
Intrinsic conductivity optimization of bi-metallic nickel cobalt selenides toward superior-rate Na-ion storage†
Chen
Wu
a,
Yuehua
Wei
a,
Qingwang
Lian
a,
Chao
Cui
a,
Weifeng
Wei
 a,
Libao
Chen
*a and
Chengchao
Li
*b
a,
Libao
Chen
*a and
Chengchao
Li
*b
aState Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, P. R. China. E-mail: lbchen@csu.edu.cn
bSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, P. R. China. E-mail: licc@gdut.edu.cn
First published on 19th October 2017
Abstract
Enhancing the conductivity of electrode materials is critically important for improving the high-rate performance of Na-ion batteries (NIBs). Herein, we report a multifaceted strategy for optimizing the conductivity and electrochemical properties of nickel cobalt selenides via the combination of fine component regulation and C coating. The electrical conductivity of C@Ni0.33Co0.67Se2/C nanofiber (CNF) (Co0.67) hybrids achieved in this study was 0.3733 S mm−1, a conductivity five-fold higher than that of selenides with a Ni/Co ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Coupled with desirable three-dimensional (3D) nanobrush morphology and the 1D conducting path of CNFs, the Co0.67 electrode achieved a superior rate performance of 413.1 mA h g−1, even at 2 A g−1. Furthermore, the Co0.67 electrode exhibited an impressive cycling performance of 499 mA h g−1 after 100 cycles (exhibiting an 89.5% capacity retention of the second cycle). Finally, electrochemical impedance spectroscopy (EIS) and cyclic voltammetry analysis at different sweep rates were conducted to demonstrate the Co0.67 electrode's fast charge/ion transport ability and increased electrode kinetics.
:1. Coupled with desirable three-dimensional (3D) nanobrush morphology and the 1D conducting path of CNFs, the Co0.67 electrode achieved a superior rate performance of 413.1 mA h g−1, even at 2 A g−1. Furthermore, the Co0.67 electrode exhibited an impressive cycling performance of 499 mA h g−1 after 100 cycles (exhibiting an 89.5% capacity retention of the second cycle). Finally, electrochemical impedance spectroscopy (EIS) and cyclic voltammetry analysis at different sweep rates were conducted to demonstrate the Co0.67 electrode's fast charge/ion transport ability and increased electrode kinetics.
Introduction
Na-ion batteries (NIBs) are distinguished by abundant resources, low cost and similar chemical characteristics to Li-ion batteries (LIBs), and have recently become increasingly attractive in the field of energy storage systems.1–3 However, as the radius and mass of Na ions are both larger than those of Li ions, the commercial graphite anode used in LIBs is unsatisfactory for use in NIBs.4 Therefore, appropriate anode materials for fast and reversible Na-ion storage are urgently needed.To pursue the goal of obtaining fast and reversible Na-ion storage, many researchers have achieved significant progress in carbonaceous materials,1,5,6 alloying materials (such as Sn, P, among others)7,8 and conversion materials (such as metal oxides, sulphides, selenides, among others).9–12 Transition metal sulfides and selenides have become the most promising candidates due to their low cost, simple synthesis and high storage capacity.13,14 For example, Chen's research group made great progress in transition metal sulfides and selenides for use in batteries, including Co-doped FeS2 nanospheres, FeSe2 microspheres comprising nanosized octahedra, CoS2 and CoSe2.15–18 It is noteworthy that the toxicity of Se is relatively lower than that of S, the radius of the Se atom is larger than that of the S atom, and the electrical conductivity of Se is higher than that of S, resulting in the rapid diffusion of Na+ and superior electrochemical performance.13,19,20
However, the commercial application of the transition metal selenides in NIBs remains limited by their low conductivity and large volume expansion during the charging/discharging process.21 Conductivity plays a vital role in the electrochemical properties because poor conductivity could affect the rapid electron migration, and increase the polarization and IR potential drop,22,23 leading to sluggish electrochemical kinetics and poor high-rate performance. Additionally, the pulverization of the active material due to large volume expansion/contraction could lead to the loss of electrical contact between the active material and the conductive additive/current collector, resulting in poor cycling stability.24 To overcome these drawbacks, significant effort has been dedicated to the development of the modification of electrode materials, including nanofabrication techniques and combination with C materials.22,25 Nanosized electrode materials have been demonstrated to significantly shorten the diffusion paths of ions and expose a larger active surface area for electrochemical reactions, which contributes to a higher rate capability.22,26 However, the decreased size of the nanosized electrode materials will also produce higher contact resistance, especially in the presence of insulating binders. Combination with C materials is an effective way to not only buffer volume expansion, but also optimize the overall conductivity of electrodes to some extent. Therefore, embedding nanosized electrode materials into a conductive three-dimensional (3D) electrode may be a promising strategy that could both effectively increase the active surface area of the materials and enhance their ionic/electric conductivities, which is highly desirable but significantly challenging.
Controlling the intrinsic conductivity of electrode materials is another way to facilitate the rapid transfer of electrons for increasing the rate performance, such as controlling the materials’ composition, which also plays an important role in the electrochemical properties. For example, many research studies have been conducted on C-based materials by doping heteroatoms to improve their electrochemical properties.5,27–29 Liu has reported nickel cobalt phosphides with different Ni/Co ratios and achieved high conductivity.23 Because transition metals such as Fe, Co and Ni have similar atomic radii, their dichalcogenides (MX2, X = O, S, Se) generally possess a similar crystalline phase.15,30,31 But the valence electrons of transition metal ions are different from each other; thus, they have different spin configurations. Introducing Ni into Co (Co2+, t62g e1g; Ni2+, t62g e2g) will lead to a change in the electronic structure and heterogeneous spin states that significantly affects the electrochemical properties.32 Finally, the electrical conductivity of transition metal selenides can be systematically optimized by tuning their components.33 In addition, the coexistence of transition metal ions combines the properties of single metals together, resulting in multiple redox reactions and a higher redox activity, thus improving the electrochemical properties.33–35 This inspired us to achieve superior electrochemical properties by constructing bimetallic compounds and tuning their compositions to control the intrinsic conductivity of electrode materials, thus facilitating the rapid transfer of electrons for increasing the rate performance.
After taking these factors into consideration, we have developed a combined strategy to enhance electron and ion transportation by regulating the components of transition metal selenides and constructing a 3D C composite electrode. The C coated selenide nanorods are separately anchored on C nanofibers (CNFs) by a two-step hydrothermal reaction and subsequent selenization process, which facilitates electron and ion transportation. Importantly, by varying the Ni/Co ratio, the electrical conductivity of the Co0.67 composite has been improved to 0.3733 S mm−1, which is five-fold higher than that of the electrode with a Ni/Co ratio of 2:1. As expected, the Co0.67 electrode exhibits better cycling stability and superior rate performance compared to electrodes made of other selenides with various Ni/Co ratios when evaluated as anodes for NIBs. Specifically, the Co0.67 electrode delivers a high discharge specific capacity, 499 mA h g−1 at 0.2 A g−1 after 100 cycles and 413.1 mA h g−1 even at 2 A g−1. This facile synthetic approach provides a new impetus for the design of NIBs with a long lifetime and high-rate performance.
Experimental
Synthesis of CNFs
The crisscross CNFs were prepared through electrospinning as described in our previous literature.30,36 Briefly, 0.3778 g of polyacrylonitrile (PAN, Mw = 150000, Sigma-Aldrich Co., Ltd, China) was dissolved in 5 mL of dimethylformamide (DMF, Sinopharm Chemical Reagent Co., Ltd, China) and mixed for 0.5 h at 60 °C to form a transparent solution. The solution was then stirred for approximately 6 h at room temperature. A syringe with a needle of an inner diameter of 0.6 mm was used as the solution container. A voltage of 12 kV was applied to generate the electrospun fibers, which were collected by a thick aluminum plate. After drying in a vacuum for 12 h, the electrospun fibers underwent a two-stage post-treatment to form CNFs: annealing was performed first at 230 °C for 2 h in air and then at 600 °C for 2 h in Ar.
Synthesis of the NixCo1−x precursor on CNFs
The facile method of hydrothermal treatment was used to fabricate the precursor. Briefly, 6(1 − x) mmol Co(NO3)2·6H2O (Sinopharm Chemical Reagent Co., Ltd, China) and 6x mmol Ni(NO3)2·6H2O (Sinopharm Chemical Reagent Co., Ltd, China) (x = 0.00, 0.33, 0.50, 0.67 or 1.00) were dissolved in 30 mL of deionized (DI) water. Subsequently, 10 mM urea (Aladdin Industrial Corporation, China) was added, and the solution was transferred into a 50 mL Teflon-lined stainless steel autoclave. In total, 10 mg of the CNFs as backbones was immersed in the homogenous solution. After a 12 h hydrothermal treatment at 120 °C, the Ni-Co precursor/CNF composites were washed with absolute ethanol and DI water several times to remove any residual precursor. C coating was performed as described in our previous paper.37 In particular, 1 g of C6H12O6·H2O in 30 mL of DI water was used to coat the C precursor (denoted “Cp”) on the composite via a 4 h hydrothermal treatment performed at 180 °C. The composites were then washed with absolute ethanol and DI water and dried overnight.Synthesis of C@NixCo1−xSe2/CNF composites
In total, 0.2 g of precursor and 0.5 g of selenium powder were placed at both ends of the crucible. The crucible was then placed in the middle of the tube. The reaction was conducted for 1 h under vacuum conditions at 550 °C at a ramping rate of 1 °C min−1. The final C@NixCo1−xSe2/CNF composites were named according to the molar ratios of Ni(NO3)2·6H2O to Co(NO3)2·6H2O as follows: Co0.0, Co0.33, Co0.5, Co0.67 and Co1.0 (x: (1 − x) (x = 0.00, 0.33, 0.50, 0.67 or 1.00, respectively)).Material characterization
X-ray powder diffraction (XRD) analysis was conducted for phase characterization (Rigaku Desktop X-ray diffractometer, Cu Kα radiation, λ = 1.54056 Å, Japan). Thermal gravimetric analysis (TGA, NETZSCH STA 449F3, Germany) was performed (air, room temperature: 1000 °C, 10 °C min−1) to determine the C content. Elemental analysis was conducted using inductively coupled plasma-optical emission spectrometry (ICP-OES, Agilent 720, Australia). The chemical states of the elements near the surface were obtained by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, USA). Scanning electron microscopy (SEM, NOVA NANO SEM230, USA) was conducted to view the morphology. A transmission electron microscope (TEM, TECNAI G2 F20, USA) equipped with an energy-dispersive X-ray analyzer (EDX) was used to observe the structural characteristics and determine the element distribution of the samples. In addition, the specific surface area was determined via a Brunauer–Emmett–Teller test (BET, Tristar 3020, USA). The electrical conductivity was measured on a powder resistivity tester (RICO, FT-300I, Ningbo RIco Instrument Co., Ltd, P. R. China) based on four-terminal measurements.Electrochemical characterization
A CR2025 half-cell was assembled to measure the electrochemical properties of the composites. First, a slurry comprising 80 wt% C@NixCo1−xSe2/CNF, 10 wt% acetylene black and 10 wt% sodium carboxymethylcellulose (CMC) was coated onto the Cu and cut into a 12 mm-diameter round sheet after drying overnight. Second, a coin-type cell that used Na metal as a counterpart and glass microfibers (Whatman GF/D) as a separator was assembled in an Ar-filled glove box (M. Braun, Germany). The electrolyte used was a solution of 1 M NaClO4 in ethylene carbonate (EC) and diethyl carbonate (DEC) (1:1, volume) containing 5% fluoroethylene carbonate (FEC). In addition, the oxygen content and hydrogen content were controlled to approximately 0.5 ppm. The cells were mainly used to test cyclic voltammetry (CV, Arbin Workstation, USA), cycling and rate performance (LANHE CT2001A, Wuhan LAND Electronics Co., P. R. China) between 0.01 V and 3 V, and electrochemical impedance spectroscopy (EIS, Princeton PARSTAT 4000, AMETEK Co. Ltd) in the range of 0.1 Hz to 106 Hz.
Results and discussion
Material synthesis and characterization
The bimetallic selenides with tuneable compositions were synthesized easily via a two-step hydrothermal reaction and subsequent heat treatment process, as presented in Fig. 1. Beginning with CNFs manufactured using an electrospinning method, NixCo1−x precursors with different compositions were grown separately on CNFs via a heterogeneous nucleation and growth process. The XRD pattern of the Co0.67 precursor is shown in Fig. S1 (see the ESI†). During the final annealing stage, the products underwent simultaneous carbonization and selenization and were then further transformed into C@NixCo1−xSe2/CNF. In addition, the red-outlined inset shows a magnified, more detailed image of the nanorods, which are similar to rhizoma nelumbinis. For further comparison, the monometallic selenides (Co1.0 and Co0.0) were also prepared in this work.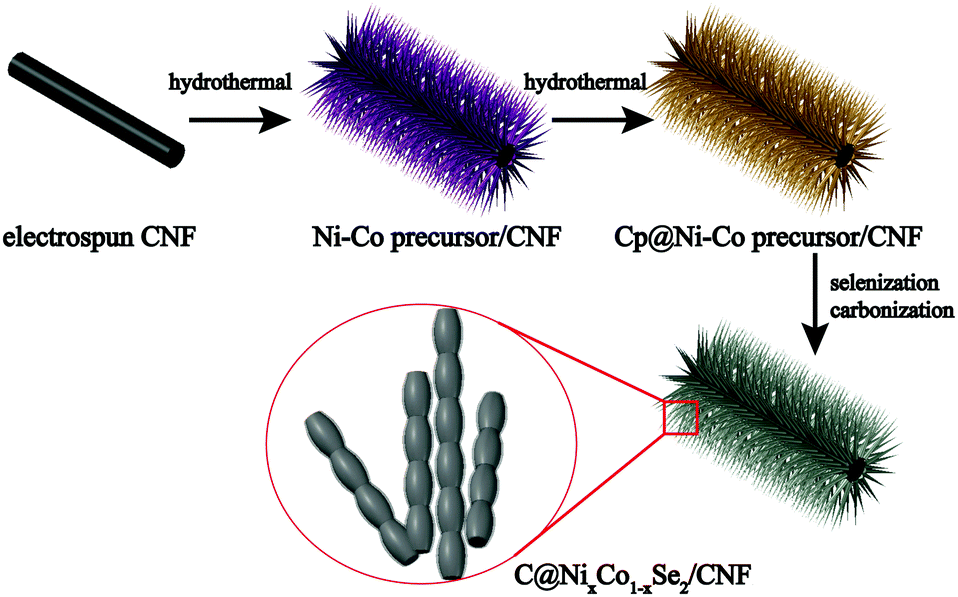 | ||
| Fig. 1 Schematic illustration of the preparation of C coated bimetallic selenides anchored on CNFs. | ||
XRD was first conducted as depicted in Fig. 2a. The XRD patterns of the prepared five composites exhibit similar characteristic peaks. When x = 0.0 or 1.0, the diffraction peaks of the composites are in complete accordance with PDF#65-3327 or PDF#65-1843, respectively, demonstrating the successful synthesis of the pure cubic phases of CoSe2 and NiSe2. As the Ni/Co ratio increases, all peaks shift to lower angles with respect to CoSe2. This phenomenon reveals that the partial substitution of cations results in no change to the pristine crystal structure, and only small changes in the lattice parameters. In addition, it indicates the formation of a solid solution. And peak shifting is observed clearly from the partial magnification of the (210) peak shown in Fig. 2b. The sharp peaks indicate the superb crystallinity of the prepared samples. No other impurity peaks are shown in Fig. 2a, demonstrating the high purity of the synthesized selenides. ICP-OES was also conducted to recognize the actual compositions of Co0.33, Co0.5 and Co0.67, respectively (see Table S1, ESI†). Thermogravimetric analysis (TGA) was implemented to obtain the C content in the temperature range of room temperature to 1000 °C using a rate of 10 °C min−1 in air. Fig. 2c indicates that two main mass losses occurred. These losses resulted from the following reactions: the combustion of C (CNFs and coated C layer), the oxidation of NixCo1−xSe2 into corresponding oxides and the sublimation of SeO2.38–41 Fig. S2 (ESI†) depicts the XRD patterns of the final products of Co0.67 after annealing at 1000 °C in air. The patterns are indexed as NiO (PDF#44-1159) and CoO (PDF#70-2855), so the C content can be calculated as 20.2 wt%. (The detailed calculations are presented in the ESI.†) The TGA curves of another two composites are displayed in Fig. S3 (ESI†) with C contents calculated as 17.5 and 24.2 wt% for Co0.5 and Co0.33, respectively.
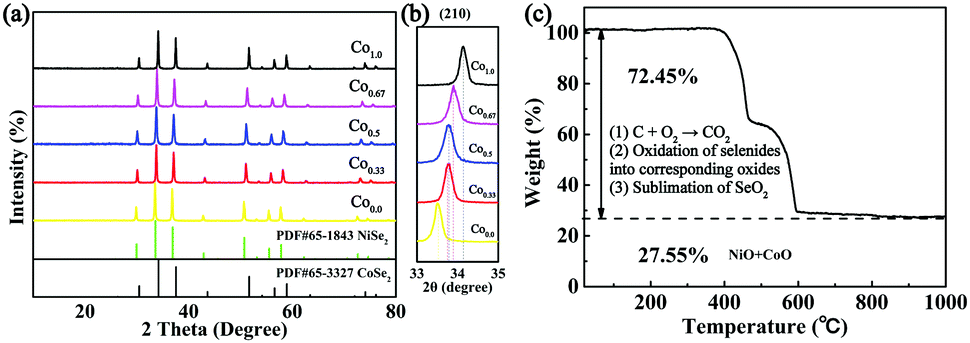 | ||
| Fig. 2 (a) XRD patterns of Co1.0, Co0.67, Co0.5, Co0.33 and Co0.0. (b) The partial magnification of the (210) peak. (c) TGA of Co0.67 from room temperature to 1000 °C at 10 °C min−1 in air. | ||
Fig. S4 (ESI†) displays the combination bond and valence states of the elements C, Ni, Co and Se near the surface of Co0.67. The high-resolution spectrum of C derived from CNFs and the C layer are fit into three peaks, which are assigned as follows: C–C (284.78 eV), C–O (286.21 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.99 eV).41,42 Fig. S4b and c (ESI†) show the elemental spectra of Ni 2p and Co 2p, which exhibit two spin–orbit doublets with separate satellites (denoted as “Sat.”) (further details are available in the ESI†). The Se peaks appear slightly complex: the peak located at 54.83 eV (Se 3d5) indicates a combined signal with Ni and Co,43 the peak located at 55.97 eV (Se 3d5) might be due to the tight integration with C,44 and the peak centered at 59.23 eV (Se 3d5) originates from SeO2, which is a result of the slight oxidization of the surface in air.33
O (287.99 eV).41,42 Fig. S4b and c (ESI†) show the elemental spectra of Ni 2p and Co 2p, which exhibit two spin–orbit doublets with separate satellites (denoted as “Sat.”) (further details are available in the ESI†). The Se peaks appear slightly complex: the peak located at 54.83 eV (Se 3d5) indicates a combined signal with Ni and Co,43 the peak located at 55.97 eV (Se 3d5) might be due to the tight integration with C,44 and the peak centered at 59.23 eV (Se 3d5) originates from SeO2, which is a result of the slight oxidization of the surface in air.33
The morphological features of the precursors and the prepared selenides were characterized by SEM as shown in Fig. 3a–e and Fig. S5 (ESI†). The low-magnification SEM images of the three bimetallic composites depict their similar morphology, which appear as nanobrushes composed of nanorods grown on interlinked CNFs (see Fig. S6, ESI†). Visibly, the morphology of the prepared selenides retains the original characteristics perfectly, except the diameters of the nanorods appear larger due to the coated C layer and subsequent selenization. Fig. S7 (ESI†) shows SEM images of Co0.0 and Co1.0 (ESI†). Most importantly, the C coating preserved the nanorod morphology extremely well. When no C coating was present, the nanoparticles fused together and were coated onto CNFs without the protection of a C coating during the selenization process (Fig. S8, ESI†). Furthermore, the coated C layer could increase the conductivity and decrease the pulverization induced by volume expansion, thus increasing structure stability and extending cycle life. Moreover, the unique nanobrush-like morphology results in bimetallic selenides with large specific areas and facilitates the infiltration of the electrolyte throughout the electrode. Several studies have indicated that constructing electrode materials with micro/nanostructures could increase the rate of the diffusion of electrons and Na+, and reduce agglomeration.18,45
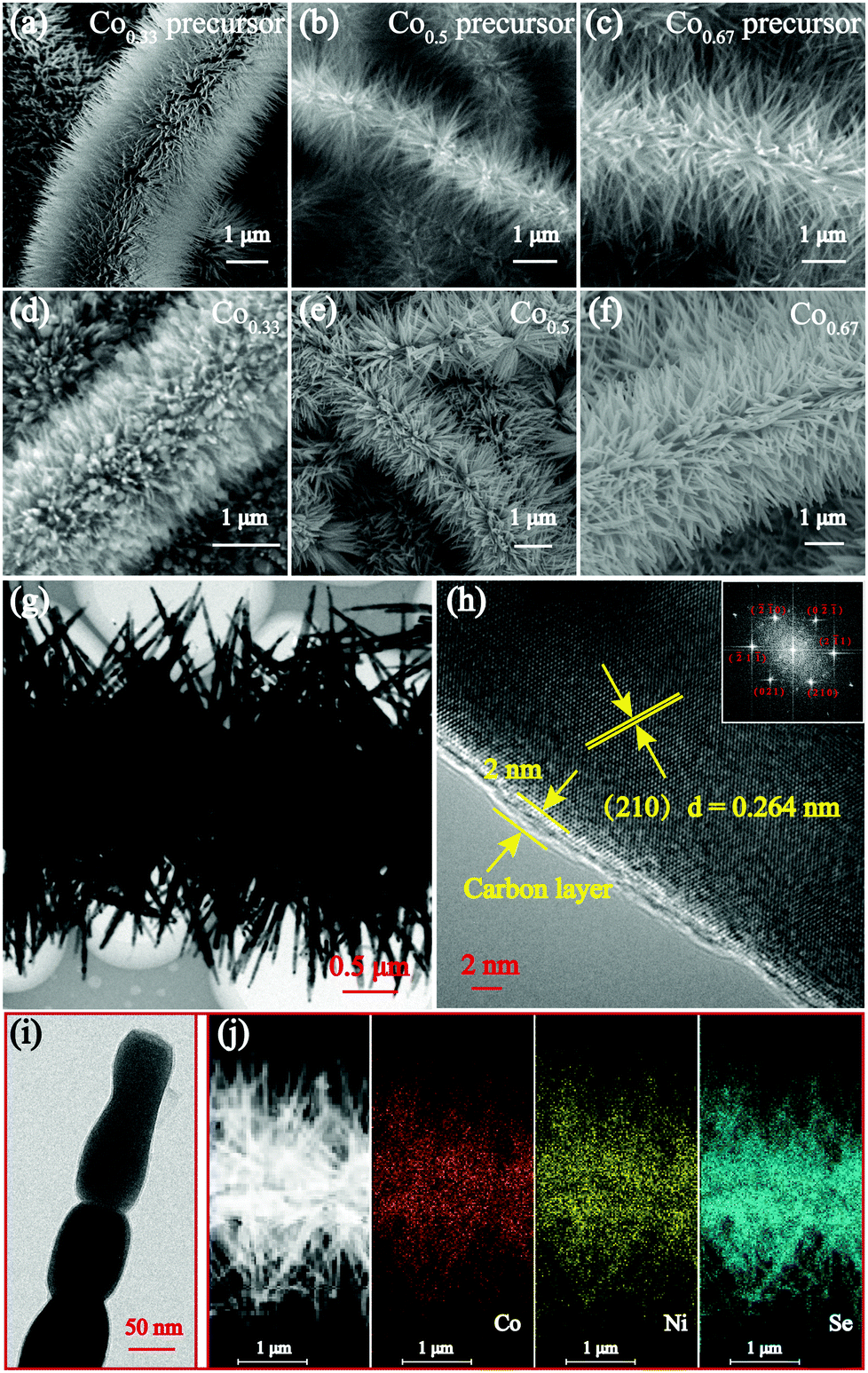 | ||
| Fig. 3 SEM images of (a) Co0.33, (b) Co0.5 and (c) Co0.67 precursors and (d) Co0.33, (e) Co0.5 and (f) Co0.67 composites. (g) TEM, (h) HRTEM, (i) partially enlarged TEM and (j) EDX mapping of the Co0.67 composite. The inset in (h) depicts the corresponding fast Fourier transform (FFT) image. | ||
TEM was also conducted to obtain the detailed morphology and structure characteristics of the samples. Fig. 3g shows the nanobrush-like morphology of Co0.67. The magnified image of the nanorods shown in Fig. 3i indicates that the nanorods composed of nanoparticles have a diameter of approximately 50 nm. Furthermore, it shows that the nanorods are coated successfully by a C layer which is 2 nm thick (Fig. 3h). In addition, the clear lattice fringe spacing is calculated to be 0.264 nm, indicative of a (210) crystal face. Fig. 3j shows the homogeneous element distribution of Ni, Co and Se. The lattice spaces near the surface and inside are the same, verifying the formation of a complete solid solution of the prepared selenide.15 The high-resolution (HR) TEM images of two other bimetallic selenides are depicted in Fig. S9 (ESI†).
The specific surface areas of the three bimetallic selenides were studied by BET analyses as shown in Fig. S10 (ESI†). The three N2 isothermal adsorption/desorption curves with one hysteresis loop are categorized as a IV-type isotherm.46 And Co0.33 exhibits the largest specific surface area of 82.67 m2 g−1. The pore diameters of the three bimetallic composites are similar, approximately 7–8 nm, as observed from the pore distribution iconograph. A high specific surface area could increase the contact area between the electrode and electrolyte but lower the Columbic efficiency, as discussed below.
The electrical conductivities of the composites were obtained via the four-terminal method, as shown in Table 1. Co0.67 exhibits the uppermost value of 0.3733 S mm−1, which is nearly five-fold that of Co0.33 (0.0755 S mm−1). The d-electron configuration of transition metal cations plays an important role in their physical properties. Notably, Co2+ in CoSe2 possesses a spin state of t62ge1g, which endows CoSe2 with metallic nature,43 while Ni2+ in NiSe2 possesses a spin state of t62ge2g. Furthermore, CoSe2 and NiSe2 possess different energy gaps47–49 and combination with C, which result in different conductivities for Co1.0 than Co0.0. However, compared with Co1.0, bimetallic selenides demonstrated superior conductivity that might be attributed primarily to the synergistic effect between Ni and Co and the introduced heterogeneous spin states to some extent. Additionally, several previous studies reported that the incorporation of Ni atoms could improve the conductivity.32,50 Moreover, electron configuration varies as the Ni/Co ratio changes; this phenomenon might be mainly responsible for the different conductivities of bimetallic selenides. In brief, the high electrical conductivity of Co0.67 is a comprehensive outcome of tuning the Ni/Co ratio in combination with the use of CNFs and C coating. High electrical conductivity is beneficial in transporting electrons, reducing polarization, accelerating the electrochemical reaction and ameliorating the rate performance.23,51
| Material | Sectional area/mm | Diameter/mm | Shape | Pressure/MPa | Electrical conductivity/S mm−1 |
|---|---|---|---|---|---|
| Co1.0 | 78.5 | 10 | Cylinder | 20 | 0.0147 |
| Co0.67 | 78.5 | 10 | Cylinder | 20 | 0.3733 |
| Co0.5 | 78.5 | 10 | Cylinder | 20 | 0.1126 |
| Co0.33 | 78.5 | 10 | Cylinder | 20 | 0.0755 |
| Co0.0 | 78.5 | 10 | Cylinder | 20 | 0.0002 |
Electrochemical performance
To verify the conjecture that nickel cobalt selenides with higher conductivities exhibit superior electrochemical properties, the composites were applied as anodes in a CR2025 half-cell configuration with Na metal as the counter electrode. Fig. 4a depicts the CV curves of Co0.67 for the first three cycles at a sweep rate of 0.2 mV s−1. According to the previous literature on selenides, two Na storage mechanisms exist in the processes of charge and discharge, intercalation and conversion. In particular, the peak at 0.99 V during the first cathodic sweep is hypothesized to originate from the intercalation of Na+ into Ni0.33Co0.67Se2 to form the intermediate NaxNi0.33Co0.67Se2. The peaks at 0.67 V and 0.33 V are believed to originate from the formation of Na2Se and solid electrolyte interface (SEI) from the irreversible decomposition of electrolytes.13,52 The prominent peak at 1.93 V in the course of anodic sweep is associated with the extraction of Na+ and the reformulation of selenide.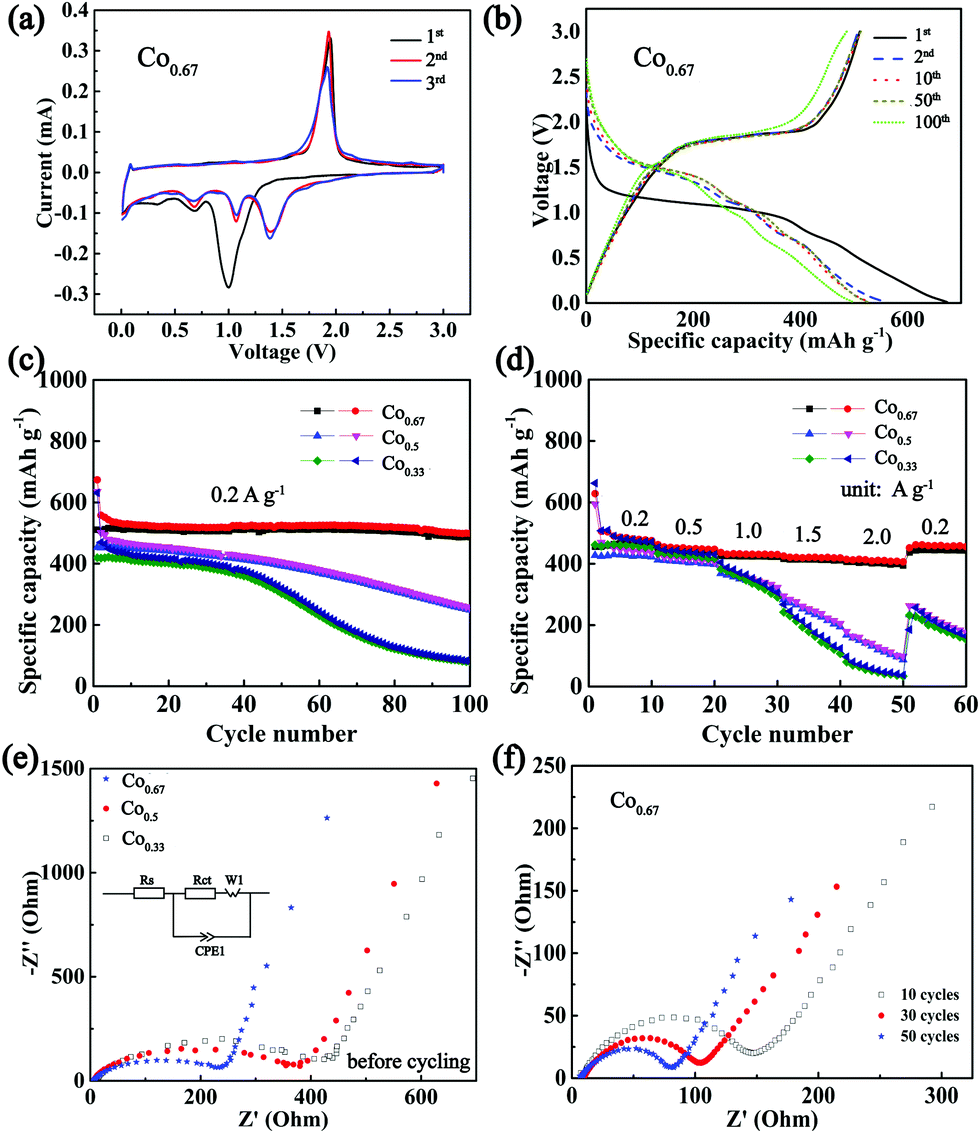 | ||
| Fig. 4 (a) The CV curves of Co0.67 at a scan rate of 0.2 mV s−1 and a voltage range of 0.01–3.0 V. (b) Charge–discharge curves for different cycles of Co0.67 at 0.2 A g−1. (c) Cycle performance of three bimetallic selenide composites at 0.2 A g−1. (d) Rate performance of three bimetallic selenides at different current densities. (e) EIS profiles of three bimetallic selenide composites before cycling. (f) EIS profile of Co0.67 during the first 50 cycles tested at 1.7 V of charge state. The inset of (e) displays the corresponding fitting circuit, where Rs, Rct, W1, and CPE1 represent the electrolyte resistance, charge-transfer resistance in the interface of the electrode and electrolyte, Warburg impedance and constant phase element, respectively. | ||
After the first cycle, the reduction peaks shift to higher voltages at 1.39 V, 1.06 V and 0.68 V, which may be ascribed to the transformation of the composite into nanoclusters (Fig. S11, ESI†).42 The subsequent cycles overlap with each other, indicating good reversibility. The CV curves of other composites are displayed in Fig. S12 (ESI†). Fig. 4b displays the 1st, 2nd, 10th, 50th and 100th charge–discharge curves of Co0.67 between 0.01 V and 3 V. With the exception of a clear and long platform at approximately 1 V and a short platform at 0.7 V in the 1st discharge curve, three platforms in other discharge curves coincide well with the CV curves. Moreover, the platforms remain obvious in the 100th curves, and the peaks in the CV curves of different cycles (Fig. S13a, ESI†) remain obvious even after 100 cycles, demonstrating its superior cyclic stability. In contrast, the redox peaks in the CV curves of other samples disappear upon cycling (Fig. S13b–e, ESI†). These phenomena demonstrate the poor reversibility and cycle stability of the four samples. Fig. 4c presents a comparison of the cycling performance of three bimetallic selenides with similar nanobrush-like morphology as anodes for NIBs. Co0.67 demonstrates a discharge capacity as high as 673.9 mA h g−1 while the charge specific capacity is 511 mA h g−1 in the first cycle, resulting in a Columbic efficiency of 75.83%. The capacity loss during the first cycle is mainly attributed to the formation of the SEI film and electrolyte decomposition.13,53 As mentioned previously, a larger specific surface area supports a larger amount of SEI film, which leads to a lower Columbic efficiency. Therefore, the Columbic efficiency of Co0.33 is only 66.45%. With the exception of the first several cycles, the Columbic efficiency is maintained at above 97% during subsequent cycling (Fig. S14, ESI†). After 100 cycles, the discharge specific capacity of Co0.67 still reaches 499 mA h g−1. In contrast, the capacities of two other materials decay upon cycling, especially the composite of Co0.33. Meanwhile, the cycling stability increases as the molar ratio of Co/Ni increases, indicating the importance of the addition of Co in improving performance, which is congruent with previous reports.54 The rate property (Fig. 4d) was tested at the current densities ranging from 0.2 to 2 A g−1 and was then returned to 0.2 A g−1. When the current density is varied from 0.2 to 2 A g−1, the retained capacity of Co0.67 is as high as 85%. Moreover, Co0.67 still demonstrates an average discharge capacity of 459.6 mA h g−1 as the current density again returns to 0.2 A g−1. However, the capacity declines sharply as the current density increases to 2 A g−1 for the other two samples. When the current density returns again to 0.2 A g−1, the two composites are unable to restore their specific capacity as well as Co0.67, implying the inferior rate performance of the two composites. The relatively poor conductivity of the two composites is a nonnegligible factor resulting in the inferior rate performance. Because low conductivity is adverse to electron transfer; it will increase polarization and lead to sluggish electrochemical kinetics, thus against the acquisition of superior electrochemical properties.23 Fig. S15 (ESI†) shows the electrochemical properties of other composites of Co0.0 and Co1.0 with worse cycling and rate properties. In our opinion, in addition to their low conductivity, their poor morphology and single component composition are major contributors to their inferior performance. The bimetallic compounds exhibit 3D and micro/nano composite morphological structure, favouring sufficient contact with the electrolyte, reducing the diffusion distance, providing sufficient space to accommodate volume expansion and impeding the tendency of agglomeration. What's more, the existence of two metals could provide multiple redox reactions and a higher redox activity, which augments the electrochemical properties.
EIS with a frequency range of 1 M Hz to 0.1 Hz was conducted to compare the kinetic differences among the three electrodes. Fig. 4e displays the spectra of these three composites and the fitting circuit. The results show similar plots with a depressed semi-circle at high frequency and an inclined line at low frequency, corresponding to charge-transfer resistance at the electrolyte/electrode interface (Rct) and diffusion of Na+, separately.55,56 The fitted data are presented in Table S2 (ESI†), which clearly reveals a smaller Rct value (225.5 Ω) of Co0.67 than those of other samples (328.5 Ω for Co0.5 and 374.0 Ω for Co0.33), suggesting the superior charge transport ability of Co0.67.57 In addition, these data also support the high conductivity of Co0.67, which most likely aids in increasing its rate performance.58 The Nyquist plot of Co0.67 during the first 50 cycles at 1.7 V of the charge state is shown in Fig. 4f. The diameter of the semi-circle (the Rct value is listed in Table S2, ESI†) decreases as the cycle number increases in the first 50 cycles, which is favourable for the fast intercalation/extraction of Na+, thus resulting in superior cycling stability.18
To further investigate the kinetics of the storage of Na+ in the composites, CV at different sweep speeds from 0.1 mV s−1 to 1 mV s−1 was performed (Fig. 5a). The three peaks formed during the cathodic sweep are denoted as R1, R2 and R3, and the peak formed during the anodic sweep is denoted as O1. As the sweep speed increases, the intensity of the peak increases. This is ascribed to the increasing polarization. The peak current (i) has a relationship with the speed (v) as eqn (1) describes:59
| i = avb, | (1) |
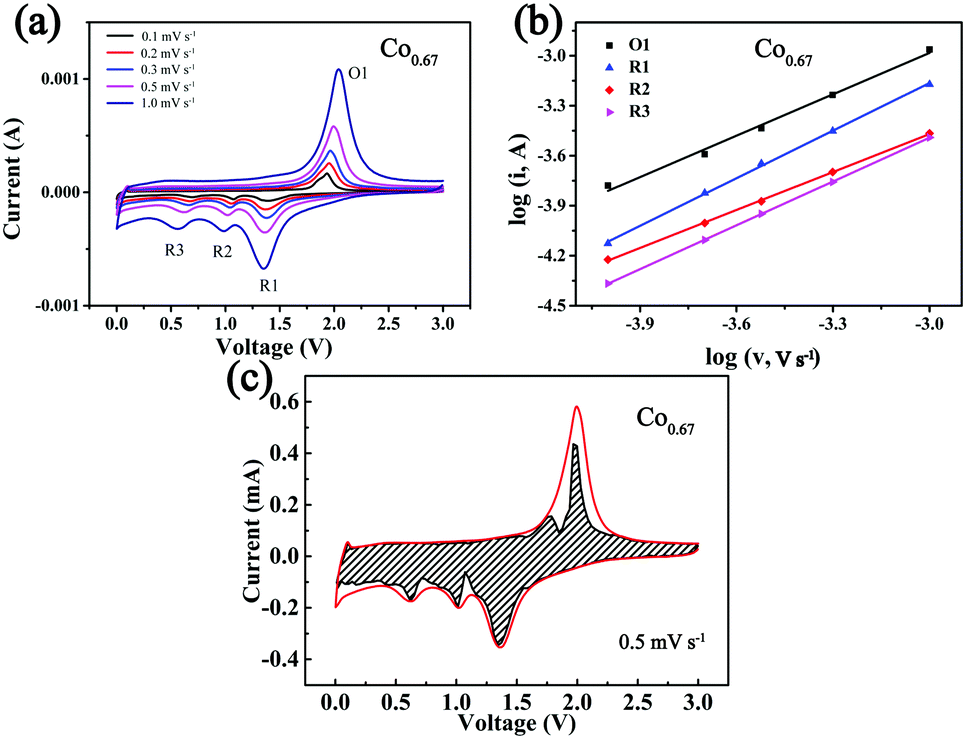 | ||
| Fig. 5 (a) CV curves at different scan rates in the voltage range of 0.01–3.0 V. (b) log(i) vs. log(v) plots at each redox peak. (c) The capacitive contribution (dashed area) shown in the CV curve at 0.5 mV s−1 for the Co0.67 composite. | ||
Eqn (1) can be further transformed into eqn (2) as follows:
| logi = loga + blogv. | (2) |
The b value reveals which mechanism controls Na+ storage. When b = 0.5, ion diffusion controls the reaction; when b = 1, the electrochemical reaction is primarily controlled by pseudocapacitance. The b value is determined from the logi versus logν plot, and it is positively correlated with rate performance.15 The corresponding results (Fig. 5b) show that the b values of the four peaks of R1, R2, R3 and O1 are 0.95, 0.76, 0.88 and 0.83, respectively, with R2 as the rate-determining step. The results also demonstrate that partial pseudocapacitance exists during the course of the redox reactions. Eqn (3) was used to calculate the contribution from capacitance, indicating that the current (i) at fixed voltage (V) is composed of two parts, one primarily from capacitance (k1v) and another from diffusion-controlled insertion (k2v0.5).59Fig. 5c displays a CV curve in which a large fraction of capacity is derived from capacitance (dashed area) at 0.5 mV s−1, which is calculated as 78.75%. This phenomenon is beneficial to the rapid insertion/desertion of Na+ and better cycling stability.18 Fig. S16 and Table S3 (ESI†) display the corresponding results of the other four composites.
| i(V) = k1v + k2v0.5. | (3) |
Therefore, the superior electrochemical properties of the Co0.67 composite could be ascribed to the following factors. First, excellent conductivity is achieved via changing the Ni/Co ratios and combination with C, which avails the rapid electron transport and reduced polarization. In addition, the C layer also plays an important role in reducing volume expansion and maintaining the structure integrity. Second, the 3D nanobrush-like morphology with the conductive CNF architecture provides a sufficient contact area between the electrode and electrolyte and prevents the agglomeration of particles. Finally, pseudocapacitance occupies a large portion of Na+ storage, which, to some extent, aids in improving reaction kinetics.
Conclusions
In conclusion, a series of nanobrush-like nickel cobalt selenides with different Ni/Co ratios on CNFs were synthesized via facile hydrothermal treatment and subsequent selenization. C coating and optimization of the Ni/Co ratio gave rise to the higher conductivity of Co0.67. When evaluated as an anode for Na-ion batteries, Co0.67 demonstrates the highest rate and cycling performance of all the composites tested. Co0.67 exhibits a high discharge specific capacity of 499 mA h g−1 after 100 cycles at 0.2 A g−1 and delivers a discharge specific capacity of 413.1 mA h g−1, even at 2 A g−1 which retains 85% of its capacity at 0.2 A g−1. Various analyses demonstrate that the nanobrush-like morphology and micro/nanocomposite structure provide more space to accommodate volume expansion and an increased contact area between the electrode and electrolyte. The superior conductivity and small charge transfer resistance derived from the tuneable ratio of Ni/Co and C coating are beneficial to electron transfer and Na+ diffusion. Furthermore, a large fraction of pseudocapacitance contributes to its superior rate property. Therefore, the excellent electrochemical properties of Co0.67 are the result of the comprehensive action of its morphology, structure, high conductivity and pseudocapacitance.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 21373081 and 51771236), the Innovation-Driven Project of Central South University (No. 2017CX002), the Program for Shenghua Overseas Talents from Central South University and the Self-established Project of State Key Laboratory of Powder Metallurgy.References
- Y. S. Yun, K. Y. Park, B. Lee, S. Y. Cho, Y. U. Park, S. J. Hong, B. H. Kim, H. Gwon, H. Kim, S. Lee, Y. W. Park, H. J. Jin and K. Kang, Adv. Mater., 2015, 27, 6914–6921 CrossRef CAS PubMed.
- J. Wan, F. Shen, W. Luo, L. Zhou, J. Dai, X. Han, W. Bao, Y. Xu, J. Panagiotopoulos, X. Fan, D. Urban, A. Nie, R. Shahbazian-Yassar and L. Hu, Chem. Mater., 2016, 28, 6528–6535 CrossRef CAS.
- Y. Lu, P. Zhou, K. Lei, Q. Zhao, Z. Tao and J. Chen, Adv. Energy Mater., 2017, 7, 1601973 CrossRef.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Energy Environ. Sci., 2015, 8, 2963–2969 CAS.
- D. Li, L. Zhang, H. Chen, J. Wang, L.-X. Ding, S. Wang, P. J. Ashman and H. Wang, J. Mater. Chem. A, 2016, 4, 8630–8635 CAS.
- D. Xu, C. Chen, J. Xie, B. Zhang, L. Miao, J. Cai, Y. Huang and L. Zhang, Adv. Energy Mater., 2016, 6, 1501929 CrossRef.
- M. Dahbi, N. Yabuuchi, M. Fukunishi, K. Kubota, K. Chihara, K. Tokiwa, X.-f. Yu, H. Ushiyama, K. Yamashita and J.-Y. Son, Chem. Mater., 2016, 28, 1625–1635 CrossRef CAS.
- B. Zhang, G. Rousse, D. Foix, R. Dugas, D. A. Corte and J. M. Tarascon, Adv. Mater., 2016, 28, 9824–9830 CrossRef CAS PubMed.
- Z. Zhang, Y. Fu, X. Yang, Y. Qu and Z. Zhang, ChemNanoMat, 2015, 1, 409–414 CrossRef CAS.
- Y. Zheng, T. Zhou, C. Zhang, J. Mao, H. Liu and Z. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- J.-L. Yue, Q. Sun and Z.-W. Fu, Chem. Commun., 2013, 49, 5868 RSC.
- F. Zhang, C. Xia, J. Zhu, B. Ahmed, H. Liang, D. B. Velusamy, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2016, 6, 1601188 CrossRef.
- H. Fan, H. Yu, X. Wu, Y. Zhang, Z. Luo, H. Wang, Y. Guo, S. Madhavi and Q. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25261–25267 CAS.
- C. Cui, X. Li, Z. Hu, J. Xu, H. Liu and J. Ma, RSC Adv., 2015, 5, 92506–92514 RSC.
- K. Zhang, M. Park, L. Zhou, G. H. Lee, J. Shin, Z. Hu, S. L. Chou, J. Chen and Y. M. Kang, Angew. Chem., 2016, 55, 12822–12826 CrossRef CAS PubMed.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS PubMed.
- X. Liu, K. Zhang, K. Lei, F. Li, Z. Tao and J. Chen, Nano Res., 2016, 9, 198–206 CrossRef CAS.
- K. Zhang, M. Park, L. Zhou, G.-H. Lee, W. Li, Y.-M. Kang and J. Chen, Adv. Funct. Mater., 2016, 26, 6728–6735 CrossRef CAS.
- C. P. Yang, Y. X. Yin and Y. G. Guo, J. Phys. Lett., 2015, 6, 256–266 CAS.
- J. Xu, J. Ma, Q. Fan, S. Guo and S. Dou, Adv. Mater., 2017, 29, 1606454 CrossRef PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- K. X. Wang, X. H. Li and J. S. Chen, Adv. Mater., 2015, 27, 527–545 CrossRef CAS PubMed.
- Y.-M. Hu, M.-C. Liu, Y.-X. Hu, Q.-Q. Yang, L.-B. Kong and L. Kang, Electrochim. Acta, 2016, 215, 114–125 CrossRef CAS.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed.
- B.-N. Yun, H. L. Du, J.-Y. Hwang, H.-G. Jung and Y.-K. Sun, J. Mater. Chem. A, 2017, 5, 2802–2810 CAS.
- D. Guo, L. Lai, A. Cao, H. Liu, S. Dou and J. Ma, RSC Adv., 2015, 5, 55856–55869 RSC.
- D. Xu, C. Chen, J. Xie, B. Zhang, L. Miao, J. Cai, Y. Huang and L. Zhang, Adv. Energy Mater., 2016, 6, 1501929 CrossRef.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef CAS PubMed.
- L. Mei, M. Mao, S. Chou, H. Liu, S. Dou, D. H. Ng and J. Ma, J. Mater. Chem. A, 2015, 3, 21699–21705 CAS.
- Y. Wei, F. Yan, X. Tang, Y. Luo, M. Zhang, W. Wei and L. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 21703–21711 CAS.
- I. H. Kwak, H. S. Im, D. M. Jang, Y. W. Kim, K. Park, Y. R. Lim, E. H. Cha and J. Park, ACS Appl. Mater. Interfaces, 2016, 8, 5327–5334 CAS.
- C. Xia, H. Liang, J. Zhu, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2017, 7, 1602089 CrossRef.
- X. Qian, H. Li, L. Shao, X. Jiang and L. Hou, ACS Appl. Mater. Interfaces, 2016, 8, 29486–29495 CAS.
- H. Chen, J. Jiang, Y. Zhao, L. Zhang, D. Guo and D. Xia, J. Mater. Chem. A, 2015, 3, 428–437 CAS.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- G. Zhou, C. Wu, Y. Wei, C. Li, Q. Lian, C. Cui, W. Wei and L. Chen, Electrochim. Acta, 2016, 222, 1878–1886 CrossRef CAS.
- Q. Lian, G. Zhou, X. Zeng, C. Wu, Y. Wei, C. Cui, W. Wei, L. Chen and C. Li, ACS Appl. Mater. Interfaces, 2016, 8, 30256–30263 CAS.
- S. Xin, Z. Liu, L. Ma, Y. Sun, C. Xiao, F. Li and Y. Du, Nano Res., 2016, 9, 3795–3811 CrossRef CAS.
- Y. J. Hong, J. H. Kim and Y. Chan Kang, J. Mater. Chem. A, 2016, 4, 15471–15477 CAS.
- Z. Zhang, X. Shi and X. Yang, Electrochim. Acta, 2016, 208, 238–243 CrossRef CAS.
- G. D. Park and Y. C. Kang, Chemistry, 2016, 22, 4140–4146 CrossRef CAS PubMed.
- J. S. Cho, S. Y. Lee and Y. C. Kang, Sci. Rep., 2016, 6, 23338 CrossRef CAS PubMed.
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- Y. Jiang, X. Ma, J. Feng and S. Xiong, J. Mater. Chem. A, 2015, 3, 4539–4546 CAS.
- P. Zheng, T. Liu and S. Guo, Sci. Rep., 2016, 6, 35620 CrossRef CAS PubMed.
- L. Zhang, L. Lu, D. Zhang, W. Hu, N. Wang, B. Xu, Y. Li and H. Zeng, Electrochim. Acta, 2016, 209, 423–429 CrossRef CAS.
- W. Maneeprakorn, M. A. Malik and P. O'Brien, J. Mater. Chem., 2010, 20, 2329 RSC.
- J.-B. Shi, P.-F. Wu, C.-T. Kao, M.-W. Lee, C.-C. Chan, P.-C. Yang, C.-L. Lin, R.-Y. Huang, Y.-J. Huang, S.-K. Lin, F.-C. Cheng, H.-S. Lin and H.-W. Lee, Cryst. Res. Technol., 2015, 50, 155–159 CrossRef CAS.
- A. Sobhani and M. Salavati-Niasari, Superlattices Microstruct., 2014, 65, 79–90 CrossRef CAS.
- C. Xia, Q. Jiang, C. Zhao, M. N. Hedhili and H. N. Alshareef, Adv. Mater., 2016, 28, 77–85 CrossRef CAS PubMed.
- G. Zhang, S. Hou, H. Zhang, W. Zeng, F. Yan, C. C. Li and H. Duan, Adv. Mater., 2015, 27, 2400–2405 CrossRef CAS PubMed.
- Y. Zhang, Z. Liu, H. Zhao and Y. Du, RSC Adv., 2016, 6, 1440–1444 RSC.
- F. Zhang, C. Xia, J. Zhu, B. Ahmed, H. Liang, D. B. Velusamy, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2016, 6, 1601188 CrossRef.
- H. Chen, S. Chen, M. Fan, C. Li, D. Chen, G. Tian and K. Shu, J. Mater. Chem. A, 2015, 3, 23653–23659 CAS.
- M. Zhen, M. Sun, G. Gao, L. Liu and Z. Zhou, Chemistry, 2015, 21, 5317–5322 CrossRef CAS PubMed.
- H. Liu, K. Cao, X. Xu, L. Jiao, Y. Wang and H. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 11239–11245 CAS.
- Y. Zhang, Z. Ma, D. Liu, S. Dou, J. Ma, M. Zhang, Z. Guo, R. Chen and S. Wang, J. Mater. Chem. A, 2017, 5, 512–518 CAS.
- Y. Zheng, T. Zhou, C. Zhang, J. Mao, H. Liu and Z. Guo, Angew. Chem., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00419b |
| This journal is © the Partner Organisations 2017 |