
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-component hybrid hydrogels – understanding the extent of orthogonal assembly and its impact on controlled release†
Vânia M. P.
Vieira
,
Laura L.
Hay
and
David K.
Smith
 *
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk; Web: http://www.york.ac.uk/chemistry/staff/academic/o-s/dsmith/
First published on 24th August 2017
Abstract
This paper reports self-assembled multi-component hybrid hydrogels including a range of nanoscale systems and characterizes the extent to which each component maintains its own unique functionality, demonstrating that multi-functionality can be achieved by simply mixing carefully-chosen constituents. Specifically, the individual components are: (i) pH-activated low-molecular-weight gelator (LMWG) 1,3;2,4-dibenzylidenesorbitol-4′,4′′-dicarboxylic acid (DBS–COOH), (ii) thermally-activated polymer gelator (PG) agarose, (iii) anionic biopolymer heparin, and (iv) cationic self-assembled multivalent (SAMul) micelles capable of binding heparin. The LMWG still self-assembles in the presence of PG agarose, is slightly modified on the nanoscale by heparin, but is totally disrupted by the micelles. However, if the SAMul micelles are bound to heparin, DBS–COOH self-assembly is largely unaffected. The LMWG endows hybrid materials with pH-responsive behavior, while the PG provides mechanical robustness. The rate of heparin release can be controlled through network density and composition, with the LMWG and PG behaving differently in this regard, while the presence of the heparin binder completely inhibits heparin release through complexation. This study demonstrates that a multi-component approach can yield exquisite control over self-assembled materials. We reason that controlling orthogonality in such systems will underpin further development of controlled release systems with biomedical applications.
Introduction
Supramolecular hydrogels have emerged as a class of soft material with wide-ranging uses in high-tech applications – from drug delivery and tissue engineering to environmental remediation and nanoscale electronics.1 One way of increasing the functionality of gels is to employ a multi-component approach.2 There has been increasing interest in systems where different units self-sort – for example, gelators programmed with different recognition pathways or which assemble in the presence of different stimuli into their own independent nanoscale networks in the presence of one another.3 Orthogonal assembly of different types of nanoscale component can also be achieved – for example gel nanofibres can assemble in the presence of vesicles and other self-assembled nanostructures.4 Hybrid hydrogels can combine the beneficial properties of responsive supramolecular gels formed by self-assembling low-molecular-weight gelators (LMWGs) with robust polymer gels formed from assembly, entanglement or crosslinking of polymer gelators (PGs).5 Hydrogels are of particular interest as a consequence of their ability to encapsulate and deliver bioactive molecules, with many reports of PGs used for drug delivery,6 and an increasing number in which LMWGs and other supramolecular materials are used.7 Heparin, which has clinical use as an anti-coagulant,8 has been incorporated in a number of polymer and silica gels for controlled release, with release rate depending on network density/swelling, interactions between heparin and the gel, or a combination of both.9 There is interest in applying these for transdermal or subcutaneous low-dose heparin delivery for long term use in hospital settings.10 Heparin has also been included in gels with potential applications in tissue engineering, to control growth factor release and/or encourage tissue growth.11 However, supramolecular gels which incorporate heparin remain very rare, and are restricted to gels based on relatively complex self-assembling peptides.12 We decided to incorporate heparin in gels based on simple, commercially-relevant LMWGs, and employ a multi-component approach to yield multi-functional materials, which combine the activities of multiple different units within a single gel. The design of our system (Fig. 1) included: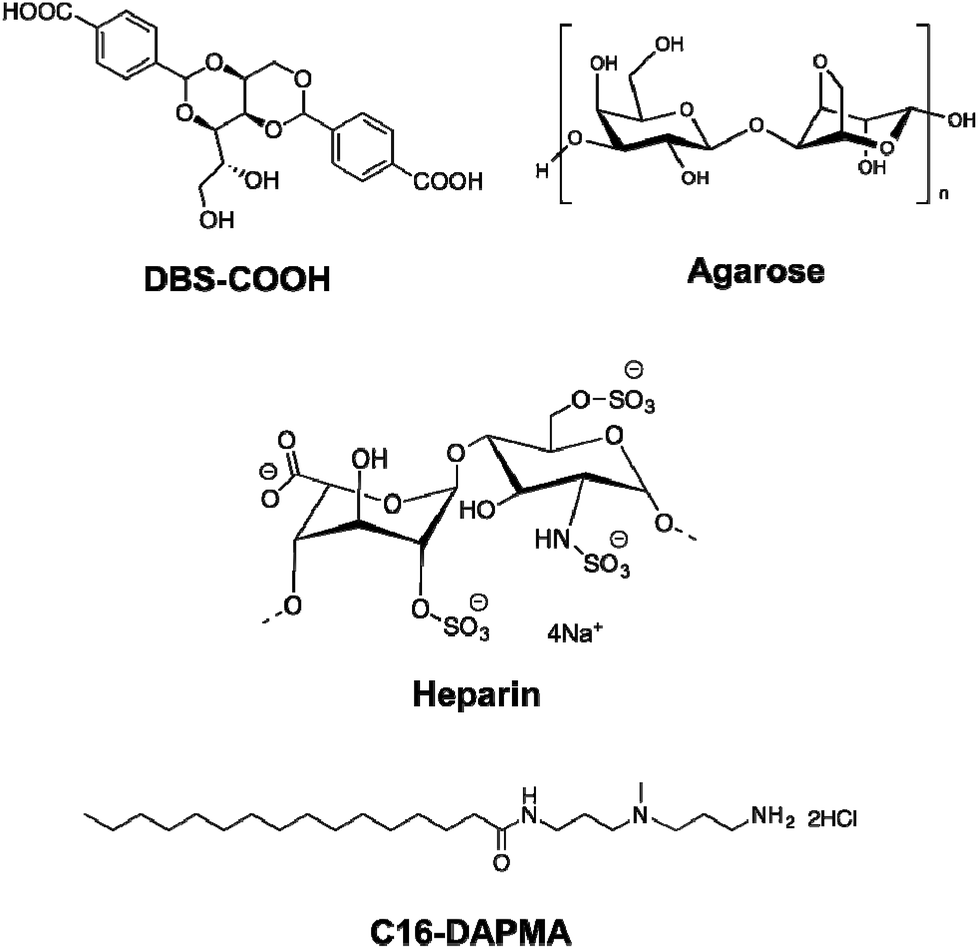 | ||
| Fig. 1 Components incorporated into the multi-component hybrid hydrogels investigated in this paper. | ||
1. Low molecular weight gelator (LMWG): DBS–COOH
This is a self-assembling pH-responsive gelator developed in our laboratory,13 based on an industrially-relevant 1,3;2,4-dibenzylidenesorbitol (DBS) framework14 modified with carboxylic acids on the aromatic ‘wings’.2. Polymer gelator (PG): agarose
This saccharide biopolymer has good biocompatibility forming a robust, stable hydrogel through a nanofibrillar mechanism.203. Bioactive polymer: heparin
This is biomedically-relevant with applications ranging from coagulation control15 to angiogenesis in transplanted cell cultures.164. Heparin binding micelles: C16-DAPMA
Cationic lipids act as nanoscale self-assembled multivalent (SAMul) heparin binders.17 We selected C16-DAPMA, which forms spherical micelles (diameter = 6.2 nm), and is an effective heparin binder.18 We previously reported full characterization of the hierarchical self-assembled nanostructures which form when cationic C16-DAPMA micelles bind anionic heparin.19Components 1 and 4 rely on self-assembly to reversibly form nanostructures, while components 2 and 3 are polymeric. The aim of this study was to characterize and fully understand multi-component systems derived from these units, determining the precise influence of each component on the others, and probing the ability to release heparin. Gel characterization is a challenging task, performed across multiple length scales – from molecular-scale to nanoscale to macro-scale,21 made more complex when multiple components are present. This study provides fundamental insights into complex multi-component self-assembled soft materials, and the extent to which the individual components can be considered to be orthogonal.
Results and discussion
Synthesis and characterization of components
LMWG DBS–COOH was synthesised on multi-gram scale according to our previously reported simple methodology.13 DBS–COOH forms gels by slow acidification induced by hydrolysis of glucono-δ-lactone (GdL)22 or by UV-initiated photoactivation with diphenyliodonium nitrate.23 On this occasion, we employed GdL as a gelation trigger – this compound could, of course, be considered as an additional fifth component in our multicomponent approach, but as it is molecular-scale rather than nanoscale, we do not focus on it as such. The thermally-stable gels formed by DBS–COOH (Tgel > 100 °C) were fully consistent with previous reports.13 C16-DAPMA was also synthesised using well-established, high yielding methods employing protecting group and amide coupling methodology.18 The heparin binding characteristics of C16-DAPMA were in-line with previous reports.18,19DBS–COOH + heparin
Initially, we aimed to understand the impact of polyanionic heparin on the self-assembly of DBS–COOH (0.2% wt/vol, 4.5 mM). We selected 38–500 μM heparin as representing meaningful concentrations.24 In all cases, macroscopic gelation was observed (Fig. S1†), and Tgel values were above the solvent boiling point (>100 °C). This indicates that heparin does not prevent DBS–COOH self-assembly. However, we wanted to determine if heparin affected LMWG assembly in subtler ways.The IR spectrum of the xerogel formed by DBS–COOH (0.2% wt/vol) and heparin (300 μM) basically corresponded to overlap of the separate IR spectra of DBS–COOH and heparin (Fig. S3–S6†). This would suggest that specific interactions between components are limited (or at least cannot be observed by IR).
1H NMR was used to investigate gelation kinetics. On pH-induced self-assembly of DBS–COOH, resonances associated with the mobile solution-phase gelator molecules disappear. Use of an internal standard allows this to be quantified.25 This experiment was performed using DBS–COOH (0.2% wt/vol) in the absence and presence of heparin (300 μM) – in both cases, results were essentially the same (Fig. S7†). For the first hour, although pH was changing no gel was formed, but after this, gel formation began, resonances associated with the gelator decreased in intensity, and self-assembly progressed over the following 7 hours until macroscopic gelation was complete.
Circular dichroism (CD) spectroscopy was similarly used to characterize assembly kinetics. However, this experiment was performed at a significantly lower concentration (0.02% wt/vol), below the minimum gelation concentration (MGC). Under these conditions, DBS–COOH nanofibers assemble, but do not entangle to form a macroscopic sample-spanning network, and hence gelation is not observed.13a Nanofibre self-assembly can be inferred from the significant increase in CD band intensity – indeed, deprotonated DBS–COO− has no CD band, whereas once the molecule becomes protonated, the CD band at 260 nm becomes very significant, consistent with π–π stacking of the aromatic rings into a nanoscale chiral environment (Fig. S8 and S9†).21 In this way, self-assembly kinetics can be followed, and the final CD spectrum gives direct insight into the nanoscale chirality of the self-assembled fibres. On self-assembly, DBS–COOH (0.02% wt/vol) developed a CD band at ca. 259 nm. A very similar band also appeared for DBS–COOH (0.02% wt/vol) in the presence of heparin (38 μM) indicating a similar mode of assembly. However, plotting the intensity of this band against time (Fig. 2) revealed that in the presence of heparin, the assembly of nanofibers is faster, and assembly starts sooner. Furthermore, in the presence of heparin, the final CD band was larger than that observed for DBS–COOH alone, suggesting that the nanoscale chirality of the resulting assembly is somewhat modified. This experiment therefore suggests that heparin can assist LMWG self-assembly at the nanoscale and may give rise to nanoscale objects with different chiralities/morphologies.
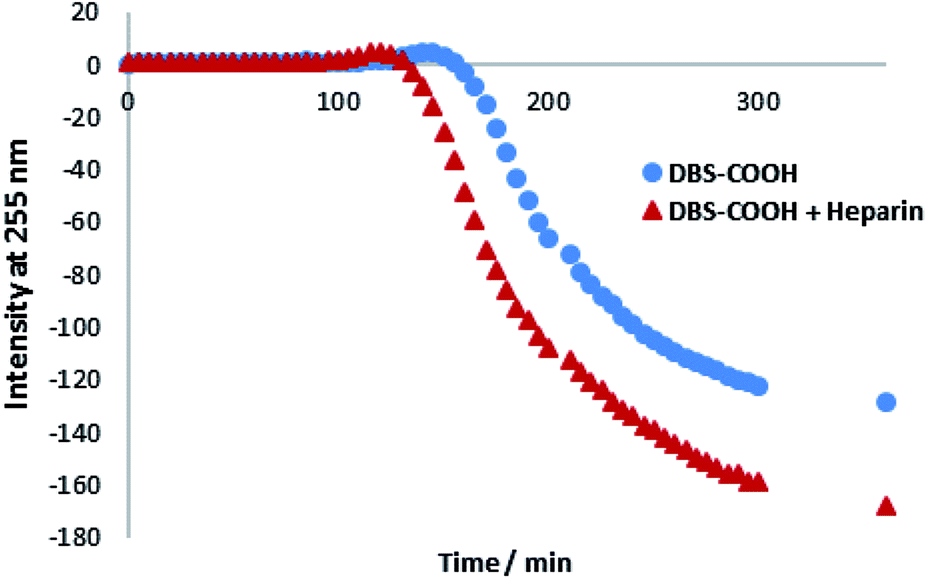 | ||
| Fig. 2 CD intensity at 255 nm plotted against time for DBS–COOH (0.02% wt/vol) in the absence and presence of heparin (38 μM). | ||
To gain greater insight into the self-assembled nanoscale morphologies, transmission electron microscopy (TEM) of the dried hydrogels (0.2% wt/vol) was performed. TEM images of DBS–COOH (Fig. 3, top) indicated intertwined long and twisted nanofibres. When heparin (38 μM) was also present (Fig. 3, bottom), long nanofibres were once again observed, but it was also possible to distinguish some differentiated structures, particularly on the tips of the nanofibres (see also Fig. S12 and S13†). We reason these are associated with or caused by the heparin – we suggest that non-specific adsorption of heparin onto the growing nanofibres modifies the morphology and gives rise to the enhanced assembly kinetics observed by CD – indeed, related effects have previously been described for soluble polymers added to gels.26 Interestingly, it is well-known that heparin can assist fibril assembly in biological peptides such as amyloids.27 Our observations here suggest heparin can also impact on simple LMWG self-assembling systems. Nonetheless, overall, TEM imaging supports the view that DBS–COOH self-assembly does still occur in the presence of heparin – although is somewhat modified in terms of nanoscale morphology.
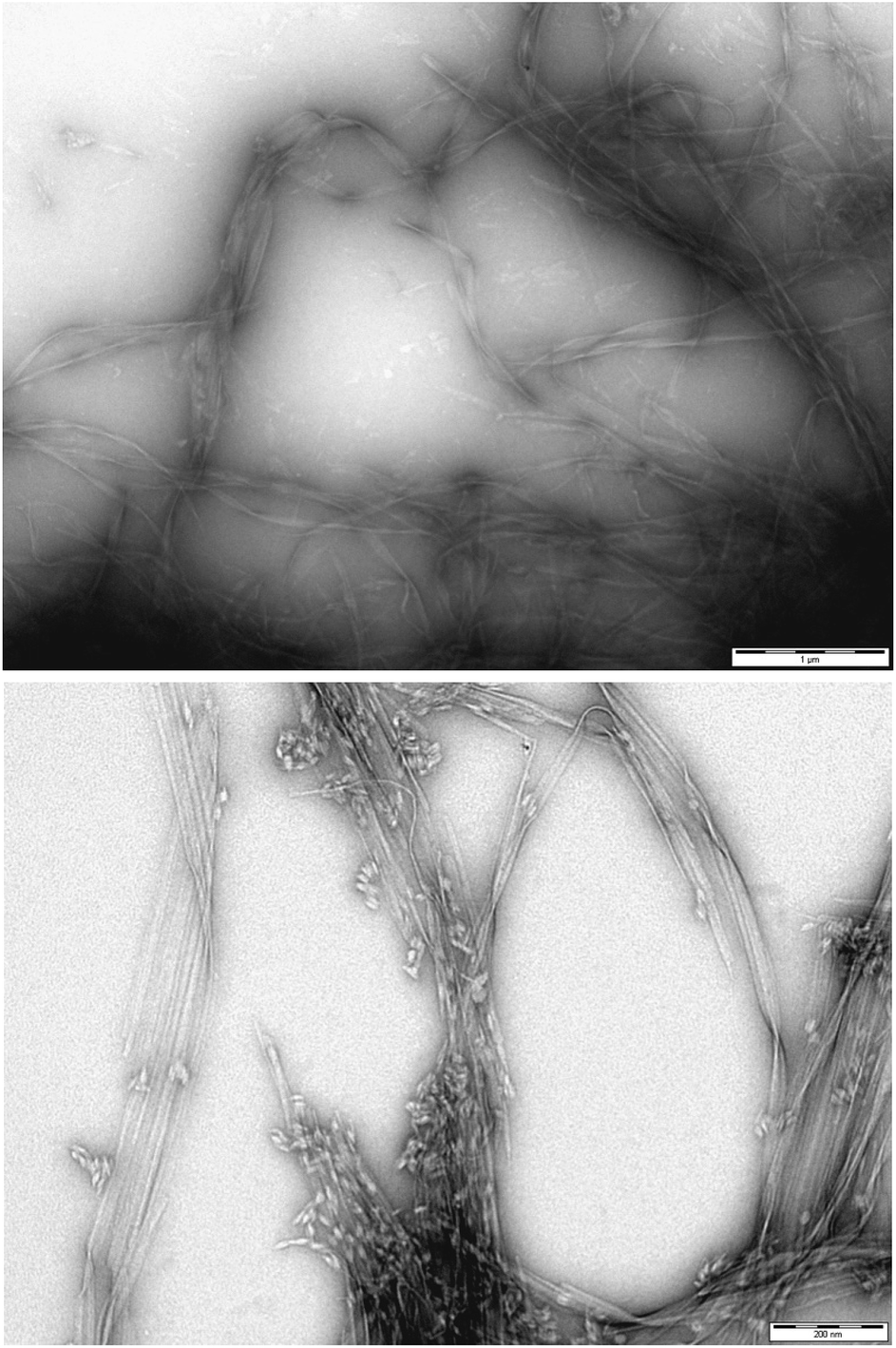 | ||
| Fig. 3 (Top) TEM image of DBS–COOH gel (0.2% wt/vol). Scale bar: 1 μM. (Bottom) TEM image of DBS–COOH gel (0.2% wt/vol) in the presence of heparin (38 μM). Scale bar: 200 nm. | ||
Scanning electron microscopy (SEM) was performed on samples prepared via freeze-drying to minimise morphological change on drying. DBS–COOH (Fig. 4, top) exhibited an expanded nanofibre mesh, with fibre diameters of ca. 80 nm – in good agreement with TEM. In the presence of heparin (Fig. 4, bottom) nanofibres were also formed, but were significantly narrower – ca. 50 nm. These smaller fibres suggest enhanced nucleation of gel nanofibers induced by heparin. In such a model, more fibres form, but can then only grow to smaller diameters. This is in agreement with time-resolved CD, which indicated enhanced nanoscale assembly kinetics, supporting the view that heparin interacts with DBS–COOH nanofibres in the growth phase and hence somewhat modifies nanoscale morphology.
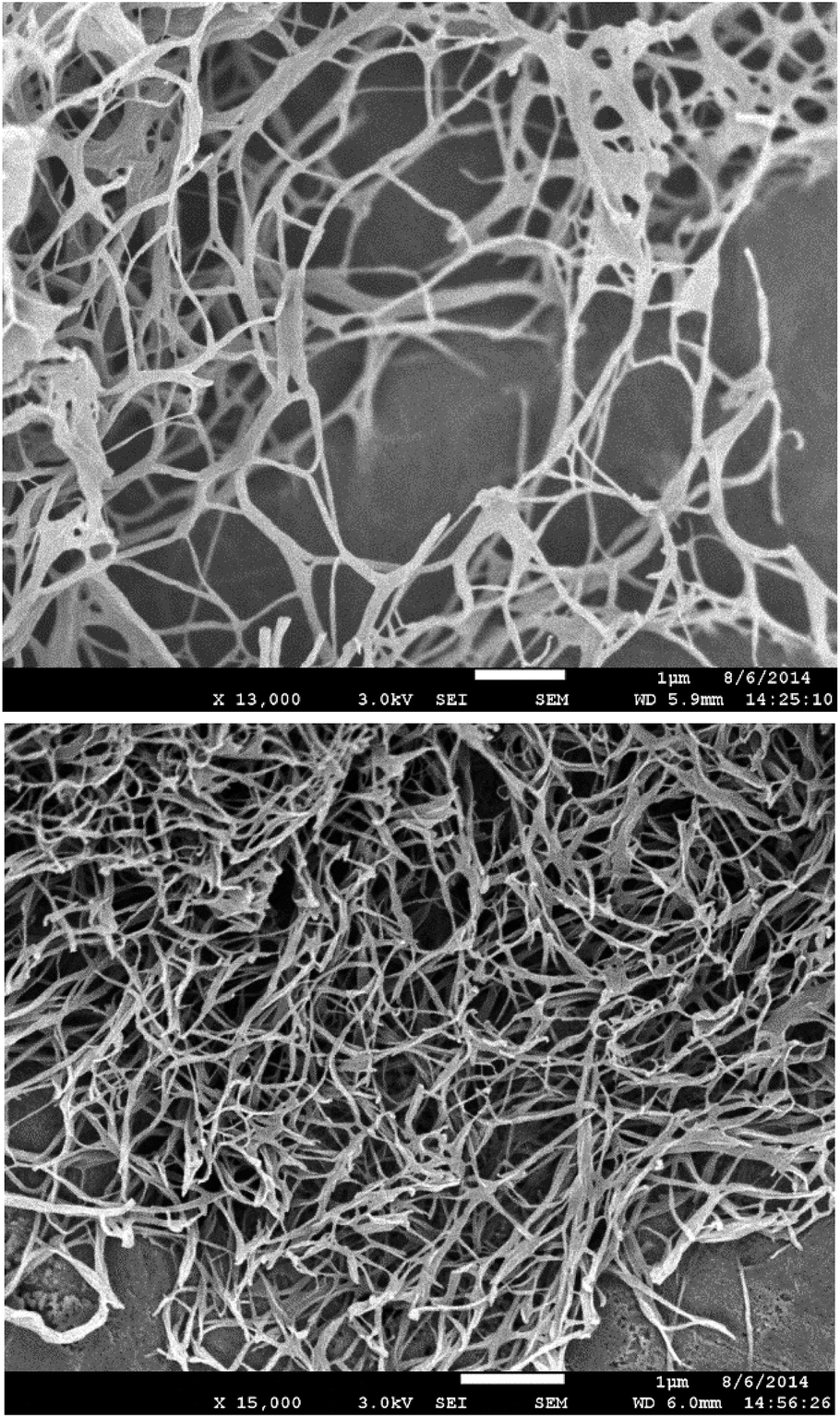 | ||
| Fig. 4 (Top) SEM image of DBS–COOH gel (0.2% wt/vol). (Bottom) SEM images of DBS–COOH gel (0.2% wt/vol) in the presence of heparin (38 μM). Scale bars: 1 μm. | ||
For macroscopic characterisation, rheology was performed on DBS–COOH (0.2% wt/vol) alone and in the presence of heparin (1 mM). This technique provides storage (elastic) modulus (G′) and loss (viscous) modulus (G′′) through application of oscillating strains. As ‘solid-like’ materials, gels exhibit G′ values ca. one order of magnitude higher than G′′ and low frequency dependence. We initially monitored strain amplitude dependence to characterize the linear viscoelastic region (LVR) over which gel-like properties persist (Fig. S17†). For DBS–COOH, gel-like behaviour was observed up to ca. 3% strain. Similar behaviour was observed in the presence of heparin. However, G′ and G′′ values were 3-fold lower in the presence of heparin – i.e., heparin makes the network less stiff. This agrees with TEM/SEM imaging of a modified nanoscale morphology, with thinner nanofibres leading to a less-stiff network on the macroscopic scale. Monitoring response to frequency led to similar conclusions (Fig. S18†) – we note that frequency was increased to high values (ca. 100 Hz) and this leads to an increase in G′ and G′′, indicative of hardening being induced by high frequencies at which gel dynamics are being studied over very short timescales – similar effects have previously been reported.28 We also characterised the 2% wt/vol DBS–COOH gel (Fig. S19 and S20†) – this system was significantly stiffer (G′ = ca. 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa in the LVR) compared with the 0.2% wt/vol gel (G′ = ca. 2600 Pa in the LVR). As might be expected these stiffer 2% wt/vol gels were less resistant to strain, and broke down at ca. 1% strain rather than ca. 3%.
000 Pa in the LVR) compared with the 0.2% wt/vol gel (G′ = ca. 2600 Pa in the LVR). As might be expected these stiffer 2% wt/vol gels were less resistant to strain, and broke down at ca. 1% strain rather than ca. 3%.
We finally explored the ability of these gels to release heparin into buffered water. Heparin release was probed by placing buffer (10 mM Tris–HCl, 150 mM NaCl, pH 7.4, 1 mL) on top of the hydrogel (3 mL) incorporating heparin (1 mM). Aliquots of buffer were removed over time, added into Mallard Blue (MalB) solution and the UV-Vis absorbance measured. MalB is a heparin sensor29 and therefore enables the quantification of heparin release via UV-Vis spectroscopy. Fig. 5 shows release of heparin (1 mM) from DBS–COOH (0.2% wt/vol and 2% wt/vol). At 0.2% wt/vol gelator loading, there is relatively rapid initial heparin release, rising to ca. 40% over the first 6 hours. There is then slower release, with a further 40–45% being released over the ensuing 42 hours, giving a total of ca. 85% release. At 2% wt/vol gelator loading, the initial rapid release over the first 6 hours still occurs, up to a total of ca. 40%, but after this, no further heparin is released. We also investigated gelator loadings of 5% and 10% wt/vol – these behaved identically to the 2% wt/vol gel (Fig. S27†).
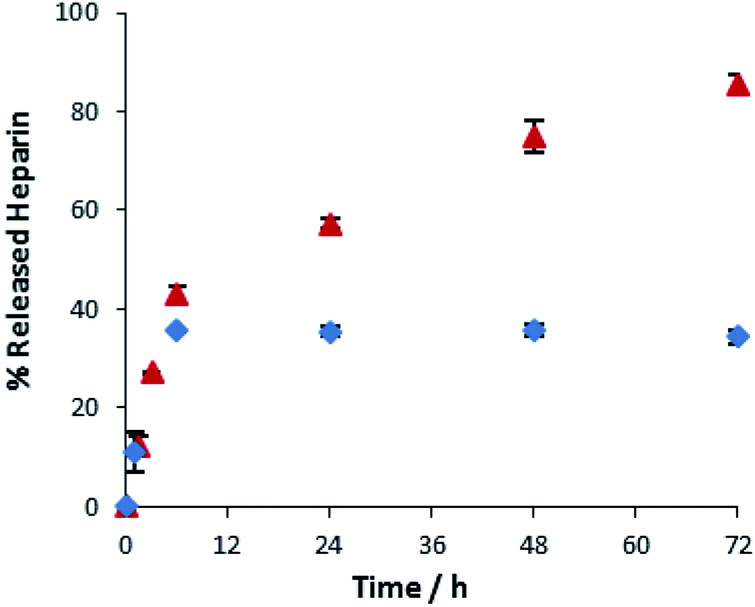 | ||
Fig. 5 Heparin release from 0.2% w/v ( ) and 2% w/v ( ) and 2% w/v ( ) of DBS–COOH hydrogels with 1 mM of heparin. ) of DBS–COOH hydrogels with 1 mM of heparin. | ||
These results suggest the gel nanofibre network plays an active role in controlling heparin release. Heparin is a relatively large biopolymer, and will have limited diffusion within a gel matrix. As the loading of gelator is increased, and rheological stiffness significantly increases (see above), the ability of the heparin to diffuse within the gel decreases. We propose that ≥2% wt/vol, only the heparin initially close to the gel surface can be released – ca. 30–40%. Heparin further away from the interface is incapable of diffusing to the interface, and is not released – further evidence for this hypothesis is presented below once agarose is also incorporated into the system. At 0.2% wt/vol loading, there is still initial rapid release of ca. 40% of the heparin, but the lower density nanofiber network then allows for some diffusion of heparin, which is slowly released over time. Alternatively, as suggested by a reviewer, it is plausible that at lower loadings, the DBS–CO2H gel is more easily damaged by the presence of buffer solution on top, enabling enhanced slow release – but we emphasise that in no case was any visible damage to the gel surface observed, and gel integrity was always tested by tube inversion at the end of the heparin release experiment. In summary, these gels can release heparin, with release rate and total amount released depending on LMWG loading.
We conclude that DBS–COOH and heparin are largely orthogonal. DBS–COOH gels still form in the presence of heparin, have good thermal stability, and macroscopic gelation kinetics are similar. However, there are subtle differences in nanoscale assembly kinetics (see CD). TEM and SEM indicate that the nanofibers formed by DBS–COOH are narrower when heparin is present, suggesting they nucleate more quickly, promoted by heparin. This has an impact on macroscopic gel performance – in the presence of heparin, the gel maintains resistance to strain, but is less stiff. Heparin release can be achieved, but there is a threshold DBS–COOH network density that prevents heparin diffusion, limiting total release. In this way, release kinetics are controlled by network density. In summary, these two components clearly tolerate one another when incorporated in a multi-component system.
DBS–COOH + C16-DAPMA
We then studied the combination of DBS–COOH and cationic lipid C16-DAPMA, which self-assembles into multivalent micelles. We tested the effect of C16-DAPMA (150 μM to 1 mM) on the gelation of DBS–COOH (0.2% wt/vol). In no case was gelation observed (Fig. S2†) – C16-DAPMA completely inhibits gelation of DBS–COOH. Either the amines on C16-DAPMA buffer the solution and limit protonation of DBS–COOH, hence preventing gelation, or direct interactions between acidic DBS–COOH and basic C16-DAPMA inhibit gel assembly. These two components are clearly not orthogonal.DBS–COOH + agarose
Inspired by the pioneering work of Yang and co-workers, who combined agarose with a LMWG,30 we previously characterized the combination of LMWG DBS–COOH and PG agarose,13 demonstrating it yields responsive but robust materials, in which each component essentially maintained its own characteristic behaviour. It is worth noting that the temperature at the outset of this experiment is higher than when DBS–COOH is studied in the absence of agarose, and that this may impact on GdL hydrolysis rate and nanofibre formation. NMR evidence suggested little difference in the kinetics of gel assembly although CD spectroscopy indicated small differences in nanofibre chirality.13Going further than previously, we performed careful SEM analysis of these gels (and their individual constituents) using our freeze drying sample preparation method to yield expanded xerogels, rather than collapsed ones. As described above, DBS–COOH exhibited a nanofibrillar morphology with fibre diameters of ca. 80 nm. As expected,20 agarose also formed a nanofibrillar gel, but with thinner fibres of ca. 10 nm (Fig. S14†). These narrower fibres are consistent with the optically transparent nature of agarose while the larger fibres of DBS–COOH are in agreement with its slightly hazy nature. Pleasingly, on imaging the hybrid gel (Fig. 6) both types of fibre appeared to be present – suggesting that the two networks do indeed assemble independently of one another. Examples where self-sorted gel nanofibres can be clearly visualised in multi-component gels remain relatively rare.31
 | ||
| Fig. 6 SEM image of DBS–COOH gel (0.2% w/v) in the presence of agarose (0.5% w/v). The two different thicknesses of fibre are labelled with white arrows (narrower agarose fibres) and black arrows (thicker DBS–COOH fibres). Scale bar: 100 nm. | ||
We also performed additional rheological characterization of the hybrid hydrogel. The addition of agarose (1% wt/vol) to DBS–COOH (0.2% wt/vol) resulted in a significant (ca. 3-fold) increase in G′ from ca. 2600 Pa to ca. 7900 Pa (Fig. S21†). The LVR remained the same, with the gel being able to resist up to ca. 3% strain, and the frequency dependence also remained similar (Fig. S22†). The impact of agarose on G′ shows that the PG contributes to the formation of a stiffer gel, reinforcing the relatively weak DBS–COOH network. Agarose alone behaves similarly in terms of rheology to the hybrid gel in terms of G′, although it breaks down at slightly lower strain (Fig. S23 and S24†). This would indicate that the polymer gel is indeed dominating the stiffness of the hybrid material, although the softer, more flexible self-assembled DBS–COOH LMWG network may be enhancing resistance to strain.
As reported previously,13 NMR methods demonstrated that addition of base to the hybrid gel deprotonated DBS–COOH, leading to disassembly of its nanoscale network – i.e., the LMWG retains its responsiveness in the presence of the PG.
In summary, these components are clearly orthogonal to one another.
DBS–COOH + heparin + C16-DAPMA
We then studied the three-component combination in which heparin and heparin binder (C16-DAPMA) are both present, alongside the LMWG (DBS–COOH). Although DBS–COOH and C16-DAPMA are a disruptive combination, we reasoned that once the C16-DAPMA micelles are bound to heparin, DBS–COOH may then self-assemble.We tested the formation of DBS–COOH gels in which a solution of C16-DAPMA and heparin was added to the sample immediately before, or immediately after, the addition of glucono-δ-lactone. The order of addition did not have any effect. A series of heparin concentrations were tested, and to each, increasing concentrations of C16-DAPMA were added (Table 1). This determined the maximum tolerance of the gel towards C16-DAPMA when heparin is also present. For each concentration of heparin, there was a threshold concentration of C16-DAPMA up to which gelation of DBS–COOH will still occur – once the amount of C16-DAPMA exceeds that, then gelation is disrupted (Table 1). For heparin concentrations of 38, 150, 300, 400 and 600 μM, the C16-DAPMA threshold concentrations are 150, 450, 800, 900 and 1400 μM respectively. As the amount of heparin increases, larger amounts of C16-DAPMA can be added without disrupting the gel. We reason (see below) that C16-DAPMA interacts with the heparin in the gel and hence does not prevent DBS–COOH assembly. Above the threshold concentration, excess C16-DAPMA causes gel breakdown. All of the gels had thermal stabilities >100 °C – equivalent to DBS–COOH on its own.
| [Heparin] (μM) | [C16-DAPMA] (μM) | Gel formation |
|---|---|---|
| 38 | ≥200 | No gel formation |
| ≤150 | Stable gel | |
| 150 | 600 | No gel formation |
| 500 | Unstable gel | |
| ≤450 | Stable gel | |
| 300 | ≥1000 | No gel formation |
| 900 | Partial gel | |
| ≤800 | Stable gel | |
| 400 | ≥1500 | No gel formation |
| 1400–1000 | Gel with aggregates | |
| ≤900 | Stable gel | |
| 600 | ≥2100 | No gel formation |
| 2000–1800 | Unstable gel | |
| 1700–1500 | Gel with aggregates | |
| ≤1400 | Stable gel |
Table 2 presents the maximum tolerated C16-DAPMA/heparin molar and charge ratios.24 At low loadings in the gel, heparin binds 4.0 molar equivalents of C16-DAPMA (a charge ratio of 2.0). However, when more heparin is present in the gel, it only binds 2.0–2.5 molar equivalents of C16-DAPMA (a charge ratio of 1.0–1.25). C16-DAPMA forms spherical micelles with a diameter of ca. 6.2 nm, while heparin is a linear polyanion. Although heparin has some flexibility and ‘adaptive’ character,32 it cannot fully wrap around these small micelles.19 At low heparin concentration therefore, some of the ligands on the micelle will be unsatisfied, and excess C16-DAPMA is required to fully bind heparin. However, as the concentration of heparin increases, the likelihood of multiple heparin chains contacting a single micelle increases – under these conditions, many more of the ligands will actively bind heparin, and the charge ratio of the complex comes closer to 1.0. As such, we have a good understanding of the complexation process between self-assembled C16-DAPMA and heparin, which occurs with the gel matrix. This is a unique study of self-assembly and binding between nanoscale systems within a self-assembled nanofibrillar gel medium.
| [Heparin] (μM) | C16-DAPMA/heparin molar ratio | Charge ratio +/− |
|---|---|---|
| 38 | 4.0:1 |
2.0 |
| 150 | 3.0:1 |
1.5 |
| 300 | 2.7:1 |
1.3 |
| 400 | 2.2:1 |
1.1 |
| 600 | 2.3:1 |
1.2 |
NMR studies of gelation kinetics in the three-component system of DBS–COOH (0.2% wt/vol), heparin (300 μM) and C16-DAPMA (800 μM) were very similar to those for DBS–COOH alone and in the presence of just heparin (described above). Gelation was effectively complete after 8–10 hours, with all of the gelator becoming immobilised (Fig. S7†).
CD spectroscopy was used to study nanoscale fibre assembly kinetics in the three component system of DBS–COOH (0.02% wt/vol), heparin (38 μM) and C16-DAPMA (150 μM). In this case, the onset of assembly was delayed (Fig. S10 and S11†), and once assembly was complete, a slightly different CD signal, with a longer wavelength peak (ca. 262 nm) was obtained. While heparin alone accelerated DBS–COOH nucleation and nanofibre assembly, heparin complexed to C16-DAPMA therefore slows and somewhat modifies chiral nanostructure assembly (see below).
Fig. 7 (and Fig. S16†) presents a TEM image of DBS–COOH (0.2% wt/vol) assembled in the presence of both heparin (38 μM) and C16-DAPMA (150 μM). Typical hierarchical aggregates between C16-DAPMA micelles and heparin as characterised previously,19 along with nanofibers of DBS–COOH were observed. This is reminiscent of the work of van Esch and co-workers visualising orthogonal assembly of gel nanofibres and vesicles.4a,b However, in our case, there are not just two, but three components present. TEM shows gel nanofibres, and also that the other two components (self-assembled C16-DAPMA micelles and heparin) are mutually interacting with one another. There is some suggestion from the imaging (Fig. S16†) that the presence of the hierarchical aggregates may somewhat hinder/disrupt DBS–COOH nanofibre assembly. TEM images with higher concentrations of heparin (300 μM) and C16-DAPMA (800 μM) were similar but with more hierarchical assembly.
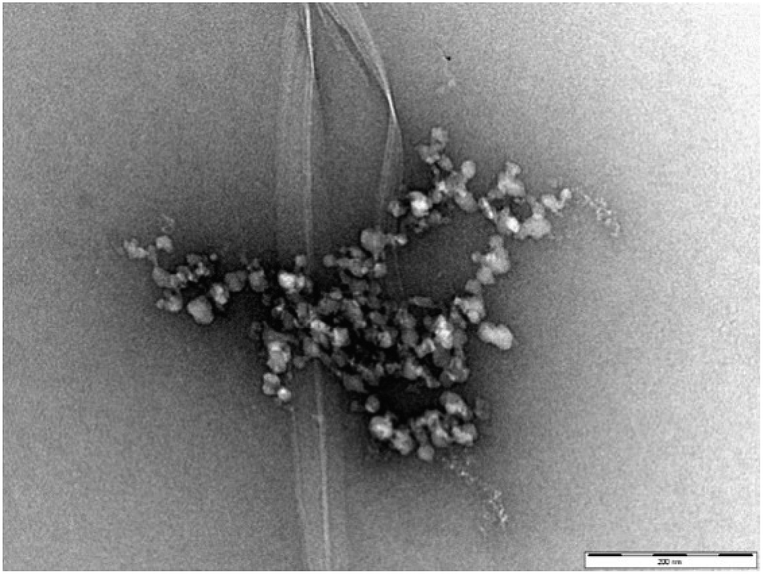 | ||
| Fig. 7 TEM image of DBS–COOH gel (0.2% w/v) in the presence of heparin (38 μM) and C16-DAPMA (150 μM). Scale bar: 200 nm. | ||
SEM of the DBS–COOH gel (0.2% wt/vol) incorporating both heparin (38 μM) and C16-DAPMA (150 μM) only showed DBS–COOH nanofibers (Fig. S15†), confirming that standard DBS–COOH nanofibres are being formed (even though the fibres visualised by TEM in Fig. 7 appeared a little different). It was not possible to observe C16-DAPMA interacting with heparin by SEM – soft self-assembled micelles are difficult to image using this technique, indeed, we have never succeeded in any of our other studies.17–19 Interestingly, the fibres were more like the larger fibres formed in the absence of heparin than the narrower fibres formed in its presence, in agreement with the view that C16-DAPMA binds heparin, hence preventing its interaction with growing gel nanofibers. SEM studies with higher concentrations of heparin and C16-DAPMA gave similar results.
Rheology was performed on these three-component gels (Fig. S17 and S18†). The gels broke down on application of 1% strain (rather than 3% as for DBS–COOH or DBS–COOH with heparin). Furthermore, the G′ value was lower than for DBS–COOH alone (and a little lower than that for DBS–COOH with heparin). As such, although SEM imaging indicated that the gel nanofibers can self-assemble intact with similar morphology to DBS–COOH alone, clearly the presence of hierarchical aggregates formed between heparin and C16-DAPMA weakens the overall network on the macroscopic level. The hierarchical aggregates are relatively large (>100 nm) and thus represent points of weakness in the overall entangled gel network.
The ability of this three-component system to release heparin was then investigated (Fig. S26†). Unlike the simple mixture of DBS–COOH and heparin described above, no release of heparin was observed over 72 hours. As a result of binding to C16-DAPMA within the gel, heparin release is completely inhibited – clearly proving that all three components are active within this multi-component gel, programming it with their individual characteristics, and hence controlling its performance.
In summary, mixing both heparin and C16-DAPMA into DBS–COOH leads to a largely orthogonal system in which DBS–COOH and C16-DAPMA both self-assemble into their own respective nanostructures, with heparin preventing the disruptive effect of C16-DAPMA by binding strongly to the micelles. On the nanoscale, as monitored by CD and TEM, the assembly of DBS–COOH is somewhat inhibited – we suggest this reflects greater difficulty in assembling nanofibres as a result of steric hindrance provided by the relatively large hierarchical heparin:C16-DAPMA aggregates. This also ultimately somewhat affects the macroscopic rheological properties of the material. The binding of C16-DAPMA to heparin completely inhibits its release from the gels. This system can therefore be considered a storage medium for bioactive heparin – only breakdown of the gel, or degradation of the heparin binder would enable heparin release. Importantly, each of the three components can fulfil its orthogonal role within this system, although they do have subtle impacts on one another.
DBS–COOH + heparin + agarose
We then studied the impact of agarose on the gels formed by DBS–COOH and heparin, in particular, their rheological performance, as the PG should make these systems more robust. As noted above, the addition of agarose (1% wt/vol) to DBS–COOH (0.2% wt/vol) resulted in a significant increase of G′ values from 2600 Pa to 7900 Pa. The addition of heparin (1 mM) to this hybrid hydrogel had no impact on rheological performance (Fig. S21 and S22†). Interestingly, this is in contrast to the impact of heparin on DBS–COOH alone (see above) in which G′ was lowered. Clearly, as noted above, the macroscopic rheological stiffness of DBS–COOH/agarose hybrid hydrogels is dominated by agarose, which is unaffected by the presence of heparin.Having demonstrated that agarose provides mechanical robustness, the pH response of DBS–COOH was checked. A hybrid gel of DBS–COOH (0.2% wt/vol) and agarose (0.5% wt/vol) was prepared in an NMR tube in the presence of heparin (1 mM) and GdL. Prior to gelation, NMR resonances associated with mobile DBS–COOH were observed, which disappeared as it is protonated and assembles into gel nanofibers. Basic aqueous NaOH was then added to the top of the gel and allowed to diffuse in. NMR spectra were recorded periodically to observe the re-appearance of the peaks associated with DBS–COOH as the compound deprotonates and the LMWG nanofibres disassemble (Fig. 8). Throughout the experiment, the sample remained as a gel, demonstrating that the agarose PG maintains overall stability. After ca. 24 hours, the gel nanofibers associated with DBS–COOH are clearly disassembling, a process complete after ca. 5 days. Diffusion into the gel in this experiment is limited by the narrow diameter of the NMR tube – in other geometries, the stimulus could be applied more rapidly. Clearly, heparin does not inhibit disassembly of the LMWG DBS–COOH network. Such hybrid multi-component systems can therefore have one of their two networks broken down – in this case, we break down the network controlling heparin release – hence leading to triggered heparin release induced by a pH change.
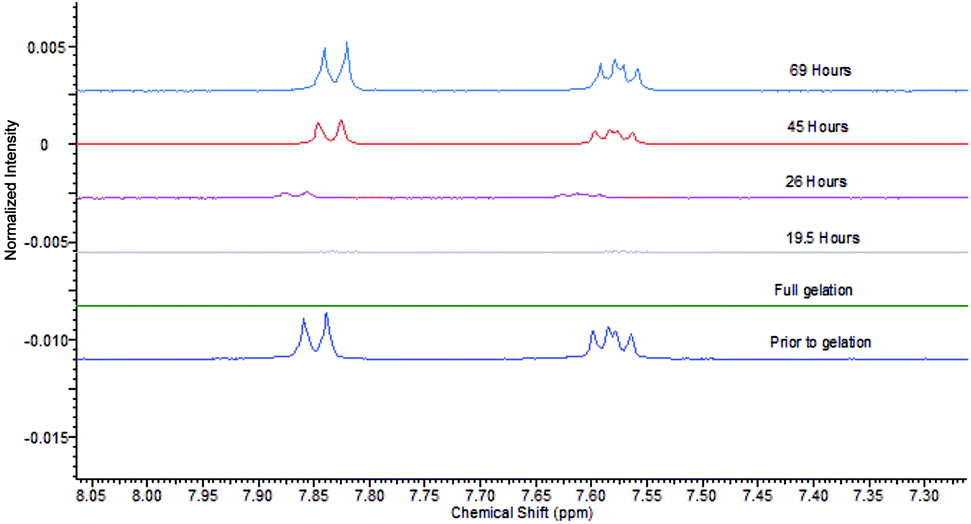 | ||
| Fig. 8 NMR spectra of aromatic protons of DBS–CO2H (0.2% wt/vol), agarose (0.5% wt/vol) and heparin (1 mM), (from bottom to top) prior to gelation, after full gelation, and after various times (19.5 h, 26 h, 45 h and 69 h) of exposure to NaOH. | ||
Heparin release was studied using hybrid hydrogels prepared using DBS–COOH (2% wt/vol) and agarose (0.5% or 1% wt/vol) and assayed with MalB as described previously. The presence of 0.5 or 1% wt/vol of agarose had no influence on heparin release – the profiles were identical to those for DBS–COOH alone (Fig. S28†).
To understand the impact of agarose PG alone on heparin release in more detail, we prepared gels at 1%, 2.5%, 5%, 7.5% and 10% wt/vol agarose with 1 mM of heparin and assayed heparin release. As agarose loading increases, heparin release slows down (Fig. 9). On increasing PG loading, the gel network will have smaller pores that will contribute to slower diffusion and thus lower release – indeed release over 4 days fell from >90% with 1% wt/vol agarose to only ca. 30% with 10% wt/vol agarose. Importantly, 1% wt/vol agarose allows maximal release of heparin, in agreement with the view that in the hybrid system of DBS–COOH, agarose and heparin, characterized above, DBS–COOH controls heparin release, while agarose simply provides the multicomponent system with rheological strength.
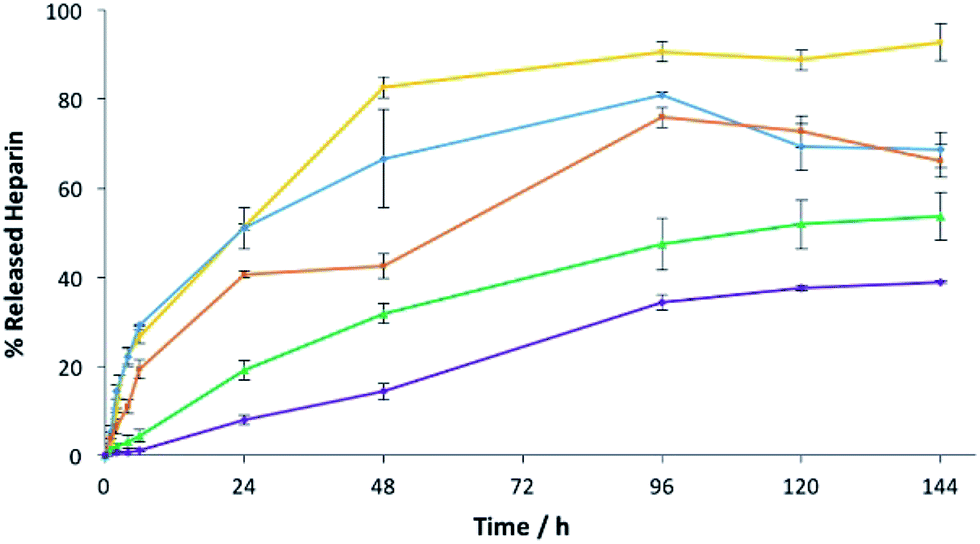 | ||
| Fig. 9 Heparin release from 1.0% w/v (yellow), 2.5% w/v (blue), 5.0% w/v (orange), 7.5% w/v (green) and 10% w/v (purple) of agarose gels with 1 mM of heparin. | ||
Interestingly, however, increasing the loading of agarose has a progressive effect on diffusion and release of heparin, while in DBS–COOH there was a threshold value beyond which diffusion appeared to stop completely. We propose that diffusion of heparin in the two different gels occurs via different mechanisms. We propose, based on all the evidence presented above, that DBS–COOH has ‘sticky’ nanofibers which interact with heparin and at high enough density (≥2% wt/vol) completely limit diffusion. In contrast, for agarose, we propose the barrier to heparin diffusion is purely steric, and heparin reptation allows some diffusion/release, even at PG loadings as high as 10% wt/vol.
A key advantage of the greater mechanical robustness of hybrid hydrogels endowed by agarose, is the ability to physically manipulate the gels. We prepared 3D gel ‘cylinders’ (Fig. S29†) containing DBS–COOH (2% wt/vol), agarose (1% wt/vol), and heparin (17 mM) and placed them into a large volume of buffer. Fig. 10 compares heparin release from these hybrid gel cylinders against gels in vials. Using gel cylinders increases heparin release from ca. 40% to ca. 80% release after 3–4 days. By decreasing the total volume of the gel, and exposing all surfaces to the receiving buffer medium, the effective surface area:volume ratio of the gel increases ca. 6.5-fold (see ESI, Section 1.10†). We suggest this enables more of the heparin to be close to the surface and hence released. This supports our view, expressed earlier in the paper, that release of heparin from DBS–COOH is limited by the heparin encapsulated more deeply within the volume of the gel being unable to diffuse to the surface as a consequence of both network density and the ability of heparin to interact with the ‘sticky’ DBS–COOH nanofibres. In addition to providing further insight into the impact of DBS–COOH on heparin release, this experiment also clearly demonstrates that the robust agarose PG enables physical formulation of these multi-component hydrogels.
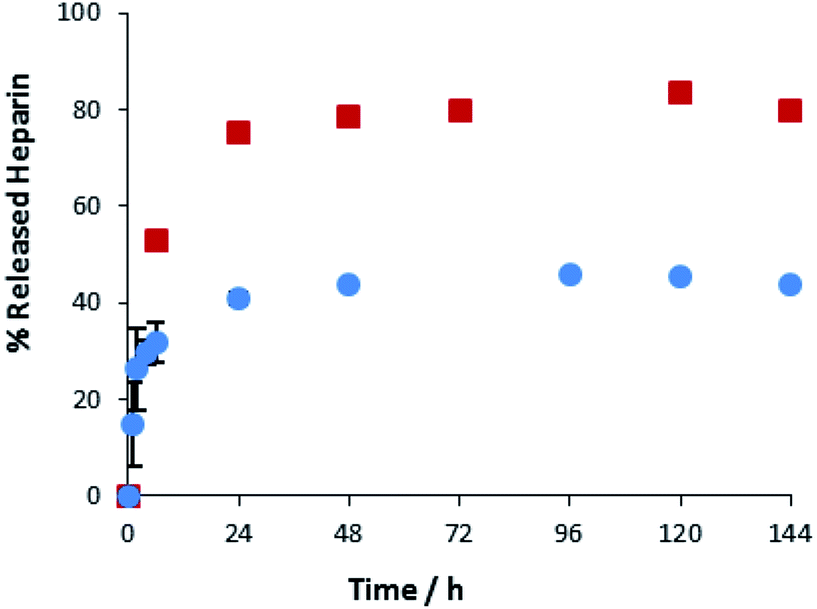 | ||
Fig. 10 Different methods of heparin release from DBS–COOH hydrogel (2% w/v) and agarose (1% w/v) containing heparin. ( ): gel cylinder. ( ): gel cylinder. ( ): buffer on top of the gel. ): buffer on top of the gel. | ||
Conclusions
We have used a simple multi-component approach to hybrid hydrogels with two self-assembling components (DBS–COOH and C16-DAPMA) and two polymeric components (heparin and agarose) and carefully characterised the impact of each component on the others. This is the first time such a detailed study has been performed in a multi-component gel of this complexity.The self-assembly of DBS–COOH is slightly modified on the nanoscale by heparin, completely disrupted by C16-DAPMA and unaffected by agarose. However, if the heparin is bound to self-assembled multivalent C16-DAPMA, then the assembly of DBS–COOH still takes place, and although the presence of the hierarchical heparin/C16-DAPMA aggregates has some minor impacts on gel performance, the components are largely orthogonal to one another. The PG reinforces all materials when present, and dominates macroscopic stiffness. DBS–COOH retains its pH sensitivity and can be disassembled in the presence of the other components, hence introducing triggered response characteristics to these hybrid hydrogels.
It is therefore possible to formulate hybrid hydrogels with different heparin release characteristics depending on:
• Loading of DBS–COOH – a threshold loading prevents heparin release from the interior of the bulk gel as a result of an interactive mechanism.
• Loading of agarose – higher agarose loadings decrease heparin release kinetics due to steric hindrance to diffusion.
• Presence of C16-DAPMA – self-assembled multivalent C16-DAPMA fully bound to heparin completely inhibits heparin release.
• Gel shape – the ratio of surface area to internal volume controls the amount of heparin released – larger relative surface areas giving rise to greater release when interactive DBS–COOH nanofibers are present.
We have gained fundamental insight into multi-component systems – this strategy is a powerful way of formulating multi-functional materials and tuning desired characteristics for bioactive release. In ongoing research, we are targeting triggered heparin release under physiologically relevant conditions and multi-component hybrid gels, which may ultimately have biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Marie Curie ITN 316656 SMARTNET for VMPV and University of York for LHH. We thank Meg Stark (Department of Biology, York) for TEM and SEM imaging and Daniel Cornwell for performing sample preparation for Fig. 6.References
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed; (c) S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC; (d) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed; (e) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed; (f) D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- (a) A. R. Hirst and D. K. Smith, Chem.–Eur. J., 2005, 11, 5496–5508 CrossRef CAS PubMed; (b) L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC; (c) J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- (a) J. R. Moffat and D. K. Smith, Chem. Commun., 2008, 316–318 Search PubMed; (b) A. Bimalendu, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC; (c) A. Das and S. Ghosh, Chem. Commun., 2011, 47, 8922–8924 RSC; (d) K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed; (e) C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC; (f) E. R. Draper, J. R. Lee, M. Wallace, F. Jackel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC; (g) N. Singh, K. Zhang, C. A. Angulo-Pachon, E. Mendes, J. H. van Esch and B. Escuder, Chem. Sci., 2016, 7, 5568–5572 RSC; (h) S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
- (a) A. Heeres, C. van der Pol, M. C. A. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed; (b) A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063–2066 CrossRef CAS PubMed; (c) H.-K. Lee, S. Soukasene, H. Jiang, S. Zhang, S. Feng and S. I. Stupp, Soft Matter, 2008, 4, 962–964 RSC; (d) M. Laupheimer, K. Jovic, F. E. Antunes, M. D. M. Miguel and C. Stubenbrauch, Soft Matter, 2013, 9, 3661–3670 RSC; (e) S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed; (f) S. Himmelein, V. Lewe, M. C. A. Stuart and B. J. Ravoo, Chem. Sci., 2014, 5, 1054–1058 RSC; (g) J. Boekhoven, A. M. Brizard, M. C. A. Stuart, L. Florusse, G. Raffy, A. Del Guerzo and J. H. van Esch, Chem. Sci., 2016, 7, 6021–6031 RSC; (h) C. Stubenrauch and F. Giesselmann, Angew. Chem., Int. Ed., 2016, 55, 3268–3275 CrossRef CAS PubMed.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- (a) T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS; (b) W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed; (c) J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS.
- (a) K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC; (b) J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed; (c) M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- (a) D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC; (b) S. Middeldorp, Thromb. Res., 2008, 122, 753–762 CrossRef CAS PubMed.
- (a) A. Gutowska, Y. H. Bae, J. Feijen and S. W. Kim, J. Controlled Release, 1992, 22, 95–104 CrossRef CAS; (b) A. Gutowska, Y. H. Bae, H. Jacobs, J. Feijen and S. W. Kim, Macromolecules, 1994, 27, 4167–4175 CrossRef CAS; (c) F. P. Bonina and L. Montenegro, Int. J. Pharm., 1994, 102, 19–24 CrossRef CAS; (d) E. R. Edelman, A. Nathan, M. Katada, J. Gates and M. J. Karnovsky, Biomaterials, 2000, 21, 2279–2286 CrossRef CAS PubMed; (e) G. McLennan, M. S. Johnson, K. R. Stookey, Z. D. Zhang and W. K. Fife, J. Vasc. Intervent. Radiol., 2000, 11, 1087–1094 CrossRef CAS; (f) M. S. Ahola, E. S. Sailynoja, M. H. Raitavuo, M. M. Vaahtio, J. I. Salonen and Y.-U. Auo, Biomaterials, 2001, 22, 2163–2170 CrossRef CAS PubMed; (g) N. Roveri, M. Morpurgo, B. Palazzo, B. Parma and L. Vivi, Anal. Bioanal. Chem., 2005, 381, 601–606 CrossRef CAS PubMed; (h) M. Radivojsa, I. Grabnar and P. A. Grabnar, Eur. J. Pharm. Sci., 2013, 50, 93–101 CrossRef CAS PubMed; (i) S. E. Sakiyama-Elbert, Acta Biomater., 2014, 10, 1581–1587 CrossRef CAS PubMed; (j) S. E. Sakiyama-Elbert and C. Werner, Drug Delivery via Heparin Conjugates, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2017 Search PubMed.
- (a) C. Loira-Pastoriza, A. Sapin-Minet, R. Diab, J. L. Grossiord and P. Maincent, Int. J. Pharm., 2012, 426, 256–262 CrossRef CAS PubMed; (b) M. R. Metanovic, I. Grabnar, M. Gosenca and P. A. Grabnar, Int. J. Pharm., 2015, 488, 127–135 CrossRef PubMed.
- (a) S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 69, 149–158 CrossRef CAS; (b) J. A. Beamish, L. C. Geyer, N. A. Haq-Siddiqi, K. Kottke-Marchant and R. E. Marchant, Biomaterials, 2009, 30, 6286–6294 CrossRef CAS PubMed.
- (a) K. Rajangam, H. A. Behanna, M. J. Hui, X. Han, J. F. Hulvat, J. W. Lomansey and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS PubMed; (b) T. Fernández-Muiños, L. Recha-Sancho, P. López-Chicón, C. Castells-Sala, A. Mata and C. E. Semino, Acta Biomater., 2015, 16, 35–48 CrossRef PubMed; (c) Y. M. Abul-Haija and R. V. Ulijn, Biomacromolecules, 2015, 16, 3473–3479 CrossRef CAS PubMed; (d) L. Recha-Sancho and C. E. Semino, J. Biomed. Mater. Res., 2016, 104, 1694–1706 CrossRef CAS PubMed; (e) M. K. Wlodarczyk-Biegun, C. J. Slingerland, M. W. T. Werten, I. A. van Hees, S. A. de Wolf, R. de Vries, M. A. C. Stuart and M. Kamperman, Biomacromolecules, 2016, 17, 2063–2072 CrossRef CAS PubMed.
- (a) D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC; (b) D. J. Corwnell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 Search PubMed.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9185 RSC.
- (a) M. Kim, J. Y. Lee, C. N. Jones, A. Revzin and G. Tae, Biomaterials, 2010, 31, 3596–3603 CrossRef CAS PubMed; (b) Y.-T. Hou, H. Ijima, T. Takei and K. Kawakami, J. Biosci. Bioeng., 2011, 112, 265–272 CrossRef CAS PubMed; (c) K. H. Hussein, K.-M. Park, K. S. Kang and H. M. Woo, Acta Biomater., 2016, 38, 82–93 CrossRef CAS PubMed.
- (a) A. C. Rodrigo, A. Barnard, J. Cooper and D. K. Smith, Angew. Chem., Int. Ed., 2011, 50, 4675–4679 CrossRef CAS PubMed; (b) S. M. Bromfield, P. Posocco, C. W. Chan, M. Calderon, S. E. Guimond, J. E. Turnbull, S. Pricl and D. K. Smith, Chem. Sci., 2014, 5, 1484–1492 RSC.
- L. Fechner, B. Albanyan, V. M. P. Vieira, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Chem. Sci., 2016, 7, 4653–4659 RSC.
- V. M. P. Vieira, V. Liljeström, P. Posocco, E. Laurini, S. Pricl, M. A. Kostiainen and D. K. Smith, J. Mater. Chem. B, 2017, 5, 341–347 RSC.
- (a) P. Serwer, Electrophoresis, 1983, 4, 375–382 CrossRef CAS; (b) B. Rahfoth, J. Weisser, F. Sternkopf, T. Aigner, K. von der Mark and R. Bräuer, Osteoarthritis Cartilage, 1998, 6, 50–65 CrossRef CAS PubMed; (c) M. Chau, S. E. Sriskandha, H. Therien-Aubin and E. Kumacheva, Adv. Polym. Sci., 2015, 68, 167–208 CrossRefR. Fan, M. Piou, E. Darling, D. Cormier, J. Sun and J. Wan, J. Biomater. Appl., 2016, 31, 684–692 CrossRef CAS PubMed.
- (a) G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC; (b) V. J. Nebot and D. K. Smith, in Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, RSC Publishing, Cambridge, 2014, pp. 30–66 Search PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- (a) J. Raeburn, T. O. McDonald and D. J. Adams, Chem. Commun., 2012, 48, 9355–9357 RSC; (b) D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS PubMed.
- Heparin concentrations are reported with respect to the most commonly occurring disaccharide repeat unit, to determine charge ratios, we assume this unit carries four negative charges under physiological conditions.
- (a) B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed; (b) A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem.–Eur. J., 2009, 15, 372–379 CrossRef CAS PubMed.
- (a) K. Hanabusa, A. Itoh, M. Kimura and H. Shirai, Chem. Lett., 1999, 767–768 CrossRef CAS; (b) X. Y. Liu and P. D. Sawant, Angew. Chem., Int. Ed., 2002, 41, 3641–3645 CrossRef CAS; (c) X. Y. Liu, P. D. Sawant, W. B. Tan, I. B. M. Noor, C. Pramesti and B. H. Chen, J. Am. Chem. Soc., 2002, 124, 15055–15063 CrossRef CAS PubMed; (d) L. Chen, S. Revel, K. Morris, D. G. Spiller, L. C. Serpell and D. J. Adams, Chem. Commun., 2010, 46, 6738–6740 RSC; (e) G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797–7802 RSC; (f) P. Chakraborty, B. Roy, P. Bairi and A. K. Nandi, J. Mater. Chem., 2012, 22, 20291–20298 RSC; (g) P. Chakraborty, S. Das, S. Mondal, P. Bairi and A. K. Nandi, Langmuir, 2016, 32, 1871–1880 CrossRef CAS PubMed.
- M. Calamai, J. R. Kumita, J. Mifsud, C. Parrini, M. Ramazzotti, G. Ramponi, N. Taddei, F. Chiti and C. M. Dobson, Biochemistry, 2006, 45, 12806–12815 CrossRef CAS PubMed; C. Iannuzzi, G. Irace and I. Sirangelo, Molecules, 2015, 20, 2510–2518 CrossRef PubMed.
- (a) E. R. Draper, L. L. E. Mears, A. M. Castilla, S. M. King, T. O. McDonald, R. Akhtar and D. J. Adams, RSC Adv., 2015, 5, 95369–95378 RSC; (b) Y. Abidine, V. M. Laurent, R. Michel, A. Duperray, L. I. Palade and C. Verdier, Europhys. Lett., 2015, 109, 38003 CrossRef.
- (a) S. M. Bromfield, A. Barnard, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 2911–2914 CrossRef CAS PubMed; (b) S. M. Bromfield, P. Posocco, M. Fermeglia, S. Pricl, J. Rodríguez-López and D. K. Smith, Chem. Commun., 2013, 49, 4830–4832 RSC.
- J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC.
- (a) J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC; (b) A. Das, M. R. Molla and S. Ghosh, J. Chem. Sci., 2011, 123, 963–973 CrossRef CAS; (c) S. Onogi, H. Shigemitsu, T. Yoshii, T. Taneda, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
- S. M. Bromfield, P. Posocco, M. Fermeglia, J. Tolosa, A. Herreros-López, S. Pricl, J. Rodriguez-López and D. K. Smith, Chem.–Eur. J., 2014, 20, 9666–9674 CrossRef CAS PubMed; B. Albanyan, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Chem.–Eur. J., 2017, 23, 6391–6397 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental methods and further data from assays. See DOI: 10.1039/c7sc03301j |
| This journal is © The Royal Society of Chemistry 2017 |