
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVapor–solid synthesis of monolithic single-crystalline CoP nanowire electrodes for efficient and robust water electrolysis†
Wei
Li
 a,
Xuefei
Gao
b,
Dehua
Xiong
a,
Fang
Xia
c,
Jian
Liu
b,
Wei-Guo
Song
b,
Junyuan
Xu
a,
Sitaramanjaneya Mouli
Thalluri
a,
M. F.
Cerqueira
d,
Xiuli
Fu
e and
Lifeng
Liu
*a
a,
Xuefei
Gao
b,
Dehua
Xiong
a,
Fang
Xia
c,
Jian
Liu
b,
Wei-Guo
Song
b,
Junyuan
Xu
a,
Sitaramanjaneya Mouli
Thalluri
a,
M. F.
Cerqueira
d,
Xiuli
Fu
e and
Lifeng
Liu
*a
aInternational Iberian Nanotechnology Laboratory (INL), Avenida Mestre José Veiga, 4715-330 Braga, Portugal. E-mail: lifeng.liu@inl.int
bBeijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
cSchool of Engineering and Information Technology, Murdoch University, Murdoch, Western Australia 6150, Australia
dCenter for Physics, University of Minho, 4710-057 Braga, Portugal
eState Key Laboratory of Information Photonics and Optical Communications, School of Science, Beijing University of Posts and Telecommunications, Beijing 100876, P. R. China
First published on 23rd January 2017
Abstract
Electrochemical water splitting into hydrogen and oxygen is a promising technology for sustainable energy storage. The development of earth-abundant transition metal phosphides (TMPs) to catalyze the hydrogen evolution reaction (HER) and TMP-derived oxy-hydroxides to catalyze the oxygen evolution reaction (OER) has recently drawn considerable attention. However, most monolithically integrated metal phosphide electrodes are prepared by laborious multi-step methods and their operational stability at high current densities has been rarely studied. Herein, we report a novel vapor–solid synthesis of single-crystalline cobalt phosphide nanowires (CoP NWs) on a porous Co foam and demonstrate their use in overall water splitting. The CoP NWs grown on the entire surface of the porous Co foam ligaments have a large aspect ratio, and hence are able to provide a large catalytically accessible surface over a given geometrical area. Comprehensive investigation shows that under the OER conditions CoP NWs are progressively and conformally converted to CoOOH through electrochemical in situ oxidation/dephosphorization; the latter serving as an active species to catalyze the OER. The in situ oxidized electrode shows exceptional electrocatalytic performance for the OER in 1.0 M KOH, delivering 100 mA cm−2 at an overpotential (η) of merely 300 mV and a small Tafel slope of 78 mV dec−1 as well as excellent stability at various current densities. Meanwhile, the CoP NW electrode exhibits superior catalytic activity for the HER in the same electrolyte, affording −100 mA cm−2 at η = 244 mV and showing outstanding stability. An alkaline electrolyzer composed of two symmetrical CoP NW electrodes can deliver 10 and 100 mA cm−2 at low cell voltages of 1.56 and 1.78 V, respectively. The CoP NW electrolyzer demonstrates exceptional long-term stability for overall water splitting, capable of working at 20 and 100 mA cm−2 for 1000 h without obvious degradation.
Introduction
Exploitation of abundant yet intermittent renewable energy sources needs efficient energy storage technologies.1,2 Electrochemical water splitting is a promising clean energy technology to convert electricity from renewable sources into hydrogen (H2) fuel.3 Water splitting consists of two half reactions: the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), occurring at the anode and cathode, respectively, both of which contribute to the overall efficiency of water electrolysis. Noble metal oxides (e.g., RuO2) and the precious metal Pt are the state-of-the-art electrocatalysts for the OER and HER, respectively. However, their high cost and scarcity restrict their widespread use in electrolyzers. Significant efforts have been devoted to developing novel, low-cost and highly efficient electrocatalysts based on earth-abundant elements, such as perovskite oxides,4 transition metal (TM) oxides,5–7 (oxy)hydroxides,8–10 and phosphates11 for the OER, and transition metal chalcogenides,12–14 phosphides,15–21 and nitrides22,23 for the HER.Among the non-precious TM-based electrocatalysts reported so far, transition metal phosphides (TMPs) have recently attracted considerable interest because of their proven outstanding electrocatalytic activity for the HER.15–21 Besides, it was recently reported that TMPs can be transformed through in situ electrochemical oxidation/dephosphorization to TM oxy-hydroxides, capable of efficiently catalyzing the OER.24–32 Moreover, the in situ transformed catalytically active species in many cases exhibit an OER performance even better than their corresponding pristine TM oxy-hydroxide counterparts with similar structures/morphologies,28,29,31,33,34 likely due to the fact that surface roughening and micro-structural reconstruction during the electrochemical cold-working enable the exposure of more active sites.
To accomplish highly-efficient and sustained overall water splitting, it is important and desirable to develop monolithic electrodes that directly integrate as many active electrocatalysts as possible in a given geometrical area. Monolithic electrodes can offer several advantages over the common powder-like catalysts.35 First, powder-like catalysts must be immobilized on a current collector using a polymeric binder. The introduction of polymer binders may not only increase the series resistance of the electrode, but also block some of active sites resulting in reduced activity. Second, the powder-like catalysts are prone to losing physical contact with the current collector if a large electrolysis current/potential is applied, owing to the violent evolution of gas bubbles that tends to push the catalysts away from the conductive support. As a consequence, long-term operational stability, which is very important for practical applications, cannot be expected. By contrast, in a monolithic electrode catalysts are directly grown on the current collector forming a monolith with the substrate, and therefore there is no need for the use of any binders to immobilize the catalysts and the electrode would be very robust upon long-term operation, particularly under high current/potential conditions. Last but not least, when casting powder-like catalysts on a current collector, a high loading of catalysts would lead to poor performance because of mass transfer restrictions and the high electrical resistance associated with a high loading of polymer binders; while a monolithic electrode would be able to overcome this limitation, providing high turnover at a high loading mass per unit area, particularly for a porous electrode on which high aspect ratio nanostructured catalysts are grown. Notwithstanding some progress on self-supported electrodes through multi-step synthetic routes,16,25,28,36,37 fabricating monolithically integrated TMP nano-catalyst electrodes in a pretty simple one-step manner and on a massive scale, which can sustain stable overall water splitting with a long lifetime at high current densities (i.e., high H2 production rate), still remains a formidable challenge.
In this work, we report the vapor–solid synthesis of monolithic electrodes covered with dense single-crystalline CoP nanowires (CoP NWs) through a one-step thermal phosphorization treatment of porous metallic Co foam (CF) using red phosphorous (P). It is worth mentioning that CF is used here as both the current collector and the precursor to grow CoP NWs, and no additional Co sources are needed. The CoP NWs grown on the ligaments of porous CF have a large aspect ratio (up to 2000), and are therefore able to provide a large catalytically accessible surface over a given geometrical area. We have carefully investigated the in situ transformation of CoP NWs to catalytically active CoOOH using comprehensive characterization techniques. The in situ oxidized CoP NW electrode shows high electrocatalytic activity for the OER (delivering 100 mA cm−2 at an overpotential of 300 mV) in 1.0 M KOH, a small Tafel slope of 78 mV dec−1 and remarkable stability at a wide range of current densities, ranking among the top-performing non-precious OER catalysts. Moreover, we demonstrate that the as-obtained CoP NW electrode exhibits outstanding HER performance in the same electrolyte. An alkaline electrolyzer prototype was constructed using two symmetrical CoP NW electrodes, which can deliver 10 and 100 mA cm−2 at small cell voltages of 1.56 V and 1.78 V at room temperature, respectively. This performance is comparable to that of an electrolyzer comprising state-of-the-art RuO2 (anode) and Pt–C (cathode) catalysts. Significantly, the CoP NW electrolyzers demonstrate extraordinary long-term stability for water splitting, capable of working at 20 and 100 mA cm−2 for up to 1000 h with little degradation.
Results and discussion
A large-area monolithic integrated CoP NW electrode (3 × 4.5 cm2) was prepared by a simple and fast one-step thermal phosphorization reaction between metallic CF and red P vapor at an optimal temperature of 600 °C (see details in ESI†). The bare CF is composed of interconnected Co ligaments with smooth surfaces (Fig. S1, ESI†). After phosphorization, the surface of CF is uniformly covered by dense CoP NWs with diameters in the range of 25–100 nm and lengths up to 50 μm (Fig. 1a and b), leading to high aspect ratios up to 2000. Fig. 1c shows the X-ray diffraction (XRD) patterns of the CoP NWs and bare CF, demonstrating that the crystal phase of the product can be indexed to orthorhombic CoP (ICDD No. 00-029-0497). The energy dispersive X-ray (EDX) spectrum (Fig. 1d) taken from the NW layer confirms that the NWs consist exclusively of Co and P, with an atomic ratio of Co/P close to 1. Calculated on the basis of the weight increment after phosphorization, the Co/P atomic ratio of the bulk foam was 2.9, indicating that the foam skeleton underneath the CoP NW layer remained as metallic Co. When the phosphorization temperature was increased to 700 °C, less dense and shorter NWs were obtained, which consisted of mixed crystal phases of CoP and Co2P (Fig. S2, ESI†). A further increase in temperature to 800 °C resulted in the growth of only sparsely distributed coarsened NWs possessing Co2P as the major phase (Fig. S2, ESI†). The Co/P atomic ratio calculated based on the weight increase is ca. 1.95, very close to the stoichiometric ratio of Co2P detected by XRD, indicating that very little metallic Co skeleton was left underneath the phosphide layer. Correspondingly, the electrodes obtained upon thermal phosphorization at 700 and 800 °C were more brittle than that obtained at 600 °C.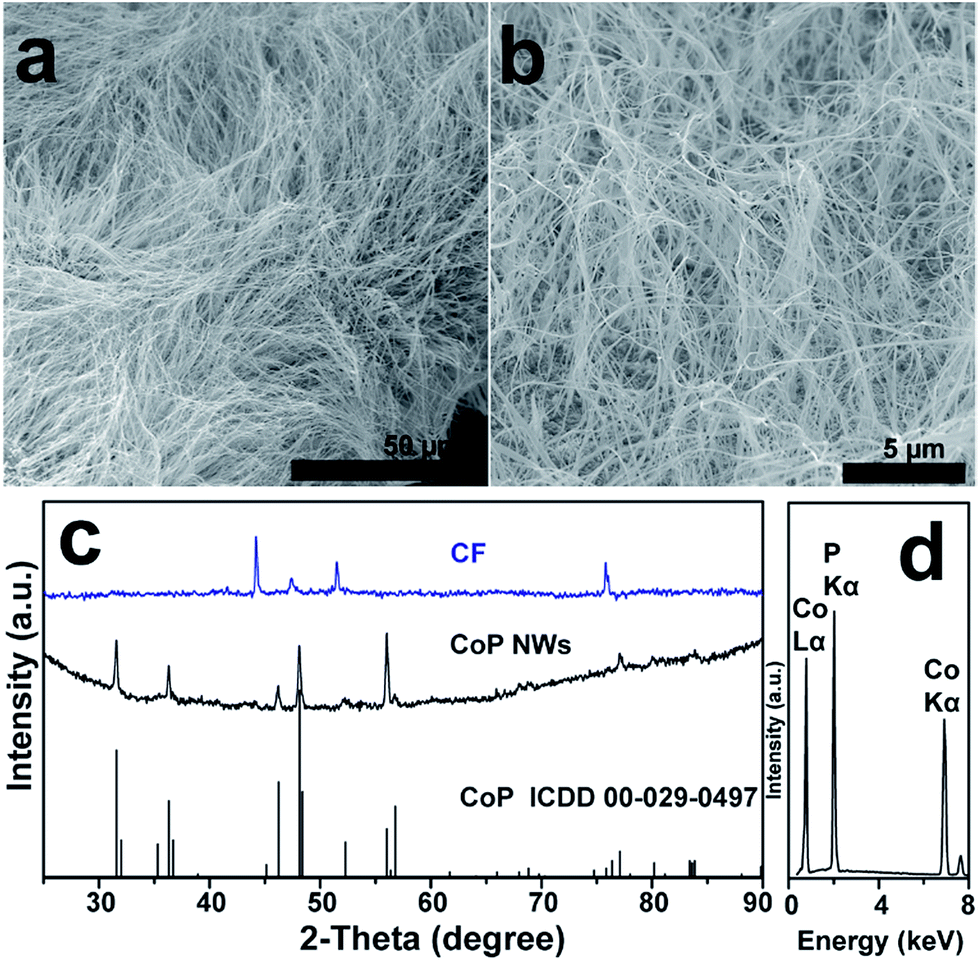 | ||
| Fig. 1 (a and b) SEM images of CoP NWs. (c) XRD patterns of the CoP NW electrode and bare Co foam (CF). The standard ICDD powder diffraction pattern of CoP (No. 00-029-0497) is also given for reference. (d) EDX spectrum of CoP NWs. The CoP NW electrode was obtained upon thermal phosphorization at 600 °C. | ||
The CoP NWs synthesized at 600 °C were characterized using transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM). Fig. 2a shows a low-magnification TEM image of many uniform CoP NWs with diameters ranging from 25 to 100 nm. The high-resolution TEM (HRTEM) image (Fig. 2b) shows a single-crystalline CoP NW with a diameter of ca. 25 nm. The well-resolved lattice fringes with an interplanar spacing of 0.254 nm reveal the (200) crystal planes of orthorhombic CoP. The corresponding fast Fourier transformation (FFT) pattern of this CoP NW demonstrates its single crystal nature (inset of Fig. 2a), that is distinct from the polycrystalline CoP nanostructures reported previously.16,17,28 The HAADF-STEM elemental mapping (Fig. 2c–f) demonstrates that Co and P are uniformly distributed over the NW. The surface chemical state of the CoP NWs was investigated by X-ray photoelectron spectroscopy (XPS), demonstrating that the peaks of Co 2p and P 2p (Fig. S3, ESI†) are consistent with previously reported CoP.16,38
 | ||
| Fig. 2 (a) TEM image of CoP NWs. Inset: FFT-electron diffraction pattern of a single NW shown in (b). (b) HRTEM image of an individual CoP NW. Inset: zoomed view of the marked area. (c) HAADF-STEM image and elemental maps of (d) Co, (e) P and (f) their overlap taken from a single CoP NW. | ||
The monolithic CoP NW foam was directly used as an integrated electrode to catalyze the OER in O2-saturated 1.0 M KOH in a three-electrode configuration. The CoP NW electrode was first conditioned by repetitive cyclic voltammetry (CV) scans between 0.8375 and 1.6375 V vs. a reversible hydrogen electrode (RHE) at a rate of 2 mV s−1 for 10 continuous cycles, after which a steady state was achieved. Fig. 3a shows the iR-corrected CV curves of the conditioned CoP NW electrode, bare CF and a commercial RuO2 nano-catalyst recorded at 2 mV s−1 on the RHE scale. The conditioned CoP NW electrode exhibits catalytic activity superior to bare CF and state-of-the-art RuO2 for the OER. The bare CF only delivers a current density of 10 mA cm−2 at an overpotential of 373 mV (η10). The RuO2 catalyst can afford 10 mA cm−2 at η10 = 318 mV. In contrast, the conditioned CoP NW electrode first shows an anodic peak at ca. 1.1 V prior to the onset of the OER, suggesting the oxidation of Co2+ to Co3+. The redox peaks are virtually reversible, consistent with previously reported CoP nanoparticles.27 The electrocatalytically accessible active sites of the conditioned CoP NWs and bare CF electrodes are estimated and compared by integrating the charge in the well-defined Co2+ ↔ Co3+ oxidation or reduction peak (Fig. S4, ESI†), assuming that the reaction occurs only on the Co exhibiting redox feature and that each electron is associated with a single surface-active Co ion.39 For the conditioned CoP NW electrode, the integrated charges of the oxidation and reduction peaks of the CV curve are 68 and 38 times higher than those of the bare CF, respectively. This indicates that the growth of CoP NWs on Co foam has significantly increased the accessible active sites of the electrode.
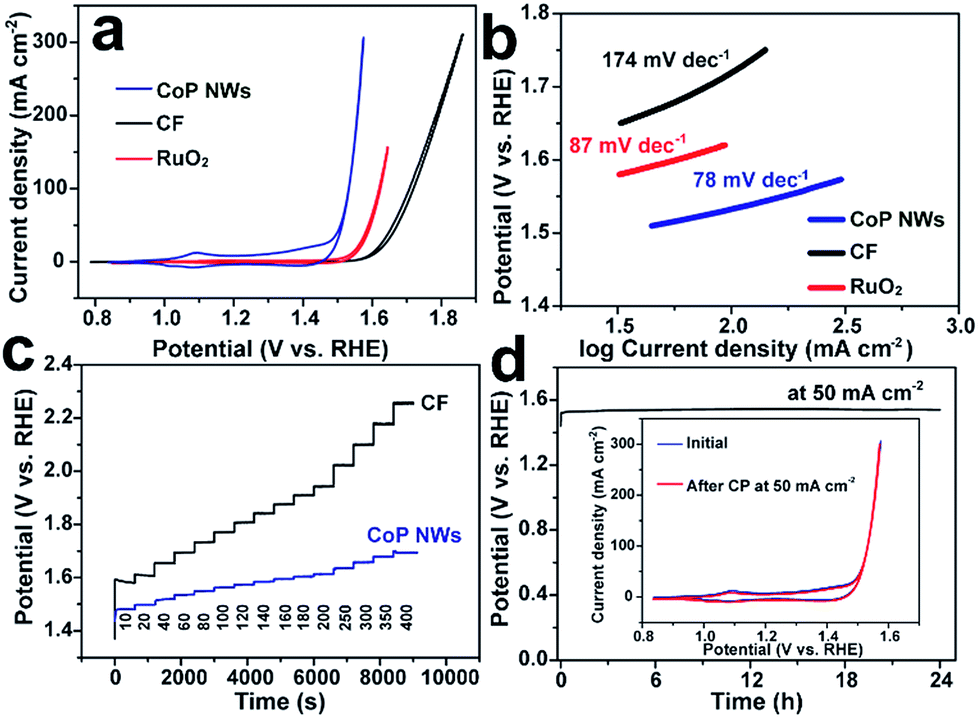 | ||
| Fig. 3 Electrocatalytic performance of electrodes for the OER measured in O2-saturated 1.0 M KOH. (a) iR-Corrected steady-state CV curves of conditioned CoP NWs, bare CF and a commercial RuO2 catalyst supported on CF. (b) The corresponding Tafel plots derived from the reduction branch of the CV curves. (c) Multi-step chronopotentiometric (CP) curves of conditioned CoP NWs and bare CF at varying current densities without iR-correction. (d) The CP curve of the conditioned CoP NW electrode recorded at a current density of 50 mA cm−2 without iR-correction. The inset shows the CV curves of the electrode before and after the CP measurement at 50 mA cm−2 for 24 h. | ||
From the reduction branch of its CV, the conditioned CoP NW electrode demonstrates OER current densities of 10, 20 and 100 mA cm−2 at overpotentials of 248 (η10), 261 (η20) and 300 mV (η100), respectively. This performance is substantially higher than that of state-of-the-art RuO2 and also better than that of many other reported non-precious OER catalysts tested under similar conditions (Table S1, ESI†), such as Co–P film (η10 = 345 mV; η100 = 413 mV),36 NiFe layered double hydroxide (LDH) (η20 = 270 mV; η100 = 470 mV) and CoSe2 (η10 = 320 mV; η20 = 370 mV).9,40 The Tafel slope of the conditioned CoP NWs is 78 mV dec−1, smaller than that of RuO2 (87 mV dec−1) and bare CF (174 mV dec−1), suggesting more favorable OER kinetics at the conditioned CoP NW electrode. The fast OER kinetics on the conditioned CoP NWs was also verified by electrochemical impedance spectroscopy (EIS) analysis. The charge transfer resistance of the conditioned CoP NWs (0.34 Ω) is substantially smaller than that of the bare CF (6.77 Ω) at 1.557 V (Fig. S5, ESI†).
It is worth mentioning that we also attempted to optimize the phosphorization conditions (e.g., the feed of red P, reaction time, and temperature) to explore if the OER catalytic performance of the conditioned CoP NW electrodes can be further improved. It turned out that increasing the loading of red P for the phosphorization treatment has little impact on the OER activity of the resulting electrodes (Fig. S6, ESI†). Similarly, prolonging the phosphorization time (e.g., to 6 h) didn't substantially change the OER activity of the obtained electrodes, either (Fig. S7, ESI†). As mentioned above, increasing the phosphorization temperature resulted in the growth of less dense and shorter NWs (Fig. S2, ESI†), and thereby slightly deteriorated the OER activity of the resulting electrodes (Fig. S8, ESI†). Nevertheless, the observed deterioration only became prominent at high overpotentials. Therefore, the CoP NW electrode obtained at 600 °C and 1 h phosphorization was demonstrated to be the optimal one for the OER.
The capability of the conditioned CoP NW electrode and bare CF to catalyze the OER at varying current densities from 10 to 400 mA cm−2 was investigated by chronopotentiometry (CP). Fig. 3c illustrates the CP curves of the electrodes without iR-correction. Upon increasing the current density, the potential of the conditioned CoP NWs rises but gets stabilized quickly, indicating their outstanding mass transfer properties and mechanical robustness.41,42 At each current density, the overpotential of the conditioned CoP NWs is significantly lower than that of the bare CF. The conditioned CoP NW electrode only needs a potential of 1.60 V (non iR-corrected) to deliver a high current density of 400 mA cm−2. The outstanding rate capability of the monolithic NW electrode is superior to that of graphene/Co0.85Se/NiFe-LDH composites and NiSe/Ni foam with a similar self-supported electrode structure.41,42 Besides, the conditioned CoP NW electrode demonstrates excellent durability with a stable potential of 1.54 V (non iR-corrected) to deliver 50 mA cm−2 for 24 h without obvious degradation. After the continuous CP test, the conditioned CoP NW electrode still exhibits a CV curve pretty similar to the initial one, implying its remarkable stability.
It is known that TMPs usually undergo structural and compositional transformation under OER conditions, forming TM oxy-hydroxides which have been proposed to be the true catalytic species for the OER.25–32,43–45 To investigate this electrochemical in situ oxidation/dephosphorization process, we performed comprehensive ex situ characterization on five as-obtained CoP NW electrodes which had undergone chronoamperometric (CA) tests at 1.54 V vs. RHE for 20 s, 5 min, 30 min, 3 h and 6 h (denoted as CA-T, T = time), respectively (Fig. S9, ESI†). While no visible changes could be found in the CA-20 s electrode using SEM, a sheath coating on the CoP NWs could be clearly resolved after 5 min CA testing (Fig. S10, ESI†). As the OER was proceeding, the NW morphology was retained, but the NW surface became roughened and coarsened. SEM-EDX spectroscopy analyses revealed the decrease of the P/Co ratio and the appearance of an O peak with ever-increasing signals over time (Fig. S11, ESI†), indicating that the in situ electrochemical oxidation/dephosphorization was ongoing during the continued CA process. Microstructural changes of the CoP NWs are seen more clearly using TEM. The HADDF-STEM image and elemental maps show that the in situ transformation had already happened in the CA-20 s electrode (Fig. S12, ESI†). This implies that the electrochemical oxidation/dephosphorization virtually occurs immediately upon the application of an oxidative potential. After the CA test for 5 min, a well-defined CoP/CoOOH core/shell structure could be observed (Fig. S13, ESI†), where O is primarily seen in the “shell”, and P is confined within the “core”, while Co is diffusively distributed across the NW. This indicates that the core of the NW remains as CoP, but that the outer layer consists of in situ generated CoOOH. Similar core/shell structures consisting of a non-oxide “core” and a metal oxy-hydroxide/oxide “shell” were also reported previously for CoP,27,32,43 Co2P,44 Co–Ni–P,25 CoN,46,47 Co2B48 and Ni2P.26 The conversion of CoP to CoOOH continued to happen as the OER electrolysis went on continuously (Fig. S14, ESI†). After 3 h, the in situ transformation was nearly completed: the core/shell structure was not visible any more, and Co and O were evenly distributed across the NW (Fig. S15 and S16, ESI†). Our Raman spectroscopy and XPS analyses support our microscopic observations (Fig. S17 and S18, ESI†), demonstrating that CoOOH starts to appear in the CA-20 s electrode, and P is leaching out as the electrolysis proceeds. This happens in a favorable way so that the integrity of the NWs is not affected. However, how this structural and compositional transformation correlates with their electrocatalytic behavior (Fig. S9, ESI†) remains unclear, and will be investigated in the future.
Aside from the OER performance, we also studied the electrocatalytic activity of the as-obtained CoP NW electrode for the HER. It was found that the electrode synthesized at 600 °C exhibits excellent catalytic performance in 1.0 M KOH with a small overpotential (−100 mA cm−2 at η = 244 mV), a Tafel slope of 105 mV dec−1, favorable rate capability as well as good stability (Fig. S19, ESI†), which are superior to those of the CoP NW electrodes obtained at 700 and 800 °C and many other non-precious HER catalysts reported in the literature (Fig. S20 and Table S2, ESI†).
Given the aforementioned remarkable catalytic performance, an electrolyzer was assembled using a conditioned CoP NW electrode as the anode and an as-obtained CoP NW electrode as the cathode, and overall water splitting was carried out in 1.0 M KOH solution. For comparison, control electrolyzers comprising RuO2 (anode) and Pt–C (cathode) noble-metal catalysts immobilized on CFs (RuO2||Pt–C) as well as a pair of bare CFs, were also fabricated.
Fig. 4a illustrates the polarization curves of the CoP NW, bare CF and RuO2||Pt–C electrolyzers measured in a two-electrode configuration. The CoP NW electrolyzer only requires cell voltages of 1.56 (V10), 1.67 (V20) and 1.78 V (V100) to deliver current densities of 10, 20 and 100 mA cm−2, respectively. These voltages are slightly larger than those of the integrated RuO2||Pt–C (V10 = 1.56 V, V20 = 1.62 V), yet much smaller than those of the bare CF electrolyzer, indicating the high electrocatalytic activity of the non-precious CoP NWs for the HER and the in situ oxidized CoP NWs for the OER. The performance of CoP NW electrolyzer outperforms that of many electrode pairs reported recently (Table S3, ESI†), such as Co–P film (V10 = 1.65 V, V20 = 1.67 V), NiFe LDH (V10 = 1.7 V, V20 = 1.78 V) and NiSe/Ni foam (V10 = 1.63 V, V20 = 1.75 V).9,36,41
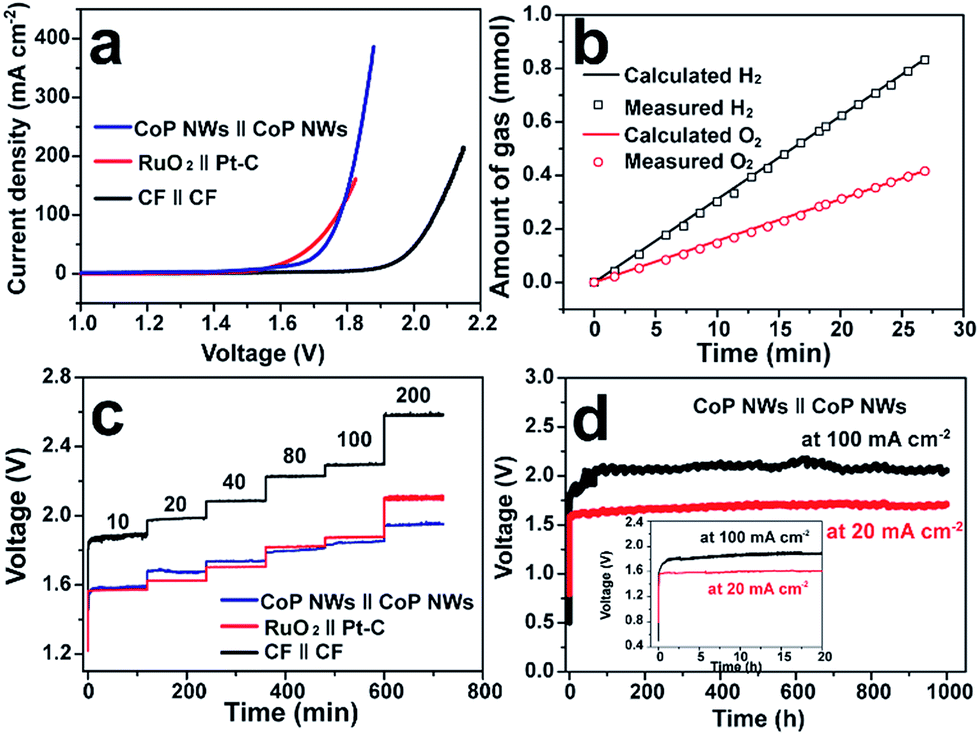 | ||
| Fig. 4 Overall water splitting performance of the two-electrode electrolyzers. (a) Polarization curves of CoP NWs, bare CF and RuO2(+)‖Pt–C(−) supported on CF. (b) Gas yield of H2 and O2 evolved over the CoP NW electrodes as a function of time at 100 mA cm−2. (c) Multi-step CP curves of the CoP NW and bare CF electrolyzers at varying current densities. (d) Long-term stability test of the CoP NW electrolyzer at 20 and 100 mA cm−2. Inset: zoomed view of the curves in the first 20 h. All experiments were conducted in 1.0 M KOH at room temperature. | ||
The evolution of H2 and O2 gases was measured during electrolysis at 100 mA cm−2 (Fig. 4b). The amounts of experimentally measured H2 and O2 match very well with theoretically calculated values, and the molar ratio of produced H2 and O2 is close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating a faradaic efficiency of nearly 100% and a H2 production rate of 1.87 mmol h−1 cm−2.
:1, indicating a faradaic efficiency of nearly 100% and a H2 production rate of 1.87 mmol h−1 cm−2.
To demonstrate the practical relevance to real water electrolysis, the rate capability and long-term stability of the CoP NW electrolyzer operating at different current densities were evaluated by galvanostatic water electrolysis. Fig. 4c presents multi-step CP curves of the CoP NW, bare CF and RuO2||Pt–C electrolyzers recorded under varying current densities without iR-correction (i.e., including real resistive loss in the electrolyte). The cell voltage of the CoP NW electrolyzer increases and becomes stabilized quickly as the current density goes up from 10 to 200 mA cm−2. The steady-state cell voltages of the CoP NW electrolyzer are 1.58, 1.68, 1.74, 1.81, 1.85 and 1.95 V, to deliver 10, 20, 40, 80, 100 and 200 mA cm−2, respectively. The energy efficiencies of these electrolyzers were calculated by dividing the thermoneutral potential of water electrolysis (i.e., 1.48 V) by the cell voltages (see details in Fig. S21, ESI†). At 10 mA cm−2, the CoP NW electrolyzer exhibits a high energy efficiency up to 93.7%. Although the energy efficiency gradually decreases with an increase in current density, the CoP NW electrolyzer still retains energy efficiencies of 80% and 76% even at 100 and 200 mA cm−2, respectively. In contrast, the energy efficiency of the bare CF electrolyzer is substantially lower than that of the CoP NW electrolyzer, indicating a greater energy loss of bare CF for full water splitting. Compared to that of RuO2||Pt–C, the energy efficiency of the CoP NW electrolyzer is slightly lower by ca. 2% at current densities below 40 mA cm−2, yet exceeds that of RuO2||Pt–C at high current densities (≥80 mA cm−2). This indicates that the cost-effective CoP and in situ oxidized CoOOH catalysts exhibit high energy efficiency for H2 production at high rates relevant to practical applications. Fig. 4d shows the long-term stability of the CoP NW electrolyzer. At 20 mA cm−2, the cell voltage of the electrolyzer rapidly increases from the open circuit voltage (OCV) to ca. 1.58 V within 10 min (inset of Fig. 4d) and finally it was stabilized at ca. 1.68 V for 1000 h with little degradation (0.1 mV h−1 increase rate in voltage). Likewise, at 100 mA cm−2, the cell voltage also undergoes a rising process in the first 2 h. Afterwards, the electrolyzer can keep working at this high current density for 1000 h with little performance decay (0.2 mV h−1 increase rate in voltage), which is of importance and relevance for industrial water splitting, showing extraordinary long-term stability. It is believed that the increase in the cell voltage at the beginning of electrolysis results from the increased internal resistance of the electrodes due to the formation of electrically more resistive CoOOH or cobalt oxide on the surface of CoP upon electrolysis. As evidence, the internal resistance of our CoP NW electrolyzer went up from the initial 0.548 Ω to 1.464 Ω after the CP test for 1000 h, according to the impedance spectroscopy measurements. The increase in cell voltage at the beginning of electrolysis was also observed previously in other metal phosphide systems for overall water splitting.24,26,30,49
The structure and morphology of the CoP NW electrodes after catalyzing overall water splitting were examined. The CoP NWs in the cathode were mostly retained, while the NWs in the anode were completely converted to layered CoOOH after full water splitting at 20 mA cm−2 for 1000 h (Fig. S22–S24, ESI†).
The outstanding electrocatalytic activity and stability of the CoP NWs and in situ converted CoP/CoOOH NWs towards water splitting can be attributed to the following aspects: (i) the unique topology of the electrode comprising a 3D porous Co foam skeleton covered with dense NWs can reduce charge transfer resistance (Fig. S5, ESI†), and facilitate the diffusion of electrolyte and release of the generated gas bubbles.16,28,50,51 (ii) The CoP NWs firmly grow over the surface of the Co skeleton and maintain strong adhesion even after long-term stability tests. In addition, the in situ oxidation/dephosphorization of CoP NWs at the anode occurs in a mild way so that the NW configuration is not damaged during the structural transformation. This helps maintain good OER activity and long-term stability. (iii) The CoP NWs and in situ oxidized CoOOH NWs with very high aspect ratios can provide a large number of exposed active sites in a given geometrical surface area. Therefore, even if deactivation happens somewhere locally (e.g., due to the degradation of CoOOH in alkaline solution),39 it would not immediately cause deterioration of the bulk catalytic activity of the electrode. This could explain why the electrode can sustain overall water splitting up to 1000 h without obvious degradation.
Conclusions
In summary, self-supported monolithic electrodes comprising ultralong single-crystalline CoP nanowires supported on Co foam have been fabricated by a simple, scalable and fast one-step thermal phosphorization of metallic Co foam via a vapor–solid growth route. The CoP NWs grown on the porous Co foam ligaments have a large aspect ratio, and hence are able to provide a large catalytically accessible surface over a given geometrical area for electrochemical water splitting. The large-area CoP NW electrode was demonstrated to be highly catalytically active for both the OER and HER in alkaline solution. Under OER conditions, the CoP NWs are in situ converted to CoOOH progressively, which then serves as the active species to catalyze the OER. The as-obtained and in situ oxidized CoP NW electrodes only need a small overpotential of 244 and 300 mV to deliver −100 and 100 mA cm−2 to catalyze the HER and OER, respectively, and show excellent stability at a wide range of current densities for both reactions. An alkaline electrolyzer composed of two CoP NW electrodes can afford 10 and 100 mA cm−2 at low cell voltages of 1.56 and 1.78 V, respectively, outperforming many non-precious electrocatalysts reported in the literature. Notably, the CoP NW electrolyzer demonstrates remarkable long-term stability for water splitting, operating at 20 and 100 mA cm−2 for 1000 h with little degradation. Given the simple and scalable one-step synthesis, single-crystalline nature and outstanding catalytic activities and stability, monolithic CoP NW electrodes hold great potential for use as efficient and robust electrodes in alkaline electrolyzers. This study also opens up a new synthesis route and opportunities in designing and fabricating monolithically integrated metal phosphide nanostructures for diverse applications.Acknowledgements
L. F. Liu acknowledges the financial support of a FCT Investigator grant (No. IF/01595/2014) and exploratory grant (No. IF/01595/2014/CP1247/CT0001) awarded by the Portuguese Foundation of Science & Technology (FCT). W. Li and D. H. Xiong are thankful for the support of the Marie Skłodowska-Curie Action COFUND programme (NanoTrainforGrowth, grant No. 600375) for postdoctoral fellowships. This work was also partly financed by the European Commission Horizon 2020 project “CritCat” (Grant No. 686053).Notes and references
- C. Xiang, K. M. Papadantonakis and N. S. Lewis, Mater. Horiz., 2016, 3, 169–173 RSC.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- C. Chen, Y. Yu, W. Li, C. Cao, P. Li, Z. Dou and W. Song, J. Mater. Chem., 2011, 21, 12836–12841 RSC.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- W. Li, X. Wang, D. Xiong and L. Liu, Int. J. Hydrogen Energy, 2016, 41, 9344–9354 CrossRef CAS.
- W. Li, D. Chen, F. Xia, J. Z. Y. Tan, J. Song, W.-G. Song and R. A. Caruso, Chem. Commun., 2016, 52, 4481–4484 RSC.
- W. Li, D. Xiong, X. Gao, W.-G. Song, F. Xia and L. Liu, Catal. Today DOI:10.1016/j.cattod.2016.09.007.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- L. Tian, X. Yan, X. Chen, L. Liu and X. Chen, J. Mater. Chem. A, 2016, 4, 13011–13016 CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- A. B. Laursen, K. R. Patraju, M. J. Whitaker, M. Retuerto, T. Sarkar, N. Yao, K. V. Ramanujachary, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2015, 8, 1027–1034 CAS.
- J. L. Lado, X. Wang, E. Paz, E. Carbó-Argibay, N. Guldris, C. Rodríguez-Abreu, L. Liu, K. Kovnir and Y. V. Kolen'ko, ACS Catal., 2015, 5, 6503–6508 CrossRef CAS.
- X. Wang, Y. V. Kolen'ko, X.-Q. Bao, K. Kovnir and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 8188–8192 CrossRef CAS PubMed.
- J. F. Xie, S. Li, X. D. Zhang, J. J. Zhang, R. X. Wang, H. Zhang, B. C. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- W. Li, C.-Y. Cao, C.-Q. Chen, Y. Zhao, W.-G. Song and L. Jiang, Chem. Commun., 2011, 47, 3619–3621 RSC.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067–4077 CrossRef CAS.
- W. Li, X. Gao, X. Wang, D. Xiong, P.-P. Huang, W.-G. Song, X. Bao and L. Liu, J. Power Sources, 2016, 330, 156–166 CrossRef CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 CAS.
- J. Ryu, N. Jung, J. H. Jang, H.-J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066–4074 CrossRef CAS.
- P. Wang, F. Song, R. Amal, Y. H. Ng and X. L. Hu, ChemSusChem, 2016, 9, 472–477 CrossRef CAS PubMed.
- X.-Y. Yu, Y. Feng, B. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 CAS.
- X. Wang, W. Li, D. Xiong and L. Liu, J. Mater. Chem. A, 2016, 4, 5639–5646 CAS.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS PubMed.
- Y. P. Zhu, Y. P. Liu, T. Z. Ren and Z. Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337–7347 CrossRef CAS.
- D. Xiong, X. Wang, W. Li and L. Liu, Chem. Commun., 2016, 52, 8711–8714 RSC.
- M. Liu and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 2158–2165 CAS.
- T. Y. Ma, S. Dai and S. Z. Qiao, Mater. Today, 2016, 19, 265–273 CrossRef CAS.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175–3180 CrossRef CAS PubMed.
- X. Yang, A.-Y. Lu, Y. Zhu, M. N. Hedhili, S. Min, K.-W. Huang, Y. Han and L.-J. Li, Nano Energy, 2015, 15, 634–641 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- C. Tang, N. Y. Cheng, Z. H. Pu, W. Xing and X. P. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 CAS.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, ACS Energy Lett., 2016, 1, 169–174 CrossRef CAS.
- M. Ledendecker, S. Krick Calderón, C. Papp, H.-P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef CAS PubMed.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., 2016, 128, 8812–8816 CrossRef.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- J. Chang, L. Liang, C. Li, M. Wang, J. Ge, C. Liu and W. Xing, Green Chem., 2016, 18, 2287–2295 RSC.
- Z. Lu, Y. Li, X. Lei, J. Liu and X. Sun, Mater. Horiz., 2015, 2, 294–298 RSC.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplemental figures and tables. See DOI: 10.1039/c6sc05167g |
| This journal is © The Royal Society of Chemistry 2017 |