
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure and dynamics of torsion-locked photoactive yellow protein chromophores†
Alice
Henley
 ,
Matus E.
Diveky
,
Anand M.
Patel
,
Michael A.
Parkes
,
James C.
Anderson
and
Helen H.
Fielding
*
,
Matus E.
Diveky
,
Anand M.
Patel
,
Michael A.
Parkes
,
James C.
Anderson
and
Helen H.
Fielding
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.h.fielding@ucl.ac.uk
First published on 16th November 2017
Abstract
The photocycle of photoactive yellow protein (PYP) begins with small-scale torsional motions of the chromophore leading to large-scale movements of the protein scaffold triggering a biological response. The role of single-bond torsional molecular motions of the chromophore in the initial steps of the PYP photocycle are not fully understood. Here, we employ anion photoelectron spectroscopy measurements and quantum chemistry calculations to investigate the electronic relaxation dynamics following photoexcitation of four model chromophores, para-coumaric acid, its methyl ester, and two analogues with aliphatic bridges hindering torsional motions around the single bonds adjacent to the alkene group. Following direct photoexcitation of S1 at 400 nm, we find that both single bond rotations play a role in steering the PYP chromophore through the S1/S0 conical intersection but that rotation around the single bond between the alkene moiety and the phenoxide group is particularly important. Following photoexcitation of higher lying electronic states in the range 346–310 nm, we find that rotation around the single bond between the alkene and phenoxide groups also plays a key role in the electronic relaxation from higher lying states to the S1 state. These results have potential applications in tuning the photoresponse of photoactive proteins and materials with chromophores based on PYP.
1 Introduction
Photoreceptor proteins play a vital role in nature facilitating various biological processes, such as ion transport, vision and signal transduction. Following photoexcitation, the small chromophores that are embedded in these proteins undergo fundamental photochemical reactions, such as isomerisation, excited-state proton transfer or excited-state electron transfer.1 The small-scale structural changes of the chromophore then propagate to large-scale structural changes in the surrounding protein framework that generate longer-lived signalling states to which the host organism may respond. It is important to understand the role the protein plays in orchestrating the photoresponse of the host organism by optimising the efficiency of the process and minimising competing pathways.Photoactive yellow protein (PYP) is the primary photoreceptor for the negative phototactic response of the Halorhodospira halophila bacterium to blue light that limits its exposure to potentially harmful UV light.3 The chromophore that lies at the centre of PYP is a deprotonated trans-para-coumaric acid anion (pCA−) covalently bound to a cysteine residue via a thio-ester linkage and stabilised by nearby residues through a hydrogen bond network (Fig. 1). Upon photoexcitation, PYP undergoes a photocycle that begins with a trans–cis isomerisation of the chromophore and is followed by restructuring of the surrounding hydrogen bond network to accommodate the cis conformation, partial unfolding of the protein and protonation of the chromophore to form the signalling state. Subsequent cis–trans isomerisation of the chromophore, refolding of the protein and deprotonation of the chromophore to form trans-pCA− in its ground electronic state, completes the photocycle. Photoisomerisation is a common initial photoresponse in photoactive proteins; for example, photoisomerisation is the initial step in the photochemistry of rhodopsins, a family of photoreceptor proteins that use the retinal chromophore for light reception.4
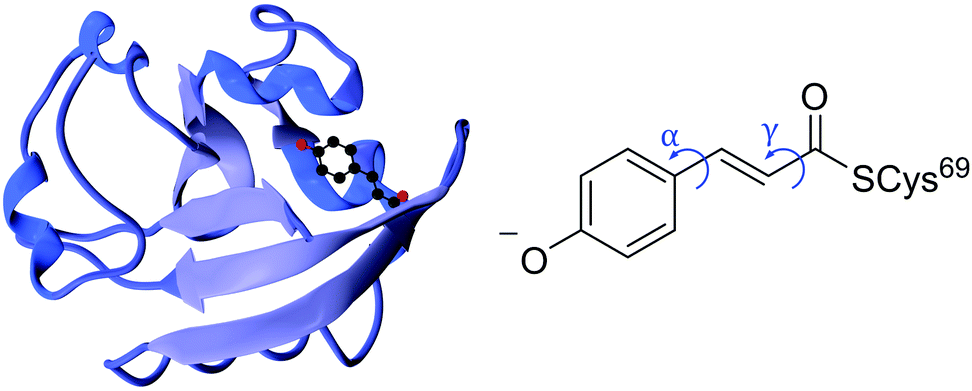 | ||
| Fig. 1 Left: PYP protein structure2 with the chromophore enclosed. Right: PYP chromophore as it exists in the protein showing the torsional rotations that are the subject of this paper. | ||
Although the excited state rotation of the double bond is of key importance in the photocycle of PYP, the trans–cis isomerisation couples with other nuclear motions within the chromophore resulting in torsional-motion about the single bonds either side of the double bond (Fig. 1).5 However, without the protein environment, it has been suggested that it is single bond, not double bond, rotation that dominates in the first excited state.6,7 It is well understood that this is the case for the methyl thio-ester analogue of pCA− in an aqueous environment, where rotation about the phenolate-ethene single bond (α rotation, Fig. 1) is the key coordinate in the relaxation pathway following photoexcitation.8,9 Theoretical studies have suggested that this single bond rotation also dominates when the chromophore is isolated in the gas phase,6 or in a mutant protein environment (R52Q).7 It was suggested that the global minimum on the excited state surface is a double-bond twisted structure with a dihedral angle across the double bond of 90°. In addition, a local minimum structure was found with a dihedral angle of 90° about the single bond joining the phenoxide and ethene moieties. It was proposed that a role of the protein scaffold is to restrict this competing single bond α rotation (Fig. 1) and hence promote trans–cis isomerisation by stabilising the twisted double bond global minimum structure and lowering the barrier to isomerisation on the excited state potential energy surface. On the other hand, some experimental studies have contradicted this. A time-resolved spectroscopy study probing the photophysics of wild-type PYP and a reconstructed PYP with a chromophore rotation-locked across the phenolate-ethene single bond by an ethane bridge10 reported that the excited state dynamics of the locked chromophore were accelerated but that the isomerisation yield was reduced by ∼60%, implying that the phenolate rotation is not an alternative to trans–cis isomerisation but that it does help facilitate the isomerisation with some coupled movement. This contrasts with the conclusions of an earlier gas-phase time-resolved photoelectron spectroscopy study of a ketone analogue of the PYP chromophore which did not show any evidence for a single-bond twisted intermediate in the excited state and revealed isomerisation times similar to those in the protein.11
Efforts have also been made to understand the importance of other single bond rotations in the chromophore. Some studies focused on the CC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–SCys bond, the connection between the chromophore and the protein.12–14 In a Fourier-transform infrared spectroscopy study it was found that PYP derivatives with locked chromophores were unable to undergo isomerisation but were still able to produce intermediates of the PYP photocycle.14 Hence, the authors deemed the trans–cis isomerisation process dispensible for photoactivity in PYP and instead suggested that rotation of the carbonyl group is more significant. Thus, it is clear that the roles of the various torsional motions in the early stages of the PYP photocycle are still not completely understood.
O–SCys bond, the connection between the chromophore and the protein.12–14 In a Fourier-transform infrared spectroscopy study it was found that PYP derivatives with locked chromophores were unable to undergo isomerisation but were still able to produce intermediates of the PYP photocycle.14 Hence, the authors deemed the trans–cis isomerisation process dispensible for photoactivity in PYP and instead suggested that rotation of the carbonyl group is more significant. Thus, it is clear that the roles of the various torsional motions in the early stages of the PYP photocycle are still not completely understood.
Non-radiative electronic relaxation pathways such as internal conversion (IC), intersystem crossing, intramolecular vibrational redistribution and electron detachment can compete with the trans–cis isomerisation process. In order to understand the role of the protein in minimising these competing processes, it is important to understand the intrinsic photochemistry of the isolated chromophores. Following excitation of a ketone analogue of pCA− in the gas-phase at 400 nm (3.1 eV), 20% of the excited state population was found to undergo autodetachment and it was deduced that one of the roles of the protein environment is to funnel excited state population through the conical intersection (CI) seam towards the cis isomer to impede radical formation.11 Electron detachment has also been observed from isolated chromophores in solution15 and from the protein,16 and in both cases the spectral signatures were those of solvated electrons, suggesting that the protein pocket provides a water-like environment for the emitted electron.
In recent work, we investigated electron detachment from different structural isomers of pCA−. We found that following excitation of high-lying excited states using UV light, the relative contributions from electron detachment and internal conversion to lower lying electronic states was different for ortho-, meta- and para- isomers of pCA−.17 We also investigated the effects of substitution at the tail end of the chromophore.18 We found that acid and methyl ester moieties had very similar relaxation dynamics but substitution by a thioester appeared to raise the electron detachment threshold and impede internal conversion to lower-lying states.
In this work, we extend our previous studies to structural analogues of pCA− designed to allow us to test the importance of torsional motions on the electronic relaxation dynamics of the PYP chromophore in the gas phase following photoexcitation of the first excited state and higher lying excited states. Specifically, we have employed anion photoelectron spectroscopy and quantum chemistry calculations to investigate the electronic structure and dynamics of four model PYP chromophores (Fig. 2): pCA−, its methyl ester pCE−, and two analogues of pCE− with aliphatic carbon bridges hindering single-bond torsional motion at either site adjacent to the unsaturated bond, denoted here as PpCE− and NpCE− due to the pyranone and (tetrahydro)napthalene motifs, respectively. We determine the electronic structure and relaxation pathways available to the chromophores and how these are influenced by locking the single bond rotations.
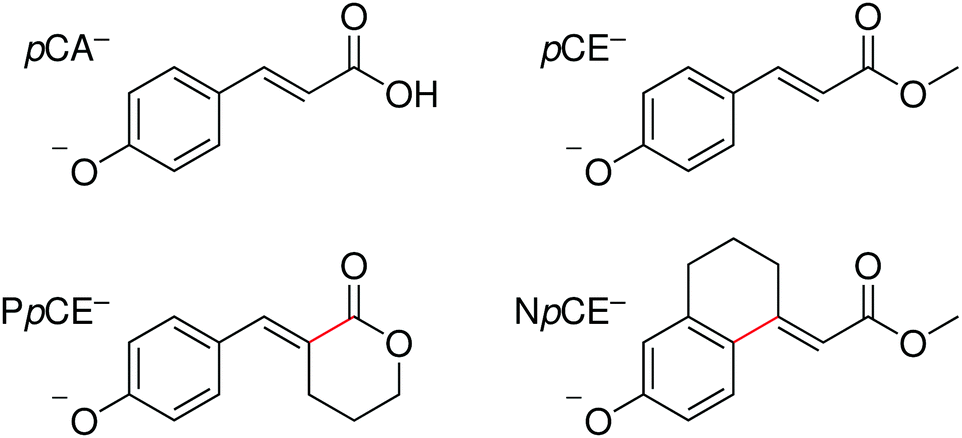 | ||
| Fig. 2 Structures of the deprotonated PYP chromophores used in this study with conformationally-locked bonds shown in red. | ||
2 Experimental and computational methods
2.1 Chromophores
pCA was purchased from Sigma-Aldrich and pCE was purchased from Tokyo Chemical Industry, both were used without further purification. PpCE and NpCE were synthesised by standard chemistry. See ESI† for further details.2.2 Photoelectron spectroscopy
Anion photoelectron spectra were recorded using our apparatus that has also been described elsewhere.19–22 In brief, we obtain gas-phase deprotonated anions of pCA, pCE, PpCE and NpCE by electrospray ionisation of ∼1 mM methanol solutions of each chromophore with a few drops of aqueous ammonia added. The anions are mass-selected by a quadrupole and guided into a hexapole ion trap. The anions are released from the ion trap at 20 Hz and focused into the source region of a collinear velocity map imaging spectrometer. Nanosecond laser pulses of wavelength 310–346 nm were generated by frequency-doubling the output of a Nd:YAG pumped dye laser. Femtosecond laser pulses (>50 fs, 250 Hz) of wavelength centred at 400 nm were produced by frequency-doubling the output of a commercial amplified Ti:Sapphire femtosecond laser system. Photoelectrons generated in the interaction region were accelerated towards a position sensitive detector and imaged using a CCD camera. Laser-only images were recorded without the pulsed ion-beam and subtracted from the overall signal to remove background counts from ionisation of residual gas or scattered laser light. Photoelectron velocity distributions were obtained using the pBasex inversion method.23 Electron kinetic energy (eKE) spectra were obtained by calibrating the radial photoelectron velocity distribution against the photoelectron spectrum of iodide24 (310–346 nm) and p-HBDI−25 (400 nm). The energy resolution of the 310–346 nm spectra is <5% and the error in eKE is around 0.05 eV.2.3 Calculations
The structures of the anionic chromophores and their corresponding neutral radicals were optimised using density functional theory with the B3LYP hybrid functional26–29 and 6-311++G(3df,3pd) basis set.30–32 Vibrational analysis was performed to confirm the optimisations resulted in minima.Vertical excitation energies (VEEs) of the singlet electronic excited states of the chromophores were calculated using time-dependent density functional theory (TD-DFT) and the algebraic diagrammatic construction method to second order (ADC(2)).33,34 TD-DFT calculations were performed with CAM-B3LYP35/6-311++G(3df,3pd), a method that has been shown to provide qualitative agreement with measured electronic excitation energies of PYP chromophores.18 ADC(2)/6-31+G** calculations have also been performed and these have shown to calculate the VEEs accurately and reliably with a reasonable computational expense (see Table S1 in ESI† for benchmarking VEE calculations).
Vertical detachment energies (VDEs) were calculated using various methods to compare to the measured values we have obtained. We have used electron propagator theory (EPT)36 with the 6-311++G(3df,3pd) basis set, a method that has proved to agree well with experimental VDEs in earlier work.18,25 Also, we have used the equation-of-motion coupled cluster method with single and double excitations for the calculation of ionisation potentials (EOM-IP-CCSD),37 performed using the aug-cc-pVDZ basis set38 which has shown previously to calculate detachment energies to higher-lying excited states of similar neutral radicals effectively.25,39 The VDEs have also been calculated as the anion-neutral radical energy difference using DFT (B3LYP/6-311++G(3df,3pd)) at the optimised geometry of the anion. Calculating the VDEs with this method gives a more direct comparison with the calculated adiabatic detachment energies (ADEs) which are found using energies from DFT (B3LYP/6-311++G(3df,3pd)) calculations. ADEs are determined by the difference in energy between the S0 minima and D0 minima (0-0 transition), accounting for zero-point energies.
Geometry optimisations, vibrational frequencies, TD-DFT and EPT calculations were performed using the Gaussian09 software suite,40 while EOM-CCSD and ADC(2) calculations were carried out using the Q-Chem software package.41
3 Results and discussion
3.1 Photoelectron spectra and computational results
Photoelectron spectra of the deprotonated chromophores (Fig. 2) were recorded as a function of electron kinetic energy (eKE) and are also presented as a function of electron binding energy, eBE = hν − eKE (Fig. 3). Lines on the spectra mark calculated VDEs and VEEs.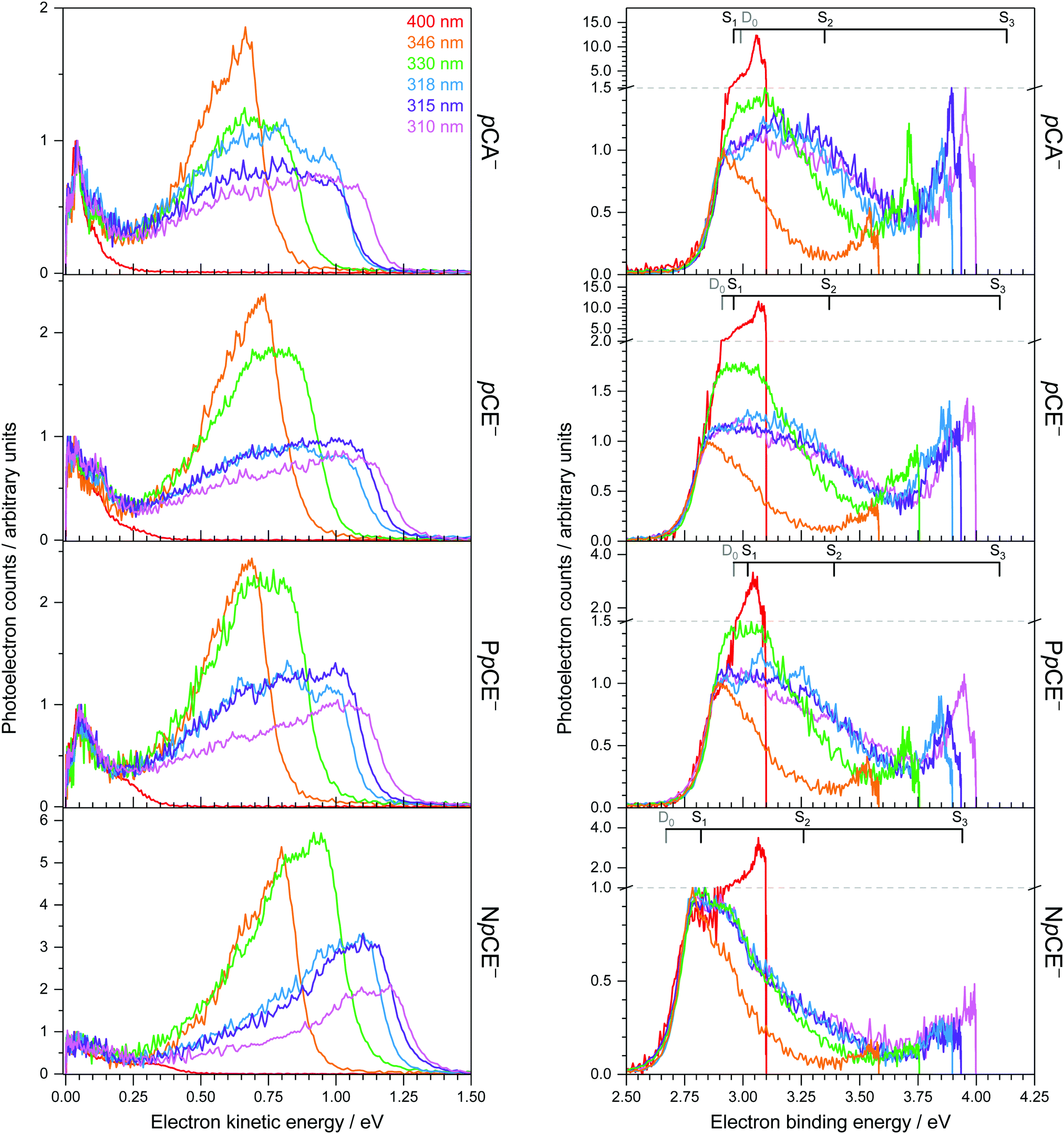 | ||
| Fig. 3 Photoelectron spectra of pCA−, pCE−, PpCE− and NpCE− recorded at 310 nm (4.00 eV), 315 nm (3.94 eV), 318 nm (3.90 eV), 330 nm (3.76 eV), 346 nm (3.58 eV) and 400 nm (3.10 eV). Intensities of the spectra plotted as a function of eKE (left) have been scaled so that the low eKE features lie on top of one another and are normalised to the maximum of the low eKE feature. Intensities of the spectra plotted as a function of eBE (right) have been scaled to align the rising edges and are normalised to the maximum of the rising edges of the 346 nm spectra. Note the scale changes used to plot the 400 nm data with the shorter wavelength data shown by the grey dashed lines. Combs mark the EPT calculated VDEs (grey) and the first three singlet excited states of the anion calculated using ADC(2)/6-31+G** (black). | ||
Possible direct and indirect photodetachment (PD) processes are illustrated on the Jablonski diagram presented in Fig. 4. The 400 nm spectra are dominated by a strong feature at eKE ≈ 0 (eBE ≈ hν). The eKE profiles are those of structured exponential decays, characteristic of thermionic emission (TE).42,43 In contrast, the 310–346 nm photoelectron spectra are dominated by broad, unresolved features at low eBEs. The sharp, low eBE edges of these broad features remain at constant eBE for all photon energies, signifying a direct PD process. There is also evidence for direct PD in the 400 nm spectra, although it is particularly weak for pCA− and pCE−. For pCA−, pCE− and PpCE−, the low eBE features broaden on their high eBE edges with increasing photon energy, indicative of an indirect PD process following resonant excitation of an excited electronic state. This broadening is less pronounced for NpCE−. The 310–346 nm spectra also have features at eBE ≈ hν (eKE ≈ 0) that increase in intensity with respect to the direct detachment features as the photon energy is increased. These eBE ≈ hν features are less intense in the NpCE− photoelectron spectra compared to those of pCA−, pCE− and PpCE−.
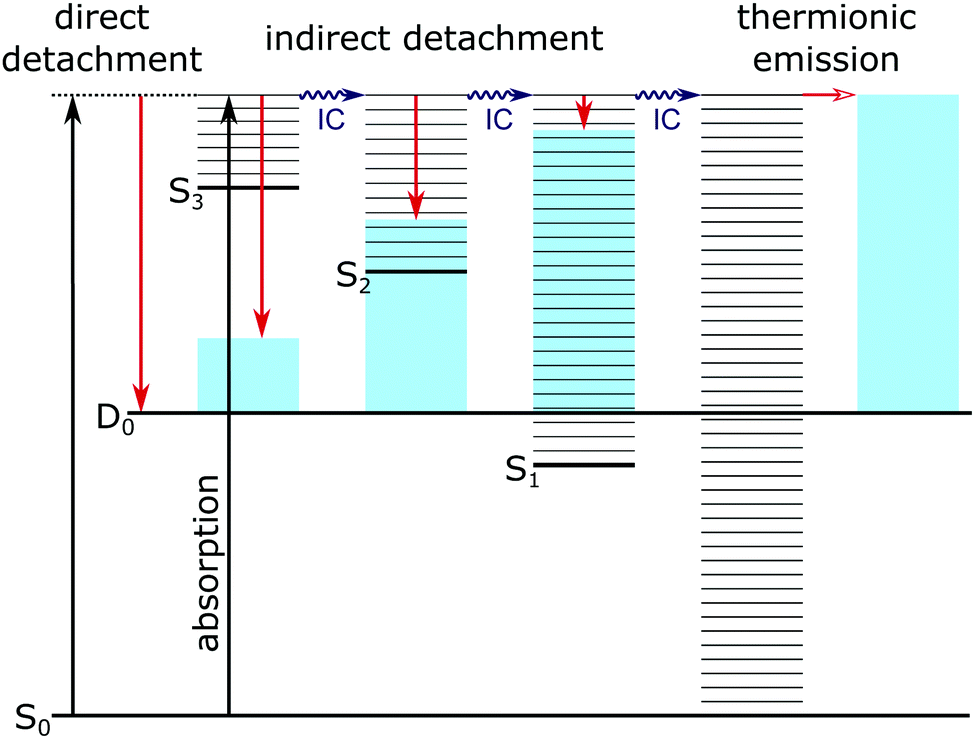 | ||
| Fig. 4 Jablonski diagram illustrating the major photodetachment pathways (red arrows) available for the model PYP chromophores employed in this work (Fig. 2) following UV photoexcitation (346–310 nm) including internal conversion (IC) and thermionic emission (horizontal red arrow). Horizontal black lines represent the vibrational levels of the excited electronic states of the anion and the blue shaded areas represent the vibrational energy left in the neutral radical upon electron detachment as a result of the propensity for conserving vibrational energy. | ||
Approximate VDEs are determined from the maxima of the direct PD features at low eBE in the 346 nm spectra since this wavelength corresponds to a minimum in the action absorption spectra of pCE−.44 These maxima are presented in Table 1 along with calculated VDEs and ADEs. For D0, VDEs and ADEs increase in the order NpCE− < pCE− < PpCE− < pCA−. The trend in the calculated and experimentally measured detachment thresholds can be explained in terms of the electronic effects of the acyl substituents and structural strain on the resonant stabilisation of the anion. The slight difference between the pCA− and pCE− detachment thresholds can be attributed to the hydroxyl group of pCA− being a slightly weaker electron donor than the OMe group. This is intuitive as the methyl group is electron releasing and in this instance can be shown by the sum of the ChelpG45 partial charges on the OH group (−0.296 e) compared to that of the OMe group (−0.275 e). In pCE− the effect of nO(sp2) → σ*CO donation will destabilise the anion. This stereoelectronic effect is not possible with PpCE− so it is surprising that the VDEs of PpCE− and pCE− are similar. The expected greater stabilising effect of the lactone ring in PpCE− may be offset by the conformation induced by the tether compromising the planarity of the system with respect to the simple ester in pCE−. The detachment threshold of NpCE− is most likely lowered by the ring strain introduced by the rotation lock (half-chair conformation) inducing a small reduction in the planarity (see Fig. S2 in ESI†) and thus hindering resonant stabilisation of the anion.
| Vertical detachment energy | ADE(0–0) | Expt. | |||
|---|---|---|---|---|---|
| EPT | EOM-IP-CCSD | DFT | |||
| pCA− | 2.99 (0.879) | 2.97 | 3.10 | 2.99 | 2.92 ± 0.05 |
| 5.25 (0.872) | 5.45 | ||||
| pCE− | 2.91 (0.879) | 2.78 | 3.01 | 2.91 | 2.85 ± 0.05 |
| 5.18 (0.873) | 4.42 | ||||
| PpCE− | 2.96 (0.878) | 2.83 | 3.03 | 2.93 | 2.89 ± 0.05 |
| 5.23 (0.871) | 4.48 | ||||
| NpCE− | 2.67 (0.879) | 2.71 | 2.93 | 2.82 | 2.78 ± 0.05 |
| 5.02 (0.868) | 4.33 | ||||
It is worth noting that pCA can deprotonate at two sites to form either the carboxylate or the phenolate tautomer. The VDE of carboxylate pCA− (4.68 eV)17 is significantly higher than that of phenolate pCA− and the photon energies used in this work (<4 eV). Additionally, the spectra of pCA− and pCE− are very similar. Hence, although both forms of pCA− could be present in our instrument, we are only sensitive to the phenolate form.
The DFT VDEs are close to the ADEs, suggesting that the geometries of the anions and their corresponding neutral radicals are similar, in agreement with our earlier work on model PYP chromophores.17,18
In terms of the calculated VDEs for D0, the EPT method has determined the VDEs of pCA−, pCE− and PpCE− to be just slightly higher than the experimental maxima (∼0.05 eV) and slightly lower for NpCE− (0.115 eV). The EPT/6-311++G(3df,3pd) method was employed to calculate the first VDE of the phenoxide anion previously46 and it was found to be within 1% of the experimental value of Gunion et al.47 Here, we find a 2% difference between the EPT calculated VDE and the experimental maxima for pCA−, pCE− and PpCE−, and a 4% difference for NpCE−. The VDEs calculated using DFT are systematically higher (∼0.15 eV) than the experimental maxima. The EOM-IP-CCSD calculations have predicted the VDEs of the chromophores to within ∼0.06 eV of the experimental maxima, although the deviations are less systematic than with DFT; for example, the EOM-IP-CCSD VDE of pCA− is higher than the experimental maximum whereas the calculated VDE of pCE− is lower than the experimental maximum. It should be noted that the accuracy of our measurement is ±0.05 eV and that the high density of vibrational transitions contributing to these vibrationally unresolved photoelectron spectra can easily shift the maxima away from the true VDE. Thus, we conclude that all the calculated VDEs are in good agreement with experiment and note that the simple, computationally inexpensive DFT method performs just as well as the other methods. Calculated VDEs for D1 are also listed in Table 1 and are all significantly higher than the photon energies used in this study suggesting that direct PD to D1 is unlikely.
The first three singlet excited states of the chromophores are 11ππ*, 11nπ* and 21ππ*, as illustrated in Fig. 5. The relevant orbitals of all the chromophores are similar to those of pCA− (shown), as one may expect from a set of structural analogues without any significant changes to functionality (see Tables S2–S5 and Fig. S3–S6, ESI†). The π and 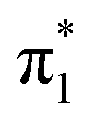 orbitals are delocalised across the whole chromophore whereas the n and
orbitals are delocalised across the whole chromophore whereas the n and 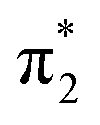 orbitals are more localised on the phenoxide end of the anion. Hence, the S3(21ππ*) and the S2(11nπ*) transitions have charge transfer character. The delocalised π orbital is the highest occupied molecular orbital and the configuration of the D0 state of the neutral radical corresponds to the removal of an electron from this orbital.
orbitals are more localised on the phenoxide end of the anion. Hence, the S3(21ππ*) and the S2(11nπ*) transitions have charge transfer character. The delocalised π orbital is the highest occupied molecular orbital and the configuration of the D0 state of the neutral radical corresponds to the removal of an electron from this orbital.
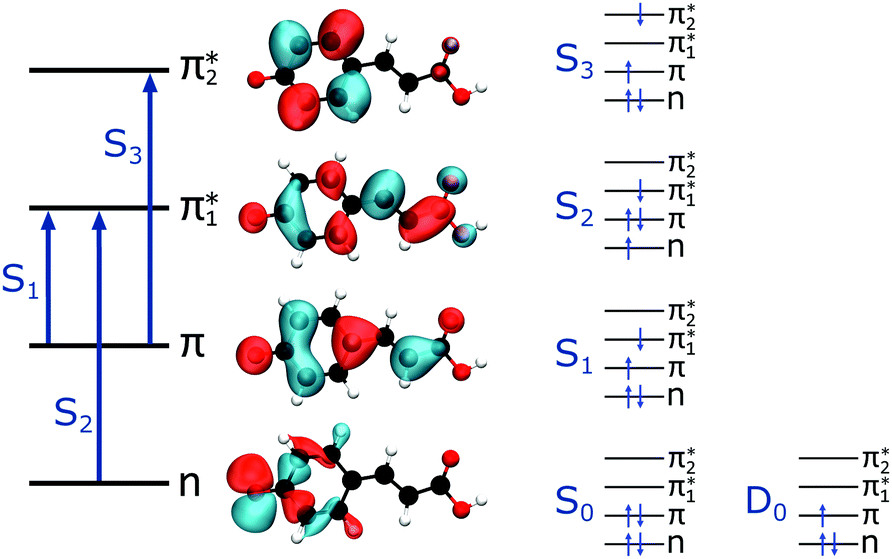 | ||
| Fig. 5 Left: The main molecular orbitals involved in the transitions to the first three singlet excited states of the anion. Although the MOs plotted are calculated for pCA−, they are similar for all the chromophores shown in Fig. 2 (see Tables S2–S5 and Fig. S3–S6, ESI†). Right: The electron configurations associated with the ground state of the anion (S0) and neutral radical (D0) and the first three singlet excited states of the anion for all the chromophores. | ||
Vertical excitation energies (VEEs) for these first three singlet excited states calculated using CAM-B3LYP/6-311++G(3df,3pd) and ADC(2)/6-31+G** methods are presented in Table 2. They are compared with SA-CASSCF(14,12)-PT2/cc-pVDZ results by Fernández García-Prieto et al.48 and experimental values recorded by Rocha-Rinza et al.49 as these have been shown to be in excellent agreement with one another and provide a benchmark for our own calculations.
| pCA− | pCE− | PpCE− | NpCE− | |||
|---|---|---|---|---|---|---|
| CAM-B3LYP | 11ππ* | S1 | 3.45 (0.910) | 3.43 (0.949) | 3.49 (0.952) | 3.36 (0.812) |
| 6-311++G(3df,3pd) | 11nπ* | S2 | 4.10 (0.000) | 4.12 (0.000) | 4.15 (0.000) | 4.10 (0.000) |
| 21ππ* | S3 | 4.33 (0.071) | 4.31 (0.110) | 4.34 (0.056) | 4.40 (0.026) | |
| ADC(2) | 11ππ* | S1 | 2.96 (1.000) | 2.96 (1.021) | 3.02 (1.032) | 2.82 (0.885) |
| 6-31+G** | 11nπ* | S2 | 3.35 (0.000) | 3.37 (0.000) | 3.39 (0.000) | 3.26 (0.000) |
| 21ππ* | S3 | 4.13 (0.091) | 4.10 (0.097) | 4.10 (0.061) | 3.94 (0.065) | |
| SA-CASSCF | 11ππ* | S1 | 2.96 | 2.94 | — | — |
| (14,12)-PT2 | 11nπ* | S2 | 3.65 | 3.65 | — | — |
| cc-pVDZ | 21ππ* | S3 | 3.82 | 3.82 | — | — |
| Experiment | 11ππ* | S1 | 2.89 | 2.88 | — | — |
The calculated oscillator strength for the S1 ← S0 transition is near unity for each of the chromophores whereas that for the S2 ← S0 transition is zero. The S3 ← S0 transition has a non-zero oscillator strength for each chromophore of approximately 0.1. From Tables 1 and 2, and the VDEs and VEEs plotted in Fig. 3, it is clear that the D0 and S1 states lie close in energy, particularly for pCA−, pCE− and PpCE−. Due to the good agreement between the literature and ADC(2) methods, the ADC(2) values are plotted on the spectra in Fig. 3. The CAM-B3LYP VEEs are similar to those calculated for PYP chromophores in earlier work using the same method;18 however, the VEEs for pCA− and pCE− are approximately 0.5 eV higher than those calculated using higher level theory48 and obtained from action absorption spectra.49 The VEEs calculated using the ADC(2)/6-31+G** method for pCA− and pCE− are in good agreement with those calculated using the SA-CASSCF(14,12)-PT2/cc-pVDZ method48 and with the available experimental measurements,49 so we use the VEEs calculated with this method to assist with the interpretation of our experimental measurements.
The electron configurations of the accessible electronically excited states of the anions and the corresponding neutral radicals (Fig. 5) are useful in the consideration of coupling strengths of the excited states of the anion to the detachment continua. Indirect PD from the S1 state to the D0 continuum involves removing a single electron from the 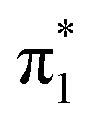 orbital to yield the D0 electron configuration. Vibrational states of S1 that lie in the D0 continuum have shape resonance character and are coupled strongly to the continuum. Indirect PD from S3 involves removing an electron from the
orbital to yield the D0 electron configuration. Vibrational states of S1 that lie in the D0 continuum have shape resonance character and are coupled strongly to the continuum. Indirect PD from S3 involves removing an electron from the 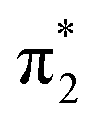 orbital. The S3 state has excited shape resonance character with respect to the D0 continuum and electron detachment through this channel is expected to be very fast. In contrast, indirect PD from the S2 state to the D0 continuum involves removing a
orbital. The S3 state has excited shape resonance character with respect to the D0 continuum and electron detachment through this channel is expected to be very fast. In contrast, indirect PD from the S2 state to the D0 continuum involves removing a 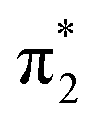 electron and making the π ← n electronic transition. Thus S2 has Feshbach resonance character with respect to the D0 continuum and indirect detachment from this state is expected to be relatively slow. Although the D1 VDEs are higher than the photon energies employed in this work, adiabatic detachment to D1 from the S2 and S3 states cannot be ruled out. The D1 state has electronic configuration
electron and making the π ← n electronic transition. Thus S2 has Feshbach resonance character with respect to the D0 continuum and indirect detachment from this state is expected to be relatively slow. Although the D1 VDEs are higher than the photon energies employed in this work, adiabatic detachment to D1 from the S2 and S3 states cannot be ruled out. The D1 state has electronic configuration 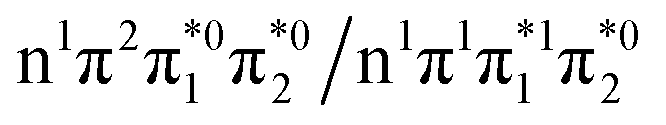 so the S3 state has Feshbach resonance character with respect to the D1 continuum suggesting that indirect PD to D1 will not be able to compete with indirect PD to D0 or ultrafast IC to lower-lying states. The S2 state is an excited shape resonance with respect to D1, suggesting that indirect PD from S2 to D1 is possible; however, there would still be competition from internal conversion to S1 or S0. Overall, it seems most likely that the photoelectron spectra are dominated by direct and indirect electron emission to the D0 continuum.
so the S3 state has Feshbach resonance character with respect to the D1 continuum suggesting that indirect PD to D1 will not be able to compete with indirect PD to D0 or ultrafast IC to lower-lying states. The S2 state is an excited shape resonance with respect to D1, suggesting that indirect PD from S2 to D1 is possible; however, there would still be competition from internal conversion to S1 or S0. Overall, it seems most likely that the photoelectron spectra are dominated by direct and indirect electron emission to the D0 continuum.
3.2 The role of torsional motion
Photoexcitation at 400 nm populates vibrationally excited states of S1 lying in the D0 detachment continuum. All the photoelectron spectra are dominated by eKE ≈ 0 (eBE ≈ hν) photoelectrons with an overall exponential decay profile, suggesting that the dominant electronic relaxation mechanism is IC to S0 followed by TE. However, the ratio of photoelectrons attributed to TE compared with those attributed to direct PD decreases in the order pCA− ≈ pCE− ≫ PpCE− > NpCE−. This suggests that torsional motions around both single bonds adjacent to the alkene group are involved in steering the PYP chromophore towards the S1/S0 CI but that rotation around the single bond between the alkene moiety and the phenoxide group plays a more important role.Photoexcitation at 346 nm results predominantly in direct detachment from S0; however, the observation of eKE ≈ 0 (eBE ≈ hν) photoelectrons with an overall exponential decay profile suggests that there is some contribution from resonant excitation of excited states of the anion followed by IC to S0 and subsequent TE. We cannot assign the resonant excitation process to any particular excited state of the anion, but the high vibrational levels of S1, low vibrational levels of S3 or the S2 state, through vibronic mixing with the bright S1 state, are all possibilities. In terms of the role of torsional motions, the ratio of photoelectrons attributed to TE compared with those from direct PD is lower for NpCE− than the other three chromophores, supporting the idea that rotation around the single bond between the alkene moiety and the phenoxide group plays an important role in IC from S1 to S0.
Increasing the photon energy to 330–310 nm results in a significant contribution from indirect PD from S1 and TE from S0 for pCA−, pCE− and PpCE−. In contrast, indirect PD from S1 is far less pronounced for NpCE− and the contribution from TE only increases slightly. The most likely explanation for these observations is that as the photon energy increases, increasing population of S3 followed by internal conversion to S1 occurs and subsequent autodetachment to D0 or IC to S0 followed by TE. Locking the rotation around the single bond between the alkene moiety and the phenoxide group in NpCE− inhibits these pathways, which suggests that rotation around the single bond between the alkene moiety and the phenoxide group also plays a role in IC from S3 to S1.
4 Conclusions
From the combination of photoelectron spectroscopy measurements and computational chemistry calculations, we have improved our understanding of the role of torsional motions around the single bonds either side of the alkene moiety in the electronic relaxation of isolated PYP chromophores in the gas-phase. We have found that single bond rotations are involved in steering the PYP chromophore towards the S1/S0 CI and that rotation around the single bond between the alkene moiety and the phenoxide group plays a particularly important role. From measurements at higher photon energies, we also find that rotation around this single bond between the alkene moiety and the phenoxide group plays a key role in IC from S3 to S1. This suggests that torsional motions around the single bonds, and particularly around the one between the alkene and phenoxide groups are not restricted in the protein. This supports the conclusion of the earlier time-resolved study by Lee et al. on the ketone analogue of the PYP chromophore in which isomerisation occurred on a similar time scale to the protein, suggesting entry into the photocycle must be unhindered by torsional motion and almost frictionless.11 These results have potential applications in biology and nanotechnology in terms of improving our understanding of how to tune the photoresponse of proteins or materials with a chromophore based on the PYP chromophore.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EPSRC grant EP/L005646/1. We are grateful to Professor Graham Worth and Dr Mariana Assmann (UCL) for advice on the computational chemistry calculations, to Dr Jörg Saßmannshausen for computational support at UCL, Dr Abil Aliev for NMR and Dr Kersti Karu (UCL) for mass spectra.References
- H. Kuramochi, S. Takeuchi, K. Yonezawa, H. Kamikubo, M. Kataoka and T. Tahara, Nat. Chem., 2017, 9, 660–666 CrossRef CAS PubMed.
- Image of 1NWZ: structure of the chromophore in the protein pocket at 0.82 Å resolution (E. D. Getzoff, K. N. Gutwin and U. K. Genick, Nat. Struct. Biol., 2003, 10, 663-668) created with VMD – Visual Molecular Dynamics (W. Humphrey, A. Dalke and K. Schulten, J. Molec. Graphics, 1996, 14, 33-38).
- K. J. Hellingwerf, J. Hendriks and T. Gensch, J. Phys. Chem. A, 2003, 107, 1082–1094 CrossRef CAS.
- W. D. Hoff, P. Düx, K. Hård, B. Devreese, I. M. Nugteren-Roodzant, W. Crielaard, R. Boelens, R. Kaptein, J. Van Beeumen and K. J. Hellingwerf, Biochemistry, 1994, 33, 13959–13962 CrossRef CAS PubMed.
- K. Pande, C. D. M. Hutchison, G. Groenhof, A. Aquila, J. S. Robinson, J. Tenboer, S. Basu, S. Boutet, D. P. DePonte, M. Liang, T. A. White, N. A. Zatsepin, O. Yefanov, D. Morozov, D. Oberthuer, C. Gati, G. Subramanian, D. James, Y. Zhao, J. Koralek, J. Brayshaw, C. Kupitz, C. Conrad, S. Roy-Chowdhury, J. D. Coe, M. Metz, P. L. Xavier, T. D. Grant, J. E. Koglin, G. Ketawala, R. Fromme, V. Šrajer, R. Henning, J. C. H. Spence, A. Ourmazd, P. Schwander, U. Weierstall, M. Frank, P. Fromme, A. Barty, H. N. Chapman, K. Moffat, J. J. van Thor and M. Schmidt, Science, 2016, 352, 725–729 CrossRef CAS PubMed.
- G. Groenhof, M. Bouxin-Cademartory, B. Hess, S. P. de Visser, H. J. C. Berendsen, M. Olivucci, A. E. Mark and M. A. Robb, J. Am. Chem. Soc., 2004, 126, 4228–4233 CrossRef CAS PubMed.
- G. Groenhof, L. V. Schäfer, M. Boggio-Pasqua, H. Grubmüller and M. A. Robb, J. Am. Chem. Soc., 2008, 130, 3250–3251 CrossRef CAS PubMed.
- A. Espagne, P. Changenet-Barret, P. Plaza and M. M. Martin, J. Phys. Chem. A, 2006, 110, 3393–3404 CrossRef CAS PubMed.
- A. Baltuška, I. H. M. van Stokkum, A. Kroon, R. Monshouwer, K. J. Hellingwerf and R. van Grondelle, Chem. Phys. Lett., 1997, 270, 263–266 CrossRef.
- A. D. Stahl, M. Hospes, K. Singhal, I. van Stokkum, R. van Grondelle, M. L. Groot and K. J. Hellingwerf, Biophys. J., 2011, 101, 1184–1192 CrossRef CAS PubMed.
- I.-R. Lee, W. Lee and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 258–262 CrossRef CAS PubMed.
- L. Mendonça, F. Hache, P. Changenet-Barret, P. Plaza, H. Chosrowjan, S. Taniguchi and Y. Imamoto, J. Am. Chem. Soc., 2013, 135, 14637–14643 CrossRef PubMed.
- A. Xie, W. D. Hoff, A. R. Kroon and K. J. Hellingwerf, Biochemistry, 1996, 35, 14671–14678 CrossRef CAS PubMed.
- R. Cordfunke, R. Kort, A. Pierik, B. Gobets, G.-J. Koomen, J. W. Verhoeven and K. J. Hellingwerf, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7396–7401 CrossRef CAS.
- D. S. Larsen, M. Vengris, I. H. van Stokkum, M. A. van der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 86, 2538–2550 CrossRef CAS PubMed.
- J. Zhu, L. Paparelli, M. Hospes, J. Arents, J. T. M. Kennis, I. H. M. Van Stokkum, K. J. Hellingwerf and M. L. Groot, J. Phys. Chem. B, 2013, 117, 11042–11048 CrossRef CAS PubMed.
- C. R. S. Mooney, M. A. Parkes, A. Iskra and H. H. Fielding, Angew. Chem., Int. Ed., 2015, 54, 5646–5649 CrossRef CAS PubMed.
- M. A. Parkes, C. Phillips, M. J. Porter and H. H. Fielding, Phys. Chem. Chem. Phys., 2016, 18, 10329–10336 RSC.
- A. R. Mckay, M. E. Sanz, C. R. S. Mooney, R. S. Minns, E. M. Gill and H. H. Fielding, Rev. Sci. Instrum., 2010, 81, 123101 CrossRef CAS PubMed.
- C. R. S. Mooney, M. E. Sanz, A. R. McKay, R. J. Fitzmaurice, A. E. Aliev, S. Caddick and H. H. Fielding, J. Phys. Chem. A, 2012, 116, 7943–7949 CrossRef CAS PubMed.
- J. Tay, M. A. Parkes, K. Addison, Y. Chan, L. Zhang, H. C. Hailes, P. C. Bulman Page, S. R. Meech, L. Blancafort and H. H. Fielding, J. Phys. Chem. Lett., 2017, 8, 765–771 CrossRef CAS PubMed.
- R. Beekmeyer, M. A. Parkes, L. Ridgwell, J. W. Riley, J. Chen, B. L. Feringa, A. Kerridge and H. H. Fielding, Chem. Sci., 2017, 8, 6141–6148 RSC.
- G. A. Garcia, L. Nahon and I. Powis, Rev. Sci. Instrum., 2004, 75, 4989–4996 CrossRef CAS.
- D. W. Arnold, S. E. Bradforth, E. H. Kim and D. M. Neumark, J. Chem. Phys., 1992, 97, 9468–9471 CrossRef CAS.
- C. McLaughlin, M. Assmann, M. A. Parkes, J. L. Woodhouse, R. Lewin, H. C. Hailes, G. A. Worth and H. H. Fielding, Chem. Sci., 2017, 8, 1621–1630 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395–2416 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299–2324 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. Linderberg and Y. Öhrn, Propagators in Quantum Chemistry, John Wiley and Sons, Hoboken, NJ, USA, 2014 Search PubMed.
- A. I. Krylov, Annu. Rev. Phys. Chem., 2008, 59, 433–462 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- J. L. Woodhouse, M. Assmann, M. A. Parkes, H. Grounds, S. J. Pacman, J. C. Anderson, G. A. Worth and H. H. Fielding, Phys. Chem. Chem. Phys., 2017, 19, 22711–22720 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, 2009 Search PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr., H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. V. Voorhis, J. M. Herbert, A. I. Krylov, P. M. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- B. Baguenard, J. C. Pinaré, C. Bordas and M. Broyer, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 63, 023204 CrossRef.
- C. E. Klots, Z. Phys. D: At., Mol. Clusters, 1991, 20, 105–109 CrossRef CAS.
- T. Rocha-Rinza, O. Christiansen, D. B. Rahbek, B. Klærke, L. H. Andersen, K. Lincke and M. Brøndsted Nielsen, Chem. – Eur. J., 2010, 16, 11977–11984 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- C. R. S. Mooney, M. A. Parkes, L. Zhang, H. C. Hailes, A. Simperler, M. J. Bearpark and H. H. Fielding, J. Chem. Phys., 2014, 140, 205103 CrossRef PubMed.
- R. F. Gunion, M. K. Gilles, M. L. Polak and W. C. Lineberger, Int. J. Mass Spectrom. Ion Processes, 1992, 117, 601–620 CrossRef CAS.
- F. Fernández García-Prieto, M. A. Aguilar, I. Fdez. Galván, A. Muñoz-Losa, F. J. Olivares del Valle, M. L. Sánchez and M. E. Martín, J. Phys. Chem. A, 2015, 119, 5504–5514 CrossRef PubMed.
- T. Rocha-Rinza, O. Christiansen, J. Rajput, A. Gopalan, D. B. Rahbek, L. H. Andersen, A. V. Bochenkova, A. A. Granovsky, K. B. Bravaya, A. V. Nemukhin, K. L. Christiansen and M. Brøndsted Nielsen, J. Phys. Chem. A, 2009, 113, 9442–9449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of chromophore synthesis and NMR data; VMI images; comparisons of the optimised structures; benchmarking excited state calculations; excited state energies and characters; coordinates of optimised geometries. See DOI: 10.1039/c7cp06950b |
| This journal is © the Owner Societies 2017 |