
Visible-light photocatalytic bicyclization of β-alkynyl propenones for accessing diastereoenriched syn-fluoren-9-ones†
Zheng-Jia
Shen
,
Hao-Nan
Shi
,
Wen-Juan
Hao
*,
Shu-Jiang
Tu
 * and
Bo
Jiang
*
* and
Bo
Jiang
*
School of Chemistry & Materials Science, Jiangsu Normal University, Xuzhou, 221116, P. R. China. E-mail: jiangchem@jsnu.edu.cn; laotu@jsnu.edu.cn
First published on 28th August 2018
Abstract
A novel visible-light photocatalytic bicyclization of β-alkynyl propenones with α-bromocarbonyls for highly diastereoselective synthesis of richly decorated syn-fluoren-9-ones is described. The reaction proceeds via a radical-triggered 5-exo-dig cyclization/1,6-H-abstraction/6-endo-trig cyclization cascade and offers a new and practical method for the assembly of 6/5/6 carbocyclic skeletons via C(sp3)–H alkenylation.
Visible light photoredox catalysis (VLPC) has been identified as a highly attractive and applicable synthetic tool for the collection of functionalized organic molecules via radical addition or cross-coupling reaction and thus has drawn considerable attention in the organic community due to its intrinsic characteristics of safety, being green and sustainability as well as availability.1–3 In particular, visible light induced cycloaddition (VLIC) reactions, such as [2+2],4 [3+2],5 [4+2]6 and [2+2+2]7 cycloaddition, have been proven to be an efficient and straightforward protocol for constructing four-, five- or six-membered cycles and polycyclic structures. For example, the group of Yoon pioneered visible light induced intramolecular [2+2] cycloaddition of bis-enones for cyclobutene synthesis.8 Later, the group of Xiao reported a visible light photocatalytic [3+2] cycloaddition of dihydroisoquinoline esters with activated alkenes toward pyrrolo[2,1-a]isoquinolines.9 Subsequently, the group of Yoon described an unusual radical cation Diels–Alder cycloaddition between electron-rich dienophiles with 1,3-butadienes by means of visible light photocatalysis.10 The same group then developed photoredox [2+2+2] cycloadditions of bis-styrenes with molecular oxygen, affording endoperoxides with high stereoselectivity.11 Moreover, Li's group achieved visible-light photocatalytic [2+2+2] cycloadditions of 1,6-enynes with arylsulfonyl chloride to access benzo[b]fluorenes.12 Recently, Xia and co-workers established a visible light photocatalytic bicyclization of 1,7-enynes with α-bromo diethyl malonates for the synthesis of cyclopenta[c]quinolines.13 Despite these significant achievements in this field, the development of new and facile visible light induced cyclizations toward installing functionalized cyclic structures is still highly desirable.
Over the years, α-allyl-α-bromocarbonyls have been found to be highly reactive reactants that are endowed with both a radical donor and a radical acceptor, thus serving as versatile building blocks for many important targets of chemical and biomedical potential.14 A great amount of cascade reactions triggered by α-allyl-α-bromocarbonyls under redox conditions have been developed, enabling radical addition-cyclizations for the synthesis of important polycyclic structures.15 Recently, Zhu's group presented an efficient visible-light photoredox-catalyzed bicyclization cascade of α-allyl-α-bromocarbonyls with 2-ethynylaldehyde hydrazones for the synthesis of various tetrahydro-2H-cyclopenta[a]naphthalenes (Scheme 1a).16 However, visible light induced bicyclization of α-allyl-α-bromocarbonyls together with their C(sp3)–H bond functionalization via 1,6-H abstraction, to the best of our knowledge, has been virtually unexplored. On the basis of the above literature survey and in continuation of our interest in radical cyclization,17 we reasoned that the preformed β-alkynyl propenones 1 were a good radical acceptor and employed them to react with α-allyl-α-bromocarbonyls to afford polyfunctionalized fluoren-9-ones via photocatalytic bicyclization. Interestingly, the reactions could work well, providing a wide range of richly decorated syn-fluoren-9-ones 3 containing two quaternary stereocenters with generally good yields and high diastereoselectivity (Scheme 1b). This reaction protocol enables successive C–C formation through C(sp3)–H bond cleavage under the neutral–redox conditions by using α-bromocarbonyls as twice radial donors, with the advantage of atom economy and extreme convergence. Here, we report these attractive transformations.
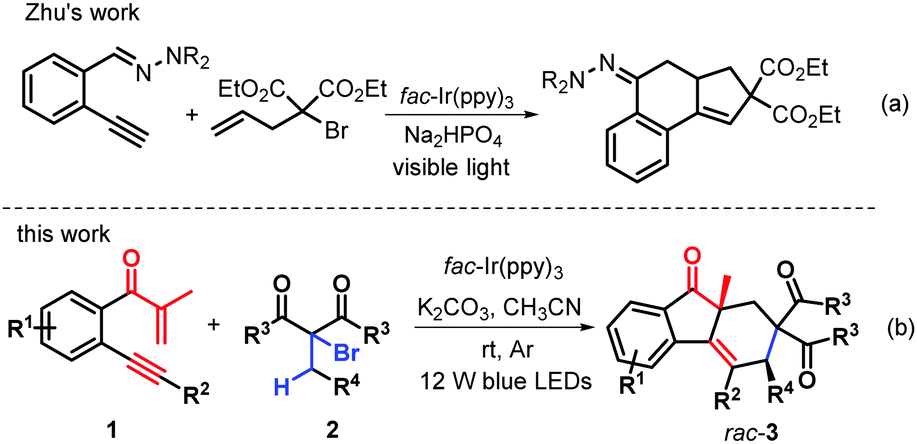 | ||
| Scheme 1 Profiles of photoinduced transformations of α-bromocarbonyls. | ||
At the outset of our studies, β-alkynyl propenone 1a as a model substrate was subjected to the reaction of diethyl α-isopentenyl-α-bromomalonate (2a) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mole ratio. As depicted in Table 1, the reaction proceeded smoothly in CH3CN in the presence of 2.0 equivalents of K2CO3 at room temperature by using 1.0 mol% of fac-Ir(ppy)3 as a photocatalyst under argon conditions and visible light irradiation (blue LEDs), enabling radical addition–cyclization to give the expected syn-fluoren-9-one 3a as a sole diastereoisomer in 51% yield (Table 1, entry 1). The stereo-structure of product 3a was assigned by 1H NMR and X-ray diffraction analysis (see the ESI†), and those of other products were assigned by analogy. The optimization of the reaction conditions was then conducted by examining visible-light photocatalysts often used in the photocatalysis, such as Ir[dF(CF3)ppy]2(dtbpy)PF6, [Ru(bpy)3](PF6)2, Ru(bpy)3Cl2·6H2O, and eosin Y, indicating that all these photocatalysts completely suppressed the reaction process (entries 2–5). Fine-tuning the amount of K2CO3 to 1.0 equivalent facilitated this photocatalysis process, delivering syn-product 3a in a higher yield of 61% (entry 6). Next, the investigation of the solvent effect revealed that other solvents, such as CH3OH, toluene, tetrahydrofuran (THF), and 1,2-dichloroethane (DCE), proved to be less effective as compared with CH3CN (entries 7–10). After systematic screening of various inorganic bases, such as Cs2CO3, K3PO4, and KOH, it was found that these tested bases were all inferior in terms of reaction yields (entries 11–13). Without visible light or fac-Ir(ppy)3 photocatalyst, the above reaction did not work (entries 14 and 15). Moreover, a lower conversion of 1a into 3a was observed when the identical reaction was carried out under air conditions (entry 16).
:2 mole ratio. As depicted in Table 1, the reaction proceeded smoothly in CH3CN in the presence of 2.0 equivalents of K2CO3 at room temperature by using 1.0 mol% of fac-Ir(ppy)3 as a photocatalyst under argon conditions and visible light irradiation (blue LEDs), enabling radical addition–cyclization to give the expected syn-fluoren-9-one 3a as a sole diastereoisomer in 51% yield (Table 1, entry 1). The stereo-structure of product 3a was assigned by 1H NMR and X-ray diffraction analysis (see the ESI†), and those of other products were assigned by analogy. The optimization of the reaction conditions was then conducted by examining visible-light photocatalysts often used in the photocatalysis, such as Ir[dF(CF3)ppy]2(dtbpy)PF6, [Ru(bpy)3](PF6)2, Ru(bpy)3Cl2·6H2O, and eosin Y, indicating that all these photocatalysts completely suppressed the reaction process (entries 2–5). Fine-tuning the amount of K2CO3 to 1.0 equivalent facilitated this photocatalysis process, delivering syn-product 3a in a higher yield of 61% (entry 6). Next, the investigation of the solvent effect revealed that other solvents, such as CH3OH, toluene, tetrahydrofuran (THF), and 1,2-dichloroethane (DCE), proved to be less effective as compared with CH3CN (entries 7–10). After systematic screening of various inorganic bases, such as Cs2CO3, K3PO4, and KOH, it was found that these tested bases were all inferior in terms of reaction yields (entries 11–13). Without visible light or fac-Ir(ppy)3 photocatalyst, the above reaction did not work (entries 14 and 15). Moreover, a lower conversion of 1a into 3a was observed when the identical reaction was carried out under air conditions (entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Base (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (0.8 mmol), cat. (1.0 mol%), base (x equiv.), solvent (6.0 mL), 12 W blue LED light, under Ar conditions. b Isolated yield based on 1a. c In the dark. d In the air. | ||||
| 1 | fac-Ir(ppy)3 | K2CO3 (2.0) | CH3CN | 51 |
| 2 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | K2CO3 (2.0) | CH3CN | N.R. |
| 3 | [Ru(bpy)3](PF6)2 | K2CO3 (2.0) | CH3CN | N.R. |
| 4 | Ru(bpy)3Cl2·6H2O | K2CO3 (2.0) | CH3CN | N.R. |
| 5 | Eosin Y | K2CO3 (2.0) | CH3CN | N.R. |
| 6 | fac-Ir(ppy)3 | K2CO3 (1.0) | CH3CN | 61 |
| 7 | fac-Ir(ppy)3 | K2CO3 (1.0) | CH3OH | Trace |
| 8 | fac-Ir(ppy)3 | K2CO3 (1.0) | Toluene | 35 |
| 9 | fac-Ir(ppy)3 | K2CO3 (1.0) | THF | 30 |
| 10 | fac-Ir(ppy)3 | K2CO3 (1.0) | DCE | 33 |
| 11 | fac-Ir(ppy)3 | Cs2CO3 (1.0) | CH3CN | 20 |
| 12 | fac-Ir(ppy)3 | K3PO4 (1.0) | CH3CN | 24 |
| 13 | fac-Ir(ppy)3 | KOH (1.0) | CH3CN | N.D. |
| 14c | fac-Ir(ppy)3 | K2CO3 (1.0) | CH3CN | N.R. |
| 15 | — | K2CO3 (1.0) | CH3CN | N.R. |
| 16d | fac-Ir(ppy)3 | K2CO3 (1.0) | CH3CN | 33 |
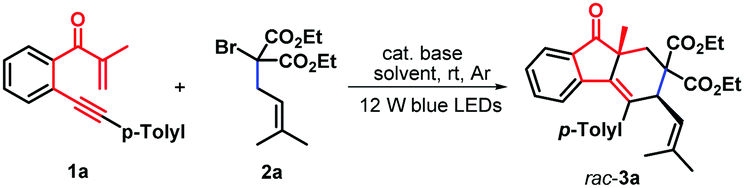
Having established the optimal reaction conditions, we next systematically investigated the scope of the photoinduced bicyclization reaction with an assortment of β-alkynyl propenone components (Scheme 2). The survey showed that the iridium-catalyzed protocol is feasible with a wide variety of β-alkynyl propenone derivatives under visible-light irradiation furnishing the corresponding cycloaddition products with good yields and excellent diastereoselectivity. At first, β-alkynyl propanones with diverse functionalities in the arylalkynyl (R2) moiety were examined in combination with diethyl α-isopentenyl-α-bromomalonate 2a. Substituents with either electronically rich, neutral, or poor properties in the arene ring completely oriented the diastereoselectivity to the formation of syn-fluoren-9-ones 3b–3g with 58–68% yields. Functional groups such as ethyl (1b), methoxy (1c, PMP = p-methoxyphenyl), fluoride (1e) chloride (1f), and bromide (1g) were well tolerated under the standard conditions Alternatively, a heterocyclic 2-thienyl analogue was proven to be effective, enabling a similar radical-induced bicyclization process to give 2-thienyl substituted syn-fluoren-9-one 3h in 59% yield and complete diastereoselectivity. The representative methyl group located at the 4-position of the internal arene ring was compatible with these photocatalytic conditions and the corresponding syn-products 3i–3n were obtained in satisfactory yields. Similarly, dimethyl α-allyl-α-bromomalonate 2b was found to show high reactivity, leading to the formation of diastereoenriched fluoren-9-ones 3o–3y with yields ranging from 56% to 66%.
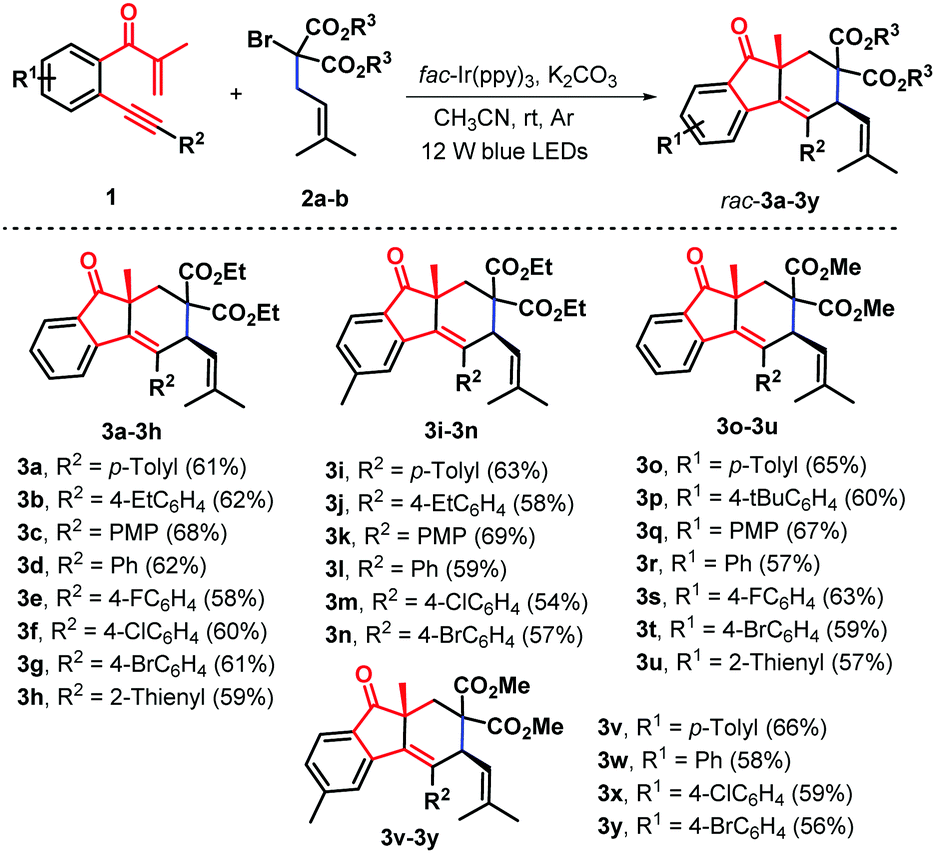 | ||
| Scheme 2 Synthesis of syn-fluoren-9-ones 3a–3y. (i) Reaction conditions: 1 (0.4 mmol), 2a–b (0.8 mmol), fac-Ir(ppy)3 (1.0 mol%), K2CO3 (1.0 equiv.) and CH3CN (6.0 mL) in the sealed reaction tube under Ar conditions at room temperature for 24.0 hours. (ii) Isolated yields in brackets based on 1. | ||
To expand the utility of this methodology, we turned our attention to exploring the feasibility of other substituted α-bromomalonates including allyl (2c and 2d), isobutenyl (2e), isobutyl (2f), and ethyl (2g). As anticipated, the reaction of b-alkynyl propenones 1 with α-bromomalonates 2c–2f under the standard conditions offered structurally diverse syn-fluoren-9-ones 3aa–3nn as sole diastereoisomers in generally good yields (Scheme 3). β-Alkynyl propenones 1 possessing electron-donating, electron-neutral, and electron-withdrawing groups linked to both the internal arene ring (R1) and the arylalkynyl (R2) moiety did not hamper the reaction process. Notably, regarding the scope of α-bromomalonates, besides the isopentenyl substrate, the allyl (2c and 2d), isobutenyl (2e) and isobutyl (2f) analogue could be successfully engaged in the current photocatalysis. Unluckily, on replacing the allyl group with an ethyl group on the malonate unit, α-bromomalonate 2g was an ineffective reaction partner for this photocatalysis (Scheme 3, 3oo), which may be ascribed to the relative instability of the methylene radical intermediate D, generated in situ from C-centered radical triggered 1,6-H- abstraction, due to its weak captodative effect18 (Scheme 5). It is noteworthy that the current photocatalytic protocol represents a new and practical pathway for the creation of highly diastereoenriched syn-fluoren-9-ones 3 through an Ir-catalyzed bicyclization cascade involving radical-triggered C(sp3)–H bond alkenylation.
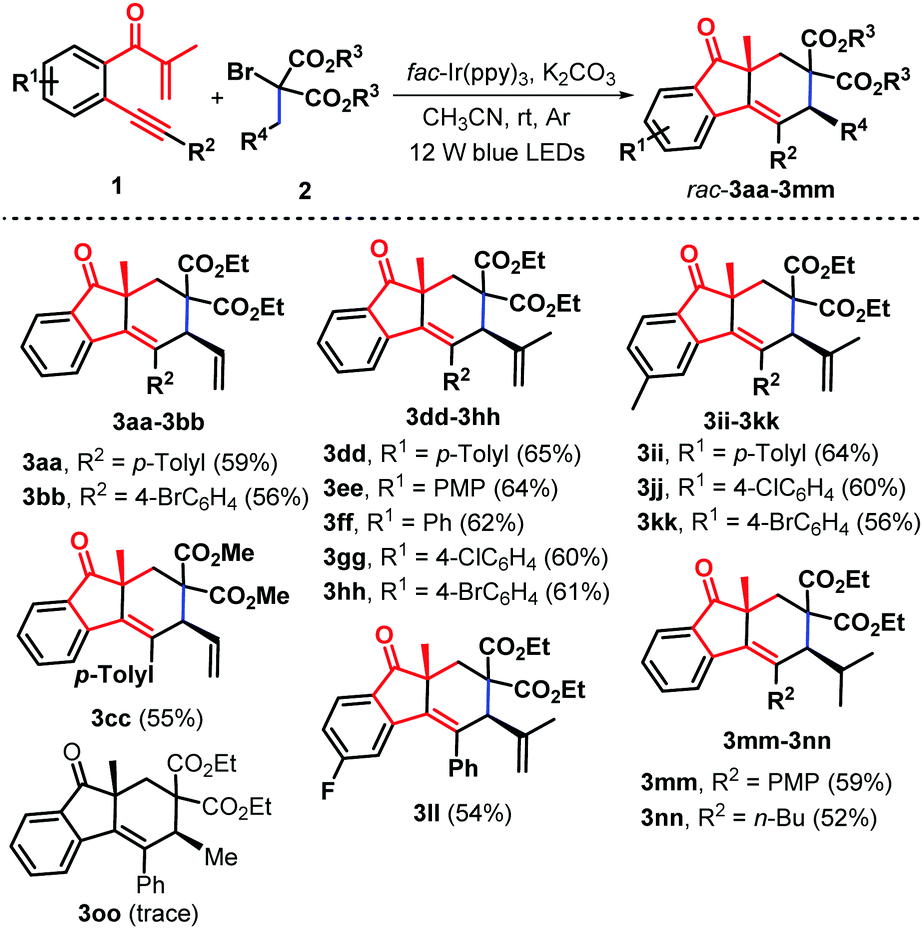 | ||
| Scheme 3 Synthesis of syn-fluoren-9-ones 3aa–3oo. (i) Reaction conditions: 1 (0.4 mmol), 2c–g (0.8 mmol), fac-Ir(ppy)3 (1.0 mol%), K2CO3 (1.0 equiv.) and CH3CN (6.0 mL) in the sealed reaction tube under Ar conditions at room temperature for 24.0 hours. (ii) Isolated yields in brackets based on 1. | ||
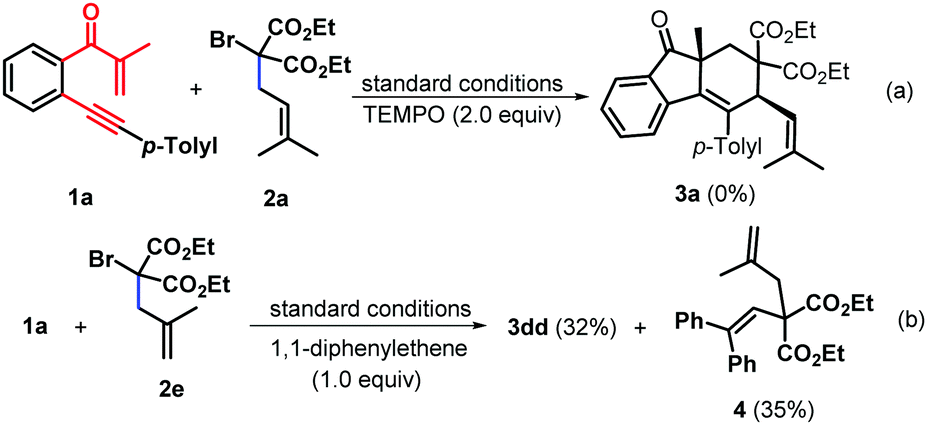
To gain further mechanistic insight into this mechanism, a preliminary controlled experiment was conducted. When 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as the radical scavenger was added into the reaction system under the standard conditions, the reaction was substantially inhibited and the starting material 1a was almost completely recovered, indicating that this transformation may include a single electron transfer (SET) process (Scheme 4a). Then, 1,1-diphenylethylene was subjected to the reaction of 1a with 2e under the standard conditions, and the product 3dd and diethyl 2-(2,2-diphenylvinyl)-2-(2-methylallyl)malonate 4 were isolated in 32% and 35% yields, respectively, suggesting that the transformation could involve the in situ-generation of a C-centered radical under visible-light photocatalytic conditions (Scheme 4b).
 | ||
| Scheme 4 Synthetic utility of the methodology. | ||
Based on the above results and literature survey,14–16 a reasonable mechanism for the bicyclization reaction was proposed in Scheme 5. Initially, visible light induces [fac-IIIIr(ppy)3] to the excited state [fac-IIIIr(ppy)3]*, which reduces α-allyl-α-bromomalonates 2 to generate a C-centered radical A along with [fac-IVIr(ppy)3] via single electron transfer (SET).19 Subsequently, radical addition of intermediate A into the C–C double bond of β-alkynyl propenones 1 yields quaternary carbon radical B, followed by 5-exo-dig cyclization and 1,6-H atom transfer (HAT)20 to afford the radical intermediate D, which undergoes 6-endo-trig cyclization to give trans-intermediate E due to the steric effect. At this stage, a key oxidization (SET) step between E and [fac-IVIr(ppy)3] occurs, thus regenerating the photocatalyst and the cation trans-intermediate F. Further deprotonation of intermediate F would give the final syn-products 3.
 | ||
| Scheme 5 Plausible reaction pathways. | ||
In summary, starting from readily available β-alkynyl propenones and α-bromomalonates, a new and visible-light photocatalytic bicyclization cascade was established, by which this photocatalysis completely oriented the high diastereoselectivity to assemble a wide range of structurally diverse syn-fluoren-9-ones with generally good yields. The present photocatalytic strategy accommodates a broad substrate scope and is distinguished by its mild conditions and high functional group compatibility. The reaction pathway including a 5-exo-dig cyclization/1,6-HAT/6-endo-trig cyclization sequence was proposed, resulting in the formation of up to three new C–C bonds and two new rings through radical-induced C(sp3)–H bond cleavage. Further application of the photocatalysis to bioactive polycycles is currently underway in our laboratory.
We are grateful for financial support from the NSFC (No. 21472071 and 21602087), PAPD of Jiangsu Higher Education Institutions, the Outstanding Youth Fund of JSNU (YQ2015003), NSF of Jiangsu Province (BK20160212), and the Qing Lan Project of Jiangsu Education Committee.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews on visible-light photoredox catalysis, see: (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) F. Teply and C. Czech, Chem. Commun., 2011, 76, 859 Search PubMed; (e) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef PubMed.
- For recent reviews on visible light photoredox catalysis, see: (a) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (b) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef PubMed.
- For other selected papers, see: (a) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef PubMed; (b) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef PubMed; (c) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef PubMed; (d) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Joergensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef PubMed; (e) G.-Q. Xu, J.-T. Xu, Z.-T. Feng, H. Liang, Z.-Y. Wang, Y. Qin and P.-F. Xu, Angew. Chem., Int. Ed., 2018, 57, 5110 CrossRef PubMed; (f) X. Huang, J. Lin, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454 CrossRef PubMed; (g) M.-M. Li, Y. Wei, J. Liu, H.-W. Chen, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 14707 CrossRef PubMed.
- For selected recent works about photocatalytic [2+2] cycloaddition reactions, see: (a) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604 CrossRef PubMed; (b) M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572 CrossRef PubMed; (c) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef PubMed; (d) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 1 CrossRef; (e) M. A. Ischay, M. S. Ament and T. P. Yoon, Chem. Sci., 2012, 3, 2807 RSC; (f) A. E. Hurtley, Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 8991 CrossRef PubMed.
- For selected recent works about photocatalytic [2+3] cycloaddition reactions, see: (a) Z. Lu, M. Shen and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 1162 CrossRef PubMed; (b) J. Xuan, X.-D. Xia, T. Zeng, Z.-J. Feng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 126, 5759 CrossRef; (c) S. Maity, M.-Z. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222 CrossRef PubMed; (d) T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 11056 CrossRef PubMed; (e) M. Rueping, D. Leonori and T. Poisson, Chem. Commun., 2011, 47, 9615 RSC.
- For selected recent works about photocatalytic [2+4] cycloaddition reactions, see: (a) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC; (b) Z.-G. Yuan, Q. Wang, A. Zheng, K. Zhang, L.-Q. Lu, Z. Tang and W.-J. Xiao, Chem. Commun., 2016, 52, 5128 RSC; (c) T.-B. Xiao, X.-C. Dong, Y.-C. Tang and L. Zhou, Adv. Synth. Catal., 2012, 354, 3195 CrossRef; (d) C. Wiegand, E. Herdtweck and T. Bach, Chem. Commun., 2012, 48, 10195 RSC.
- For selected recent reviews about photocatalytic [2+2+2] cycloaddition reactions, see: (a) V. Gandon, C. Aubert and M. Malacria, Chem. Commun., 2006, 2209 RSC; (b) G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2011, 40, 3430 RSC; (c) N. Weding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525 RSC.
- M. A. Ischay, M. E. Anzoino, J. Dum and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef PubMed.
- Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171 CrossRef PubMed.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350 CrossRef PubMed.
- J. D. Parrish, M. A. Ischay, Z. Lu, S. Guo, N. R. Peters and T. P. Yoon, Org. Lett., 2012, 14, 1640 CrossRef PubMed.
- G.-B. Deng, Z.-Q. Wang, J.-D. Xia, P.-C. Qian, R.-J. Song, M. Hu, L.-B. Gong and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 1535 CrossRef PubMed.
- F. Gao, C. Yang, N. Ma, G.-L. Gao, D. Li and W.-J. Xia, Org. Lett., 2016, 18, 600 CrossRef PubMed.
- (a) C. Che, Q. Huang, H. Zheng and G. Zhu, Chem. Sci., 2016, 7, 4134 RSC; (b) N. Zhou, Y.-X. Cheng, J. Xie and C.-J. Zhu, Chem. Commun., 2017, 53, 10707 RSC; (c) M.-H. Huang, W.-J. Hao and B. Jiang, Chem. – Asian J., 2018 DOI:10.1002/asia.201801119.
- (a) D. Lu, Y. Wan, L. Kong and G. Zhu, Chem. Commun., 2016, 52, 13971 RSC; (b) H. Zhu, X. Nie, Q. Huang and G. Zhu, Tetrahedron Lett., 2016, 57, 2331 CrossRef; (c) D. Lu, Y. Wan, L. Kong and G. Zhu, Org. Lett., 2017, 19, 2929 CrossRef PubMed; (d) W. Jin, Y. Zhou, Y. Zhao, Q. Ma, L. Kong and G. Zhu, Org. Lett., 2018, 20, 1435 CrossRef PubMed; (e) Y. Yu and U. K. Tambar, Chem. Sci., 2015, 6, 2777 RSC; (f) A. A. S. Gietter, P. G. Gildner, A. P. Cinderella and D. A. Watson, Org. Lett., 2014, 16, 3166 CrossRef PubMed; (g) R. Ding, Z.-D. Huang, Z.-L. Liu, T.-X. Wang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2016, 52, 5617 RSC; (h) Y. Noda and T. Nishikata, Chem. Commun., 2017, 53, 5017 RSC; (i) B. Liu, J.-X. Yu, Y. Li, J.-H. Li, D.-L. He, B. Liu, J.-X. Yu, Y. Li, J.-H. Li and J.-H. Li, Org. Lett., 2018, 20, 2129 CrossRef PubMed.
- N. Zhou, J. Liu, Z.-F. Yan, Z.-K. Wu, H.-L. Zhang, W.-P. Li and C.-J. Zhu, Chem. Commun., 2017, 53, 2036 RSC.
- (a) J.-K. Qiu, B. Jiang, Y.-L. Zhu, W.-J. Hao, D.-C. Wang, J. Sun, P. Wei, S.-J. Tu and G. Li, J. Am. Chem. Soc., 2015, 137, 8928 CrossRef PubMed; (b) J. Sun, J.-K. Qiu, Y.-N. Wu, W.-J. Hao, C. Guo, G. Li, S.-J. Tu and B. Jiang, Org. Lett., 2017, 19, 754 CrossRef PubMed; (c) J. Li, W.-W. Zhang, X.-J. Wei, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Org. Lett., 2017, 19, 4512 CrossRef PubMed; (d) F. Liu, J.-Y. Wang, P. Zhou, G.-G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570 CrossRef PubMed; (e) Z.-J. Shen, Y.-N. Wu, C.-L. He, L. He, W.-J. Hao, A.-F. Wang, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 445 RSC; (f) M.-H. Huang, Y.-L. Zhu, W.-J. Hao, A.-F. Wang, D.-C. Wang, F. Liu, P. Wei, S.-J. Tu and B. Jiang, Adv. Synth. Catal., 2017, 359, 2229 CrossRef.
- (a) H. Viehe, Z. Janousek and R. Merenyi, Acc. Chem. Res., 1985, 18, 148 CrossRef; (b) Y.-L. Zhu, C.-F. Zhu, P. Zhou, W.-J. Hao, D.-C. Wang, S.-J. Tu and B. Jiang, J. Org. Chem., 2018 DOI:10.1021/acs.joc.8b00994.
- M.-H. Huang, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Chem. Commun., 2018 10.1039/C8CC04618B.
- H. Zhu, J. C. T. Leung and G. M. Sammis, J. Org. Chem., 2015, 80, 965 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1850405 (3a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc06086j |
| This journal is © The Royal Society of Chemistry 2018 |