
Total synthesis of conosilane A via a site-selective C–H functionalization strategy†
Ziyun
Yuan
a,
Xiaojun
Hu
a,
Hao
Zhang
a,
Lin
Liu
a,
Peng
Chen
a,
Min
He
a,
Xingang
Xie
 a,
Xiaolei
Wang
a and
Xuegong
She
*ab
a,
Xiaolei
Wang
a and
Xuegong
She
*ab
aState Key Laboratory of Applied Organic Chemistry, Department of Chemistry, Lanzhou University, Lanzhou, 730000, China. E-mail: shexg@lzu.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin, 30071, China
First published on 2nd January 2018
Abstract
The first total synthesis of conosilane A, a densely oxygenated nonisoprenoid sesquiterpene, has been achieved through stereoselective intramolecular radical cyclization and double utilization of site-selective C–H functionalization as key steps, thus simplifying the synthetic endeavors in natural product synthesis.
Members of the genus Conocybe are a rich source of biologically active natural products. In the past ten years, a series of novel tremulane-type sesquiterpenes from the culture of mushroom C. siliginea have been reported by Liu and co-workers.1 Among these, conosilane A, a novel highly oxygenated nonisoprenoid sesquiterpene2 with an unprecedented carbon skeleton, was isolated from the cultures of the basidiomycete Conocybe siliginea in 2012.1d The plausible biogenetic pathway for the synthesis of conosilane A was that nature uses its two phases (cyclase and oxidase phase)3 to synthesize the natural product (Scheme 1), and its structure was elucidated by extensive spectroscopic methods and further confirmed by single crystal X-ray diffraction analysis. Structurally, conosilane A consists of bicycle[3.3.0] frameworks bearing three continuous stereocenters including a quaternary one, and it is comprised of a unique cyclopentenone fused with both a cyclohexenone unit and a tetrahydrofuran moiety.
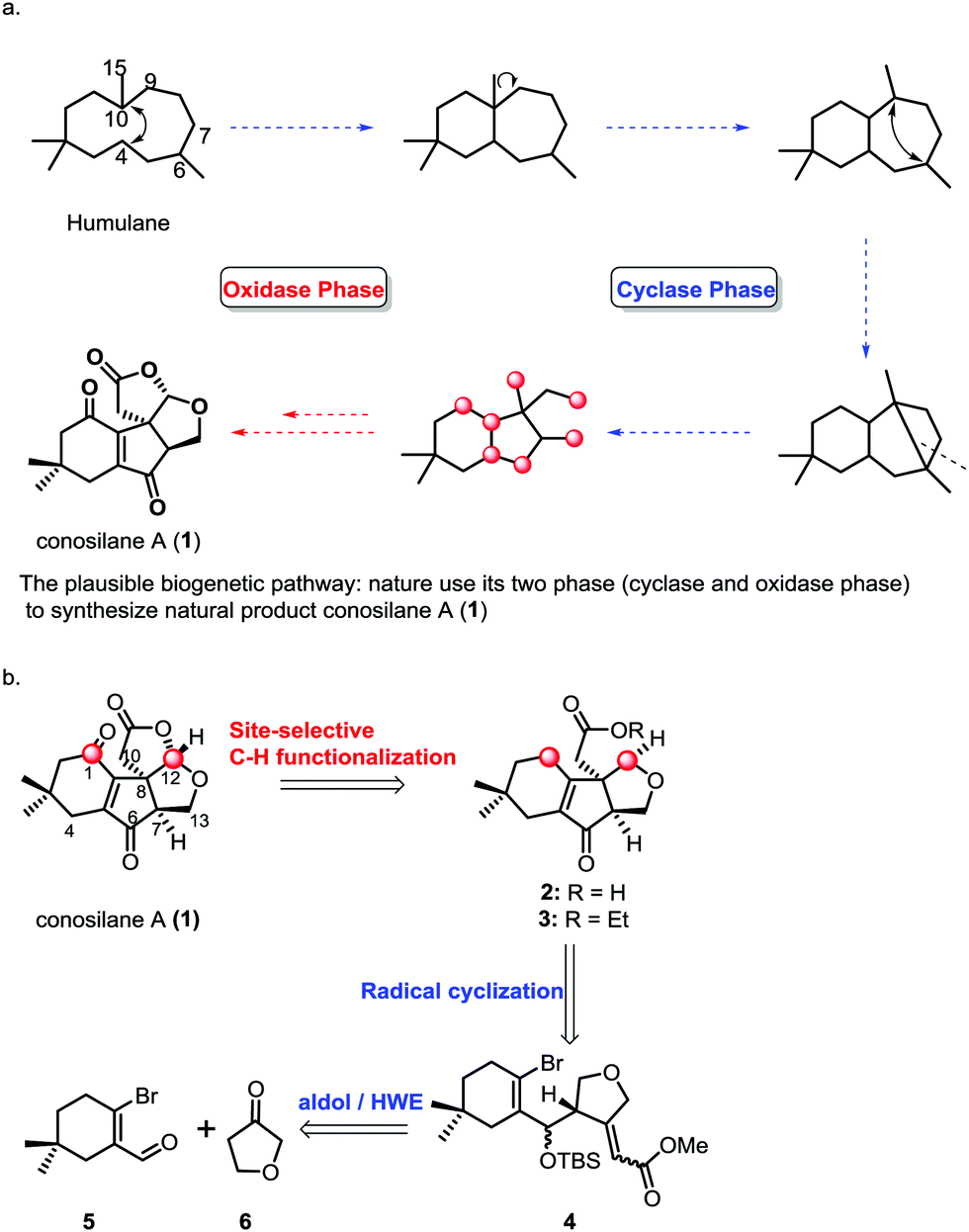 | ||
| Scheme 1 Retrosynthetic analysis of conosilane A (1). | ||
Due to our long-term interest in the total syntheses of sesquiterpenes4 with a novel skeleton, the impressive structural complexity together with potential bioactivities has rendered conosilane A as an attractive target for our synthetic endeavor. Herein, we reported the first total synthesis of conosilane A in a stereocontrol manner.
C–H functionalization,3,5 especially, the C–H oxidation has been gaining tremendous interest in organic syntheses since it can dramatically increase synthetic efficiency in terms of step, atom and redox economy when incorporated into retrosynthetic analyses of complex natural products.6 Logically, conosilane A (1) (Scheme 1) was envisioned to ultimately arise from a late-stage C–H (C1–H and C12–H) oxidation of 2 or 3 to install the 1, 4-dienone unit and the bicycle[3.3.0] framework, respectively. 3 could be achieved from 4 by a 5-exo-trig radical cyclization reaction to obtain a unique cyclopentenone core with the C-8 quaternary stereocenter and subsequential functional group interconversion. Finally, the essential precursor α,β-unsaturated ester 4 could be regioselectively prepared from known aldehyde 57 and ketone 6 followed by the Horner–Wadsworth–Emmons reaction.
Our synthetic study commenced with the coupling reaction of the known compound aldehyde 5 and ketone 6 (Scheme 2). Inspired by the work of Kraus,8 the predominant kinetic enolate prepared from 6 and lithium diisopropyl amide (LDA) reacted with 5 to afford the desired hydroxyl ketone 7, presumably via the kinetic deprotonation of the non-oxygenated methylene of the tetrahydrofuran ring and subsequential nucleophilic addition using the Zimmerman–Traxler principle. As an anticipated process, 8 was accessed by a sequential regioselective aldol reaction and TBS-protection in 62% yield over two steps (anti/syn = 2.8/1).9 Subsequently, the condensation of anti-8 with ethyl (diethoxyphosphoryl)acetate via a Horner–Wadsworth–Emmons reaction10 generated the chromatographically inseparable α,β-unsaturated ester 4 in 82% combined yields with (Z)-configuration of C8–C10 double bond as the major isomer. Notably, compound 4 was light and base-sensitive11 since the ratio of aromatization by-product 9 increased when the reaction was conducted at higher temperature such as 0 °C or room temperature.
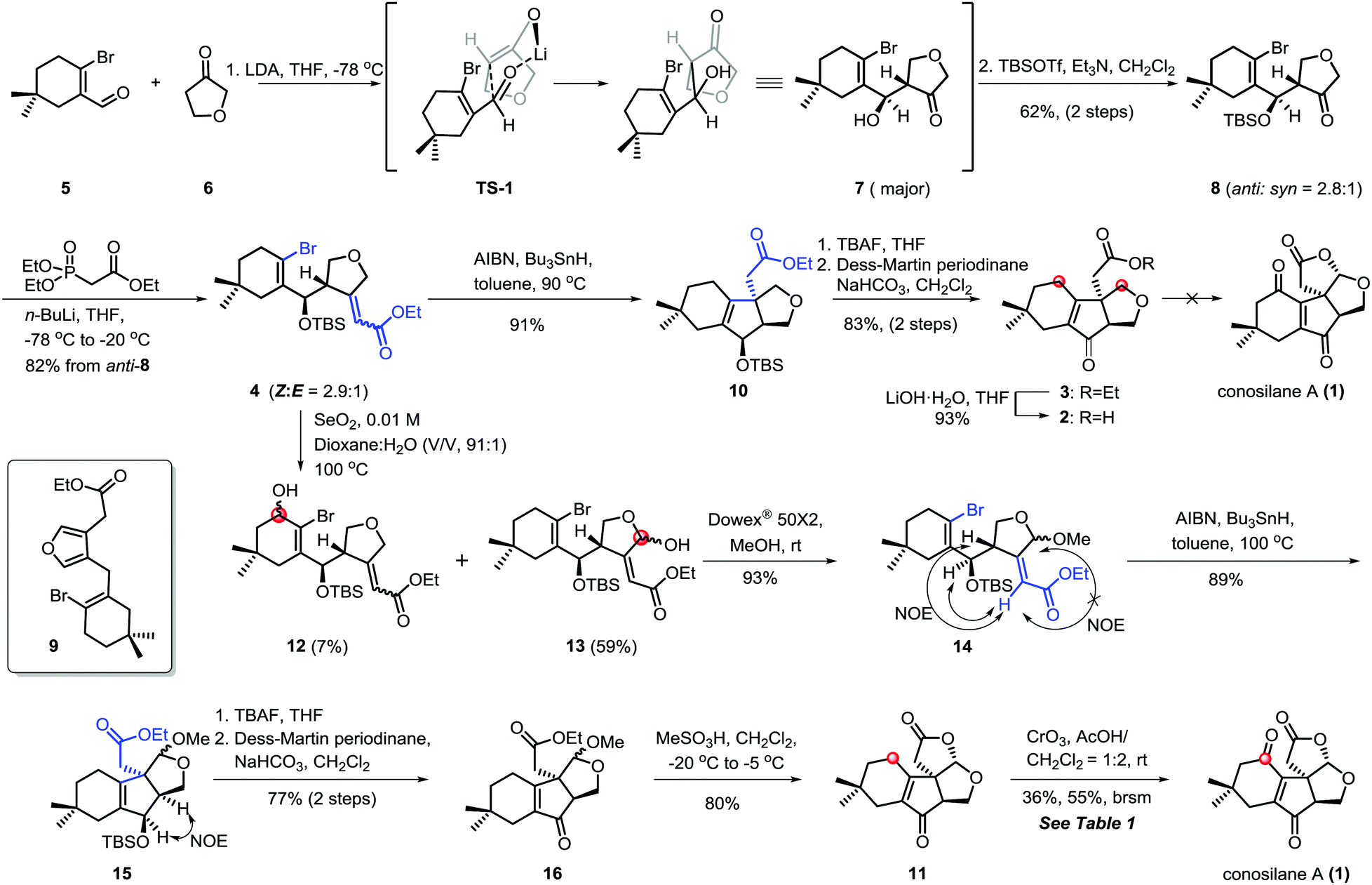 | ||
| Scheme 2 Synthetic efforts toward conosilane A using the late-stage C–H functionalization/cyclization strategy. | ||
Having developed an approach capable of efficiently yielding 4 with all carbon atoms for natural conosilane A, we next focused on the key cyclization reaction to furnish the essential cyclopentenone skeleton. Initially, we attempted the [Ni(cod)2]-mediated cyclization under typical reaction conditions12 and improved conditions developed by the Ma group.13 We could recover only the starting materials under both conditions. While ester 4 was treated with the Pd-catalyzed reductive Heck reaction, a complex mixture was formed under variant conditions.10,14,15 Fortunately, ester 10 bearing a quaternary stereocenter was obtained in 91% yield via the classic radical cyclization.16 Ester 3, the precursor of the natural product conosilane A, was obtained through two simple reactions in 83% yield.
To complete the synthesis of conosilane A, a bold strategy involving late-stage C–H functionalization/cyclization was explored. To our disappointment, no desired product was detected under various conditions for substrate 2 or 3,17 including DDQ, PCC, PDC, CrO3,17i,j DMDO (prepared or in situ)17f–h and peroxide combined with metals17d,e (Scheme 2).
Due to the negative results for substrate ester 3 and acid 2, we modified our synthetic route wherein the C–H hydroxylation occurred before the key radical cyclization reaction (Scheme 2). As for ester 4, we have to deal with the site-selectivity in the presence of four allylic oxidizable positions. To our delight, we could successfully obtain the desired hemiacetal 13 with high selectivity under the SeO2 conditions. The good selectivity in allylic C–H hydroxylation might is due to the steric hindrance and the electronic effect. Ester 15 was formed when acetal 14 was subjected to the previously optimized conditions for the key radical cyclization in 89% yield. Subsequently, a two step protocol, the removal of TBS protection of 15 and oxidation of the resulting allylic alcohol with Dess–Martin periodinane, generated 16 in 77% yield. Upon treatment of 16 with methanesulfonic acid18 at low temperature, the lactonization proceeded smoothly generating lactone 11 in 80% yield which consisted of the essential tetra-cyclic framework of conosilane A.
To accomplish the synthesis of conosilane A, conditions for a late-stage and site-selective C–H oxidation was screened (Table 1). Lactone 11 was recovered in most of the conditions (entries 1–3) or converted to the desired product in a low yield (entries 4 and 5). To our delight, when lactone 11 was subjected to CrO3 in a solvent mixture (AcOH/CH2Cl2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), conosilane A was generated in a moderate yield (36%, 55% brsm) which was further structurally identified by X-ray crystallographic analysis (entry 6). However, the reaction mixture was totally decomposed under harsher conditions (entries 7 and 8).
:2), conosilane A was generated in a moderate yield (36%, 55% brsm) which was further structurally identified by X-ray crystallographic analysis (entry 6). However, the reaction mixture was totally decomposed under harsher conditions (entries 7 and 8).
|
|
||
|---|---|---|
| Entry | Conditions | Yield |
| 119 | Pd(OH)2/C, K2CO3, CH2Cl2, rt | N.R. |
| 217f | DMDO (∼0.1 M, in acetone), 0 °C to rt | N.R. |
| 317i | CrO3/3,5-DMP, CH2Cl2, −15 °C to rt | N.R. |
| 417e | RuCl3·xH2O, tBuOOH, cyclohexane, rt | <5% |
| 520 | PCC, pyridine, Celite, benzene, reflux | <5% |
| 617j | CrO3, AcOH/CH2Cl2 = 1/2, rt | 36%, 55% (brsm) |
| 717j | CrO3, AcOH, rt | Decomposed |
| 821 | K2CrO4, AcOH/Ac2O, toluene, rt to 60 °C | Decomposed |
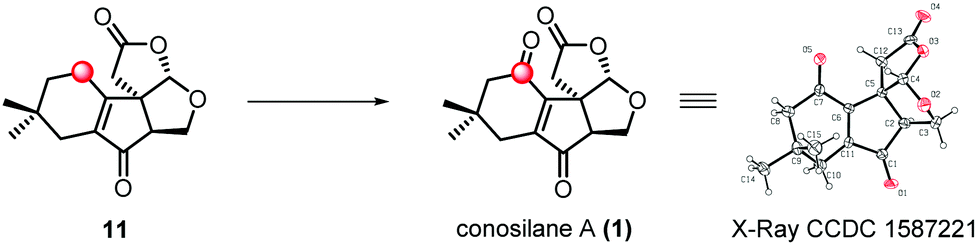
In summary, the first concise total synthesis of the nonisoprenoid sesquiterpene conosilane A was accomplished in 10 steps. The synthesis features: (1) a sequential regioselective aldol reaction and subsequent Horner–Wadsworth–Emmons reaction to assemble all the skeletal carbons, (2) a radical cyclization to stereo-specifically assemble the unique cyclopentenone core bearing a quaternary stereocenter, and (3) double manipulation of allylic C(sp3)–H functionalization to furnish the natural product. The synthetic strategies developed in the total synthesis renders the power of C–H functionalization and would be complementary for the synthetic repositories of highly oxygenated natural products.
This work was supported by the National Science Foundation of China (21372103, 21472079, 21572088 and 21732001), program 111, PCSIRT (IRT_15R28) and the Fundamental Research Funds for the Central Universities (lzujbky-2016-ct02).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D.-Z. Liu, F. Wang and J.-K. Liu, J. Nat. Prod., 2007, 70, 1503 CrossRef CAS PubMed; (b) Z.-Y. Zhou, J.-G. Tang, F. Wang, Z.-J. Dong and J.-K. Liu, J. Nat. Prod., 2008, 71, 1423 CrossRef CAS PubMed; (c) X. Yang, T. Feng, X. Yin, Z. Li, L. Zhang and J. Liu, Chin. J. Chem., 2012, 30, 1231 CrossRef CAS; (d) X.-Y. Yang, T. Feng, Z.-H. Li, Y. Sheng, X. Yin, Y. Leng and J.-K. Liu, Org. Lett., 2012, 14, 5382 CrossRef CAS PubMed.
- (a) A. Fukuzawa, M. Aye, M. Nakamura, M. Tanura and A. Murai, Tetrahedron Lett., 1990, 31, 4895 CrossRef CAS; (b) P. A. Evans, V. S. Murthy, J. D. Roseman and A. L. Rheingold, Angew. Chem., Int. Ed., 1999, 38, 3175 CrossRef CAS; (c) K. S. Madden, F. A. Mosa and A. Whiting, Org. Biomol. Chem., 2014, 12, 9029 RSC.
- The concept of two-phase synthesis was developed by P. S. Baran. For selected reviews see: (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) H. Chu, J. M. Smith, J. Felding and P. S. Baran, ACS Cent. Sci., 2017, 3, 47 CrossRef CAS PubMed.
- (a) Q. Wang, Q. Huang, B. Chen, J. Lu, H. Wang, X. She and X. Pan, Angew. Chem., Int. Ed., 2006, 45, 3651 CrossRef CAS PubMed; (b) Q. Huang, Q. Wang, J. Zheng, J. Zhang, X. Pan and X. She, Tetrahedron, 2007, 63, 1014 CrossRef CAS; (c) S. Tang, Y. Xu, J. He, Y. He, J. Zheng, X. Pan and X. She, Org. Lett., 2008, 10, 1855 CrossRef CAS PubMed; (d) X. Wang, J. Zheng, Q. Chen, H. Zheng, Y. He, J. Yang and X. She, J. Org. Chem., 2010, 75, 5392 CrossRef CAS PubMed; (e) H. Zhang, S. Ma, Z. Xing, L. Liu, B. Fang, X. Xie and X. She, Org. Chem. Front., 2017, 4, 2211 RSC.
- For selected reviews of C–H functionalization, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (c) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC; (d) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (e) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (f) S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937 RSC; (g) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed; (h) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923 CrossRef CAS PubMed; (i) P. Tao and Y. Jia, Sci. China: Chem., 2016, 59, 1109 CrossRef CAS.
- For leading examples of strategic late-stage C–H functionalization in total synthesis, see: (a) K. Chen and P. S. Baran, Nature, 2009, 459, 824 CrossRef CAS PubMed; (b) E. M. Stang and M. C. White, Nat. Chem., 2009, 1, 547 CrossRef CAS PubMed; (c) B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J.-Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317 CrossRef CAS PubMed; (d) C. M. Rasik and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 14522 CrossRef CAS PubMed; (e) D. A. Siler, J. D. Mighion and E. J. Sorensen, Angew. Chem., Int. Ed., 2014, 53, 5332 CrossRef CAS PubMed; (f) N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4909 CrossRef CAS PubMed; (g) M. E. McCallum, C. M. Rasik, J. L. Wood and M. K. Brown, J. Am. Chem. Soc., 2016, 138, 2437 CrossRef CAS PubMed; (h) K. L. White and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 11383 CrossRef CAS PubMed; (i) J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706 RSC; (j) S. A. Loskot, D. K. Romney, F. H. Arnold and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 10196 CrossRef CAS PubMed.
- C.-M. Park, M. Bruncko, J. Adickes, J. Bauch, H. Ding, A. Kunzer, K. C. Marsh, P. Nimmer, A. R. Shoemaker, X. Song, S. K. Tahir, C. Tse, X. Wang, M. D. Wendt, X. Yang, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2008, 51, 6902 CrossRef CAS PubMed.
- G. A. Kraus and L. Chen, J. Am. Chem. Soc., 1990, 112, 3464 CrossRef CAS.
- The stereochemistry of the major diastereomer was elucidated by nuclear Overhauser effect (NOE) studies of compound 15, in which the correlation was found between C6–H and C7–H. More information of the NOE study is in the ESI†.
- J.-Q. Chen, J.-H. Xie, D.-H. Bao, S. Liu and Q.-L. Zhou, Org. Lett., 2012, 14, 2714 CrossRef CAS PubMed.
- It should be noted that compound 4 was stored as the petroleum ether solution at low temperature (−20 °C) otherwise decomposed slowly when it was stored in the flame-dried flask as its pure form.
- (a) D. Sole, Y. Cancho, A. Llebaria, J. M. Moreto and A. Delgado, J. Am. Chem. Soc., 1994, 116, 12133 CrossRef CAS; (b) J. Bonjoch, D. Sole and J. Bosch, J. Am. Chem. Soc., 1995, 117, 11017 CrossRef CAS; (c) D. Solé, J. Bonjoch and J. Bosch, J. Org. Chem., 1996, 61, 4194 CrossRef; (d) J. Bonjoch, D. Solé, S. García-Rubio and J. Bosch, J. Am. Chem. Soc., 1997, 119, 7230 CrossRef CAS; (e) J. Ma, W. Yin, H. Zhou and J. M. Cook, Org. Lett., 2007, 9, 3491 CrossRef CAS PubMed; (f) F. Yu, B. Cheng and H. Zhai, Org. Lett., 2011, 13, 5782 CrossRef CAS PubMed.
- M. Teng, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2014, 53, 1814 CrossRef CAS PubMed.
- (a) V. H. Rawal and S. Iwasa, J. Org. Chem., 1994, 59, 2685 CrossRef CAS; (b) D. Solé, J. Bonjoch, S. García-Rubio, E. Peidró and J. Bosch, Chem. – Eur. J., 2000, 6, 655 CrossRef.
- A. B. Dounay, L. E. Overman and A. D. Wrobleski, J. Am. Chem. Soc., 2005, 127, 10186 CrossRef CAS PubMed.
- K. A. Parker and H. J. Kim, J. Org. Chem., 1992, 57, 752 CrossRef CAS.
- (a) L. Chen, E. Shi, Z. Liu, S. Chen, W. Wei, H. Li, K. Xu and X. Wan, Chem. – Eur. J., 2011, 17, 4085 CrossRef CAS PubMed; (b) T. Dohi, N. Takenaga, A. Goto, A. Maruyama and Y. Kita, Org. Lett., 2007, 9, 3129 CrossRef CAS PubMed; (c) S. Sathyamoorthi and J. Du Bois, Org. Lett., 2016, 18, 6308 CrossRef CAS PubMed; (d) J. Zhao, H. Fang, W. Zhou, J. Han and Y. Pan, J. Org. Chem., 2014, 79, 3847 CrossRef CAS PubMed; (e) E. McNeill and J. Du Bois, J. Am. Chem. Soc., 2010, 132, 10202 CrossRef CAS PubMed; (f) R. Mello, M. Fiorentino, O. Sciacovelli and R. Curci, J. Org. Chem., 1988, 53, 3890 CrossRef CAS; (g) W. G. Shuler, S. L. Johnson and M. K. Hilinski, Org. Lett., 2017, 19, 4790 CrossRef CAS PubMed; (h) C. J. Pierce and M. K. Hilinski, Org. Lett., 2014, 16, 6504 CrossRef CAS PubMed; (i) T. C. Johnson, M. R. Chin, T. Han, J. P. Shen, T. Rana and D. Siegel, J. Am. Chem. Soc., 2016, 138, 6068 CrossRef CAS PubMed; (j) C. Zheng, I. Dubovyk, K. E. Lazarski and R. J. Thomson, J. Am. Chem. Soc., 2014, 136, 17750 CrossRef CAS PubMed.
- (a) E. J. Corey and K. Kamiyama, Tetrahedron Lett., 1990, 31, 3995 CrossRef CAS; (b) C. Kim, R. Hoang and E. A. Theodorakis, Org. Lett., 1999, 1, 1295 CrossRef CAS; (c) T. P. Brady, S. H. Kim, K. Wen, C. Kim and E. A. Theodorakis, Chem. – Eur. J., 2005, 11, 7175 CrossRef CAS PubMed.
- J.-Q. Yu and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 3232 CrossRef CAS PubMed.
- E. Alvarez-Manzaneda, R. Chahboun, E. Alvarez, R. Tapia and R. Alvarez-Manzaneda, Chem. Commun., 2010, 46, 9244 RSC.
- F. Jiménez, A. Fernández, E. Boulifa, A. I. Mansour, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, J. Org. Chem., 2017, 82, 9550 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1587221 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc09367e |
| This journal is © The Royal Society of Chemistry 2018 |