
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced energy- and electron-transfer from a photoactive coordination cage to bound guests†
Jerico R.
Piper
a,
Lewis
Cletheroe
a,
Christopher G. P.
Taylor
a,
Alexander J.
Metherell
a,
Julia A.
Weinstein
a,
Igor V.
Sazanovich
b and
Michael D.
Ward
 *a
*a
aDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: m.d.ward@sheffield.ac.uk
bCentral Laser Facility, STFC Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire OX11 0QX, UK
First published on 6th December 2016
Abstract
In a coordination cage which contains an array of twelve naphthyl chromophores surrounding a central cavity, photoinduced energy or electron-transfer can occur from the chromophore array to the bound guest in supramolecular host/guest complexes.
The ability to perform multiple photoinduced energy- or electron-transfer processes in a single assembly, with several light-harvesting units interacting with a single energy- or electron-accepting unit, is of fundamental importance for applications from solar energy harvesting to photocatalysis. For photoinduced energy-transfer (PEnT), the antenna effect is a well-understood way of ensuring that (i) as much light as possible is absorbed, and (ii) the resulting excitation energy is channelled in a predictable way down an energy gradient to the ultimate acceptor, which may be a photosynthetic reaction centre1 or a dye-sensitised solar cell.2 For photoinduced electron transfer (PET), being able to induce multiple electron-transfer events in a single assembly is key to the ongoing search for artificial catalysis for both photochemical water splitting and harvesting the energy from sunlight to make highly reduced solar fuels.3
There is accordingly a need for simple synthetic strategies that will allow a single component – the ultimate user of the absorbed light energy – to interact with multiple chromophores. Covalent connection of multiple components in one molecule may allow precise control of structure, but each individual compound is necessarily limited in scope and probably challenging to prepare. Nature exploits supramolecular methods to achieve self-assembled arrays of the necessary components for photo-synthesis,4 and the use of the methodology of supramolecular chemistry to assemble photophysically active components into arrays with desirable properties is now well-established.5
Against this background the recent emergence of self-assembled coordination cages offers possibilities that are only just beginning to be exploited. Many such cages are photophysically unremarkable and are of interest purely for their structures and ability to act as containers for small-molecule guests.6 However, a minority have been prepared containing photophysically active components, and these provide possibilities for interaction of a large number of chromophoric units – either at the metal vertices7 or in the organic ligands8 – in the cage assembly with a single bound guest. Such cages present an ideal way to prepare an assembly in which a very high local concentration of chromophores in a known orientation can be brought into close proximity to a substrate bound in the cage cavity, which can therefore interact with the chromophore array via photoinduced energy- or electron-transfer.9 It is becoming clear from these recent examples that the relatively mature field of coordination cage self-assembly offers barely-exploited possibilities for photoinduced reactions between bound guests and the surrounding array of chromophores in a nice fusion of self-assembly and photophysics.9
Here we report studies on the photophysical consequences of binding different guest types in the cavity of an octanuclear cubic coordination cage in which an array of twelve naphthyl chromophores surrounds the central cavity.10,11 We demonstrate examples of both PET and PEnT between cage and guest, including the dynamic behaviour of a short-lived charge-separated cage˙+/guest˙− excited state. The cage that we use in this work is [Cd8L12](NO3)16 (hereafter abbreviated as ‘H’ for ‘Host’) which has an approximately cubic structure with a Cd(II) ion at each vertex, and a bis(pyrazolyl-pyridine) bridging ligand incorporating a fluorescent naphthyl spacer spanning each edge (Fig. 1a). It is isostructural to the [Co8L12]16+ analogue which we have studied for its outstanding guest binding properties,10 but it contains non-quenching Cd(II) ions at the vertices to allow the fluorescence from the array of naphthyl units to be retained. We reported the preparation of [Cd8L12](NO3)16 recently.11
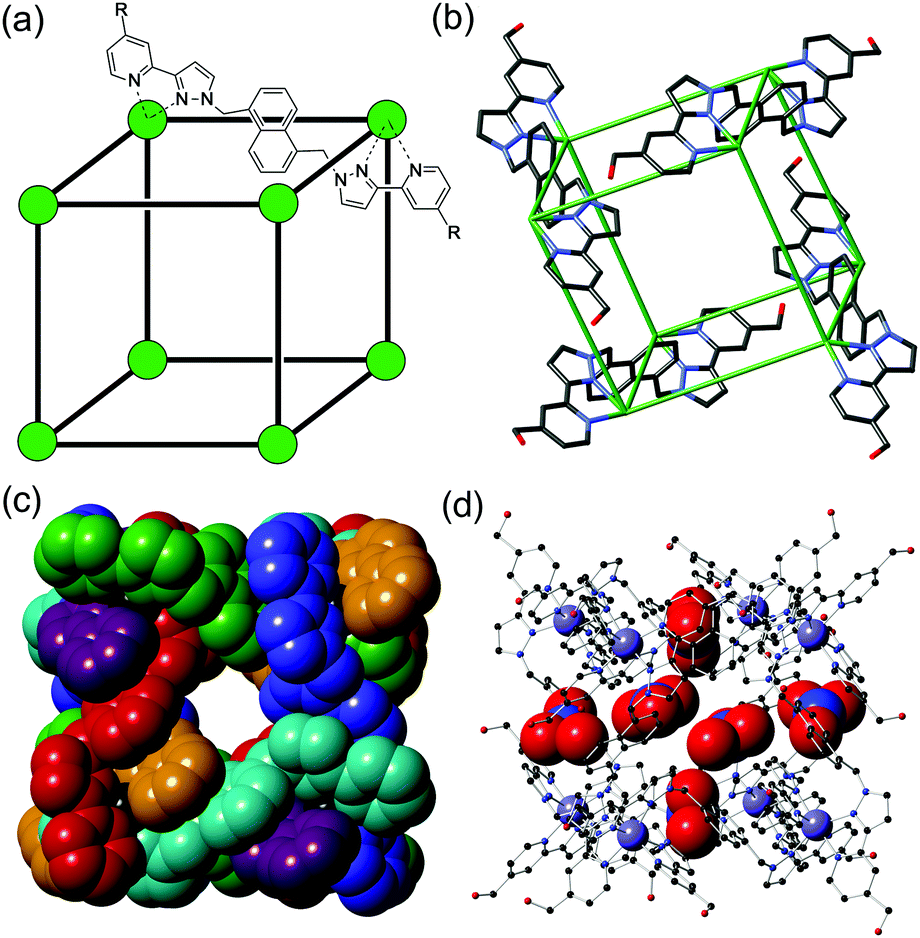 | ||
| Fig. 1 The host cage [Cd8L12](NO3)24, H. (a) A sketch emphasising the cubic array of Cd(II) ions and the disposition of a bridging ligand L (R = CH2OH); (b) a view of the complex cation from the crystal structure showing only four of the bridging ligands; (c) space-filling view of the complex cation with ligands coloured separately for clarity; (d) view of the complex emphasising the positions of some of the nitrate anions (shown space-filling) which occupy the portals on the faces of the cube. Additional figures showing the inclusion of dmf guest molecules and their interactions with the interior cage surface are in ESI.† | ||
We start by reporting the crystal structure of H (Fig. 1b–d; see also Fig. S1, ESI†); it is, as expected, essentially identical to that of the Co(II) analogue reported earlier.10b Despite slightly longer metal–N distances associated with the larger ionic radius of Cd(II) compared to Co(II), the Cd⋯Cd separations along the edges are comparable to those of Co⋯Co separations in the Co(II)-based cubes.10d,b The central cavity contains two dmf guest molecules whose C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups are directed into the H-bond donor pockets that are associated with the internal surface of the cage around each of the two fac tris-chelate metal vertices (Fig. S1, ESI†).12 Every face of the cube has a nitrate anion associated with the window in the face centre, with the anion forming a set of CH⋯O interactions with the cage surface (Fig. 1d).
O groups are directed into the H-bond donor pockets that are associated with the internal surface of the cage around each of the two fac tris-chelate metal vertices (Fig. S1, ESI†).12 Every face of the cube has a nitrate anion associated with the window in the face centre, with the anion forming a set of CH⋯O interactions with the cage surface (Fig. 1d).
H has the ability to act as an energy or electron-donor from its excited state. The singlet excited state of naphthalene has an energy content of ca. 4 eV13 and can therefore act as an energy-donor to any species with lower energy excited states. The relative ease of oxidation of naphthalene to its radical cation13a means that its excited state is also a powerful electron donor, capable of effecting PET to a range of electron-acceptor quenchers.14 The steady-state fluorescence spectrum of host cage H is shown in Fig. 2; the properties of the naphthyl units are perturbed from those of free naphthalene due to their π-stacking interactions with electron-deficient pyrazolyl-pyridine units on either side of them.8a,10aH displays, in solution, a broad and unstructured fluorescence centred at 400 nm from the array of twelve naphthyl groups around the cage periphery, which resembles that of a naphthalene excimer.15 At 77 K in MeOH/EtOH glass this is blue-shifted to 350 nm, indicating an available excited-state energy of ca. 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1.
500 cm−1.
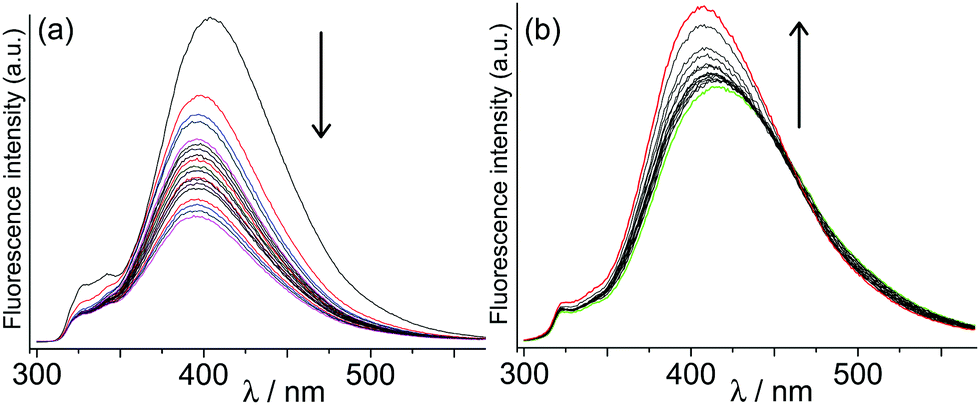 | ||
| Fig. 2 Effect of non-chromophoric guests on fluorescence of the host. (a) Decrease in fluorescence of H on titration with the flexible guest 1 to form H·1; (b) increase in fluorescence of H on titration with the rigid guest 2 to give H·2. | ||
The guests (generically denoted G) that we investigated for their ability to bind in the cavity of H are shown in Scheme 1. These include some aliphatic ketones (1 and 2) which are non-chromophoric and therefore not expected to participate in PET or PEnT with the naphthyl groups: these can be considered as controls. The remaining guests are rigid aromatic species whose ability to be accommodated in the cage cavity was confirmed by molecular modelling and/or a calculation using molecular docking software.10c These are the substituted pyrene 3 which is expected to be a good energy acceptor from the cage based on relative excited state energies of H and 3; and three good electron-acceptors (quinones 4 and 5, and tetracyanobenzene 6).
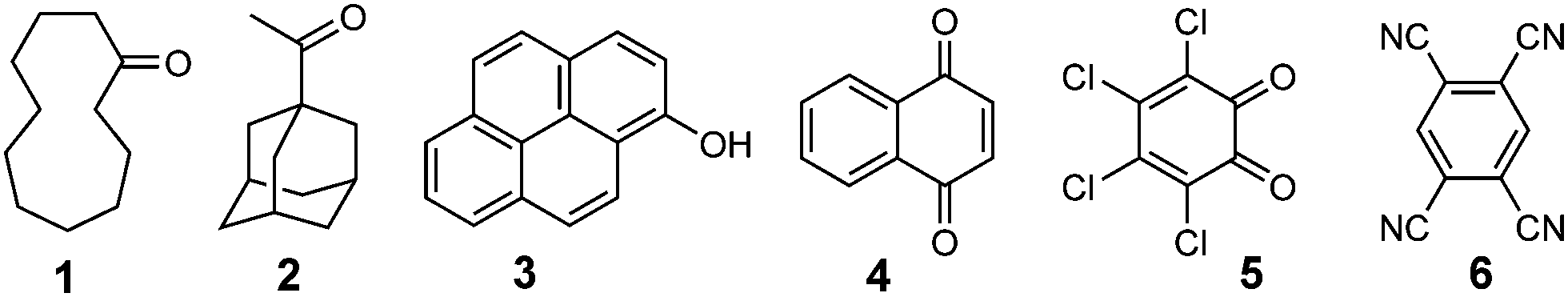 | ||
| Scheme 1 Structural formulae of guests used in this work. | ||
Initially we investigated the effect on cage fluorescence of binding the non-chromophoric and non redox-active guests 1–2, by conventional luminescence titrations in which portions of guest were added stepwise to a 0.01 mM aqueous solution of H. In each case a change in luminescence intensity was observed, with the cyclic ketone 1 resulting in a partial loss of luminescence intensity, but the more rigid guest 2 affording a slight increase in intensity (Fig. 2). We ascribe these changes to mechanical effects associated with differing degrees of rigidity of these guests: the flexible guest 1 provides sufficient additional vibrational modes to facilitate non-radiative deactivation, whereas the much more rigid guest 2 has the opposite effect by stiffening the cage and making non-radiative deactivation less likely. Importantly, the binding constants for these guests calculated from fitting the observed intensity changes to a 1:1 binding isotherm [1, 1.1(1) × 106 M−1; 2, 3(1) × 104 M−1] agree well with what we previously observed for binding the same guests in the isostructural Co(II) cage measured by NMR titrations.10d
Next, we investigated the effect of guest 3 on the cage fluorescence. Given that naphthyl → pyrenyl photoinduced energy transfer has been demonstrated,16 we would expect the H·3 complex to provide an example of PEnT from the array of chromophores in the host to the bound pyrene guest, possibly resulting in sensitised luminescence. This was confirmed by a luminescence titration in which small portions of 3 were added to a solution of H in water (1 μM). The high binding constant for formation of H·3 in water [1.3(1) × 106 M−1, from this data, in good agreement with previous observations]10c means that by the end of the titration, H·3 should be ca. 80% formed under the conditions used. As 3 binds in the cavity of H the normal broad fluorescence band centred at 400 nm reduces in intensity and is gradually replaced by the characteristic sharp, structured features characteristic of pyrene fluorescence (Fig. 3).
 | ||
| Fig. 3 Main figure: changes in fluorescence during addition of 3 to a solution of cage H (1 μM) in water resulting in formation of H·3: the cage fluorescence (green) is progressively quenched, and sensitised fluorescence from 3 (red) grows in. Inset: Restoration of fluorescence of cage H, and loss of sensitised pyrene fluorescence, on addition of cycloundecanone (3 equiv.) which displaces guest 3 from the cavity of H. | ||
Three pieces of evidence confirm that the pyrene fluorescence is sensitised by PEnT from H, and does not just arise by direct excitation of pyrene as its concentration grows during the titration. Firstly, we see progressive quenching of the naphthyl-based fluorescence from free H. Secondly, excitation was at 280 nm, where the array of naphthyl fluorophores in the cage absorbs strongly (ε = 55000 M−1) but 1-hydroxypyrene absorbs weakly (ε = 1400 M−1; see Fig. S2, ESI†); the intensity of fluorescence from 3 is too high to be accounted for by direct excitation. Thirdly, addition of the competing guest cycloundecanone (1) – which is photophysically innocent under these conditions – results in loss of sensitised pyrene-based fluorescence and restoration of cage-based fluorescence as 1 displaces 3 from the cage cavity (Fig. 3, inset). This unambiguously confirms that cage-to-guest PEnT is occurring in the H·3 assembly.
Next, we examined the effect of strongly electron-accepting guests 4–6. All of these guests caused strong quenching of the cage luminescence on binding (Fig. S3 and S4, ESI†); these titrations yielded similar binding constants of 1.54(1) × 104 M−1, 2.9(5) × 104 M−1, and 1.3(5) × 104 M−1, respectively. Given the facile one-electron reductions of these guests, this quenching can be tentatively ascribed to PET from the chromophore array to the guest bound in the cage cavity. Note that the quenching in each case (i) follows a 1:1 binding curve and (ii) is far higher than could occur by bimolecular collisional quenching, so must be associated with guest binding in the cavity.‡ Such PET would generate a charge-separated H˙+/G˙− pair, and the characteristic spectroscopic signature of the radical species involved should permit transient absorption (TA) measurements to probe the formation and decay kinetics of the charge-separated state.
We therefore used femtosecond TA spectroscopy17 to monitor the behaviour of a system containing H (0.01 mM) and guest 6 (4.0 mM) in water. Under these conditions, the large excess of 6 should ensure that the host is >99% bound, i.e. concentration of the bound H·6 complex should be close to 0.01 mM based on the binding constant reported above. In a control experiment, we monitored the behaviour of H on its own, and of guest 6 on its own, under the same conditions.
Excitation of a 0.01 mM solution of H in water (with no guest) at 290 nm with a 40 fs pulse,17 followed by measurement of the absorption spectra at a series of time delays up to 3 ns, produced the differential TA spectra shown in Fig. 4(a). There is a broad positive signal across the entire region of interest, from 320 nm to 550 nm, with a pronounced band with subtle vibronic structure at around 400 nm. This spectral shape closely resembles that of singlet–singlet excited-state absorption of naphthalene, which could be further broadened by delocalisation over the π-stacked array expected in the cage.18 The presence of a distribution of environments for 1Naph*, and the influence of π-stacking, results in complex decay behaviour which can be satisfactorily described by a bi-exponential decay with time constants of 32(±11) ps, and 1130(±150) ps.
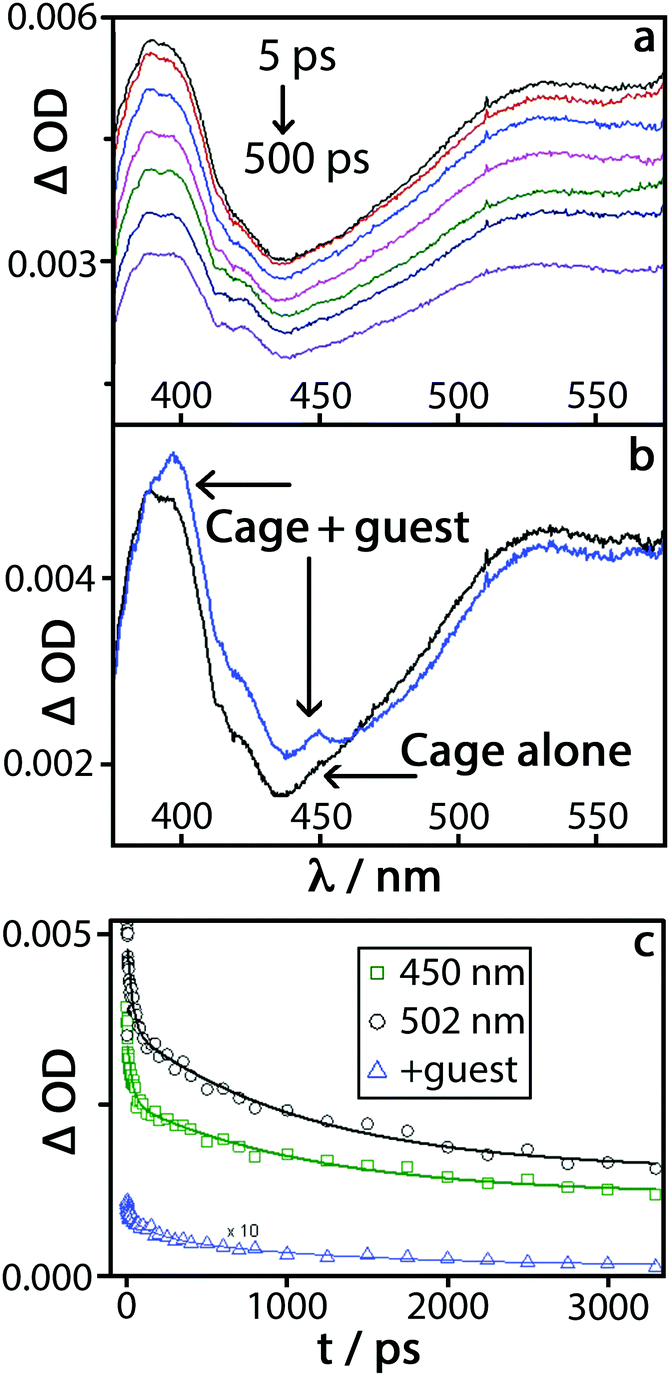 | ||
| Fig. 4 Transient absorption spectra at a series of time delays following 290 nm, 40 fs excitation. (a) Cage H; (b) an overlay of transient absorption spectra of H and H·6 at the time delay of 20 ps (the spectra are normalised to OD at 385 nm); (c) decay of transient signal at 450 nm and 502 nm (H alone). Solid lines represent bi-exponential fit to the data with time constants of 32 and 1130 ps. A differential signal “(H·6)–H” at 450 nm is shown as –△–. The solid line represents a tri-exponential fit to the data; the values of two time constants were fixed as 32 and 1130 ps, the third time constant was obtained as 160 ps. | ||
Excitation of the complex H·6 under the same conditions produced the set of difference spectra shown in Fig. S5 (ESI†). These spectra look superficially similar to those observed for free H, as (i) there is only one guest per many naphthalene units and (ii) there is a small amount of free H in the equilibrium mixture. However, there are clear differences in the shape of the TA spectra of H·6vs.H: namely, an increased absorbance in the 380–460 nm region; an additional narrow band at ca. 450 nm; and pronounced additional fine structure in the ∼400 nm band (Fig. 4b). The region of new absorbance in the range 380–410 nm matches what is expected from the presence of both tetracyanobenzene radical anion, 6˙−,19 and a naphthyl radical cation from H˙+.14b,20 The naphthyl radical cation has a characteristic comparatively sharp absorption band at 382 nm with a shoulder at 366 nm, which explains the increased absorbance in the TA spectrum of H·6 compared to H. The clear, relatively sharp feature at 450 nm in the TA spectrum of H·6 matches the most intense feature in the absorption spectrum of 6˙−;19 this feature did not appear in a control experiment using just 6.
The features in the TA spectrum of H·6 that are absent in the spectrum of H alone (Fig. 4b) are therefore consistent with the occurrence of cage → guest PET to generate the H˙+/6˙− charge-separated pair. Both of the excited state features attributed to H˙+/6˙− decay synchronously with a first-order time constant of ≈160(25) ps, which can be ascribed to charge recombination by back electron transfer. The features of the transient spectrum of H·6 which correspond to 1Naph* persist over longer time scales, decaying with the same dynamics as the free cage.
In conclusion, we have demonstrated that the array of 12 naphthyl fluorophores built into the self-assembled cage H is capable of acting as an antenna group, effecting photoinduced energy or electron transfer to a guest that is bound in the central cavity in supramolecular H·G complexes. This opens up extensive possibilities for use of these cages in both single- and multi-electron photocatalysis.
We thank EPSRC for financial support (studentships to J. R. P. and C. G. P. T.); ‘Shine’ CDT and the Grantham Centre for Sustainable Futures at the University of Sheffield (studentship to L. C.); and STFC for award of access time to the ps-TA facility.
Notes and references
- (a) H. Zuber, Trends Biochem. Sci., 1986, 11, 414 CrossRef CAS; (b) P. Horton and A. Ruban, J. Exp. Bot., 2005, 56, 365 CrossRef CAS PubMed; (c) Y.-C. Cheng and G. R. Fleming, Annu. Rev. Phys. Chem., 2009, 60, 241 CrossRef CAS PubMed; (d) R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492 CrossRef CAS PubMed; D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 Search PubMed.
- (a) C. A. Bignozzi, R. Argazzi and C. J. Kleverlaan, Chem. Soc. Rev., 2000, 29, 87 RSC; (b) C. Y. Lee and J. T. Hupp, Langmuir, 2010, 26, 3760 CrossRef CAS PubMed; (c) J. Warnan, F. Buchet, Y. Pellegrin, E. Blart and F. Odobel, Org. Lett., 2011, 13, 3944 CrossRef CAS PubMed; (d) J. Warman, Y. Pellegrin, E. Blart and F. Odobel, Chem. Commun., 2012, 48, 675 RSC.
- (a) S. Karlsson, J. Boixel, Y. Pellegrin, E. Blart, H.-C. Becker, F. Odobel and L. Hammarström, Faraday Discuss., 2012, 155, 233 RSC; (b) L. Hammarström, Acc. Chem. Res., 2015, 48, 840 CrossRef PubMed; (c) K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503 CrossRef CAS PubMed.
- R. Kouril, J. P. Dekker and E. J. Boekema, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 2 CrossRef CAS PubMed.
- (a) M. D. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC; (b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed; (c) F. D’Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (d) G. Calzaferri and K. Lutkouskaya, Photochem. Photobiol. Sci., 2008, 7, 879 RSC; (e) A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC; (f) H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Rev., 2015, 115, 7502 CrossRef CAS PubMed.
- (a) R. J. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (b) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed; (c) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (d) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (e) M. D. Ward, Chem. Commun., 2009, 4487 RSC.
- (a) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (b) A. B. Wragg, A. J. Metherell, W. Cullen and M. D. Ward, Dalton Trans., 2015, 44, 17939 RSC; (c) K. Li, L.-Y. Zhang, C. Yan, S.-C. Wei, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef CAS PubMed; (d) B. El Aroussi, L. Guénée, P. Pal and J. Hamacek, Inorg. Chem., 2011, 50, 8588 CrossRef CAS PubMed.
- (a) N. K. Al-Rasbi, C. Sabatini, F. Barigelletti and M. D. Ward, Dalton Trans., 2006, 4769 RSC; (b) P. D. Frischmann, V. Kunz and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 7285 CrossRef CAS PubMed; (c) P. K. Neelakandan, A. Jiménez and J. R. Nitschke, Chem. Sci., 2014, 5, 908 RSC; (d) Z. Li, N. Kishi, K. Hasegawa, M. Akita and M. Yozhizawa, Chem. Commun., 2011, 47, 8605 RSC.
- (a) D. M. Dalton, S. R. Ellis, E. M. Nichols, R. A. Mathies, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 10128 CrossRef CAS PubMed; (b) Z. Jing, C. He, Y. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 3967 CrossRef PubMed; (c) R. Gera, A. Das, A. Jha and J. Dasgupta, J. Am. Chem. Soc., 2014, 136, 15909 CrossRef CAS PubMed; (d) Y. Furutani, H. Kandori, M. Kawano, K. Nakabayashi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2009, 131, 4764 CrossRef CAS PubMed; (e) Y. Yang, J.-S. Chen, J.-Y. Liu, G.-J. Zhao, L. Liu, K.-L. Han, T. R. Cook and P. J. Stang, J. Phys. Chem. Lett., 2015, 6, 1942 CrossRef CAS PubMed.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC; (c) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 2790 RSC; (d) S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475 CrossRef CAS PubMed.
- C. G. P. Taylor, J. R. Piper and M. D. Ward, Chem. Commun., 2016, 52, 6225 RSC.
- (a) A. J. Metherell and M. D. Ward, Dalton Trans., 2016, 45, 16096 RSC; (b) S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122 CrossRef CAS PubMed.
- (a) E. S. Pysh and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 2124 CrossRef CAS; (b) G. George and G. Morris, J. Mol. Spectrosc., 1968, 26, 67 CrossRef CAS; (c) R. Dabestani and I. N. Ivanov, Photochem. Photobiol., 1999, 70, 10 CAS.
- (a) K. K. Mentel, R. M. D. Nunes, C. Serpa and L. G. Arnaut, J. Phys. Chem. B, 2015, 119, 7571 CrossRef CAS PubMed; (b) H. Kotani, K. Ohkubo and S. Fukuzumi, Faraday Discuss., 2012, 155, 89 RSC; (c) G. Accorsi, F. Barigelletti, A. Farrán, F. Herranz, R. M. Claramunt, M. Marcaccio, G. Valenti, F. Paolucci, E. Pinilla and M. R. Torres, J. Photochem. Photobiol., A, 2006, 203, 166 CrossRef; (d) T. H. Ghaddar, J. F. Wishart, D. W. Thompson, J. K. Whitesell and M. A. Fox, J. Am. Chem. Soc., 2002, 124, 8285 CrossRef CAS PubMed; (e) S. Hashimoti and M. Yamaji, Phys. Chem. Chem. Phys., 2008, 10, 3124 RSC.
- (a) J. R. Froines and P. J. Hagerman, Chem. Phys. Lett., 1969, 4, 134 CrossRef; (b) P. F. Jones and M. Nicol, J. Chem. Phys., 1968, 48, 5440 CrossRef CAS; (c) R. J. Locke and E. C. Lim, J. Phys. Chem., 1989, 93, 6017 CrossRef CAS.
- D. F. Anghel, V. Alderson, F. M. Winnik, M. Mizusaki and Y. Morishima, Polymer, 1998, 39, 3035 CrossRef CAS.
- G. M. Greetham, P. Burgos, Q. A. Cao, I. P. Clark, P. S. Codd, R. C. Farrow, M. W. George, M. Kogimtzis, P. Matousek, A. W. Parker, M. R. Pollard, D. A. Robinson, Z.-J. Xin and M. Towrie, Appl. Spectrosc., 2010, 64, 1311 CrossRef CAS PubMed.
- (a) R. Bonneau, J. Faure and J. Joussot-Dubien, Chem. Phys. Lett., 1968, 2, 65 CrossRef CAS; (b) R. McNeil, J. T. Richards and J. K. Thomas, J. Phys. Chem., 1970, 74, 2290 CrossRef CAS.
- M. Fagnoni, S. Protti, D. Ravelli and A. Albi, Beilstein J. Org. Chem., 2013, 9, 800 CrossRef CAS PubMed.
- (a) T. Fushimi, Y. Fujita, H. Ohkita and S. Ito, J. Photochem. Photobiol., A, 2004, 165, 69 CrossRef CAS; (b) J. Szczepanski, D. Roser, W. Personette, M. Eyring, R. Pellow and M. Vala, J. Phys. Chem., 1992, 96, 7876 CrossRef CAS; (c) A. Moissette, H. Vezin, I. Gener and C. Bremard, J. Phys. Chem. B, 2003, 107, 8935 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental information (luminescence measurements, TA spectroscopy, crystallography); additional figures; full crystallographic details in CIF format. CCDC 1518317. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc09298e |
| ‡ For collisional (dynamic) quenching a standard Stern–Volmer analysis applies: I0/I = 1 + k[guest]τ0, where τ0 ≈ 2 ns (lifetime of naphthalene excited state). Even assuming a diffusion-limited rate constant for collisions of 1010 s−1, and a huge excess of guest (4 mM), the maximum possible fluorescence quenching would be just 8% – so quenching is associated with guest binding in the cage cavity. |
| This journal is © The Royal Society of Chemistry 2017 |