
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective synthesis of unsymmetric dibenzo[a,e]pentalenes by a rhodium-catalysed stitching reaction†
Keisuke
Takahashi
 ,
Shingo
Ito
*,
Ryo
Shintani
* and
Kyoko
Nozaki
*
,
Shingo
Ito
*,
Ryo
Shintani
* and
Kyoko
Nozaki
*
Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ito_shingo@chembio.t.u-tokyo.ac.jp; shintani@chembio.t.u-tokyo.ac.jp; nozaki@chembio.t.u-tokyo.ac.jp; Fax: +81-3-5841-7263; Tel: +81-3-5841-7261
First published on 14th November 2016
Abstract
A rhodium-catalysed stitching reaction between 2-(silylethynyl)arylboronates and 2-(silylethynyl)aryl bromides has been developed for the synthesis of unsymmetric dibenzo[a,e]pentalenes. The introduction of appropriately sized silyl groups on the starting substrates led to a high crossover selectivity without using an excess amount of either substrate. The present stitching reaction could produce a variety of unsymmetric dibenzo[a,e]pentalene derivatives, including those with electronically different substituents on the fused benzene rings as well as heteroarene fused compounds. Desilylative halogenation was also demonstrated to synthesise the corresponding halogenated dibenzo[a,e]pentalenes, which can be used as building blocks for further chemical transformations.
Introduction
Pentalene has attracted much attention because of its antiaromatic character, which is derived from its planar structure with 8π-electrons.1 However, due to its instability,2 the construction of a pentalene framework often requires fusion of arene rings. Thus, dibenzo[a,e]pentalenes have been prepared and isolated as stable π-conjugated molecules bearing narrow energy gaps,3 which are expected to be utilised in optical and electronic device applications.4 For the construction of a dibenzo[a,e]pentalene framework, a variety of synthetic methods have been developed: intramolecular cyclisation of 5,6,11,12-tetradehydrodibenzo[a,e]cyclooctene derivatives,5 reductive cyclisation of 1,4-diiodo-2,3-diaryl-1,3-butadienes6 or di(2-aroylphenyl)acetylenes,7 and B(C6F5)3-induced cyclisation of 1,2-bis(phenylethynyl)benzenes.8 Transition metal complexes are also often used to synthesise dibenzo[a,e]pentalenes as exemplified by the nickel-mediated4a,d,9 or palladium-catalysed4b,10 reductive dimerisation of 2-alkynylaryl halides, palladium-catalysed oxidative dimerisation of arylacetylenes,11 and palladium-catalysed cross annulations of 2-alkynylaryl halides.4c,10a,12 Despite these significant advances, the synthesis of dibenzo[a,e]pentalene derivatives that are unsymmetrically fused by two different (hetero)arenes or ones that are unsymmetrically substituted have been less studied.4c,5e,f,10,12 This could be mainly due to difficulties in selective cross annulation between two different substrates. For example, Tilley and co-workers found that a palladium-catalysed reaction of 2-alkynylphenyl bromide and (2-alkynylphenyl)tributylstannane afforded the corresponding cross annulation product in moderate yields accompanied by the formation of homo-dimerisation products (Scheme 1a).10a As another example, Jin and co-workers reported palladium-catalysed cross annulations of 2-alkynylaryl chlorides and diarylacetylenes, which offer a general synthetic method to prepare various unsymmetric dibenzo[a,e]pentalene derivatives including heteroarene-fused pentalenes (Scheme 1b).12 However, the use of an excess amount of diarylacetylenes is required in this catalyst system. To achieve a highly selective cross annulation to synthesise unsymmetric pentalene derivatives without using an excess amount of either substrate, the development of a new catalytic system would therefore be desired.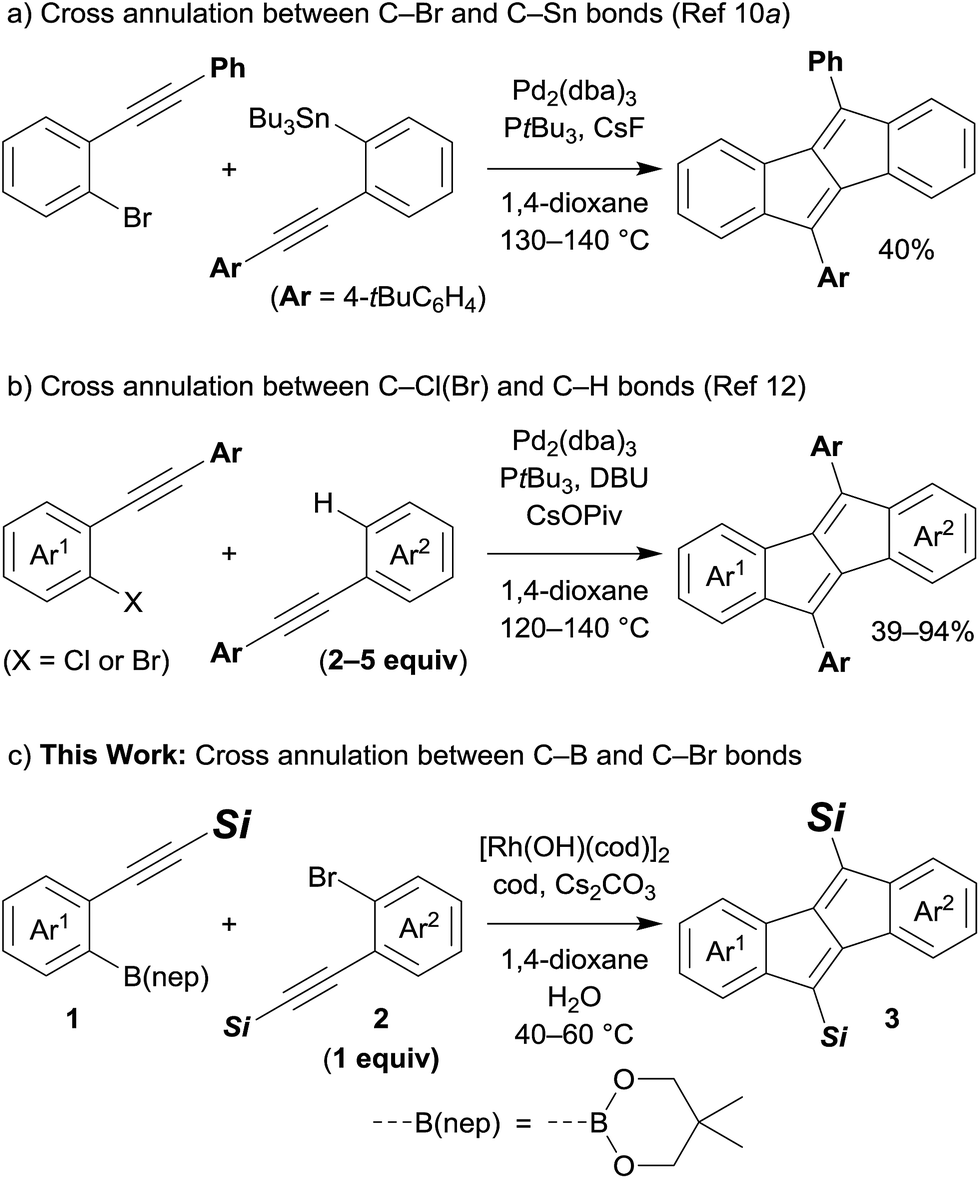 | ||
| Scheme 1 Transition-metal-catalysed cross annulations to form unsymmetric dibenzo[a,e]pentalene derivatives. | ||
We recently developed a rhodium-catalysed stitching reaction, as a novel strategy for the intermolecular synthesis of polycyclic π-conjugated ladder-type compounds via successive insertion of alkynes.13,14 This method allowed for the synthesis of quinoidal fused oligosiloles for the first time through the reaction of an oligo(silylene-ethynylene) containing an arylmetal moiety and a haloarene moiety at each end with another oligo(silylene-ethynylene) of an appropriate length. On the basis of this strategy, we envisaged that the construction of a dibenzo[a,e]pentalene structure could be achieved through a stitching reaction between alkyne 1 bearing an arylboron moiety and alkyne 2 bearing a bromoarene moiety in the presence of a rhodium catalyst (Scheme 1c). Herein we describe the successful synthesis of various unsymmetric dibenzo[a,e]pentalenes using this strategy. High crossover- and regioselectivities could be achieved through proper differentiation of the size of silyl groups on each reaction component.
Results and discussion
Synthesis of dibenzo[a,e]pentalenes through the rhodium-catalysed stitching reaction
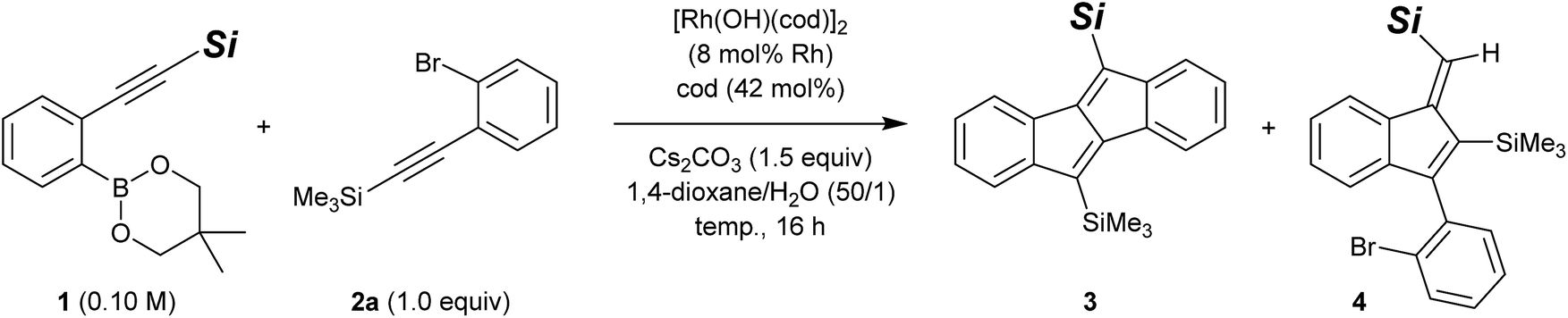
Considering the ease of post-functionalisation of the synthesised dibenzo[a,e]pentalenes, we chose 2-(silylethynyl)arylboronates (1) and 2-(silylethynyl)aryl bromides (2) as the substrate combination for the rhodium-catalysed stitching reaction. We initially employed equimolar amounts of 2-[(trimethylsilyl)ethynyl]phenylboronate (1a)15 and [(2-bromophenyl)ethynyl]trimethylsilane (2a) and conducted the reaction in the presence of di-μ-hydroxidobis[(1,5-cyclooctadiene)rhodium] (8 mol% Rh), 1,5-cyclooctadiene (42 mol%), and cesium carbonate (1.5 equiv.) in 1,4-dioxane/water (50/1) at 60 °C (Table 1, entry 1).13 Under these conditions, 83% of 1a and 53% of 2a were consumed after 16 h, and the desired dibenzo[a,e]pentalene 3aa was obtained in 43% yield along with 2% yield of side-product 4aa (vide infra). We subsequently found that the yield of 3 could be significantly improved through the use of boronate 1b bearing a bulkier tert-butyldimethylsilyl group (3ba: 87% yield, 3ba/4ba = 90/10; entry 2), but no further improvement was observed using boronate 1c with an even bulkier triisopropylsilyl group (3ca: 48% yield; entry 3). For the reaction of 1b and 2a, raising the reaction temperature to 80 °C resulted in slightly lower yield and selectivity of 3ba (79% yield, 3ba/4ba = 89/11; entry 4). By lowering the reaction temperature to 40 °C, in contrast, the selectivity of 3ba/4ba was improved to 92/8 and the yield of 3ba reached 90% (78% isolated yield; entry 5). An essentially identical result was obtained when the reaction was scaled up to a 0.50 mmol scale (entry 6). It is worth noting that the present rhodium-catalysed stitching reaction for the synthesis of dibenzo[a,e]pentalene efficiently proceeds at a low temperature of 40 °C, which is in stark contrast to reported palladium-catalysed reactions that usually require high reaction temperatures of 120–140 °C.10,12|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | Temp. (°C) | Product | 3 (%) | 4 (%) | Recovery of 1b (%) | Recovery of 2ab (%) |
| a Reaction conditions: a mixture of 1 (0.10 mmol), 2a (0.10 mmol), [Rh(OH)(cod)]2 (8 μmol Rh), cod (42 μmol), and Cs2CO3 (0.15 mmol) in 1,4-dioxane/H2O (50/1; 1.0 mL) was stirred for 16 h at the indicated temperature. b Yields were determined using 1H NMR analysis against an internal standard (CH2Br2). The isolated yields are shown in parentheses. c The reaction was conducted on a 0.50 mmol scale. | |||||||
| 1 | 1a (Si = SiMe3) | 60 | 3aa | 43 | 2 | 17 | 47 |
| 2 | 1b (Si = SitBuMe2) | 60 | 3ba | 87 | 10 | 0 | 0 |
| 3 | 1c (Si = SiiPr3) | 60 | 3ca | 48 | 6 | 18 | 31 |
| 4 | 1b | 80 | 3ba | 79 | 10 | 0 | 0 |
| 5 | 1b | 40 | 3ba | 90 (78) | 8 | 0 | 0 |
| 6c | 1b | 40 | 3ba | 87 (77) | 8 | 0 | 0 |
A proposed catalytic cycle for the present reaction of 1b with 2a to form 3ba is illustrated in Scheme 2, left. Initially, the transmetalation of arylboronate 1b with rhodium(I) complex A forms arylrhodium intermediate B. Alkenylrhodium species C is then generated through the intermolecular insertion of 2a into the carbon–rhodium bond of B16 in an orientation that forms a carbon–carbon bond at the silylated carbon of 2a. Successive intramolecular insertion of the alkyne into the carbon–rhodium bond of C gives intermediate D.17 Finally, the intramolecular oxidative addition of bromoarene gives E and subsequent carbon–carbon bond-forming reductive elimination produces dibenzo[a,e]pentalene 3ba along with the regeneration of rhodium(I) complex A.18 Side-product 4ba is presumably formed through the insertion of 2a into the carbon–rhodium bond of B in the opposite regioselectivity, forming a carbon–carbon bond at the arylated carbon of 2a to give intermediate C′. This then undergoes intramolecular alkyne insertion to give intermediate D′, protonolysis of which leads to 4ba with the regeneration of rhodium(I) complex A. The formation of undesired 4ba could be suppressed to some extent by conducting the reaction at a lower temperature (Table 1, entries 2, 4, and 5).
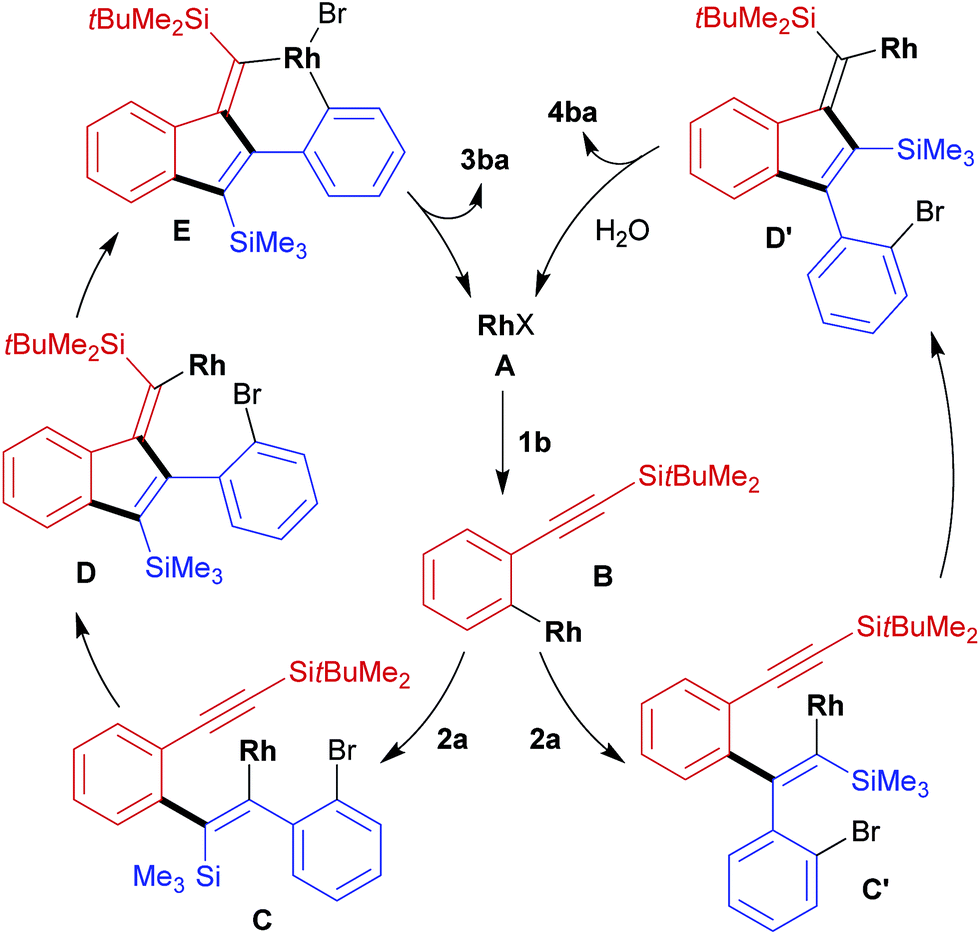 | ||
| Scheme 2 Proposed catalytic cycles for the reaction of 1b with 2a to form dibenzo[a,e]pentalene 3ba and side-product 4ba (Rh = Rh(cod) and X = OH or Br). | ||
The high crossover selectivity of the present stitching reaction led us to examine the scope using various 2-[(tert-butyldimethylsilyl)ethynyl]arylboronates (1) and 2-[(trimethylsilyl)ethynyl]aryl bromides (2) (Scheme 3). In addition to the synthesis of 3ba, the present method was also effective for the synthesis of π-extended pentalenes such as benzo[a]naphtho[2,3-e]pentalene 3bb (74% yield) and dinaphtho[2,3-a:2′,3′-e]pentalene 3db (63% yield). Both electron-rich aryl bromide 2c and electron-deficient aryl bromide 2d were also applied to afford the corresponding dibenzopentalenes 3bc and 3bd in 41% yield and 69% yield, respectively, although both reactions had to be carried out at 60 °C for 32 h to reach full conversion. In this catalysis, bromides at other positions in compounds 2 are tolerated because the reaction is initiated by the transmetalation of an arylboronate rather than the oxidative addition of an aryl bromide (see Scheme 2). For example, trimethyl[(2,4,5-tribromophenyl)ethynyl]silane 2e could be used in the reaction with 1b to give dibromodibenzopentalene 3be in 74% yield. With the aim of obtaining donor–acceptor type pentalenes, the use of electron-rich arylboronate 1e bearing a dimethylamino group was also examined. The reaction of 1e with 2a successfully gave 3ea, albeit in a moderate yield of 34% with full conversion of both substrates. Similarly, by combining arylboronate 1e and aryl bromide 2d, donor–acceptor type pentalene 3ed, bearing a dimethylamino group and a nitro group on each aromatic ring, was obtained in 36% yield. In addition, the present method could be extended to the synthesis of the pyridine-fused pentalene, benzo[a]pyrido[2,3-e]pentalene 3bf, in a high yield (81% yield).
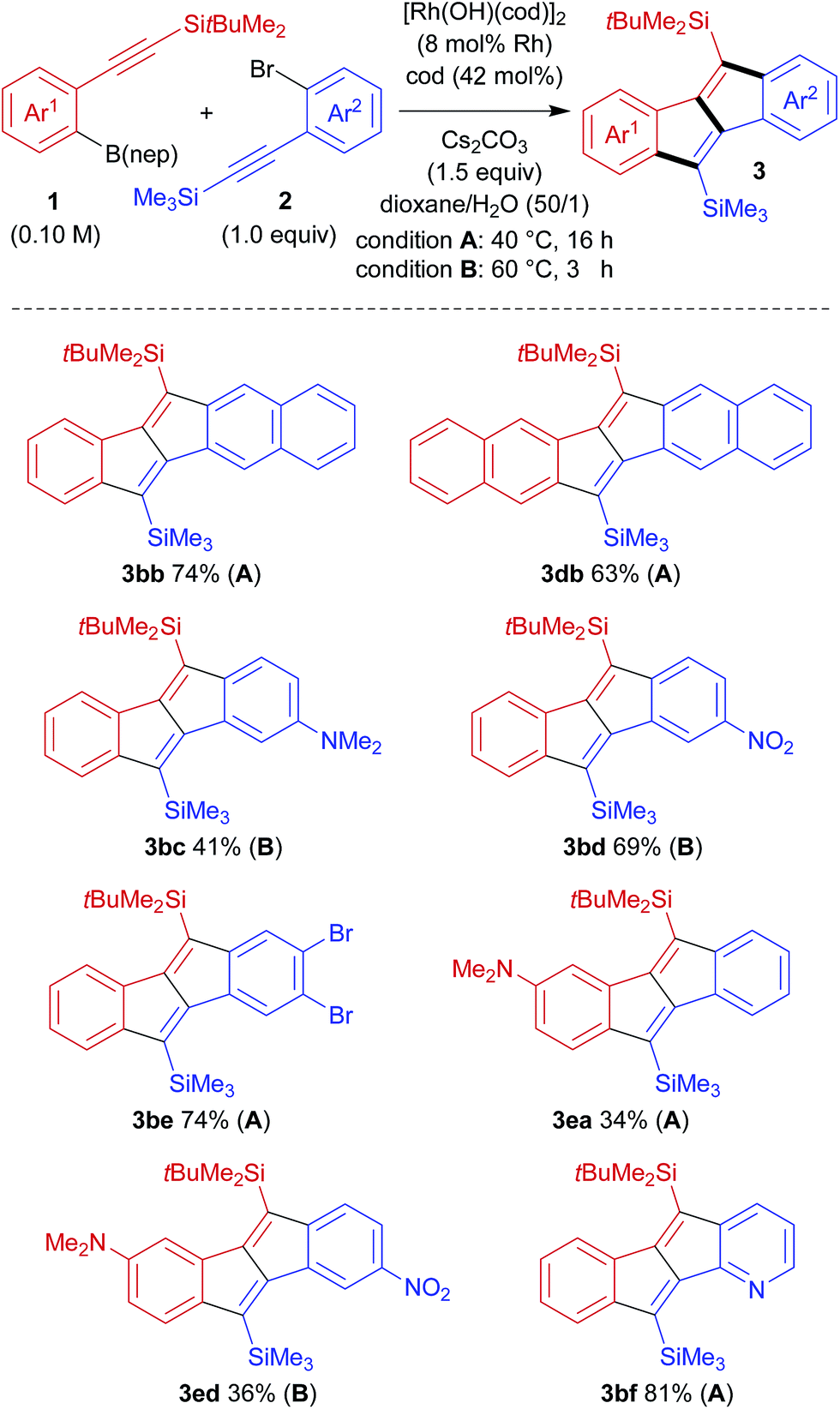 | ||
| Scheme 3 Scope of the rhodium-catalysed stitching reaction for the synthesis of dibenzo[a,e]pentalene derivatives. | ||
The present catalysis could also be applied to the synthesis of benzothienopentalenes, although the yields are not particularly high (Scheme 4). Thus, benzothienopentalenes 3bg and 3bh were isolated in 20% yield and 23% yield, respectively, by employing corresponding thienyl bromides 2g and 2h in the reaction with arylboronate 1b at 40 °C for 16 h. Somewhat higher yields were achieved by conducting these reactions at 60 °C for 32 h, giving 3bg in 50% 1H NMR yield and 3bh in 43% 1H NMR yield along with the generation of inseparable side products including 4. The synthesis of benzothienopentalenes could also be achieved using thienylboronates 1f and 1g with aryl bromide 2a, giving 3fa in 10% isolated yield and 3ga in 41% 1H NMR yield with inseparable side-products. Pyrido[2,3-a]thieno[3′,2′-e]pentalene 3ff and pyrido[2,3-a]thieno[2′,3′-e]pentalene 3gf were prepared in 48% yield and 20% yield, respectively, using pyridyl bromide 2f as the reaction partner. This represents the first synthesis of pentalenes that are fused by two different heteroarenes, although the yields are still low to moderate.
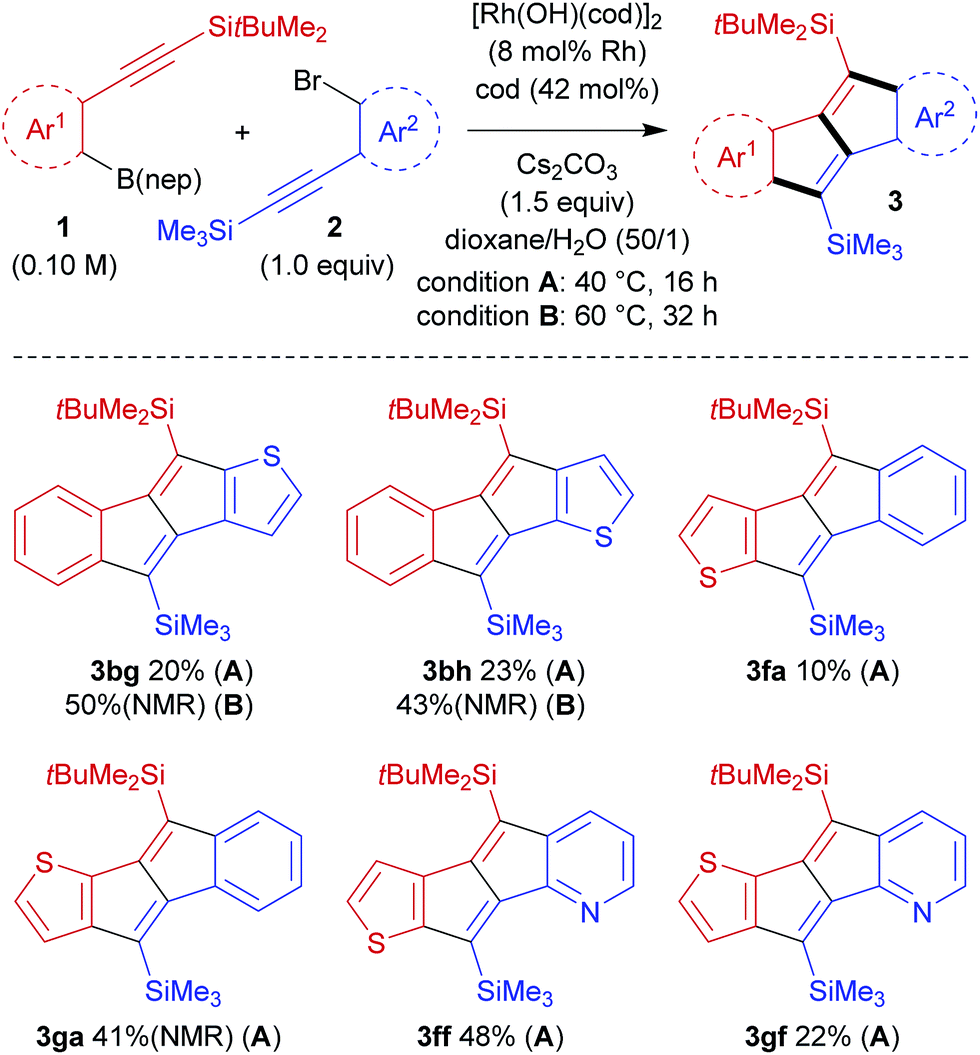 | ||
| Scheme 4 Rhodium-catalysed stitching reaction to synthesise thienopentalenes. | ||
Selective desilylative halogenation
For the purpose of utilizing the obtained pentalenes as building blocks for further extended π-conjugated systems, the desilylative halogenation of compounds 3 was examined (Scheme 5). Thus, the treatment of benzonaphthopentalene 3bb with bromine in diethyl ether at room temperature smoothly converted both of the silyl groups into bromides without brominating other positions, to give 5,12-dibromobenzo[a]naphtho[2,3-e]pentalene 5 in 71% yield. On the other hand, the reaction of 3bb with iodine in tetrahydrofuran at 40 °C selectively converted only the trimethylsilyl group to an iodide, keeping the tert-butyldimethylsilyl group intact to give compound 6 in 77% yield. These halogenated pentalene derivatives could be of further use with the rich chemistry of organo halides, such as in cross-coupling reactions5b and nucleophilic substitution reactions.19 This demonstrates that the present rhodium catalysis for the synthesis of unsymmetric dibenzo[a,e]pentalenes and the subsequent desilylative halogenation would be an efficient method for the preparation of pentalene-based functional molecules.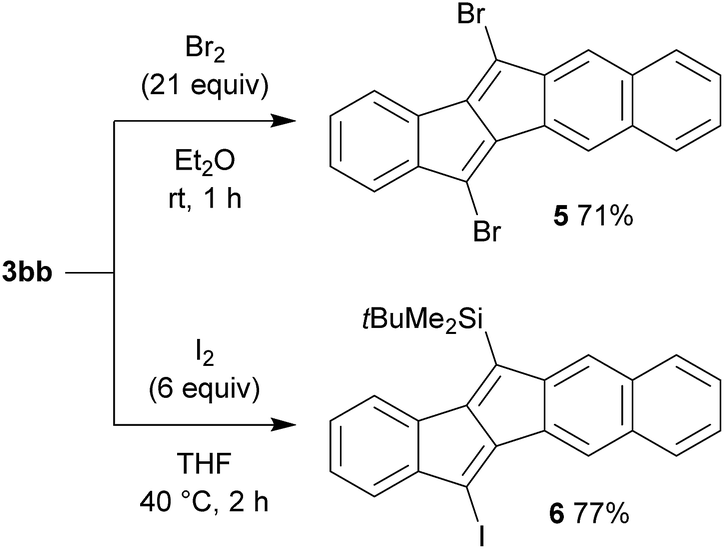 | ||
| Scheme 5 Desilylative halogenation of benzonaphthopentalene 3bb. | ||
Optical and electronic properties
With various dibenzopentalene derivatives (3) in hand, we evaluated their optical and electronic properties by UV-vis absorption spectroscopy and cyclic voltammetry (Table 2). UV-vis absorption spectra of the obtained pentalenes were obtained in dichloromethane (Fig. 1). Dibenzopentalene 3ba exhibited an absorption maximum (λmax) at 444 nm (εmax = 1.0 × 104 L mol−1 cm−1), and λmax was shifted bathochromically and εmax increased as the number of fused rings increased in the order of 3bb (λmax = 478 nm and εmax = 1.7 × 104 L mol−1 cm−1) and 3db (λmax = 500 nm and εmax = 1.9 × 104 L mol−1 cm−1), owing to the extension of the π-conjugation (Fig. 1a). Time-dependent density functional theory (TD-DFT) calculations at the B3LYP/6-31G(d) level of theory suggested that the absorption band of 3ba in the 350–550 nm region can be attributed to the HOMO−1 ⇒ LUMO and HOMO ⇒ LUMO+1 transitions (Fig. S1 and Table S1†). In the case of 3ba, the oscillator strength (f) of the HOMO–LUMO transition was calculated to be zero (f = 0.0000), which is consistent with the fact that the corresponding absorption was not observed.9a| Compound | λ max [nm] (εmaxa [×104 L mol−1 cm−1]) | E onsetox [eV] | E onsetred [eV] | E HOMO,CV [eV] (EHOMO,cald [eV]) | E LUMO,CV [eV] (ELUMO,cald [eV]) | E g,CV [eV] (Eg,calf [eV]) |
|---|---|---|---|---|---|---|
| a Measured in CH2Cl2 (1.0 × 10−4 M). b CV measured with Bu4NPF6 in CH2Cl2 (1.0 × 10−3 M) with a scan rate of 100 mV s−1 under argon containing Ag/Ag+ as the reference electrode, Pt as the working electrode, and Pt wire as the counter electrode. Values are against Fc/Fc+. c E HOMO(LUMO),CV = −(Eonsetox(red) + 4.8). d Values calculated at the B3LYP/6-31G(d) level of theory. e E g,CV = ELUMO,CV − EHOMO,CV. f E g,cal = ELUMO,cal − EHOMO,cal. | ||||||
| 3ba | 444 (1.0) | 0.82 | −1.69 | −5.62 (−5.31) | −3.11 (−2.36) | 2.51 (2.95) |
| 3bb | 478 (1.7) | 0.80 | −1.72 | −5.60 (−5.31) | −3.08 (−2.30) | 2.52 (3.01) |
| 3db | 500 (1.9) | 0.71 | −1.78 | −5.51 (−5.16) | −3.02 (−2.26) | 2.48 (2.90) |
| 3ea | 613 (0.24) | 0.05 | −1.82 | −4.85 (−4.65) | −2.98 (−2.12) | 1.87 (2.52) |
| 3bd | 455 (1.5) | 1.08 | −1.33 | −5.88 (−5.79) | −3.47 (−2.92) | 2.41 (2.87) |
| 3ed | 676 (0.28) | 0.16 | −1.42 | −4.96 (−5.00) | −3.38 (−2.68) | 1.58 (2.32) |
| 3fa | 600 (0.045) | 0.48 | −1.60 | −5.28 (−5.00) | −3.20 (−2.45) | 2.08 (2.55) |
| 3bf | 434 (0.68) | 0.91 | −1.59 | −5.71 (−5.50) | −3.21 (−2.51) | 2.50 (2.99) |
| 3ff | 595 (0.053) | 0.61 | −1.50 | −5.41 (−5.18) | −3.30 (−2.61) | 2.11 (2.58) |
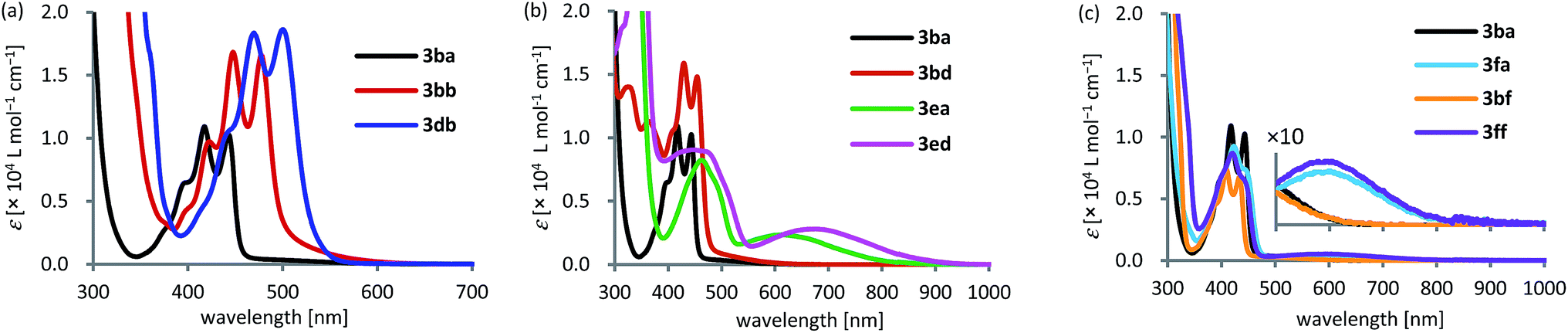 | ||
| Fig. 1 UV-vis absorption spectra of (a) 3ba, 3bb, and 3db, (b) 3ba, 3bd, 3ea, and 3ed, and (c) 3ba, 3fa, 3bf, and 3ff in CH2Cl2 (1.0 × 10−4 M) at 25 °C. | ||
The effect of functional groups, such as dimethylamino and nitro groups, was also investigated (Fig. 1b). The shape of the absorption spectrum of dibenzo[a,e]pentalene 3bd bearing a nitro group resembled that of 3ba with a small bathochromic shift of 11 nm (λmax = 455 nm and εmax = 1.5 × 104 L mol−1 cm−1). This phenomenon could be explained by charge transfer absorption, similar to the relationship between benzene and nitrobenzene.20 As was the case for 3ba, the oscillator strength corresponding to the HOMO ⇒ LUMO transition of 3bd was calculated to be nearly zero (f = 0.0012), and the corresponding absorption was not observed. In contrast, the introduction of a dimethylamino group dramatically changed the shape of the absorption spectrum and generated a new broad absorption band in the longer wavelength region (3ea: λmax = 613 nm and εmax = 0.24 × 104 L mol−1 cm−1). TD-DFT calculations suggested that the broad absorption at 613 nm corresponds to the HOMO ⇒ LUMO transition (f = 0.0355). Furthermore, donor–acceptor type pentalene 3ed bearing both dimethylamino and nitro groups exhibited a further red shift of λmax to 676 nm (εmax = 0.28 × 104 L mol−1 cm−1).
A similar phenomenon was observed for the heteroarene-fused pentalenes (Fig. 1c). The absorption spectrum of benzopyridopentalene 3bf resembled that of dibenzopentalene 3ba, while benzothienopentalene 3fa and pyridothienopentalene 3ff showed new broad absorption bands at around 600 nm, albeit with small εmax values (3fa: εmax = 4.5 × 102 L mol−1 cm−1 and 3ff: εmax = 5.3 × 102 L mol−1 cm−1; Fig. 1c, inset). TD-DFT calculations indicated the positive oscillator strengths corresponding to the HOMO ⇒ LUMO transitions for these compounds (3fa: f = 0.0066 and 3ff: f = 0.0061), while the oscillator strengths of 3ba and 3bf were calculated to be zero (f = 0.0000).
Cyclic voltammetry measurement of the obtained pentalenes (3) was performed to estimate the energy levels of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) (Fig. 2 and Table 2). Symmetric pentalenes, such as dibenzopentalene 3ba and dinaphthopentalene 3db, exhibited one reversible oxidation process and one reversible reduction process, whereas an unsymmetric pentalene, benzonaphthopentalene 3bb, showed one irreversible oxidation and one reversible reduction (Fig. 2a). Compared with the parent dibenzopentalene 3ba, dibenzopentalene 3ea bearing a dimethylamino group exhibited a lower oxidation potential (Fig. 2b), indicating that the introduction of an electron-donating dimethylamino group increased the HOMO level (EHOMO,CV = −4.85 eV for 3eavs. −5.62 eV for 3ba). On the other hand, the nitro group decreased the LUMO level (ELUMO,CV = −3.47 eV for 3bdvs. −3.11 eV for 3ba) judging from the higher reduction potential of 3bd. The donor–acceptor type pentalene 3ed exhibited one reversible oxidation process similar to that of 3ea and two reversible reduction processes similar to those of 3bd (EHOMO,CV = −4.96 eV and ELUMO,CV = −3.38 eV). This corresponds to a HOMO–LUMO gap of 1.58 eV, which is the narrowest among donor–acceptor-type dibenzo[a,e]pentalene derivatives ever reported.5b,12 With regard to the heteroarene-fused derivatives, benzothienopentalene 3fa exhibited relatively high HOMO and low LUMO energy levels (EHOMO,CV = −5.28 eV and ELUMO,CV = −3.20 eV; Fig. 2c), indicating that the replacement of one of the fused benzene rings of dibenzo[a,e]pentalene by a thiophene ring results in a narrower HOMO–LUMO energy gap. The introduction of a pyridine ring, on the other hand, shows a different electronic effect. Thus, compared with 3ba and 3fa, benzopyridopentalene 3bf and pyridothienopentalene 3ff showed lower LUMO and HOMO energy levels with similar energy gaps, respectively.
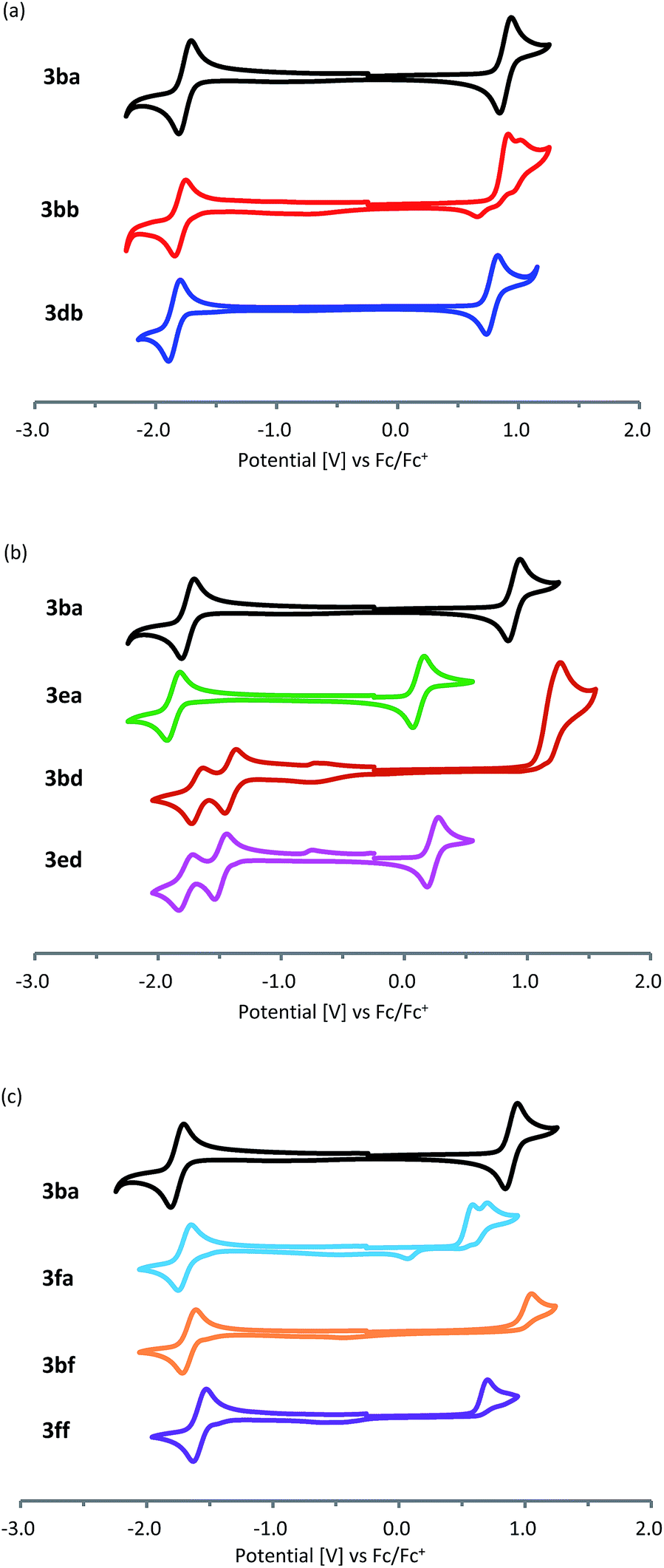 | ||
| Fig. 2 Cyclic voltammograms of (a) 3ba, 3db, and 3bb, (b) 3ba, 3ea, 3bd, and 3ed, and (c) 3ba, 3fa, 3bf, and 3ff measured with Bu4NPF6 in CH2Cl2 (0.1 M) with a scan rate of 100 mV s−1 under argon containing Ag/Ag+ as the reference electrode, Pt as the working electrode, and Pt wire as the counter electrode. The potential was externally calibrated against Fc/Fc+. | ||
The origin of the electronic effects caused by the introduction of thiophene and pyridine rings was assessed by comparing the calculated nucleus-independent chemical shift (NICS) values of compounds 3ba, 3fa, and 3ff (B3LYP/6-31G(d); Fig. 3). The NICS(0) values of the central five-membered rings indicate that the antiaromaticity of thiophene-fused 3fa and 3ff is higher than that of dibenzopentalene 3ba, which leads to the narrower HOMO–LUMO gaps. Pentalenes that are fused by heteroles, such as thiophene and pyrrole, via the b bond are known to have higher antiaromaticity, because the contribution of the resonance structure with isolated 8π-electrons becomes more dominant.4d,9b,10b On the other hand, the negligible effect of the pyridine ring on the NICS(0) values can explain the similar HOMO–LUMO gaps of 3fa and 3ff.
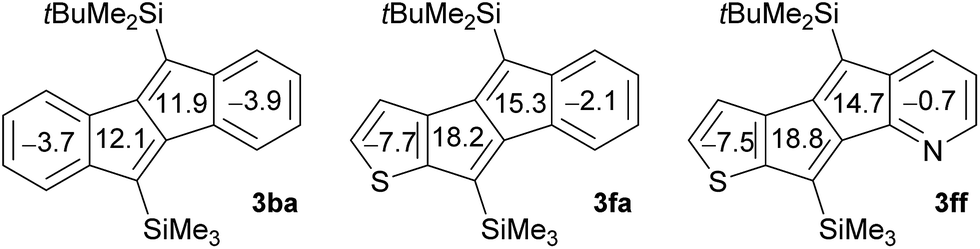 | ||
| Fig. 3 NICS(0) values of 3ba, 3fa, and 3ff (B3LYP/6-31G(d)). | ||
Conclusions
In summary, we have developed a novel method for the synthesis of dibenzo[a,e]pentalene derivatives using a rhodium-catalysed stitching reaction. The introduction of appropriately sized silyl groups on the starting substrates led to a high crossover selectivity without using an excess amount of either substrate. Various unsymmetric dibenzo[a,e]pentalene derivatives, including those with electronically different substituents on the fused benzene rings as well as heteroarene fused compounds, could be synthesised using the present catalysis. The obtained pentalenes can also be transformed into halogenated pentalenes by desilylative halogenation reactions. The optical and electronic properties of these dibenzo[a,e]pentalene derivatives were also examined, and the novel donor–acceptor type pentalene 3ed bearing dimethylamino and nitro groups exhibited a small HOMO–LUMO gap of 1.58 eV.Acknowledgements
This work was supported in part by a Grant-in-Aid for Young Scientists (A) (S.I.: No. 16H06030), a Grant-in-Aid for Challenging Exploratory Research (R.S.: 15K13688), and “Nanotechnology Platform” (project No. 12024046), MEXT, Japan, and in part by JGC-S Scholarship Foundation (R.S.) and Asahi Glass Foundation (R.S.). K. T. would like to thank the Japan Society for the Promotion of Science (JSPS) for their Program for Leading Graduate Schools (MERIT), and for a Research Fellowship for Young Scientists. The theoretical calculations were performed using computational resources provided by Research Center for Computational Science, National Institutes of Natural Sciences, Okazaki, Japan.Notes and references
- (a) H. Hopf, Angew. Chem., Int. Ed., 2013, 52, 12224 CrossRef CAS PubMed; (b) H. Hopf, Classics in Hydrocarbon Chemistry, Wiley-VCH, Weinheim, 2000, p. 277 Search PubMed.
- (a) K. Hafner, R. Dönges, E. Goedecke and R. Kaiser, Angew. Chem., Int. Ed. Engl., 1973, 12, 337 CrossRef; (b) R. Dönges, K. Hafner and H. J. Lindner, Tetrahedron Lett., 1976, 17, 1345 CrossRef.
- For reviews: (a) T. Kawase and J.-i. Nishida, Chem. Rec., 2015, 15, 1045 CrossRef PubMed; (b) M. Saito, Symmetry, 2010, 2, 950 CrossRef CAS; (c) M. Rabinovitz, I. Willner and A. Minsky, Acc. Chem. Res., 1983, 16, 298 CrossRef CAS.
- (a) T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Angew. Chem., Int. Ed., 2010, 49, 7728 CrossRef CAS PubMed; (b) G. Dai, J. Chang, W. Zhang, S. Bai, K.-W. Huang, J. Xu and C. Chi, Chem. Commun., 2015, 51, 503 RSC; (c) C. Li, C. Liu, Y. Li, X. Zhu and Z. Wang, Chem. Commun., 2015, 51, 693 RSC; (d) G. Dai, J. Chang, X. Shi, W. Zhang, B. Zheng, K.-W. Huang and C. Chi, Chem.–Eur. J., 2015, 21, 2019 CrossRef CAS PubMed.
- (a) G. Babu, A. Orita and J. Otera, Chem. Lett., 2008, 37, 1296 CrossRef CAS; (b) F. Xu, L. Peng, A. Orita and J. Otera, Org. Lett., 2012, 14, 3970 CrossRef CAS PubMed. See also: (c) M. Chakraborty, C. A. Tessier and W. J. Youngs, J. Org. Chem., 1999, 64, 2947 CrossRef CAS PubMed; (d) F. Xu, L. Peng, K. Wakamatsu, A. Orita and J. Otera, Chem. Lett., 2014, 43, 1548 CrossRef CAS; (e) S. Nobusue, A. Yoshizaki, M. Miki, H. Miyoshi, A. Shimizu and Y. Tobe, Tetrahedron, 2014, 70, 8474 CrossRef CAS; (f) F. Xu, L. Peng, K. Shinohara, T. Nishida, K. Wakamatsu, M. Uejima, T. Sato, K. Tanaka, N. Machida, H. Akashi, A. Orita and J. Otera, Org. Lett., 2015, 17, 3014 CrossRef CAS PubMed.
- (a) M. Saito, M. Nakamura, T. Tajima and M. Yoshioka, Angew. Chem., Int. Ed., 2007, 46, 1504 CrossRef CAS PubMed; (b) H. Li, B. Wei, L. Xu, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 10822 CrossRef CAS PubMed; (c) H. Li, X.-Y. Wang, B. Wei, L. Xu, W.-X. Zhang, J. Pei and Z. Xi, Nat. Commun., 2014, 5, 4508 CAS.
- H. Zhang, T. Karasawa, H. Yamada, A. Wakamiya and S. Yamaguchi, Org. Lett., 2009, 11, 3076 CrossRef CAS PubMed.
- (a) C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992 CrossRef CAS PubMed; (b) C. Chen, M. Harhausen, A. Fukazawa, S. Yamaguchi, R. Fröhlich, C. G. Daniliuc, J. L. Petersen, G. Kehr and G. Erker, Chem.–Asian J., 2014, 9, 1671 CrossRef CAS PubMed.
- (a) T. Kawase, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, Chem.–Eur. J., 2009, 15, 2653 CrossRef CAS PubMed; (b) A. Konishi, T. Fujiwara, N. Ogawa, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, C. Kitamura and T. Kawase, Chem. Lett., 2010, 39, 300 CrossRef CAS.
- (a) Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 2796 CrossRef CAS PubMed; (b) X. Yin, Y. Li, Y. Zhu, Y. Kan, Y. Li and D. Zhu, Org. Lett., 2011, 13, 1520 CrossRef CAS PubMed.
- T. Maekawa, Y. Segawa and K. Itami, Chem. Sci., 2013, 4, 2369 RSC.
- J. Zhao, K. Oniwa, N. Asao, Y. Yamamoto and T. Jin, J. Am. Chem. Soc., 2013, 135, 10222 CrossRef CAS PubMed.
- R. Shintani, R. Iino and K. Nozaki, J. Am. Chem. Soc., 2016, 138, 3635 CrossRef CAS PubMed.
- For review articles on related rhodium-catalyzed carbon–carbon bond-forming reactions, see: (a) M. M. Heravi, M. Dehghani and V. Zadsirjan, Tetrahedron: Asymmetry, 2016, 27, 513 CrossRef CAS; (b) M. Shiotsuki, F. Sanda and T. Masuda, Polym. Chem., 2011, 2, 1044 RSC; (c) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093 RSC; (d) T. Aoki, T. Kaneko and M. Teraguchi, Polymer, 2006, 47, 4867 CrossRef CAS; (e) M. G. Mayershofer and O. Nuyken, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5723 CrossRef CAS; (f) Modern Rhodium-Catalyzed Organic Reactions, ed. P. Andrew Evans, Wiley-VCH, Weinheim, 2005 Search PubMed; (g) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed; (h) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS PubMed.
- The use of the corresponding arylboronic acid or its pinacol ester instead of the neopentyl glycol ester produced dibenzo[a,e]pentalenes in comparable yields. See ESI† for details.
- (a) T. Hayashi, K. Inoue, N. Taniguchi and M. Ogasawara, J. Am. Chem. Soc., 2001, 123, 9918 CrossRef CAS PubMed; (b) M. Lautens and M. Yoshida, Org. Lett., 2002, 4, 123 CrossRef CAS PubMed; (c) T. Fujii, T. Koike, A. Mori and K. Osakada, Synlett, 2002, 295 CrossRef CAS; (d) Y. Nakao, M. Takeda, J. Chen and T. Hiyama, Synlett, 2008, 774 CrossRef CAS.
- (a) T. Miura, M. Yamauchi and M. Murakami, Synlett, 2007, 2029 CAS; (b) L. Artok, M. Kuş, B. N. Ürer, G. Türkmen and Ö. Aksn-Artok, Org. Biomol. Chem., 2010, 8, 2060 RSC.
- (a) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2005, 7, 2229 CrossRef CAS PubMed; (b) R. Shintani, T. Yamagami and T. Hayashi, Org. Lett., 2006, 8, 4799 CrossRef CAS PubMed; (c) S. D. Timpa, C. M. Fafard, D. E. Herbert and O. V. Ozerov, Dalton Trans., 2011, 40, 5426 RSC; (d) Y. J. Jang, H. Yoon and M. Lautens, Org. Lett., 2015, 17, 3895 CrossRef CAS PubMed.
- F. Xu, L. Peng, K. Wakamatsu, A. Orita and J. Otera, Chem. Lett., 2014, 43, 1548 CrossRef CAS.
- S. Nagakura and J. Tanaka, J. Chem. Phys., 1954, 22, 236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General, experimental, spectral, and theoretical information. CCDC 1511836–1511838. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04560j |
| This journal is © The Royal Society of Chemistry 2017 |