
Asymmetric total synthesis of pleurospiroketals A and B†
Toyoharu
Kobayashi
 a,
Konomi
Tanaka
a,
Masako
Ishida
a,
Natsumi
Yamakita
a,
Hideki
Abe
ab and
Hisanaka
Ito
*a
a,
Konomi
Tanaka
a,
Masako
Ishida
a,
Natsumi
Yamakita
a,
Hideki
Abe
ab and
Hisanaka
Ito
*a
aSchool of Life Sciences, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan
bDepartment of Chemical and Biological Sciences, Faculty of Science, Japan Women's University, 2-8-1 Mejirodai, Bunkyo-ku, Tokyo 112-8681, Japan
First published on 20th August 2018
Abstract
The first asymmetric total synthesis of pleurospiroketals A and B has been accomplished in 16 steps from 5-methyl-5-hexenoic acid. Key features of the synthesis are the highly syn-selective Evans aldol reaction, ring-closing metathesis, highly diastereoselective dihydroxylation and acid-mediated spiroketalization.
In 2013, Liu and co-workers reported the isolation of new sesquiterpenoids, pleurospiroketals A (1) and B (2), from the edible mushroom Pleurotus cornucopiae, along with pleurospiroketals C–E.1 The structures of these terpenoids were established through analysis of 2D NMR spectra, single-crystal X-ray diffraction, and CD data analysis as depicted in Fig. 1. Pleurospiroketals A and B are epimers at the C2 position and possess a unique perhydrobenzannulated 5,5-spiroketal skeleton bearing four contiguous stereocenters. Compounds 1 and 2 possess inhibitory activities against nitric oxide production in lipopolysaccharide-activated macrophages and cytotoxicity against the HeLa cell line.1 A structurally closed sesquiterpenoid, pleurospiroketal F (3), was also isolated from the solid-state fermentation of Pleurotus citrinopile.2 Although no total syntheses or synthetic studies of these unique sesquiterpenoids have been reported, the total synthesis of pleurolactone (4),3 which has a perhydrobenzofuran skeleton, as a racemate was achieved by our group4 and the Mehta group.5 The structural features and biological activities of these terpenoids attracted our interest, and a synthetic study of pleurospiroketals A (1) and B (2) in optically active form was initiated.
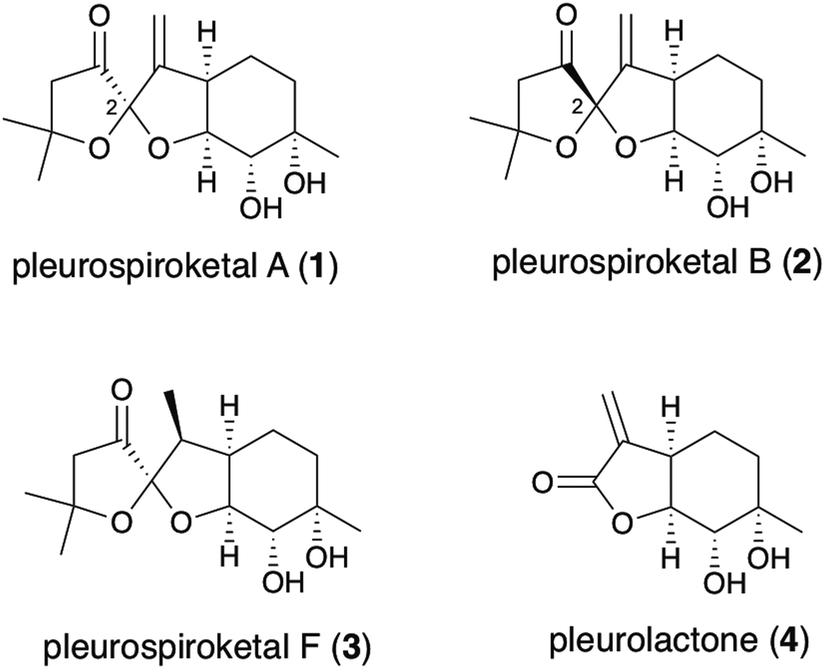 | ||
| Fig. 1 Structures of pleurospiroketals A (1) and B (2) and related natural products 3 and 4. | ||
Herein, we describe the first asymmetric total synthesis of pleurospiroketals A (1) and B (2) in 16 steps using the highly syn-selective Evans aldol reaction, ring-closing metathesis, highly diastereoselective dihydroxylation and acid-mediated spiroketalization as key steps.
The synthetic strategy for pleurospiroketals A (1) and B (2) is outlined in Scheme 1. The target molecules 1 and 2 could be synthesized by deprotection of silyl ethers and acetonide groups and construction of the 5,5-spiroketal moiety from diketone 5. Compound 5 would be obtained using nucleophilic addition of an acyl anion equivalent onto unsaturated aldehyde 6 and conversion of the resulting adduct in a few steps. Compound 6 could be obtained using an established procedure for pleurolactone synthesis,4 which includes highly diastereoselective dihydroxylation of 7 to construct four contiguous stereocenters. Ketone 7 would be synthesized by the conversion of the chiral auxiliary of compound 8 to a methyl ketone and protection of the alcohol group. Compound 8 would be constructed using ring-closing metathesis of compound 9, which could be obtained by the asymmetric syn-selective aldol reaction of compound 10 with acrolein.
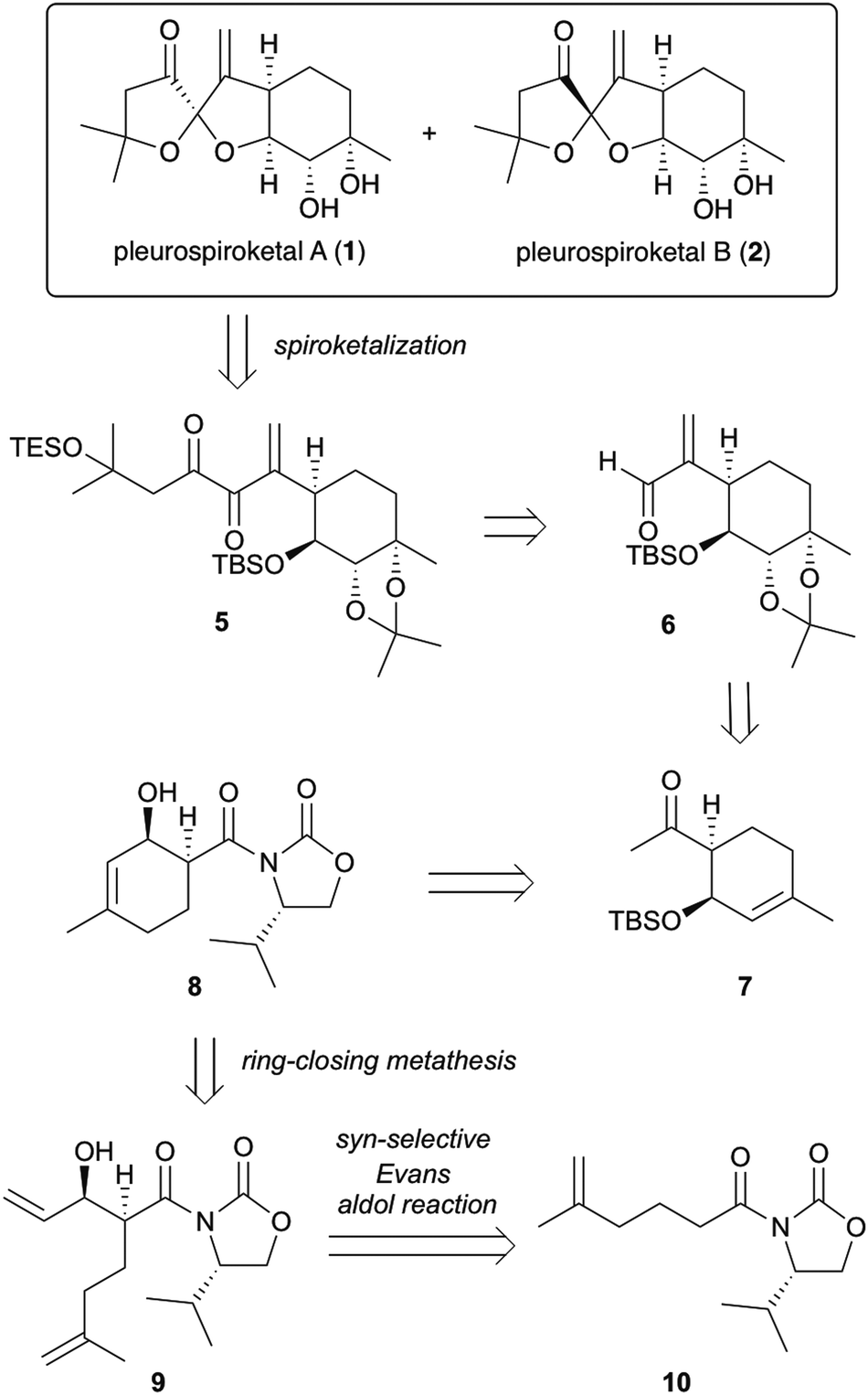 | ||
| Scheme 1 Retrosynthesis of pleurospiroketals A (1) and B (2). | ||
Our investigation started with the synthesis of compound 10 with a chiral auxiliary for the subsequent syn-selective Evans aldol reaction as shown in Scheme 2. Installation of (S)-4-isopropyloxazolidinone as the chiral auxiliary into the known carboxylic acid 116 was achieved by a two-step operation: transformation of the carboxylic acid to the mixed anhydride, followed by addition of the lithiated oxazolidinone into the resulting mixed anhydride to afford 10 in 93% yield. The syn-selective Evans aldol reaction7 of 10 and acrolein using di-n-butylboryl trifluoromethanesulfonate and diisopropyl(ethyl)amine provided the desired Evans syn product (+)-9 in 77% yield as a single isomer. The absolute stereochemistries of the two newly formed stereocenters of (+)-9 were confirmed by X-ray crystallographic analysis of compound (−)-16. The RCM8 of diene compound (+)-9 with a Grubbs second-generation reagent afforded the desired cyclohexenol derivative (−)-8 in 77% yield. Conversion of the chiral auxiliary to methyl ketone was achieved via a Weinreb amide derivative. Thus, treatment of compound (−)-8 with N,O-dimethylhydroxylamine hydrochloride and trimethylaluminum provided the Weinreb amide (−)-12 in 90% yield. Compound (−)-13 was obtained by the addition of methylmagnesium bromide to the Weinreb amide (−)-12 in 79% yield. Protection of the hydroxy group of compound (−)-13 with a tert-butyldimethylsilyl group gave compound (−)-7 in 87% yield.
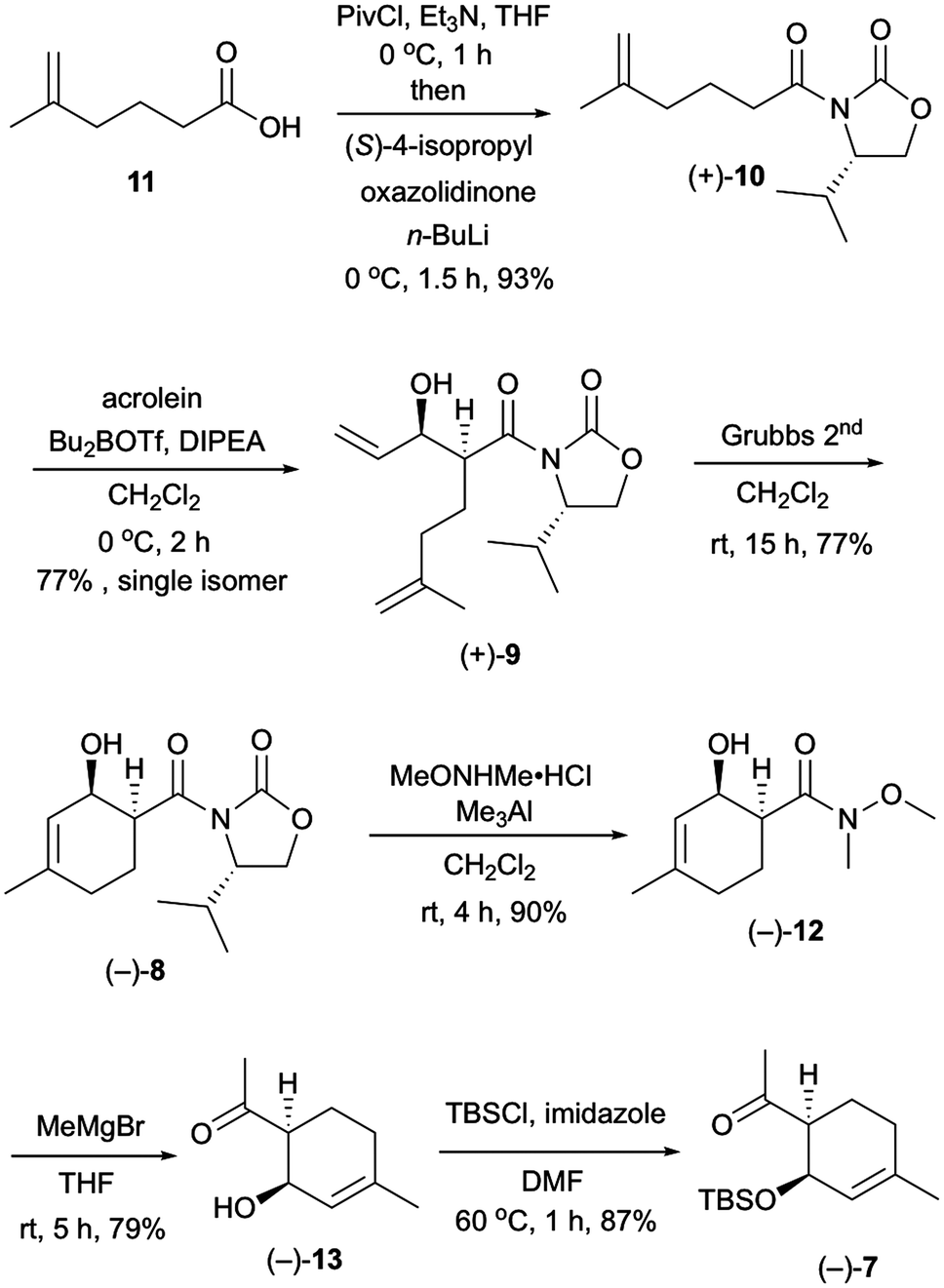 | ||
| Scheme 2 Synthesis of compound (−)-7 in an optically active form. | ||
The stereocontrolled synthesis of (−)-16 from compound (−)-7 was achieved using a procedure established by our group for pleurolactone synthesis4 (Scheme 3). Thus, diastereoselective dihydroxylation of (−)-7 with potassium osmate gave the desired diol (+)-14 in 97% yield as the sole product. This dihydroxylation with potassium osmate occurred on the opposite side of the TBS-protected hydroxyl group. Protection of the dihydroxyl group using 2,2-dimethoxypropane in N,N-dimethylformamide afforded the acetonide (−)-15 in 89% yield. Treatment of (−)-15 with N-phenyltrifluoromethanesulfonimide and potassium hexamethyldisilazide provided the corresponding vinyl triflate. The unsaturated ester (−)-16 was obtained in 93% yield (2 steps) using 2,4,6-trichlorophenyl formate as the carbon monoxide equivalent,9 palladium catalyst [Pd(OAc)2, Xantphos], and triethylamine in toluene. The determination of the absolute configuration of compound (−)-16, which included three chlorine atoms, was accomplished by single-crystal X-ray crystallographic analysis.10 Therefore, the absolute configuration of (−)-16 was established as 3aR, 4S, 5S, 7aS on the basis of the value of the Flack absolute structure parameter, –0.01(3).11 Thus, the stereocontrolled synthesis of the cyclohexane core with four contiguous stereocenters in the chiral form was achieved successfully.
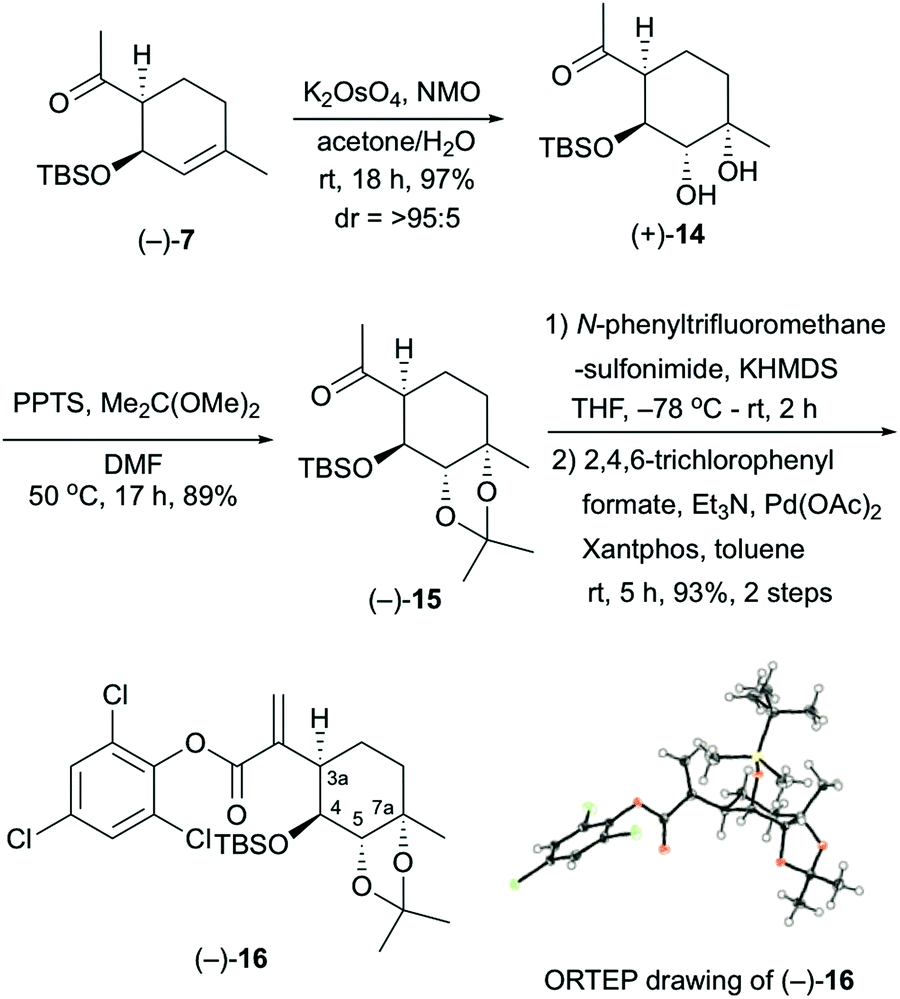 | ||
| Scheme 3 Synthesis of compound (−)-16 and determination of absolute configuration. | ||
With the stereocontrolled synthesis of compound (−)-16 in hand, we next focused on the construction of the 5,5-spiroketal moiety (Scheme 4). Unsaturated aldehyde (−)-6 was obtained via a two-step operation. Reduction of ester (−)-16 with diisobutylaluminum hydride in dichloromethane provided the allyl alcohol (−)-17 in 50% yield, and then oxidation of allyl alcohol (−)-17 with manganese dioxide gave the unsaturated aldehyde (−)-6 in 80% yield. After several attempts at the nucleophilic addition of the acyl anion equivalent to the unsaturated aldehyde (−)-6, we found that dithiane derivative 1812 was suitable as an acyl anion equivalent. Treatment of unsaturated aldehyde (−)-6 with the lithiated dithiane derivative, which was derived from dithiane 18 and n-BuLi, in THF at 0 °C provided the desired adduct in 84% yield as a single diastereomer. Removal of the dithiane protecting group was achieved by treatment of the resulting adduct with iodine to give the corresponding ketone in 60% yield. Oxidation of the resulting compound with 2-iodoxybenzoic acid (IBX) provided the spiroketalization precursor (+)-5 in 99% yield.13 Finally, upon treatment of (+)-5 with conc. HCl in MeOH for 3 h at room temperature, deprotection of the TBS, TES and acetonide groups and construction of the 5,5-spiroketal moiety proceeded simultaneously, and the target molecules 1 and 2 were obtained in 68% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture. These compounds could easily be separated by HPLC. Both 1H and 13C NMR spectra of the synthetic compounds 1 and 2 were identical to those of natural pleurospiroketals A and B. The optical rotations of synthetic 1 and 2 had the same rotations as those reported for the natural products [synthetic 1: [α]D +94.0 (c 1.0, MeOH); natural product 1: [α]D +104.8 (c 1.0, MeOH);1 synthetic 2: [α]D +17.1 (c 1.1, MeOH); natural product 2: [α]D +15.0 (c 1.0, MeOH)1]. Additionally, the structure of compound 1 was unambiguously confirmed by X-ray crystallographic analysis.14
:1 mixture. These compounds could easily be separated by HPLC. Both 1H and 13C NMR spectra of the synthetic compounds 1 and 2 were identical to those of natural pleurospiroketals A and B. The optical rotations of synthetic 1 and 2 had the same rotations as those reported for the natural products [synthetic 1: [α]D +94.0 (c 1.0, MeOH); natural product 1: [α]D +104.8 (c 1.0, MeOH);1 synthetic 2: [α]D +17.1 (c 1.1, MeOH); natural product 2: [α]D +15.0 (c 1.0, MeOH)1]. Additionally, the structure of compound 1 was unambiguously confirmed by X-ray crystallographic analysis.14
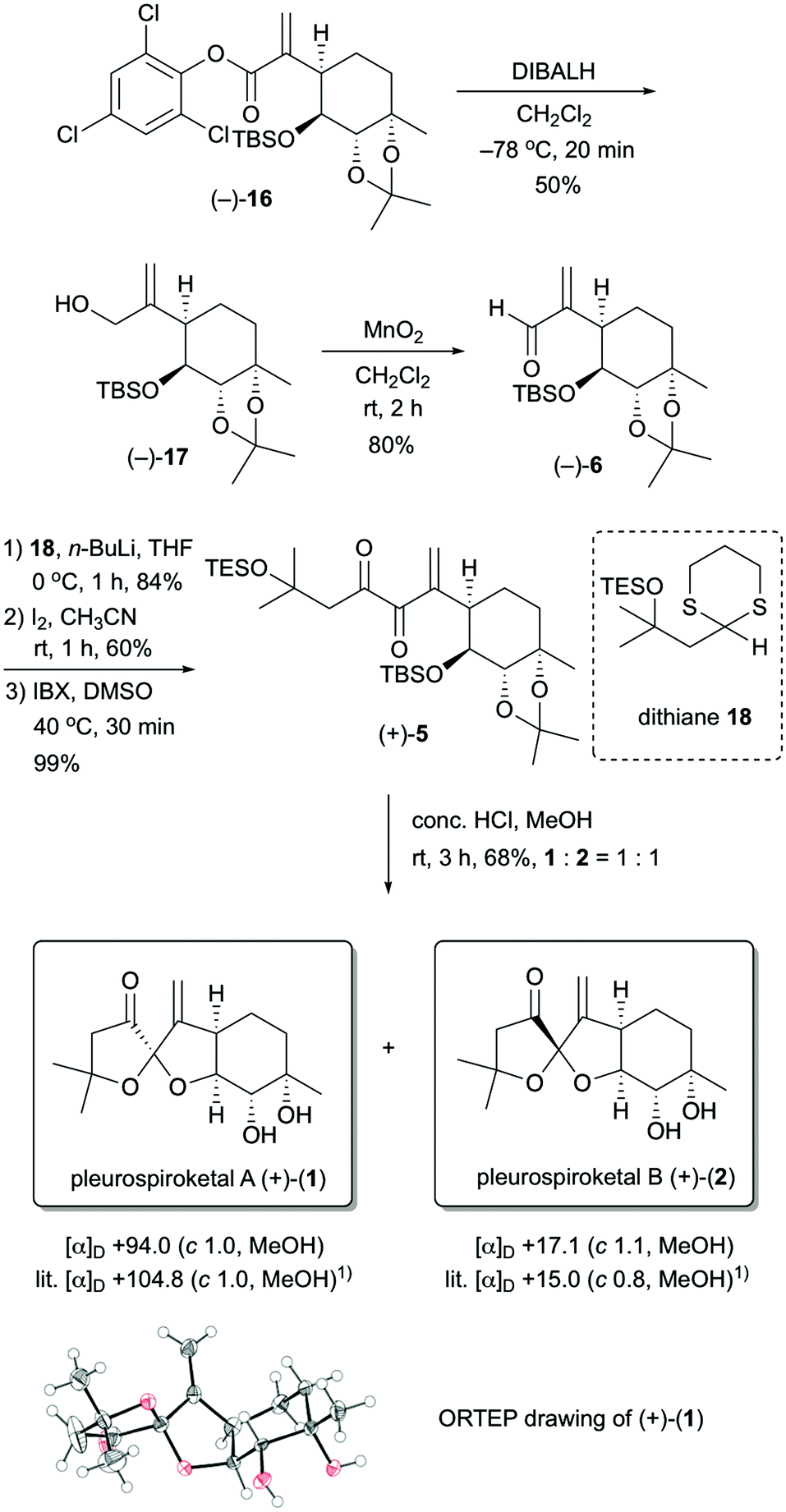 | ||
| Scheme 4 Asymmetric synthesis of pleurospiroketals A (1) and B (2). | ||
In conclusion, the first asymmetric total synthesis of pleurospiroketals A (1) and B (2) was accomplished in 16 steps from known carboxylic acid 11. This synthesis featured the highly syn-selective Evans aldol reaction of compound (+)-10 with acrolein, the synthesis of cyclohexenol derivative (−)-8 by ring-closing metathesis of Evans aldol adduct (+)-9, the highly diastereoselective dihydroxylation of compound (−)-7 and the acid-mediated spiroketalization of diketone (+)-5. Our methodology can be extended to the synthesis of other pleurospiroketals and structurally related terpenoids. Further investigations are now in progress in our laboratory.
This work was supported by the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S.-J. Wang, L. Bao, J.-J. Han, Q.-X. Wang, X.-L. Yang, H.-A. Wen, L.-D. Guo, S.-J. Li, F. Zhao and H.-W. Liu, J. Nat. Prod., 2013, 76, 45 CrossRef PubMed.
- Q.-Q. Tao, K. Ma, L. Bao, K. Wang, J.-J. Han, W.-Z. Wang, J.-X. Zhang, C.-Y. Huang and H.-W. Liu, Planta Med., 2016, 82, 639 CrossRef PubMed.
- (a) L. Y. Liu, Z. H. Li and J. K. Liu, Chin. J. Nat. Med., 2013, 11, 71 CrossRef; (b) S. J. Wang, L. Bao, F. Zhao, Q. X. Wang, S. J. Li, J. W. Ren, L. Li, H. Wen, L. D. Guo and H. W. Liu, J. Agric. Food Chem., 2013, 61, 5122 CrossRef PubMed.
- T. Kobayashi, M. Ishida, K. Imaida, H. Abe and H. Ito, Tetrahedron Lett., 2017, 58, 3294 CrossRef.
- S. Rashid, B. A. Bhat and G. Mehta, Tetrahedron Lett., 2017, 58, 3800 CrossRef.
- E. E. Lee and T. Rovis, Org. Lett., 2008, 10, 1231 CrossRef PubMed.
- (a) D. A. Evans, J. V. Nelson, E. Vogel and R. Taber, J. Am. Chem. Soc., 1981, 103, 3099 CrossRef; (b) S. Kriening, A. Evagelou, B. Claasen, A. Baro and S. Laschat, Eur. J. Org. Chem., 2014, 6720 CrossRef.
- For reviews, see: (a) A. Gradillas and J. Pérez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086 CrossRef PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490 CrossRef PubMed; (c) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef PubMed; (d) R. H. Grubbs, Tetrahedron, 2004, 60, 7117 CrossRef.
- (a) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 5370 CrossRef PubMed; (b) H. Konishi, T. Ueda and K. Manabe, Org. Synth., 2015, 90, 39 Search PubMed.
- CCDC 1858657 contains the supplemental crystallographic data of compound (−)-16†.
- H. D. Flack, Acta Crystallogr., 1983, A39, 876 CrossRef.
- The synthetic procedure of dithiane derivative 18 is described in the ESI†.
- In the case of using manganese dioxide, compound (+)-5 was obtained in 10–20% yield, and when Parikh−Doering oxidation was carried out, a complex mixture was obtained.
- CCDC 1858659 contains the supplemental crystallographic data of compound (+)-1†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1858657 and 1858659. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc06185h |
| This journal is © The Royal Society of Chemistry 2018 |