 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–activity relationship for the solid state emission of a new family of “push–pull” π-extended chromophores†
Andrea
Nitti
a,
Francesca
Villafiorita-Monteleone
b,
Aurora
Pacini
a,
Chiara
Botta
b,
Tersilla
Virgili
c,
Alessandra
Forni
d,
Elena
Cariati
e,
Massimo
Boiocchi
f and
Dario
Pasini
 *a
*a
aDepartment of Chemistry and INSTM Research Unit, University of Pavia, Viale Taramelli, 10, 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it
bISMAC-CNR, Via Corti 12, 20133 Milano, Italy
cIFN-CNR, Dipartimento di Fisica, Politecnico di Milano, Piazza Leonardo Da Vinci, 32, 20132 Milano, Italy
dISTM – CNR, c/o Dipartimento di Chimica, Università Degli Studi di Milano, Via Venezian, 21, 20133 Milano, Italy
eUniversità Degli Studi di Milano, Dipartimento di Chimica, via Golgi 19, 20133 Milano, Italy
fCentro Grandi Strumenti, University of Pavia, Via Bassi, 21 – 27100 – Pavia, Italy
First published on 22nd July 2016
Abstract
We report the design, synthesis, molecular optical properties, and solid state emissive behaviour of a series of novel compounds, which, similar to the archetypal AIE luminogen tetraphenylethene, are formed of a central olefin stator and decorated with either three or four rotors. These rotors, being either electron-rich substituted benzenes, or electron-withdrawing functional groups (esters, ketones, cyano groups) confer a “push–pull” character to the overall molecular structure. Building on both new and already published contributions, a comprehensive picture of the properties and the potential of these compounds is provided.
Introduction
Strong emission in the aggregated state for organic compounds has long been considered counterintuitive: aggregation-caused quenching (ACQ) takes place in the condensed phase for most π-extended, emitting chromophores. ACQ severely inhibits their application in real-world devices, such as light-emitting diodes, optical waveguides and lasers.1 In recent years, Tang and other groups have demonstrated that selected organic dyes can indeed behave in such a counterintuitive way: they are non-emissive when in diluted solutions, and become highly emissive in the solid state.2Many molecular systems have been proven to be efficient AIE luminogens; one prototypical AIE emissive chromophore, and the first to have been reported, is hexaphenyl-substituted silole (HPS), published by Tang and coworkers in 2001.3 Aggregation-induced emission (AIE) is frequently ascribed to restricted internal rotations (RIR):4 for example, when HPS is in solution, the rotation of its phenyl rings dissipates the excitation energy, whereas aggregate formation in crystals does not allow the phenyl ring rotations and a radiative decay with a nanosecond lifetime is activated. Apart from silole systems, a great deal of work has been done with other AIE luminogens, aimed at confirming and exploiting the RIR principle. To date, many other families of compounds have since been proposed, featuring a variety of scaffold functionalities and different “switching on” mechanisms for emission in the solid state. The field has been recently and comprehensively reviewed.5
The most widely used of prototypical AIE chromophores is tetraphenylethene (TPE), which has been extensively investigated. The central olefin stator of the molecule is surrounded by four peripheral aromatic rotors (the phenyl rings). Its high solid state quantum yield, and its relatively simple synthesis have prompted its inclusion in a wide variety of complex architectures, such as linear6 and hyperbranched polymers,7 and metal–organic frameworks.8 In most cases, the high solid state efficiency of the molecular scaffold is fully preserved with the introduction of the organic functionalities needed for the its covalent or non-covalent bridging to macromolecular/supramolecular architectures. In other words, the TPE scaffold is able to withhold AIE activity even if major modifications are inserted within its chemical structure.
D–π–A dyes usually exhibit unique emissive properties due to their intramolecular charge transfer (ICT) transitions, and they are of great potential interest since ground and excited electronic states can be tuned under various conditions.9 We have serendipitously discovered a new class of AIE luminogens with a “push–pull” structure. They possess similarities to TPE, since the central stator is still a carbon–carbon double bond, which is decorated with three (not four, as in TPE) substituents, two carboxylate esters and a 4-dialkylaminophenyl branch (Fig. 1, compounds series 1).10 Studying one of the AIE active compounds in detail using ultrafast pump–probe spectroscopy and combining with calculations, we have given direct evidence that RIR is the key process for switching on the AIE properties.11
 | ||
| Fig. 1 Chemical structures of compound series 1–5. | ||
More recently, compound 2b (Fig. 1) was effective in unravelling important features of the twisted intramolecular charge transfer mechanism, a peculiar characteristic of such “push–pull” structures.12
The potential of “push–pull” molecular scaffolds for the design of innovative AIE luminogens is huge, since the molecular optical properties (absorption and emission λmax), and consequently the solid state emission, can in principle be tuned using “push” and “pull” substituents with variable electron-donating and electron-withdrawing characters. In fact, we have recently reported that one of these compounds (5a in Fig. 1) crystallizes in four different polymorphs all showing different emission colors, and that the emission color can be tuned via heating and grinding in the solid state, highlighting the potential application for stimuli responsive solid-state materials.13
In this paper, we report our further achievements into the fine tuning of the previously proposed molecular “push–pull” structures in order to investigate their potential in terms of AIE, to match requirements for optoelectronic and sensing applications. We will discuss the optical properties and the AIE and solid state behavior of a series of compounds in which the original molecular structure has been systematically varied (Fig. 1). Together with the electronic characteristics of the rotor groups around the stator mentioned above, which are necessary to give a “push–pull” character, given the importance of the RIR mechanism, variations in the steric hindrance of the rotor functional groups around the stator have also been addressed.
Results and discussion
Design and synthesis of the molecular modules
The compounds presented and discussed in this study are shown in Fig. 1. For the sake of clarity we have divided them into five different classes (compounds 1–5). The compounds in series 1 have been already reported, and possess ester derivatives as the “pull” components in a trisubstituted ethylene molecular scaffold; they bear minimal differences between them, either in the nature of the alkyl ester substituents, or in the nature of the dialkyl amino substituents. In the compounds in series 2, the “pull” ester moieties have been changed to ketones and trifluoromethylketones, both possessing a similar degree of steric hindrance with respect to esters, but with substantially different electron-withdrawing characteristics, as testified by their σp Hammett parameters14 (0.45 for COOCH3, 0.50 for COCH3, and 0.80 for COCF3). Furthermore, for the compounds in series 2, the “push” dimethylamino group has been changed in two complementary ways: (a) it has been substituted with a methoxy group, and (b) it has been changed with linear or cyclic dialkylamino groups, addressing differences in the packing properties in the solid state. The importance of the length of the dialkylamino substituent has already been demonstrated in the compounds in series 1, where a switching off of the AIE properties was observed on changing the dimethylamino to diethylamino “pull” moieties.10aThe compounds in series 3 have been designed with cyano substituents: linear functional groups with a low steric hindrance, possessing different electronic characteristics with respect to esters or ketones (σp = 0.66). Variable dialkylamino substituents have also been synthesized in this series. The introduction of a further dimethylaminoaryl “pull” moiety into the molecular skeleton to form tetrasubstituted ethylene, “cruciform-like” derivatives15 has been synthetically achieved (series 4 and 5).
The synthesis of series 2–3 was carried out following adaptations of reported procedures starting from the appropriate aldehyde and the 1,3-dicarbonyl compound or malononitrile. An initial screening of the optimal conditions was performed for the synthesis of compound 2a, which has been previously reported. Yields using the reported procedure,16 with piperidinium acetate as the catalyst, were somewhat disappointing (17%). Other published methods (with CuCl2 as catalyst)17 for unsubstituted benzaldehyde were equally unsatisfactory (17%), whereas the use of an excess of acetic anhydride (procedure published for the synthesis of the previously known 2d)18 gave improved yields (25% for 2a). This last methodology was then applied to all compounds in series 2 and 3, with yields ranging, after purification using column chromatography, from 25% to 76% (Scheme 1, top).
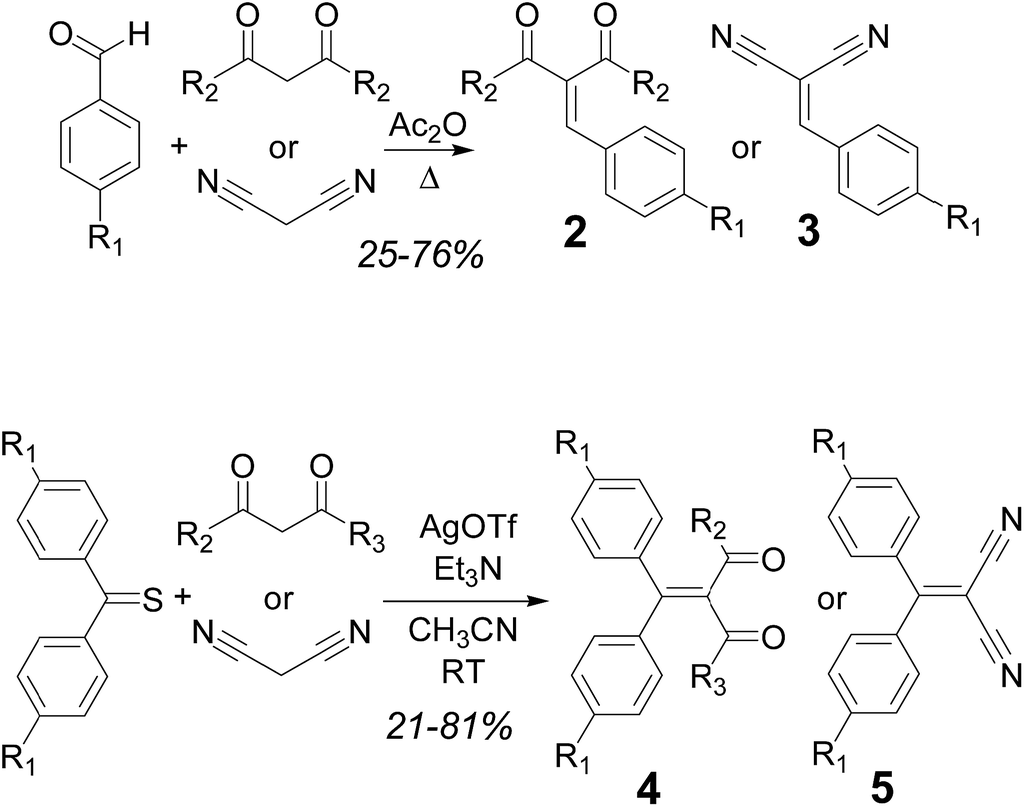 | ||
| Scheme 1 Synthesis of compounds in series 2–5. | ||
The compounds in series 4 and 5 were synthesized via silver triflate mediated condensation19 of the appropriate 4,4′-disubstituted thiobenzophenone (either commercially available or ad hoc synthesized from the corresponding dibenzophenone with Lawesson's reagent20), and the appropriate 1,3-dicarbonyl compound or malononitrile (Scheme 1, bottom). Yields after purification using column chromatography ranged between 21 and 81%. All new compounds were fully characterized using NMR spectroscopy and mass spectrometric techniques. Some of the yields are rather low: however, monitoring of the reaction mixture using TLC in these cases revealed essentially a spot-to-spot conversion of the starting aldehyde or ketone into the product. It is likely, therefore, that collateral reactions between the starting material and the catalysts or reaction solvents generate byproducts, presumably removed during the work-up.
The room temperature 1H NMR spectra of all the compounds showed the expected simple patterns and the presence of only one set of signals for each group of symmetry-related proton resonances, revealing that all possible dynamic processes are fast on the NMR timescale at this temperature. A comparison between the relevant signals of the 1H and 13C NMR spectra is presented in Tables S1–S4 of the ESI† section. No evidence of enol structures could be found in the spectra of compounds 2a and 2c, and 4b, 4c, 4e, 4f, which bear hydrogen atoms α to a carbonyl to thus support the possibility of enolization: signals related to the CH3 groups integrate correctly with respect to the rest of the proton resonances, and no vinyl signals in the appropriate region (5–6 ppm) could be detected.
The locking of the rotation of the “pull” moieties (for example, the ester and ketones in compounds 1a and 2b, respectively) and of the aryl moiety around its own axis are both key elements responsible for the molecular rigidification and the activation of the RIR mechanism in solid-state emission.10a,11,12 Compounds 3a and 5a possess linear cyano moieties, for which any lateral steric interaction is not possible as the “pull” components, and thus they can be ideal models to investigate the “pirouetting” movements of the aryl moieties. Variable temperature NMR studies performed on compound 3a revealed that this molecular rotation becomes slow on the NMR timescale (Fig. 2) upon freezing the sample in d8-THF. At low temperatures, the Hb protons are split into two different signals as a result of the loss of the local symmetry around the aryl main axis, and coalesce at 213 K. A free energy barrier for such a dynamic process (9.6 kcal mol−1) could be calculated with the coalescence method.21
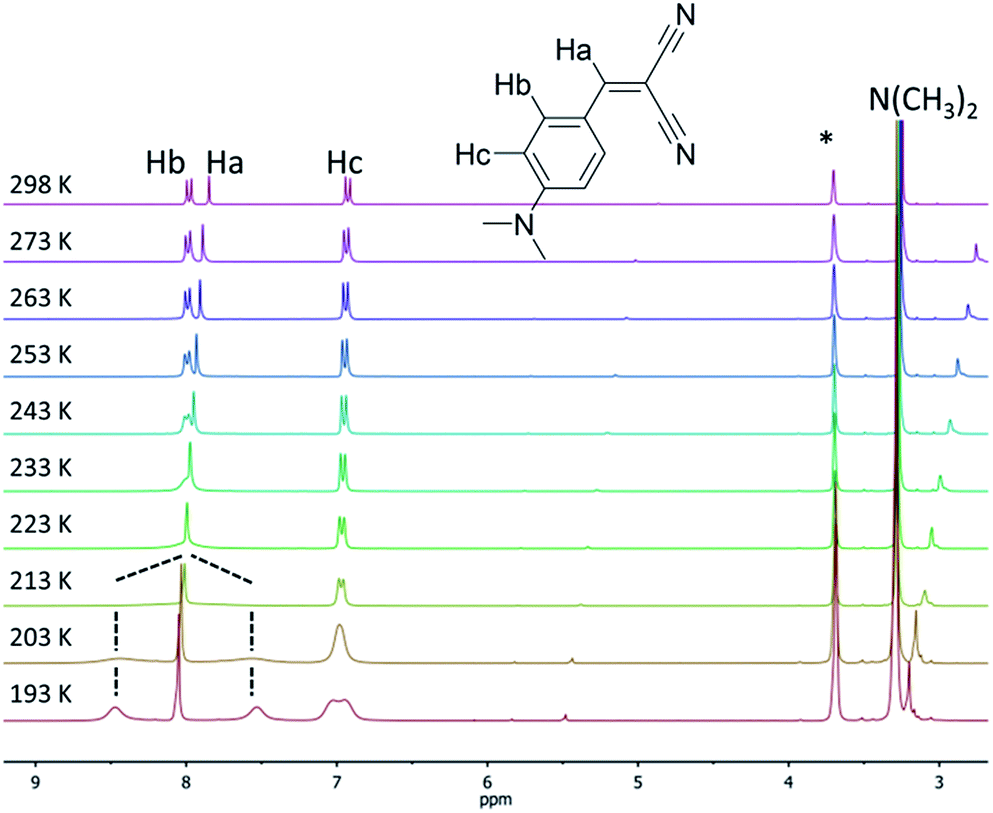 | ||
| Fig. 2 Variable temperature 1H NMR spectra (300 MHz, d8-THF) of compound 3a. The asterisk denotes residual the solvent peak. | ||
For compound 5a, such signal splitting could not be observed down to 193 K (Fig. S1†), meaning that a precise value for the energy barrier of the rotation of the aryl rings around their axes, to compare with 3a, could not be calculated.
Molecular optical properties
The relevant optical properties for the compounds discussed are reported in Table 1 in order to rationalize their optical–structural relationship.| Compound | λ abs (nm) | λ em (nm), solutionb | λ em (nm), powder | PL QY (%), solution | PL QY (%), powder | Physical appearance |
|---|---|---|---|---|---|---|
| a In MeCN solution (1–5 10−5 M). b Emission maximum, 10−6 M solution. c Data taken from ref. 10a, in toluene. d Max PL QY of the polymorphs. | ||||||
| 1a | 378 | 429c | 468 | <0.1 | 38 | Solid |
| 1b | 380 | 440c | 473 | <0.1 | 38 | Solid |
| 1c | 380 | 445c | 522 | <0.1 | 1 | Solid |
| 2a | 382 | 462 | 520 | 0.33 | 1 | Solid |
| 2b | 461 | 553 | 625 | <0.1 | 11 | Solid |
| 2c | 274 | n.a. | n.a. | n.a. | n.a. | Oil |
| 2d | 360 | n.a. | n.a. | n.a. | n.a. | Oil |
| 2e | 467 | 528 | 630 | <0.1 | <0.1 | Solid |
| 2f | 390 | 456 | 540 | 1 | <0.1 | Solid |
| 3a | 430 | 488 | 615 | 0.9 | 3 | Solid |
| 3b | 436 | 491 | 550, 630 | 1.4 | 5 | Solid |
| 3c | 436 | 491 | 630 | 0.87 | 3 | Solid |
| 4a | 360 | n.a. | n.a. | n.a. | n.a. | Oil |
| 4b | 385 | 495 | 500 | <0.1 | <0.1 | Solid |
| 4c | 384 | 495 | 512 | 0.1–1 | 1 | Solid |
| 5a | 432 | 511 | 530, 535, 595 | <0.1 | 11d | Solid |
| 5b | 337 | 395 | 480 | <0.1 | 5 | Solid |
The λmax, corresponding to the HOMO–LUMO energy, for a homologous series of compounds bearing the same electron-donating substituent was found to be modulated by the nature of the electron-withdrawing substituent. In fact, a linear correlation between the σp Hammett's parameters of the “pull” moiety (Fig. 3) and the λmax for compounds 1a, 2a, 2b, 3a, all bearing dimethylamino groups, was found. Such a correlation indicates a “through bond” effect of the substituents, and demonstrates an effective conjugation through the π-systems, formally composed of a styrene-like moiety. A similar linear correlation (based on compounds 4a, 4b and 5a) could be verified for cruciform-like compounds (Fig. S2†). Such correlations could also be useful in predicting the properties of analogous compounds utilizing different “push–pull” substituents. The direct comparison, where possible, between monoaryl systems (series 2 and 3) and cruciform-like systems (series 4 and 5) demonstrates (Table 1) that the introduction of a further aryl branch does not enhance the λmax and the “push–pull” character of the system (compare for example 3avs.5a, and 1avs.4a).
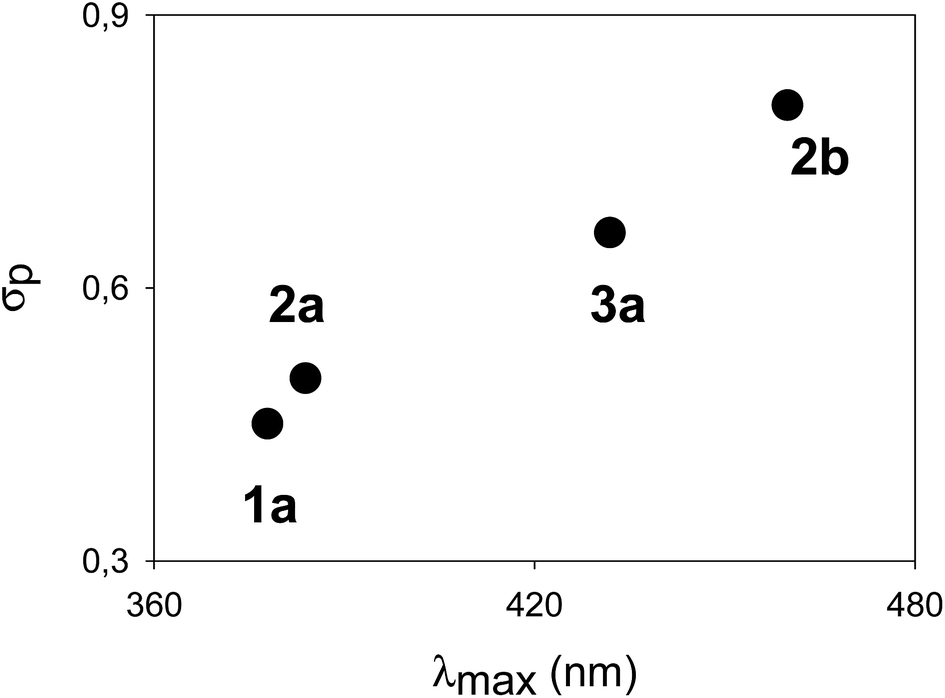 | ||
| Fig. 3 Correlation between λmax and σp Hammett's parameters for the dimethylamino-terminated compounds 1a, 2a, 2b and 3a (fitting coefficient r2 = 0.99). | ||
All the compounds reported in Table 1 show a very low PL QY in solution, which does not depend much on the polarity of the solvent. Compounds 1a,10a2b,122e, 3a, 3b and 5a13 show solvatochromic behaviour related to their push–pull molecular structure (see ESI†) without any relevant variation in emission intensity.
However, for compounds 1a and 2b, a strong increase in PL intensity on increasing the solvent viscosity has been observed,10a,11 indicating that the rigidity of the environment, rather than its polarity, plays an important role in their emissive process. On freezing the solutions or on adding a non-solvent to the solutions, we have reported an increase in the PL intensity for compounds 1a and 5b.10a,12 In diluted solutions below the solidification point of the solvent, molecular motions are blocked by the rigidified solvent. Upon adding a non-solvent at room temperature to diluted solutions, molecular aggregation into nanoparticles blocks intramolecular motion. Their AIE properties have therefore been ascribed to the commonly observed RIR effect.
As shown in Table 1, some of the compounds display a strong enhancement in PL QY in the solid state, while others maintain nearly the same low value as in solution. Compounds 1a and 1b display the strongest PL enhancement, the series 3 and 5 a moderate one, while in the case of series 4 and 2 (with the exception of 2b) no relevant variation is observed in the PL intensity between solution and the solid state. Changes in the lateral alkyl chains in the “push” moieties bring about substantial changes in the AIE properties: on changing from dimethylamino to diethylamino aryl substituents the AIE behaviour is either strongly reduced (from 1a to 1c) or switched off (from 2b to 2e). In the case of 3a (dimethylamino, already without AIE behaviour) the change to diethylamino (3b) confirms the absence of AIE behaviour.
Among the other compounds, 3b and 5a display quite interesting features with solvatochromism in solution, good solid state QYs and the presence of different components in the solid state emission, already studied in detail in the case of compound 5a.12 The optical absorption, PL excitation profiles (PLE) and emission spectra of 3b in acetonitrile solution and as powders are reported in Fig. 4. For the powder, a strong red-shift in the PL and the presence of two peaks (495 and 580 nm) in the PLE spectra are observed. The latter are very probably associated to the presence of two different species which can be separated thanks to their different solubility in pentane. Upon pentane extraction, two main contributions are observed in the emission spectrum, a shoulder at 550 nm and the main peak at 630 nm, while the insoluble portion of the powder displays a main emission at 635 nm. The relative intensity of the two contributions can be changed upon manual grinding of the powder (see ESI†).
 | ||
| Fig. 4 Optical absorption and PL spectra of compound 3b in acetonitrile solution (dotted lines). PLE and PL spectra of solid 3b: pentane soluble component (solid line) and pentane insoluble component (dashed-dotted line). | ||
Since, for RIR materials, the emissive properties strictly depend on the type of aggregation (crystalline vs. amorphous) and on the crystal rigidity, in the next section we analyse the crystal structures of the compounds in detail.
X-ray crystal structures
Crystal structures of the series 1,10a and compounds 2b12 and 5a13 were previously reported by us, together with a thorough discussion, supported with DFT and TDDFT calculations,11–13 of the structure–optical property relationships governing their emissive behaviour. It was found that their AIE properties were strictly related to their crystal structure, which is able to activate a RIR process. Moreover, by comparing the crystal structures and the solid state optical behaviours of the different compounds, we have ascribed the high PL QY (up to 38%) observed for compounds 1a and 1b to the formation of J-dimers.Unlike the molecular structures of the previously reported compounds, those of series 3 (3a22 and 3b23 being previously reported) are essentially planar. Crystals of 3c belong to the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with 6 molecules in the asymmetric unit (see Fig. 5 for its crystal packing). A subtle, not previously highlighted, feature shared by these structures, which could have some role in their emissive behaviour, was the slightly greater degree of conjugation of the trans CN group with the benzene ring with respect to the cis group. The angles between the CN bond and the normal to the plane through the benzene ring were in fact 88.9(1)° (3a22c) and in the range 85.9(2)–89.5(2)° (3c) for the trans CN groups, and 79.5(1)° (3a) and in the range 77.6(2)–85.3(2)° for the cis groups. In the case of 3b,23b the two angles were comparable (84.8(1) and 86.7(1)°, respectively).
space group with 6 molecules in the asymmetric unit (see Fig. 5 for its crystal packing). A subtle, not previously highlighted, feature shared by these structures, which could have some role in their emissive behaviour, was the slightly greater degree of conjugation of the trans CN group with the benzene ring with respect to the cis group. The angles between the CN bond and the normal to the plane through the benzene ring were in fact 88.9(1)° (3a22c) and in the range 85.9(2)–89.5(2)° (3c) for the trans CN groups, and 79.5(1)° (3a) and in the range 77.6(2)–85.3(2)° for the cis groups. In the case of 3b,23b the two angles were comparable (84.8(1) and 86.7(1)°, respectively).
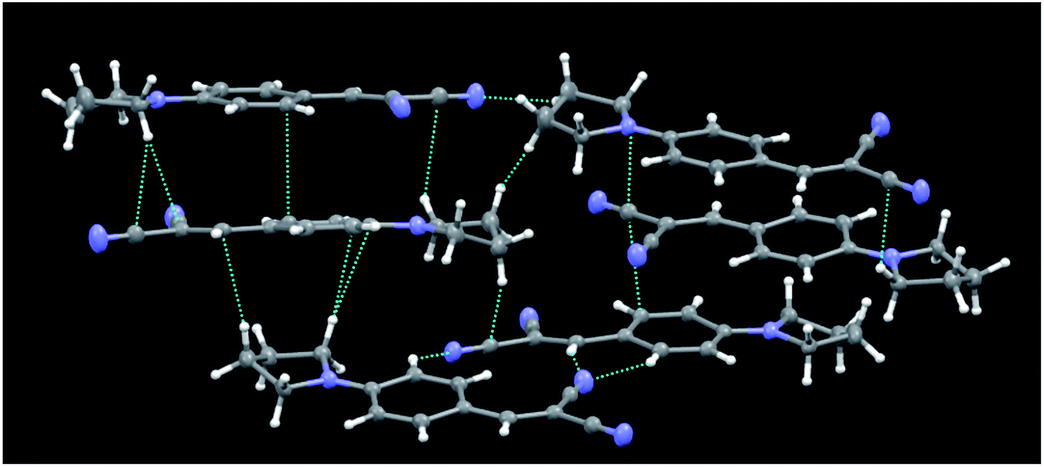 | ||
| Fig. 5 Partial view of the crystal packing of 3c, where contacts shorter than the sum of the van der Waals radii are included. Ellipsoids are drawn at the 50% probability level. | ||
The crystal structures of 3 share a pseudo layered structure, but 3a and 3b do not reveal the presence of significant π–π stacking interactions owing to the too long interplanar distance between adjacent benzene rings. Only C–H⋯N (3a) or C–H⋯N and C–H⋯π (3b) intermolecular interactions stabilize their crystal structures. In the case of 3c, besides C–H⋯N and C–H⋯π interactions, we also observe short CC contacts (3.337 and 3.317 Å) which involve, however, only 4 of the 6 independent molecules. As a result, the presence of weak interactions in the crystal phase explains the AIE behaviour of the series of compounds 3, while their rather low PL QY can be ascribed to the absence of strong aggregation (Fig. 5).
Crystals of 5b belong to the C2/c space group with half a molecule in the asymmetric unit (see Fig. 6). Unlike 5a, for which four kinds of crystals were obtained, characterized by different morphologies and absorption and emission colours, only one phase was obtained for 5b. A distinctive feature of these molecular structures is their twisted conformation due to the steric hindrance both between the CN and the dimethylamino-phenyl substituents and between the phenyl rings. As previously evidenced,12 three geometrical factors can act in a concerted way to reduce such hindrance, that is, the (N)C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C(Ph) torsion angle, the reciprocal tilting of the phenyl rings (quantified through the dihedral angle between the least-squares planes through the phenyl carbon atoms) and the central double bond, which in the present structures is significantly elongated with respect to the value of 1.331(9) Å reported for (C2)–CC–(C2) unconjugated bonds,24 denoting a high degree of conjugation. It is to be pointed out that, owing to their cross-conjugated architecture,25 the phenyl rings, connected with each other via two single bonds, are separately conjugated to each CN group, as well as the CN groups being separately conjugated to each phenyl ring. The conformational differences observed in the four crystals of 5a, though small, were found to be associated with a different degree of conjugation between the molecular moieties connected through the CC double bond. In particular, the lower the dihedral angle between the phenyl rings, the larger the distortion around the double bond and the greater the cross-conjugation. In the case of 5b, we observe a large dihedral angle between the phenyl rings (71.6(1) vs. 71.8(1)–58.0(1)° found in 5a), a lower (N)C–CC–C(Ph) torsion angle (9.0(1) vs. 11.8(1)–19.9(1)° of 5a) and a shorter CC bond length with respect to 5a (1.366(3) vs. 1.376(3)–1.390(2) Å). All these features indicate a lower cross-conjugation for 5b with respect to 5a.
C–C(Ph) torsion angle, the reciprocal tilting of the phenyl rings (quantified through the dihedral angle between the least-squares planes through the phenyl carbon atoms) and the central double bond, which in the present structures is significantly elongated with respect to the value of 1.331(9) Å reported for (C2)–CC–(C2) unconjugated bonds,24 denoting a high degree of conjugation. It is to be pointed out that, owing to their cross-conjugated architecture,25 the phenyl rings, connected with each other via two single bonds, are separately conjugated to each CN group, as well as the CN groups being separately conjugated to each phenyl ring. The conformational differences observed in the four crystals of 5a, though small, were found to be associated with a different degree of conjugation between the molecular moieties connected through the CC double bond. In particular, the lower the dihedral angle between the phenyl rings, the larger the distortion around the double bond and the greater the cross-conjugation. In the case of 5b, we observe a large dihedral angle between the phenyl rings (71.6(1) vs. 71.8(1)–58.0(1)° found in 5a), a lower (N)C–CC–C(Ph) torsion angle (9.0(1) vs. 11.8(1)–19.9(1)° of 5a) and a shorter CC bond length with respect to 5a (1.366(3) vs. 1.376(3)–1.390(2) Å). All these features indicate a lower cross-conjugation for 5b with respect to 5a.
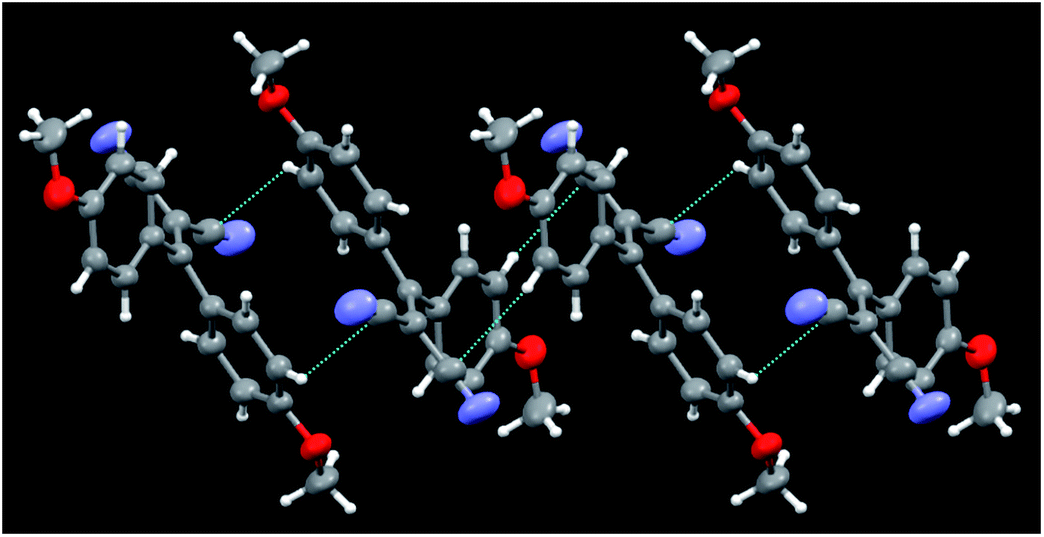 | ||
| Fig. 6 Partial view of the crystal packing of 5b, where contacts shorter than the sum of the van der Waals radii are included. Ellipsoids are drawn at the 50% probability level. | ||
The twisted conformations of compounds 5a and 5b rule out the presence of strong intermolecular π–π stacking interactions, excluding the formation of H- or J-aggregates. On the other hand, as noted for the series of compounds 3, the weak C–H⋯N and C–H⋯π intermolecular interactions found in the structures of 5a and 5b are enough to fix the molecular conformations in the crystal structures to activate the RIR mechanism. In both series of compounds 3 and 5, the absence of strong intermolecular interactions and the conformational freedom associated with the presence of several single bonds explain the formation of different polymorphs, as demonstrated from X-ray investigation for 5a and suggested from spectroscopic evidence for 3b.
Pump–probe experiments
A deep understanding of the role of the intramolecular torsional mobility in AIE molecules is of crucial importance in order to design new organic compounds with improved optoelectronic properties. We reported the spectral evolution of the stimulated emission of the series of compounds 1 dissolved in solution, showing that the torsional relaxation toward the equilibrium geometry of the excited state takes place on a time scale that depends on the solvent viscosity.11 Pump–probe ultrafast dynamics have also been employed to study the time evolution of the excited states of compound 2b, whose long-living optical gain was detected only when the twisted intramolecular charge transfer mechanism is inhibited on increasing the solvent viscosity.12Here we report ultrafast pump–probe measurements on two solutions of compound 5a displaying different viscosities, namely polyethylene glycol (PEG) and acetonitrile (ACN). In this way it is possible to temporally resolve the spectral evolution of the excited states created after the pump excitation of the molecule dispersed in viscous and non-viscous solvents.
In this experiment, the transmission of a white light pulse (probe) is detected at different time delays with respect to the pump excitation (400 nm). The differential transmission spectra of the probe pulse ΔT/T (where ΔT = Ton − T, and Ton is the transmission of the probe light with pump excitation and T is the transmission of the probe light without pump excitation) taken at different probe delays is then obtained. A positive ΔT/T signal (transmission increasing after pump excitation) is an indication of bleaching of the ground state when the signal spectrally overlaps the absorption spectrum, or of Stimulated Emission (SE) from the excited state when the signal overlaps the PL spectrum of the molecule. The time-resolved spectra (Fig. 7) recorded in both solvents show a positive broad band that can be associated with Stimulated Emission (SE peak at around 530 nm). In ACN solution after around 1 ps the formation of a negative Photoinduced Absorption (PA) band centered at 520 nm is observed, showing a fast decay on a time scale of a few ps (red line, inset Fig. 7, top panel). On the contrary, in PEG solution there is no formation of a PA band in this time (black line, inset Fig. 7, top panel).
 | ||
| Fig. 7 Differential transmission spectra at 0 fs (black line), 2 ps (red line) and 20 ps (blue line) probe delays for PEG (top panel) and ACN (bottom panel) solutions. Dynamics at 530 nm are shown for both solutions in the inset of the top panel. | ||
Conclusions
We have reported and compared the design, synthesis, molecular optical properties, and solid state emissive behaviour of several series of novel compounds with “push–pull” character. The compounds bear three or four substituents around a central olefin stator. While all the structures are non-emissive in solution, a handful of them possess solid-state emission with quantum yields over 10%. With the exception of compound 5a, all AIE compounds are characterized by having three substituents around the stator. As a general trend, the use of 4-methoxyaryl substituents as the “push” component does not bring any useful solid-state emissive behavior.Our classes of compounds are efficient in terms of tunability of emission response, but the translation of molecular design into efficient solid state emission is far from straightforward. The nice correlations with Hammett's parameters demonstrate the possibility of predicting and rationally tuning the energy gaps of these structures. However, good solid state emission is activated or deactivated with subtle changes in the molecular structures, sometimes in unpredictable ways. As such, the potential incorporation of the most promising of the structures presented here into complex covalent scaffolds (polymers, covalent organic frameworks) for functional applications cannot at present be considered immediately as occurs with other AIE scaffolds.
Experimental
General experimental for synthesis
All available compounds were purchased from commercial sources and used as received. Compounds 2b12 and 5a13 were previously reported by us. THF (Na, benzophenone), Et2O (Na, benzophenone) and CH2Cl2 (CaH2) were dried and distilled before use. 1H and 13C NMR spectra were recorded from solutions of CDCl3 on a Bruker 200 or AMX300 with the solvent residual proton signal as a standard. Analytical thin layer chromatography was performed on silica gel, chromophore loaded, commercially available plates. Flash chromatography was carried out using silica gel (pore size 60 Å, 230–400 mesh). 1H and 13C NMR spectra were recorded from solutions of CDCl3 on 200, 300 or 500 MHz spectrometers with the solvent residual proton signal or tetramethylsilane as a standard. Mass spectra were recorded using an electrospray ionization instrument (ESI). Melting points are uncorrected.General procedure for the synthesis of series 2 and 3
A solution of the appropriate aldehyde (1 equiv.) and ketone or malononitrile (1 equiv.) in Ac2O (5–10 mL) was heated under reflux (140 °C) for 18 h. After cooling to room temperature, the reaction mixture was poured into water/ice, and the aqueous suspension extracted with CH2Cl2. The organic phase was washed with a saturated solution of NaHCO3 and then dried (Na2SO4). The product was isolated after purification using column chromatography.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOEt = 7:3) and obtained as a yellow-orange solid (572 mg, 25%). Rf = 0.3 (hexane:AcOEt = 7:3). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.40 (s, 1H; vinyl CH), 7.31 (d, 2H; J = 9 Hz, ArH), 6.65 (d, 2H; J = 9 Hz, ArH), 3.05 (s, 6H; –N(CH3)2), 2.39 (s, 3H; –COCH3), 2.37 (s, 3H; –COCH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 207.0 (Cq), 196.3 (Cq), 151.8 (Cq), 140.9 (vinyl CH), 137.8 (Cq), 132.2 (Ar CH), 120.1 (Cq), 111.8 (Ar CH), 40.0 (N(CH3)2), 31.6 (CH3), 26.1 (CH3). MS-ESI m/z (%) = 232 [M + H]+ (100), 485 [2M + Na]+ (70). The crystal structure of this compound has been previously reported.26 The 1H and 13C NMR spectra matched those previously reported.27
:AcOEt = 9:1) and obtained as a yellow oil (255 mg, 25%). Rf = 0.14 (hexane:AcOEt = 9:1). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.63 (s, 1H; vinyl CH), 7.47 (d, 2H; J = 9 Hz, ArH), 6.93 (d, 2H; J = 9 Hz; ArH), 3.83 (s, 3H; –OCH3), 2.12 (m, 6H; 2CH3). The 1H NMR spectrum matched that previously reported.28
:CH2Cl2 = 7:3) and obtained as a yellow oil (925 mg, 28%). Rf = 0.3 (hexane:CH2Cl2 = 7:3). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.99 (s, 1H; vinyl CH), 7.42 (d, 2H; J = 9 Hz; ArH), 6.97 (d, 2H; J = 9 Hz; ArH), 3.87 (s, 3H; –OCH3). This compound was previously reported, but no NMR information was given.18
:CH2Cl2 = 1:1) and obtained as a pink waxy solid (185 mg, 50%). Rf = 0.5 (hexane:CH2Cl2 = 1:1). Mp = 60–62 °C. 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.83 (s, 1H; vinyl CH), 7.34 (d, 2H; J = 9 Hz; ArH), 6.65 (d, 2H; J = 9 Hz; ArH), 3.48 (q, 4H; J = 7 Hz; 2CH2), 1.25 (t, 6H; J = 7 Hz; 2CH3).
:AcOEt = 8:2) and obtained as an orange-brown solid (421 mg, 37%). Rf = 0.3 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.40 (s, 1H; vinyl CH), 7.30 (d, 2H; J = 9 Hz; ArH), 6.52 (d, 2H; J = 9 Hz; ArH), 3.36 (m, 4H; 2CH2), 2.38 (s, 3H; CH3), 2.37 (s, 3H; CH3), 2.04 (m, 4H; 2CH2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 207.2 (Cq; –CO), 196.1 (Cq; –CO), 149.4 (Cq), 141.3 (CH), 137.1 (Cq), 132.4 (CH), 119.3 (Cq), 111.7 (CH), 47.4 (CH2), 31.6 (CH3), 26.1 (CH3), 25.3 (CH2).
:CH2Cl2 = 1:1) and obtained as a dark red solid (512 mg, 28%). Rf = 0.4 (hexane:CH2Cl2 = 1:1). Mp = 89–91 °C. 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.85 (s, 1H; vinyl CH), 7.35 (d, 2H; J = 9 Hz; ArH), 6.45 (d, 2H; J = 9 Hz; ArH), 3.45 (t, 4H; 2CH2), 2.08 (m, 4H; 2CH2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 187.6 (q, Cq; J = 35 Hz), 177.5 (q, Cq; J = 35 Hz), 152.1 (Cq), 151.8 (CH), 135.2 (CH), 119.2 (Cq), 118.1 (Cq), 117.1 (q, Cq; J = 300 Hz), 115.3 (q, Cq; J = 300 Hz), 112.4 (CH), 47.8 (CH2), 25.2 (CH2).
:CH2Cl2 = 2:8) and obtained as an orange solid (388 mg, 40%). Rf = 0.7 (hexane:CH2Cl2 = 2:8). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.83 (d, 2H; J = 9 Hz; ArH), 7.48 (s, 1H; vinyl CH), 6.68 (d, 2H; J = 9 Hz; ArH), 3.15 (s, 6H; –N(CH3)2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 157.9 (CH), 154.1 (Cq), 133.7 (CH), 119.2 (Cq), 115.9 (Cq; –CN), 114.8 (Cq; –CN), 111.5 (CH), 71.9 (Cq), 40.0 (CH3). The 1H NMR spectrum matched the one previously reported.29
:CH2Cl2 = 2:8) and obtained as a dark pink solid (851 mg, 76%). Rf = 0.4 (hexane:CH2Cl2 = 2:8). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.81 (d, 2H; J = 9 Hz; ArH), 7.44 (s, 1H; vinyl CH), 6.68 (d, 2H; J = 9 Hz; ArH), 3.48 (q, 4H; J = 7 Hz; 2CH2), 1.25 (t, 6H; J = 7 Hz; 2CH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 157.7 (CH), 152.3 (Cq), 134.0 (CH), 118.8 (Cq), 116.1 (Cq; –CN), 114.9 (Cq; –CN), 111.2 (CH), 71.0 (Cq), 44.8 (CH2), 12.4 (CH3). The 1H and 13C NMR spectra matched those previously reported.30 The crystal structure of this compound has been previously reported.23b
:CH2Cl2 = 2:8) and obtained as a brown-orange solid (316 mg, 29%). Rf = 0.55 (hexane:CH2Cl2 = 2:8). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.80 (d, 2H; J = 9 Hz; ArH), 7.44 (s, 1H; vinyl CH), 6.57 (d, 2H; J = 9 Hz; ArH), 3.45 (m, 4H; 2CH2), 2.09 (m, 4H; 2CH2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 157.9 (CH), 151.8 (Cq), 133.9 (CH), 119.0 (Cq), 116.1 (Cq), 115.1 (Cq), 111.9 (CH), 70.9 (Cq), 47.8 (CH2), 25.2 (CH2). This compound was previously reported.31
:AcOEt = 7:3) and obtained as a yellow-orange solid (572 mg, 25%). Rf = 0.3 (hexane:AcOEt = 7:3). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.40 (s, 1H; vinyl CH), 7.31 (d, 2H; J = 9 Hz, ArH), 6.65 (d, 2H; J = 9 Hz, ArH), 3.05 (s, 6H; –N(CH3)2), 2.39 (s, 3H; –COCH3), 2.37 (s, 3H; –COCH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 207.0 (Cq), 196.3 (Cq), 151.8 (Cq), 140.9 (vinyl CH), 137.8 (Cq), 132.2 (Ar CH), 120.1 (Cq), 111.8 (Ar CH), 40.0 (N(CH3)2), 31.6 (CH3), 26.1 (CH3). MS-ESI m/z (%) = 232 [M + H]+ (100), 485 [2M + Na]+ (70). The crystal structure of this compound has been previously reported.26 The 1H and 13C NMR spectra matched those previously reported.27
:AcOEt = 9:1) and obtained as a yellow oil (255 mg, 25%). Rf = 0.14 (hexane:AcOEt = 9:1). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.63 (s, 1H; vinyl CH), 7.47 (d, 2H; J = 9 Hz, ArH), 6.93 (d, 2H; J = 9 Hz; ArH), 3.83 (s, 3H; –OCH3), 2.12 (m, 6H; 2CH3). The 1H NMR spectrum matched that previously reported.28
:CH2Cl2 = 7:3) and obtained as a yellow oil (925 mg, 28%). Rf = 0.3 (hexane:CH2Cl2 = 7:3). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.99 (s, 1H; vinyl CH), 7.42 (d, 2H; J = 9 Hz; ArH), 6.97 (d, 2H; J = 9 Hz; ArH), 3.87 (s, 3H; –OCH3). This compound was previously reported, but no NMR information was given.18
:CH2Cl2 = 1:1) and obtained as a pink waxy solid (185 mg, 50%). Rf = 0.5 (hexane:CH2Cl2 = 1:1). Mp = 60–62 °C. 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.83 (s, 1H; vinyl CH), 7.34 (d, 2H; J = 9 Hz; ArH), 6.65 (d, 2H; J = 9 Hz; ArH), 3.48 (q, 4H; J = 7 Hz; 2CH2), 1.25 (t, 6H; J = 7 Hz; 2CH3).
:AcOEt = 8:2) and obtained as an orange-brown solid (421 mg, 37%). Rf = 0.3 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.40 (s, 1H; vinyl CH), 7.30 (d, 2H; J = 9 Hz; ArH), 6.52 (d, 2H; J = 9 Hz; ArH), 3.36 (m, 4H; 2CH2), 2.38 (s, 3H; CH3), 2.37 (s, 3H; CH3), 2.04 (m, 4H; 2CH2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 207.2 (Cq; –CO), 196.1 (Cq; –CO), 149.4 (Cq), 141.3 (CH), 137.1 (Cq), 132.4 (CH), 119.3 (Cq), 111.7 (CH), 47.4 (CH2), 31.6 (CH3), 26.1 (CH3), 25.3 (CH2).
:CH2Cl2 = 1:1) and obtained as a dark red solid (512 mg, 28%). Rf = 0.4 (hexane:CH2Cl2 = 1:1). Mp = 89–91 °C. 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.85 (s, 1H; vinyl CH), 7.35 (d, 2H; J = 9 Hz; ArH), 6.45 (d, 2H; J = 9 Hz; ArH), 3.45 (t, 4H; 2CH2), 2.08 (m, 4H; 2CH2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 187.6 (q, Cq; J = 35 Hz), 177.5 (q, Cq; J = 35 Hz), 152.1 (Cq), 151.8 (CH), 135.2 (CH), 119.2 (Cq), 118.1 (Cq), 117.1 (q, Cq; J = 300 Hz), 115.3 (q, Cq; J = 300 Hz), 112.4 (CH), 47.8 (CH2), 25.2 (CH2).
:CH2Cl2 = 2:8) and obtained as an orange solid (388 mg, 40%). Rf = 0.7 (hexane:CH2Cl2 = 2:8). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.83 (d, 2H; J = 9 Hz; ArH), 7.48 (s, 1H; vinyl CH), 6.68 (d, 2H; J = 9 Hz; ArH), 3.15 (s, 6H; –N(CH3)2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 157.9 (CH), 154.1 (Cq), 133.7 (CH), 119.2 (Cq), 115.9 (Cq; –CN), 114.8 (Cq; –CN), 111.5 (CH), 71.9 (Cq), 40.0 (CH3). The 1H NMR spectrum matched the one previously reported.29
:CH2Cl2 = 2:8) and obtained as a dark pink solid (851 mg, 76%). Rf = 0.4 (hexane:CH2Cl2 = 2:8). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.81 (d, 2H; J = 9 Hz; ArH), 7.44 (s, 1H; vinyl CH), 6.68 (d, 2H; J = 9 Hz; ArH), 3.48 (q, 4H; J = 7 Hz; 2CH2), 1.25 (t, 6H; J = 7 Hz; 2CH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 157.7 (CH), 152.3 (Cq), 134.0 (CH), 118.8 (Cq), 116.1 (Cq; –CN), 114.9 (Cq; –CN), 111.2 (CH), 71.0 (Cq), 44.8 (CH2), 12.4 (CH3). The 1H and 13C NMR spectra matched those previously reported.30 The crystal structure of this compound has been previously reported.23b
:CH2Cl2 = 2:8) and obtained as a brown-orange solid (316 mg, 29%). Rf = 0.55 (hexane:CH2Cl2 = 2:8). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.80 (d, 2H; J = 9 Hz; ArH), 7.44 (s, 1H; vinyl CH), 6.57 (d, 2H; J = 9 Hz; ArH), 3.45 (m, 4H; 2CH2), 2.09 (m, 4H; 2CH2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 157.9 (CH), 151.8 (Cq), 133.9 (CH), 119.0 (Cq), 116.1 (Cq), 115.1 (Cq), 111.9 (CH), 70.9 (Cq), 47.8 (CH2), 25.2 (CH2). This compound was previously reported.31
General procedure for the synthesis of series 4 and 5
:AcOEt = 8:2) and obtained as a brown oil (89 mg, 23%). Rf = 0.1 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.12 (d, 4H; J = 9 Hz; ArH), 6.54 (d, 4H; J = 9 Hz; ArH), 3.76 (s, 6H; –OCH3), 2.85 (s, 12H; –N(CH3)2). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 168.0 (Cq; CO), 158.9 (Cq), 151.0 (Cq), 131.5 (Cq), 127.9 (CH), 118.8 (Cq), 111.6 (CH), 51.8 (OCH3), 40.1 (N(CH3)2).
:AcOEt = 9:1) and obtained as a green emerald solid (109 mg, 31%). Rf = 0.4 (CH2Cl2:AcOEt = 9:1). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.07 (d, 4H; J = 9 Hz; ArH), 6.65 (d, 4H; J = 9 Hz; ArH), 3.03 (s, 12H; –N(CH3)2), 1.92 (s, 6H; 2COCH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 204.5 (Cq; CO), 154.7 (Cq), 151.6 (Cq), 132.5 (Cq), 127.4 (CH), 126.3 (Cq), 111.4 (CH), 40.1 (N(CH3)2), 31.1 (CH3). MS-ESI m/z (%) = 351 [M + H]+ (100), 723 [2M + Na]+ (40).
:AcOEt = 8:2) and obtained as a yellow solid (190 mg, 52%). Rf = 0.2 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.08 (m, 4H; ArH), 6.63 (m, 4H; ArH), 3.62 (s, 3H; –OCH3), 3.01 (m, 12H; 2N(CH3)2), 1.88 (s, 3H; CH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 201.6 (Cq; CO), 169.5 (Cq; COCH3), 156.9 (Cq), 151.7 (Cq), 132.6 (Cq), 131.6 (CH), 127.1 (Cq), 111.1 (CH), 51.7 (OCH3), 40.0 (2N(CH3)2), 30.2 (CH3). MS-ESI m/z = 367 [M + H]+ (100), 755 [2M + Na]+ (70).
:AcOEt = 8:2) and obtained as a white solid (26 mg, 21%). Rf = 0.2 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.12 (d, 2H; J = 9 Hz; ArH), 6.88 (d, 2H; J = 9 Hz; ArH), 3.84 (s, 6H; –2OCH3), 1.93 (s, 6H; 2CH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 204.0 (Cq; CO), 161.1 (Cq), 150.3 (Cq), 141.3 (Cq), 131.9 (CH), 113.96 (CH), 55.3 (OCH3), 31.1 (CH3). The compound has been previously reported.32
:AcOEt = 8:2) and obtained as a white solid (26 mg, 21%). Rf = 0.2 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.12 (d, 2H; J = 9 Hz; ArH), 6.88 (d, 2H; J = 9 Hz; ArH), 3.84 (s, 6H; –2OCH3), 1.93 (s, 6H; 2CH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 204.0 (Cq; CO), 161.1 (Cq), 150.3 (Cq), 141.3 (Cq), 131.9 (CH), 113.9 (CH), 55.3 (OCH3), 31.1 (CH3).
:AcOEt = 8:2) and obtained as a white solid (60 mg, 34%). Rf = 0.5 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.12 (m, 4H; ArH), 6.86 (m, 4H; ArH), 3.88 (m, 6H; –ArOCH3), 3.61 (s, 3H; COOCH3), 1.93 (s, 3H; CH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 201.3 (Cq; CO), 168.2 (Cq; COOCH3), 160.7 (Cq), 153.6 (Cq), 132.4 (Cq), 131.9 (CH), 131.8 (Cq), 113.9 (CH), 55.3 (OCH3), 51.9 (OCH3), 30.3 (CH3). MS-ESI m/z = 340 [M]+.
:AcOEt = 8:2) and obtained as a white solid (77 mg, 47%). Rf = 0.2 (hexane:AcOEt = 8:2). 1H NMR (CDCl3, 200 MHz, 25 °C) δ = 7.43 (d, 2H; J = 9 Hz; ArH), 6.98 (d, 2H; J = 9 Hz; ArH), 3.89 (s, 6H; –2OCH3). 13C NMR (CDCl3, 75 MHz, 25 °C) δ = 173.8 (Cq), 163.3 (Cq), 132.9 (CH), 128.3 (Cq), 114.9 (CN), 114.1 (CH), 77.4 (Cq), 55.5 (OCH3). The compound has been previously reported.33
Spectroscopical measurements
UV-Vis absorption spectra were obtained with a Perkin Elmer Lambda 900 spectrometer and PL spectra with a SPEX 270 M monochromator equipped with a N2 cooled charge-coupled device exciting with a monochromated 450 W Xe lamp. Spectra were corrected for the instrument response. Photoluminescence quantum yields (PL QYs) of solutions were obtained using quinine sulfate or coumarine 153 as a standard. The PL QYs of solid state samples were obtained using a home-made integrating sphere, as previously reported.34X-ray crystal structures
Single crystals of 3c suitable for X-ray analysis were obtained using CH2Cl2/pentane. Diffraction data were collected on a Bruker Smart Apex II CCD area detector using graphite monochromated Mo-Kα radiation. Data reduction was made using SAINT programs; absorption corrections based on multiscans were obtained using SADABS.35 The structures were solved using SHELXS-97 and refined on F2 by full-matrix least-squares using SHELXL-14.36 All the non-hydrogen atoms were refined anisotropically, hydrogen atoms were included as ‘riding’ and not refined. Crystal data and results of the refinement: orange prism 0.35 × 0.27 × 0.25 mm, C14H13N3, Mr = 223.27; triclinic, P; a = 10.8613(6) Å, b = 13.0319(7) Å, c = 26.5818(15) Å, α = 89.750(1)°, β = 82.163(1)°, γ = 72.238(1)°, V = 3546.9(3) Å3; Z = 12; T = 120(2) K; μ(Mo) = 0.077 mm−1. 58049 measured reflections, 15850 independent reflections, 11640 reflections with I > 2σ(I), 1.55 < 2θ < 54.54°, Rint = 0.0386. Refinement on 15850 reflections, 928 parameters. Final R = 0.0642, wR = 0.1649 for data with F2 > 2σ(F2), S = 1.071, (Δ/σ)max = 0.001, Δρmax = 0.668, Δρmin = −0.462 e Å−3.
Single crystals of 5b suitable for X-ray analysis were obtained via slow evaporation from a CHCl3 solution. Diffraction data were collected on a conventional Enraf-Nonius CAD4 four circle diffractometer, working at ambient temperature with graphite monochromated Mo-Kα Mo Kα X-radiation (λ = 0.7107 Å). Data reduction was performed with the WinGX package.37 Absorption effects were evaluated with the ψ-scan method38 and absorption correction was applied to the data. The crystal structure was solved by direct methods (SIR 97)39 and refined by full-matrix least-squares procedures on F2 using all reflections (SHELXL-14).36 Anisotropic displacement parameters were refined for all non-hydrogen atoms; hydrogens were placed at calculated positions with the appropriate AFIX instructions and refined using a riding model. Crystal data and results of the refinement: prism 0.75 × 0.55 × 0.2 mm, C18H14N2O2, Mr = 290.31; monoclinic, C2/c; a = 17.518(4) Å, b = 8.666(2) Å, c = 10.772(3) Å, β = 110.164(5)°, V = 1535.1(6) Å3; Z = 4; T = 293(2) K; μ(Mo) = 0.083 mm−1. 2340 measured reflections, 2234 independent reflections, 1378 reflections with I > 2σ(I), 4.96 < 2θ < 59.96°, Rint = 0.0182. Refinement on 2234 reflections, 129 parameters. Final R = 0.0491, wR = 0.1127 for data with F2 > 2σ(F2), S = 1.007, (Δ/σ)max = 0.000, Δρmax = 0.170, Δρmin = −0.175 e Å−3.†
Pump–probe experiments
Time-resolved measurements were performed using a home-built femtosecond pump–probe setup. A Ti:sapphire regenerative amplifier (Libra, Coherent) was used as a laser source, delivering 100 fs pulses at a central wavelength of 800 nm with a 4 mJ pulse energy at a repetition rate of 1 kHz. For the excitation pulses, we used the second harmonic of the fundamental beam at 400 nm. In order to minimize bimolecular effects, the excitation density was kept at ≈6 mJ cm−2. White light generated with a 2 mm-thick sapphire plate was used as a probe in the visible region from 490 to 700 nm. For a spectrally resolved detection of the probe light, a spectrograph and CCD array were used. The chirp in the white light pulse was carefully taken into account during the analysis and evaluation of the obtained two-dimensional (wavelength and time) ΔT(λ,t)/T maps before extraction of the spectral and temporal data with homemade software. Overall, a temporal resolution of at least 150 fs was achieved for all excitation wavelengths.Acknowledgements
DP acknowledges grants from MIUR (PRIN 2009-A5Y3N9) and INSTM-Regione Lombardia for partial support of this work. We thank Sara Benedini for early involvement in this work, and Prof. Mariella Mella for assistance in the VT NMR studies.Notes and references
- (a) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., 2014, 126, 2151 CrossRef; (b) X. Wang, Y. Zhou, T. Lei, N. Hu, E. Q. Chen and J. Pei, Chem. Mater., 2010, 22, 3735 CrossRef CAS; (c) W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS PubMed; (d) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed; (e) Q. Liao, H. B. Fu and J. N. Yao, Adv. Mater., 2009, 21, 4153 CrossRef CAS; (f) J. Y. Zheng, Y. L. Yan, X. P. Wang, Y. S. Zhao, J. X. Huang and J. N. Yao, J. Am. Chem. Soc., 2012, 134, 2880 CrossRef CAS PubMed; (g) X. F. Duan, Y. Huang, R. Agarwal and C. M. Lieber, Nature, 2003, 421, 241 CrossRef CAS PubMed.
- (a) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) Z. Y. Zhang, B. Xu, J. H. Su, L. P. Shen, Y. S. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654 CrossRef CAS PubMed; (c) B. Wang, Y. C. Wang, J. L. Hua, Y. H. Jiang, J. H. Huang, S. X. Qian and H. Tian, Chem.–Eur. J., 2011, 17, 2647 CrossRef CAS PubMed; (d) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- (a) X. Y. Qi, H. Li, J. W. Y. Lam, X. T. Yuan, J. Wei, B. Z. Tang and H. Zhang, Adv. Mater., 2012, 24, 4191 CrossRef CAS PubMed; (b) Z. Li, Y. Q. Dong, J. W. Y. Lam, J. X. Sun, A. J. Qin, M. Häußler, Y. P. Dong, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Adv. Funct. Mater., 2009, 19, 905 CrossRef CAS; (c) F. Mahtab, Y. Yu, J. W. Y. Lam, J. Z. Liu, B. Zhang, P. Lu, X. X. Zhang and B. Z. Tang, Adv. Funct. Mater., 2011, 21, 1733 CrossRef CAS; (d) G. Yu, S. W. Yin, Y. Q. Liu, J. S. Chen, X. J. Xu, X. B. Sun, D. G. Ma, X. W. Zhan, Q. Peng, Z. G. Shuai, B. Z. Tang, D. B. Zhu, W. H. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- R. Hu, J. L. Maldonado, M. Rodriguez, C. Deng, C. K. W. Jim, J. W. Y Lam, M. M. F. Yuen, G. Ramos-Ortiz and B. Z. Tang, J. Mater. Chem., 2012, 22, 232–240 RSC.
- J. Wang, J. Mei, W. Yuan, P. Lu, A. Qin, J. Sun, Y. Ma and B. Z. Tang, J. Mater. Chem., 2011, 21, 4056–4059 RSC.
- N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef CAS PubMed.
- (a) X. Sun, Y. Liu, X. Xu, C. Yang, G. Yu, S. Chen, Z. Zhao, W. Qiu, Y. Li and D. Zhu, J. Phys. Chem. B, 2005, 109, 10786 CrossRef CAS PubMed; (b) Y. Shirota, J. Mater. Chem., 2005, 15, 75 RSC; (c) X. Y. Shen, W. Z. Yuan, Y. Liu, Q. Zhao, P. Lu, Y. Ma, I. D. Williams, A. Qin, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 10541 CrossRef CAS; (d) X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334 CrossRef CAS; (e) F. S. Kim, X. Guo, M. D. Watson and S. A. Jenekhe, Adv. Mater., 2010, 22, 478 CrossRef CAS PubMed; (f) T. C. Lin, G. S. He and Q. Zheng, J. Mater. Chem., 2006, 16, 2490 RSC.
- (a) E. Cariati, V. Lanzeni, E. Tordin, R. Ugo, C. Botta, A. Giacometti Schieroni, A. Sironi and D. Pasini, Phys. Chem. Chem. Phys., 2011, 13, 18005 RSC; (b) C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666 RSC.
- T. Virgili, A. Forni, E. Cariati, D. Pasini and C. Botta, J. Phys. Chem. C, 2013, 117, 27161 CAS.
- M. M. Mróz, S. Benedini, A. Forni, C. Botta, D. Pasini, E. Cariati and T. Virgili, Phys. Chem. Chem. Phys., 2016, 18, 18289–18296 RSC.
- C. Botta, S. Benedini, L. Carlucci, A. Forni, D. Marinotto, A. Nitti, D. Pasini, S. Righetto and E. Cariati, J. Mater. Chem. C, 2016, 4, 2979–2989 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- A. J. Zucchero, P. L. McGrier and U. H. F. Bunz, Acc. Chem. Res., 2010, 43, 397 CrossRef CAS PubMed.
- Y. M. Issa and W. H. Hegazy, Synth. React. Inorg. Met.-Org. Chem., 2000, 30, 1731–1746 CrossRef CAS.
- O. Attanasi, P. Filippone and A. Mei, Synth. Commun., 1983, 13, 1203–1208 CrossRef CAS.
- S. Zhu, B. Xu and J. Zhang, J. Fluorine Chem., 1995, 74, 167–170 CrossRef CAS.
- I. Shibuya, Y. Taguchi, T. Tsuchiya, A. Oishi and E. Katoh, Bull. Chem. Soc. Jpn., 1994, 67, 3048–3052 CrossRef CAS.
- B. A. Burkett, J. M. Kane-Barber, R. J. O'Reilly and L. Shi, Tetrahedron Lett., 2007, 48, 5355–5358 CrossRef CAS.
- The free energy barrier for the dynamic process (ΔG‡c) could be calculated using the coalescence method, where values for the rate constant kc at the coalescence temperature (Tc) were calculated from the approximate expression kc = π(Δν)/20.5, where Δν is the limiting chemical shift difference (Hz) between the coalescing signals in the absence of exchange, and the Eyring equation was subsequently employed. See: (a) I. O. Sutherland, Annu. Rep. NMR Spectrosc., 1971, 4, 71–235 CrossRef CAS. See also: (b) P. R. Ashton, S. E. Boyd, S. Menzer, D. Pasini, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White, D. J. Williams and P. G. Wyatt, Chem.–Eur. J., 1998, 4, 299–310 CrossRef CAS.
- (a) M. Yu. Antipin, T. V. Timofeeva, R. D. Clark, V. N. Nesterov, M. Sanghadasa, T. A. Barr, B. Penn, L. Romero and M. Romero, J. Phys. Chem. A, 1998, 102, 7222–7232 CrossRef CAS; (b) K. Wang, Z. Wang and C. Yan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o214–o215 CAS; (c) R. D. Gandour and F. R. Fronczek, CSD Communication (Private Communication), 2015 Search PubMed; (d) V. K. Gupta and R. A. Singh, RSC Adv., 2015, 5, 38591–38600 RSC.
- (a) W. Ma, S.-Y. Zhang, J.-Y. Wu, Y.-P. Tian and H.-K. Fun, Chin. J. Appl. Chem., 2003, 20, 862 CAS; (b) Y. Jing and L.-T. Yu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1556 CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- N. F. Phelan and M. Orchin, J. Chem. Educ., 1968, 45, 633 CrossRef CAS.
- M. H. Habibi, K. Barati, H. Etedali Habibadi, R. W. Harrington and W. Clegg, Anal. Sci., 2008, 24, x285–x286 CAS.
- E. Solcaniova, P. Hrnciar and T. Liptaj, Org. Magn. Reson., 1982, 18, 55–57 CrossRef CAS.
- J. S. Yadav, D. C. Bhunia, V. K. Singh and P. Srihari, Tetrahedron Lett., 2009, 50, 2470–2473 CrossRef CAS.
- A. Szłap, S. Kula, U. Błaszkiewicz, M. Grucela, E. Schab-Balcerzak and M. Filapek, Dyes Pigm., 2016, 129, 80–89 CrossRef.
- G. B. Kharas, S. M. Russell, V. Tran, Q. L. Tolefree, D. M. Tulewicz, A. Gora, J. Bajgoric, M. T. Balco, G. A. Dickey and G. Kladis, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 5–8 CrossRef CAS.
- J. S. A. Brunskill, A. De and G. M. Vas, Synth. Commun., 1978, 8, 1–7 CrossRef CAS.
- M. G. Wood, A. R. Smith and D. Judd, US Pat. No. 20030000130, 2003.
- G. Charles, Bull. Soc. Chim. Fr., 1963, 8–9, 1559–1565 Search PubMed.
- J. Moreau, U. Giovanella, J.-P. Bombenger, W. Porzio, V. Vohra, L. Spadacini, G. Di Silvestro, L. Barba, G. Arrighetti, S. Destri, M. Pasini, M. Saba, F. Quochi, A. Mura, G. Bongiovanni, M. Fiorini, M. Uslenghi and C. Botta, ChemPhysChem, 2009, 10, 647 CrossRef CAS PubMed.
- SMART, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 1997.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. C. T. North, D. C. Phillips and F. S. Mathews, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1968, 24, 351–359 CrossRef.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphs with molecular optical properties, tables with NMR shifts, copies of NMR spectra for new compounds. CCDC 1487807 and 1487808. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6fd00161k |
| This journal is © The Royal Society of Chemistry 2017 |