 CrossRef; (d) E. Manoni and M. Bandini, Eur. J. Org. Chem., 2016, 3135 CrossRef.
- For selected examples of asymmetric cycloaddition of the terminal CC bond of N-allenamides, see:
(a) S. Suárez-Pantiga, C. Hernández-Díaz, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2012, 51, 11552 CrossRef PubMed;
(b) J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Sigüenza, E. Díez, I. Alonso, R. Fernández, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322 CrossRef PubMed;
(c) G.-H. Li, W. Zhou, X.-X. Li, Q.-W. Bi, Z. Wang, Z.-G. Zhao, W.-X. Hu and Z. Chen, Chem. Commun., 2013, 49, 4770 RSC;
(d) H. Faustino, I. Alonso, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526 CrossRef PubMed;
(e) M. Jia, M. Monari, Q.-Q. Yang and M. Bandini, Chem. Commun., 2015, 51, 2320 RSC;
(f) Y. Wang, P. Zhang, Y. Liu, F. Xia and J. Zhang, Chem. Sci., 2015, 6, 5564 RSC;
(g) H. Hu, Y. Wang, D. Qian, Z.-M. Zhang, L. Liu and J. Zhang, Org. Chem. Front., 2016, 3, 759 RSC;
(h) I. Varela, H. Faustino, E. Díez, J. Iglesias-Sigüenza, F. Grande-Carmona, R. Fernández, J. M. Lassaletta, J. L. Mascareñas and F. López, ACS Catal., 2017, 7, 2397 CrossRef;
(i) V. Pirovano, M. Borri, G. Abbiat, S. Rizzato and E. Rossi, Adv. Synth. Catal., 2017, 359, 1912 CrossRef.
- Enantioselective cycloaddition involving the internal CC bond of N-allenamides, see:
(a) Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849 CrossRef PubMed;
(b) R.-R. Liu, J.-P. Hu, J.-J. Hong, C.-J. Lu, J.-R. Gao and Y.-X. Jia, Chem. Sci., 2017, 8, 2811 RSC;
(c) W.-F. Zheng, G.-J. Sun, L. Chen and Q. Kang, Adv. Synth. Catal., 2018, 360, 1790 CrossRef.
- T. Nada, Y. Yoneshige, Y. Li, T. Matsumoto, H. Fujioka, S. Shuto and M. Arisawa, ACS Catal., 2016, 6, 3168 CrossRef.
- For a review of methylenecyclobutane, see: A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef PubMed.
- For reviews on chiral N,N′-dioxides, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef; (e) X. M. Feng, Z. Wang and X. H. Liu, Chiral Lewis Acid Rare-Earth Metal Complexes in Enantioselective Catalysis, in Chiral Lewis Acids Top. Organomet. Chem., ed. K. Mikami, Springer, 2017, vol. 62, p. 147 Search PubMed.
- The BArF4− ion might act as a large and noncoordinating counterion in the reaction. For a review, see: (a) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef PubMed . For selected examples using BArF4− as a counterion, see: ; (b) W.-J. Shi, Q. Zhang, J.-H. Xie, S.-F. Zhu, G.-H. Hou and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 2780 CrossRef PubMed; (c) B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834 CrossRef PubMed; (d) X. H. Zhao, X. H. Liu, H. J. Mei, J. Guo, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 4032 CrossRef PubMed.
- CCDC 1831461 (4d) and CCDC 1848226 (rac-8a), ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1831461 and 1848226. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc06416d |
| This journal is © The Royal Society of Chemistry 2018 |