
Checkpoints for preliminary identification of small molecules found enriched in autophagosomes and activated mast cell secretions analyzed by comparative UPLC/MSe†
Chad P.
Satori
a,
Marzieh
Ramezani
a,
Joseph S.
Koopmeiners
b,
Audrey F.
Meyer
a,
Jose A.
Rodriguez-Navarro
c,
Michelle M.
Kuhns
a,
Thane H.
Taylor
a,
Christy L.
Haynes
a,
Joseph J.
Dalluge
a and
Edgar A.
Arriaga
*a
aUniversity of Minnesota, Department of Chemistry, 207 Pleasant Street SE, Minneapolis, MN 55455-0431, USA. E-mail: arriaga@umn.edu; Tel: +1-612-624-8024
bUniversity of Minnesota, Division of Biostatistics, 420 Delaware Street SE, Minneapolis, MN 55455, USA
cAlbert Einstein College of Medicine, Institute for Aging Studies, Marion Besin Liver Research Center, Department of Developmental and Molecular Biology, 1300 Morris Park Avenue, Bronx, NY 10461, USA
First published on 11th October 2016
Abstract
We report the use of ultra high performance liquid chromatography (UPLC) coupled with acquisition of low- and high-collision energy mass spectra (MSe) to explore small molecule compositions that are unique to either enriched-autophagosomes or secretions of chemically activated murine mast cells. Starting with thousands of features, each defined by a chromatographic retention time, m/z value and ion intensity, manual examination of the extracted ion chromatograms (XIC) of chemometrically selected features was essential to eliminate false positives, occurring at rates of 33, 14 and 37% in samples of three biological systems. Forty-six percent of features that passed the XIC-based checkpoint, had IDs in compound databases used here. From these, 19% of IDs had experimental high-collision energy MSe spectra that were in agreement with in silico fragmentation. The importance of this second checkpoint was highlighted through validation with selected commercially available standards. This work illustrates that checkpoints in data processing are essential to ascertain reliability of unbiased metabolomic studies, thereby reducing the risk of generating ‘false identifications’ which is a major concern as ‘omics’ data continue to proliferate and be used as platforms to launch novel biological hypotheses.
Introduction
Liquid chromatography coupled to mass spectrometry (LC/MS) employing high resolution mass analyzers is often the method of choice for the analysis of compounds in biological systems due to its high sensitivity, selectivity, and mass accuracy.1–3 Specifically, UPLC/MSe is a rapid and robust method for biological chemical profiling studies.2,4–6 UPLC/MSe employing an orthogonal quadrupole-time-of-flight mass spectrometer provides rapid, high peak-capacity separations along with simultaneous acquisition of both low- and high-collision energy mass spectra with excellent mass accuracy for both parent and fragment ions. Thus, the resulting data sets are comprised of data features, each characterized by a chromatographic retention time, m/z and relative intensities of ions in both low and high-energy dissociation spectra.Such data sets require chemometric approaches to expedite the determination of system-specific chemical features.7 While chemometric approaches in concert with database searches have become essential to improving efficiency in mass-spectrometry related studies, there is the likelihood that some selected features may be false positives and/or misidentified due to limitations of the chemometric strategy. Additionally, database IDs based only on mass accuracy measurements of detected precursor ions are often erroneous.8 As such, scrutiny of chemometrics-determined features and subsequent database-identified compounds becomes imperative to assigning reliable preliminary identifications. A suitable chemometric approach for processing of UPLC/MSe datasets is orthogonal partial least squares with discriminate analysis (OPLS-DA) that classifies data features into one of two comparative groups.9–12 Disadvantages of OPLS-DA include the failure to detect selected features that are not well grouped into one of the two groups due to group-averaging effects from discriminate analysis.13 Alternate approaches to using OPLS-DA for detecting selected features include the t-test and linear mixed model (LMM) which compare the average intensity of extracted-ion chromatograms (XICs) between samples.14 However, the LMM is susceptible to model misspecification, which can result in inaccurate choice of selected features and potential omission of some selected features unique to a sample that has increased variation.13 Thus, applying multiple chemometric approaches to a given data set can compensate for the limitations of a single chemometric approach and increase coverage of features unique to a comparative group.
Despite the advantages of LC/MS-based approaches combined with chemometrics for metabolite profiling studies, there are other potential experimental pitfalls.8,15,16 For instance, slight changes in chromatographic retention times can cause misalignment of sample-specific features, resulting in variations in ion suppression of chemical species due to co-eluting compounds, which ultimately leads to inaccurate assessment of the relative abundances of analytes.
The numerous caveats regarding chemometric analysis, database searching, and experimental variations summarized above make additional evaluation of both chemometric-selected features and database IDs imperative. As such, here we demonstrate the importance of validation of (1) chemometric selected features using XICs, and (2) comparison of the experimental high-collision energy MSe spectra with in silico fragmentation of tentative ID's from database searches. Application of these two checkpoints to define preliminary identifications from comparative samples were 100% correct when compared against commercially available standards. Here, we used these checkpoints in the processing of data resulting from the UPLC/MSe analysis of enriched autophagosomes and secretions of activated mast cells, which lack prior reports on unsupervised metabolomic analysis. Autophagosomes are organelles involved in autophagy, a process that fails in multiple diseases including Parkinson's disease,17–19 Huntington's disease,17 and Alzheimer's disease.19,20 Chemically stimulated mast cells play an important role in allergic/inflammatory response.21 Thus, having passed two orthogonal checkpoints, the preliminary identifications described here, constitute potentially important molecules in autophagosomes and secretions from activated mast cells. Overall, we recommend use of these checkpoints in the initial testing of (unsupervised) analysis of mass spectrometric datasets, which ultimately results in improved molecular characterization of biological systems.
Materials and methods
Reagents
Metrizamide was purchased from Amresco (Solon, OH). Vinblastine sulfate, sucrose, gentamycin, Trizma-hydrochloric acid, potassium chloride, sodium chloride, D-glucose, calcium chloride, protease inhibitor cocktail (for mammalian cell and tissue extracts, in DMSO solution), sodium hydroxide, and magnesium chloride were from Sigma Aldrich (St. Louis, MO). Glycyl-L-phenylnaphthylamine was from Bachem (Torrance, CA). Methanol was from Fisher Scientific (Fairlawn, NJ). Dichloromethane was from Mallinkrodt (Phillipsburg, NJ). Ultra LC/MS-grade methanol, acetonitrile, and water were from JT Baker (Center Valley, PA). Formic acid was from EMD (Darmstadt, Germany). Penicillin-streptomycin solution (PS) was from Life Technologies (Grand Island, NY). Anti-TNP IgE antibody was from BD Biosciences (San Jose, CA). CXCL10 was from Shenandoah Biotechnology (Warwick, PA). Dubelco's Modified Eagle Medium (DMEM) high glucose cell medium, bovine calf serum, and fetal bovine serum were from Thermo Scientific (Waltham, MA). Standards including: 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine, 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine, 1-hexadecanoyl-sn-glycero-3-phosphoethanolamine, and D-erythro-sphingosine (C17 base) were purchased from AvantiPolarLipids (Alabaster, AL).Sample preparation and extraction
For the detailed preparation of biological fractions of interest, please refer to the ESI.† Briefly, enriched autophagosome fractions from rat liver were prepared by density gradient centrifugation.22,23 The second fraction of autophagosomes was prepared from rat myoblast cell culture by first increasing the number of autophagosomes by a vinblastine treatment to increase the number of autophagosomes,24 differential centrifugation to remove unwanted organelles, immuno-depletion of remaining mitochondria, and osmotic lysis with glycyl-L-phenylnaphthylamine (GPN) to remove remaining lysosomes.24 Each autophagosome-enriched sample was separately extracted for their polar and nonpolar contents and concentrated by vacuum centrifugation to remove extraction solvents.25 Murine peritoneal mast cells were stimulated with Tris buffer, CXCL-10, or TNP-ova creating four unique biological systems that model different inflammatory conditions. Species secreted during mast cell stimulation were filtered and concentrated prior to analysis.UPLC/MSe analysis
Samples were resuspended prior to UPLC/MSe analysis as previously described.25 Briefly, extracts were resuspended in 200 μL 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v methanol:deionized water. Samples were vortexed for 30 s to resuspend the pellet followed by sonication for 30 min. The samples were centrifuged at 16100 × g for 10 min to remove any non-resuspended materials.
:1 v/v methanol:deionized water. Samples were vortexed for 30 s to resuspend the pellet followed by sonication for 30 min. The samples were centrifuged at 16100 × g for 10 min to remove any non-resuspended materials.
Four pure chemical standards were purchased from Avanti Polar Lipids. 1-Palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine and 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine were prepared in 1:1 HPLC-grade MeOH:H2O to the final concentration of 50 μg mL−1 and vortexed for 1 minute. 1-Hexadecanoyl-sn-glycero-3-phosphoethanolamine, was dissolved in CHCl3/MeOH/H2O (8:4:1) mixture (C = 10 mg mL−1). One μl of the solution was transferred into 200 μL 1:1 HPLC-grade MeOH:H2O to the final concentration of 50 μg mL−1 and vortexed for 1 minute. D-erythro-sphingosine (C17 base) was transferred into 1:1 HPLC-grade MeOH:H2O so that the final concentration of the sample was 50 μg mL−1 and vortexed for 1 minute. Standards samples were added to mass spectrometry sample tubes (MS conditions: polar positive).
Standards were then spiked from their stocks into post-nuclear fraction of L6 cells (25000–100000 cells) prepared with nitrogen cavitation. Standards were spiked at a final concentration of 50 μg mL−1. Samples were treated with 1.0 mL MeOH:H2O. (1:1 v/v) and the pellet was resuspended and vortexed for 1 min. Samples were centrifuged for 10 min, at 16100 ×g. The supernatant was removed and dried with speed vac. Remaining pellet was treated with 1.0 mL DCM:MeOH (1:3 v/v), and the pellet was resuspended and vortexed for 1 min. Samples were then centrifuged for 10 min, at 16100 ×g. Supernatant was removed and dried with vacuum centrifugation at 24 °C for ∼36 h. Samples were resuspended in 225 μL HPLC-grade MeOH:H2O. (1:1 v/v), vortexed for 1 min, sonicated for 10 min, and centrifuged for 10 min at 16100 ×g. Supernatant was collected, and the samples were injected for mass spectrometry analysis.
A Waters Acquity UPLC coupled to a Waters Synapt G2 HDMS quadrupole orthogonal acceleration time of flight mass spectrometer was used for UPLC/MSe analysis. The reversed-phase column used was a Waters HSS T3 C18 2.1 mm × 100 mm column (1.7 μm diameter particles) operated at 35 °C. The following 28 min linear gradient separations were employed at a flow rate of 0.40 mL min−1 using a mobile phase consisting of A: water containing 0.1% formic acid and B: acetonitrile containing 0.1% formic acid. The gradient profile for samples from polar extractions was: 3% B, 0 min to 5 min; 3% B to 97% B, 5 min to 18 min; 97% B, 18 min to 21 min; 97% B to 3% B, 21 min to 23 min; 3% B 23 min to 28 min. The gradient profile for nonpolar extractions was: 30% B, 0 min to 5 min; 30% B to 97% B, 5 min to 18 min; 97% B, 18 min to 21 min; 97% B to 3% B, 21 min to 23 min; 3% B, 23 min to 28 min. Dead time was 0.68 min for the polar separation and 0.54 min for the non-polar separation as determined by injection of acetone. Simultaneous low- and high-collision energy (CE) mass spectra were collected in centroid mode over the range m/z 50–1200 every 0.1 s during the chromatographic separation. MSe parameters in positive electrospray ionization mode were as follows: capillary, 2.0 kV; sampling cone, 35.0 V; extraction cone, 5.0 V; desolvation gas flow, 800 L h−1; source temperature, 100 °C; desolvation temperature, 350 °C; cone gas flow, 20 L h−1; trap CE, off (low CE collection), trap CE ramp 15–65 V (high CE collection); lockspray configuration used the average of three m/z measurements (0.2 s scan, m/z 100–1200, every 10 s) of protonated leucine-enkephalin (m/z 556.2771) formed from infusion of a 5 μg mL−1 solution; this configuration typically yields mass accuracies <2 ppm. All MSe parameters were identical in negative ionization mode except the following: capillary, 2.5 kV; sampling cone, 30.0 V; extraction cone, 4.0 V.
UPLC/MSe data analysis & workflow
Waters MarkerLynx™ was used for mining chromatographic and mass spectral data using three-dimensional peak integration and data set alignment (m/z and separation time) to determine data features (Fig. 1). The parameters were set to achieve determination of 1000–2000 data features specific to a given biological system. Data extraction parameters within Markerlynx™ were set as follows: extracted ion chromatogram tolerance was ±20 mDa, peak width assessment was done at half-height of the peak, peak-to-peak baseline noise was 8.0 counts, marker intensity threshold was 100 counts (S/N > 12), mass window was ±20 mDa, and the retention time window was ±0.10 min.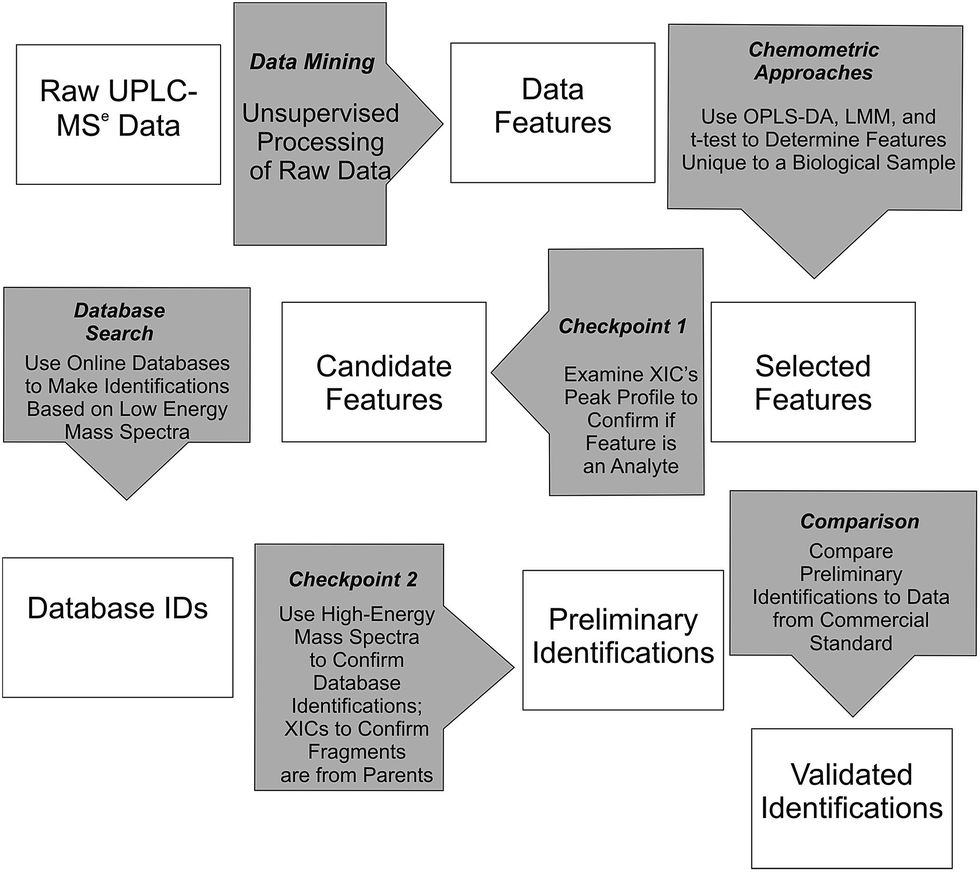 | ||
| Fig. 1 Workflow for determination, confirmation, and preliminary identification of features from UPLC/MSe data. | ||
Chemometrics analysis was used for determination of candidate, system-specific features from UPLC/MSe data (Fig. 1). The LMM and t-test approaches were implemented in the R programming language using a home-written script and OPLS-DA was included in MarkerLynx™ from Waters.26 Comparison of the relative abundance of ions, as measured by XIC intensity, in either the control or enriched fraction for the autophagosome samples or in either the control or stimulated fractions for the mast cell sample was used to select features with LMM or t-tests (Fig. 1). Experiments with no biological replicates but with three instrumental replicates (autophagosomes from rat liver tissue) were analyzed using the two-sample t-test with unequal variances. Experiments with both three biological replicates and three instrumental replicates each (autophagosomes from rat myoblast skeletal muscle cell culture and activated mast cells) were analyzed using LMM.14 Analysis for the t-test and LMM was performed on the log-transformed scale, and differences between biological samples were described by the ratio of geometric means. Multiple comparisons were accounted for by controlling the false discovery rate, which was calculated using the approach described in Benjamini and Yekutieli.27 For autophagosome-enriched samples, data features with a false discovery rate of 10% or less were deemed selected features. For activated mast cells, which had at least 10-fold greater number of features, features with a false discovery rate of 1% or less were deemed selected features for Checkpoint 1 (Fig. 1).
Selected features with OPLS-DA were those at the edges of the OPLS-DA-generated S plot (Fig. 2). An S plot is one option available for visualization of OPLS-DA data.28 The enrichment of the preliminary feature in a biological system is plotted on the x-axis and correlation of enrichment is plotted on the y-axis. Features that were >|0.001| for coefficient 2 (x-axis) and >|0.90| for correlation (y-axis) were deemed selected features (Fig. 1). The number of selected features in each sample is summarized in Table S1.†
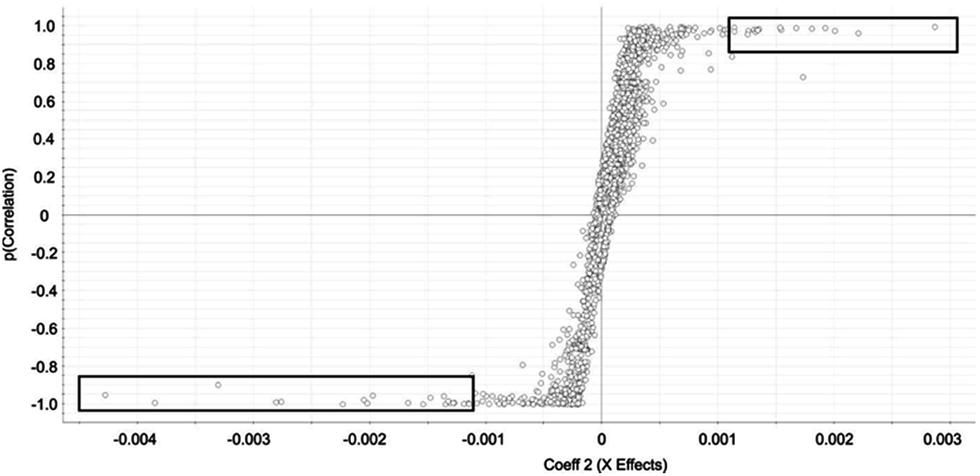 | ||
| Fig. 2 OPLS-DA-generated S-Plot from OPLS-DA chemometric approach for identification of rat liver autophagosome-specific selected features. Selected features are indicated by circles. Features approaching x = −0.004 are specific to autophagosome-enriched while features approaching x = 0.003 are specific to the control. Squares indicate the features selected in the autophagosome-enriched and control samples. | ||
All selected features by any of the chemometric approaches described above were examined at Checkpoint 1 (Fig. 1). At this checkpoint, the XICs from the low-collision energy mass spectra for a given m/z value with a 5 ppm mass tolerance were examined. A selected feature was rejected when the XIC did not have true chromatographic peak profile. Candidate features were those that passed Checkpoint 1 (Fig. 1). The number of candidate features that passed this checkpoint in each sample is summarized in Table S1 (ESI†).
Online database searches of candidate features made it possible to assign Database IDs to such features (Fig. 1). Online databases included the Chemical Entities of Biological Interest, Human Metabolome Database, Lipid MAPS, and ChemSpider. Although other databases are available (e.g. MyCompoundID, MZmine, Massbank, METLIN, mz Cloud),29–31 we focused on using those who were immediately accessible to us. Searches for the neutral mass values corresponding to [M + H]+ and [M − H]− ions were done with mass error <10 mDa (or <11 ppm). This is an acceptable mass accuracy based on recently published metabolomics-based reports,32–35 and would be a small concern because of the subsequent checkpoint described below. Due to the large number of candidate features for activated mast cells, the top 250 selected features for the LMM and all 65 selected features for the OPLS-DA were selected for database searches. Results from the database searches are summarized in Table S1 (ESI†).
Features with database IDs were evaluated at Checkpoint 2 (Fig. 1). Checkpoint 2 consists of (1) XIC alignment of co-eluting ions to verify the presence of putative precursor ions corresponding to the database-identified species; (2) corroboration of identity by comparison of fragmentation patterns observed in the high-collision energy mass spectra of each precursor with simulated fragmentation patterns calculated in silico using Waters MassFragment™ software; and (3) manual precursor-product XIC alignment of precursor and fragment ions. The MassFragment score system to predict fragmentation (i.e. low score) used the default scoring system as follows: the 20 most intense m/z species from the high-collision energy mass spectra were compared to theoretical fragments with a tolerance of 10 mDa. Double bond equivalence values were between −10 to 50, electron count was set to “both”, maximum H deficit was 6, fragment number of bonds was 4, and scoring parameters were for aromatic (6), multiple (4), ring (2), phenyl (8), other (1), H-deficit (0), hetero modifier (0.5), alpha penalty (5), and maximum score (16). Preliminary identifications supported by the evaluation criteria described here are reported using their database ID number36 and are summarized in Tables S1 and S2 (ESI†).
Results
We used UPLC/MSe analysis to carry out preliminary identifications of unique small molecules in biological samples resulting from subcellular enrichment (autophagosome samples) or biological mimics of inflammation (activation of mast cells). In particular, the samples included rat liver autophagosomes, from rat myoblast cell culture autophagosomes, and secretions from mouse mast cells under different activation conditions. For subcellular enrichment, the reference controls were the respective post nuclear subcellular fraction. For secretions from mast cell activation, the reference controls were those from non-activated mast cells (negative control) and those from TNP-ova-activated mast cells (positive control). A summary of the results is shown in Tables S1 and S2 (ESI†).Preliminary identifications from rat liver
Using the OPLS-DA and t-test, the combined results included 114 selected features (Table S1, ESI†). Confirmation of selected features via evaluation of XICs (Checkpoint 1, Fig. 1) yielded 76 candidate features (67% of detected selected features). Database searching yielded 46 IDs (61% of the candidate features). Checkpoint 2 scrutinized the online database IDs by manual examination of low- and high-collision energy mass spectra and matching observed fragmentation patterns with simulated fragmentation patterns for the identified compound (determined using Waters MassFragment™), as well as precursor-product XIC alignment. Overall, this process yielded 20 preliminary identifications (46% of the database IDs). All preliminary identifications had mass errors ≤2 ppm and molecular structures compatible with their expected subcellular environment (Table S2, Fig. S7a, ESI†).Among the 20 preliminary identifications was the compound 1-octadecanoyl-glycero-3-phosphocholine (LMGP01050026). The compound's parent [M + H]+ ion (Fig. 3A) shows aligned peaks in the low- and high-collision energy extracted ion chromatograms (Fig. 3B and C, respectively). A trend plot for the observed precursor ion indicates the relative abundance of this compound in the autophagosome-enriched fraction (Aps, Fig. 3D) versus the non-enriched fraction (control, ctl, Fig. 3D). Trend plot data was used to calculate the fold-enrichment reported in Table S2.† Despite the complex nature of the low- and high-collision energy mass spectra (Fig. 3E and F, respectively) caused by other molecules with overlapping retention times to this compound, a theoretical fragment ion generated in silico using MassFragment™ matched an observed fragment ion (Fig. 3G and F). Other ions present in the spectra that could confound the assignment did not interfere, and the XIC of this fragment matched the XIC of the parent ion, confirming the preliminary identification of the parent ion (Checkpoint 2, Fig. 1).
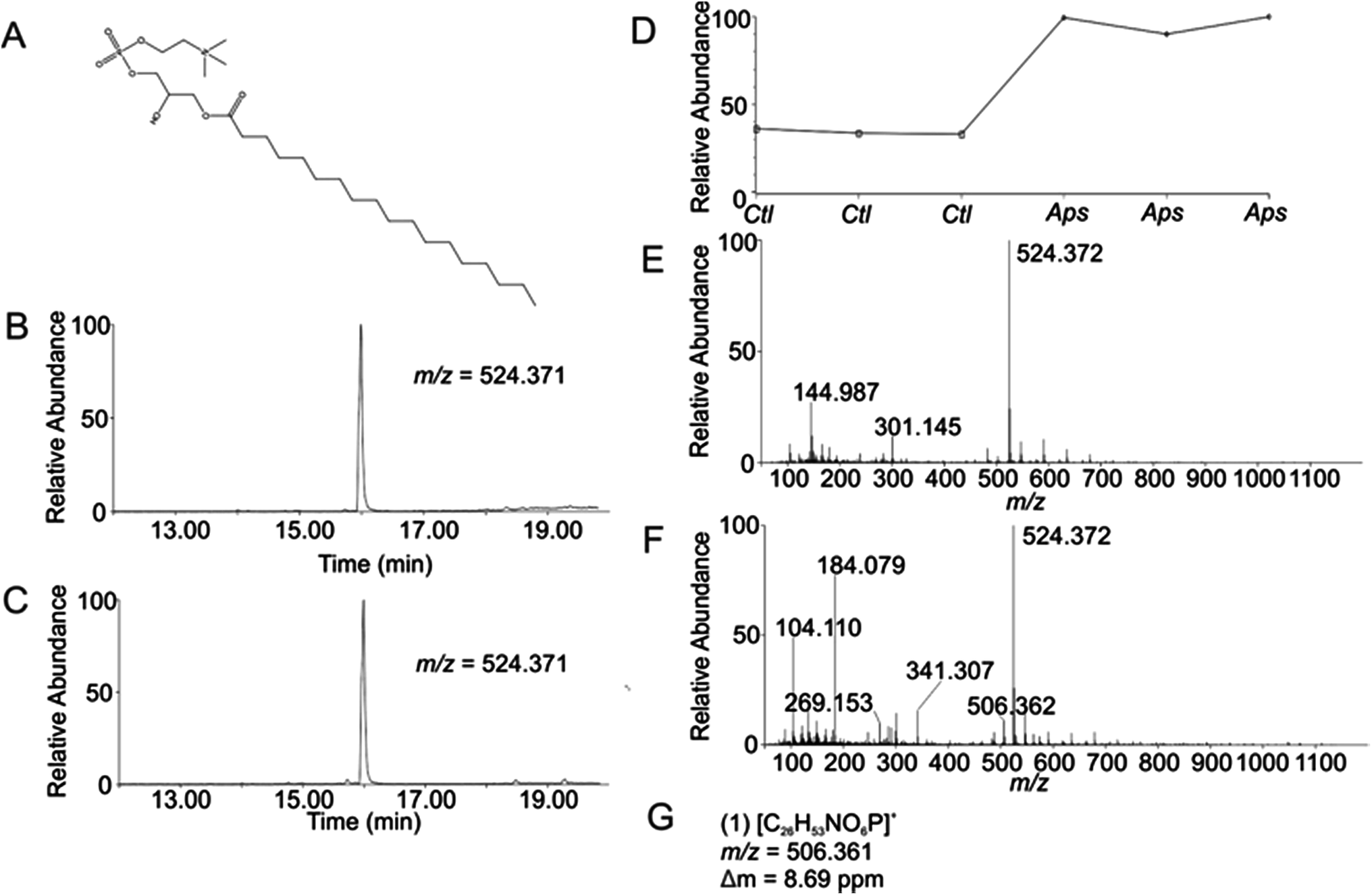 | ||
| Fig. 3 Preliminary Identification of m/z 524.371 (1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine, LMGP01050026) in autophagosome enriched fractions of rat liver tissue (TR = 16.00 min, p-value = 2.58 × 10−5). (A) Structure of LMGP01050026; (B) low-collision energy XIC for m/z = 524.371; (C) high-collision energy XIC for m/z = 524.371; (D) trend plot for m/z = 524.371 in control (Ctl) versus autophagosome-enriched (Aps) samples; (E) low-collision energy mass spectrum at TR = 16.00 min; (F) high-collision energy mass spectrum at TR = 16.00 min; (G) elemental compositions, m/z values, and mass error of observed fragment ions corresponding to theoretical fragment ions generated in silico using Mass Fragment™. | ||
To validate the identification of LMGP01050026 described in the previous paragraph we also ran a commercial standard and the liver autophagosome-enriched sample spiked with the standard (Fig. 4). The parent ion mass (m/z 523.3638), the match between the XIC (m/z 523.3638; TR = 15.98 min) of the sample/spiked-in standard and the standard alone (Fig. 4B and C, respectively) and match the main peaks in the low and high-collision energy mass spectra of the sample/spiked-in standard and the standard alone (Fig. 4D and E, respectively) are strong evidence for identification of LMGP01050026 in the liver autophagosome-enriched sample. Furthermore, a peak with m/z 506.36 related to a fragment formed by loss of a water molecule, used for the high-collision energy XIC was also present in the spectra obtained with the use of standards (Fig. 4D and E). Lastly, other fragments supported the structural identification (m/z 184.07 defines the loss of C21H40O3 group; m/z 104.10 represents a fragment formed by losing a C21H41O6P group).
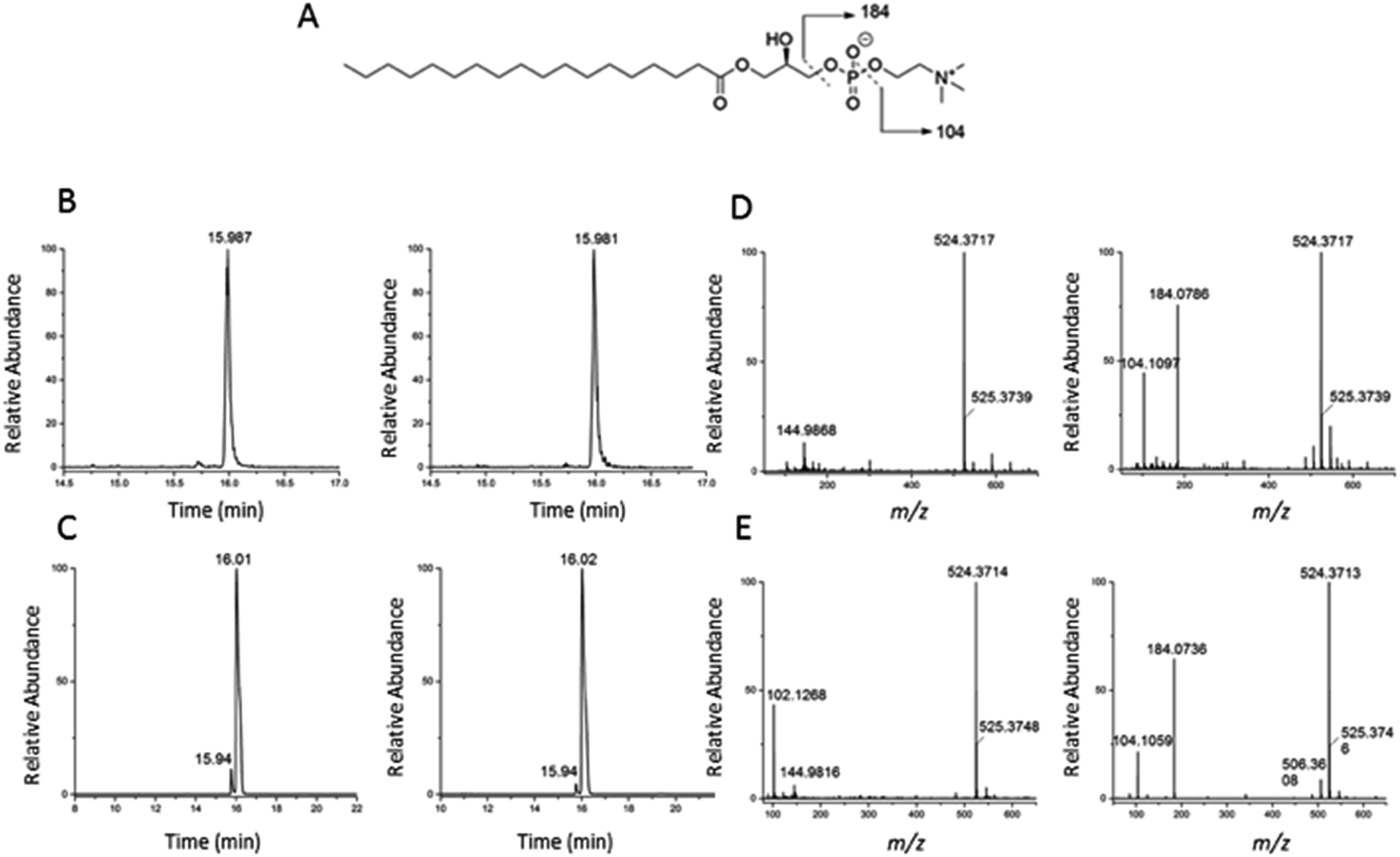 | ||
| Fig. 4 Confirmation of identification of m/z 523.3638 (1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine) described in Fig. 3. (A) Chemical structure; (B) low and high-collision energy XIC for m/z = 523.3638; (C) low and high-collision energy XIC for the standard m/z = 523.3638; (D) low and high-collision energy mass spectrum at TR = 15.98 min; (E) low and high-collision energy mass spectrum of the standard at TR = 15.98 min. | ||
Identification of LMGP01050026 as an enriched glycerophospholipid in autophagosomes is also consistent with other preliminary identifications which included lysophospholipids such as LysoPC (22:5(7Z,10Z,13Z,16Z,19Z)) (HMDB10403), and LysoPC (18:2(9Z,12Z)) (HMDB10386) (Table S2, ESI†). These types of lipids are involved in membrane fusion and elongation in macroautophagy37 and alteration of these lysophospholipids causes disruption of autophagy-related organelle membranes by modifying lipid biosynthesis.38
Beyond LMGP01050026, among the other 19 preliminary identifications that passed Checkpoint 2 (Table S2 and Fig. S7a, ESI†), two had commercially available standards: 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (HMDB10382) and 1-hexadecanoyl-sn-glycero-3-phosphoethanolamine (LMGP02050002). For validation of their respective preliminary identifications we also ran separately the commercial standards and the liver autophagosome-enriched sample spiked with each of standards (Fig. S8 and S9, ESI†). Their parent ion masses, the match between the XICs of the sample/spiked-in standard and the standard alone, match the main peaks in the low and high-collision energy mass spectra of the sample/spiked-in standard and the standard alone, and the fragmentation patterns (Fig. S8 and S9, ESI†) support the identification of HMDB10382 and LMGP02050002 as enriched compounds in the liver autophagosome-enriched sample.
The degree of confidence for preliminary identifications, sans validation with standards, increases when there are reports on the roles that such compounds play in autophagy. Preliminary identification of PE (16:0/0:0) (LMGP02050002) is of interest because it is a member of the phosphatidylethanolamine (PEA) family. PEA is a critical factor in autophagy due to its conjugation with Atg8 in the protein complex required for autophagosome formation.39,40 Sphinganine (HMDB00269), a sphingolipid base, represents another compelling preliminary identification as sphingolipids have previously been shown to stimulate macroautophagy41,42 and accumulate in biological systems such as Niemman Pick C disease that also accumulate autophagosomes.43,44 Autophagy may also play a role in vitamin D regulation, which makes intriguing the preliminary identification of 1α,23-dihydroxy-24,25,26,27-tetranorvitamin D3 (LMST03020020), a vitamin D3 metabolite.45
Preliminary identifications from rat myoblast cell culture
Using the OPLS-DA and LMM, the combined results included 130 selected features (Table S1, ESI†). Confirmation of selected features via evaluation of XICs (Checkpoint 1, Fig. 1) yielded 112 candidate features. Database searches of the candidate features resulted in 69 potential database IDs. Checkpoint 2 scrutinized the online database IDs by manual examination of low- and high-collision energy mass spectra and matching observed fragmentation patterns with simulated fragmentation patterns for the identified compound (determined using Waters MassFragment™), as well as precursor-product XIC alignment. Overall, 8 preliminary identifications passed Checkpoint 2 (Table S1 and Fig. S7b, ESI†). Due to the lack standards when this work was completed, preliminary validations were not further validated.Features from activated mast cells
To explore the use of the workflow on other systems of biological significance, we applied it to the analysis of aqueous secretions of mast cells activated using different treatments (untreated, CXCL10, TNP-ova, CXCL10 + TNP-ova). The combined results from the OPLS-DA and LMM included 2655 selected features because this is a more complex biological system, (Table S1, ESI†). Confirmation of selected features via evaluation of XICs (Checkpoint 1, Fig. 1) yielded 1661 candidate features (63% of selected features). Because the use of Checkpoint 2 to manually scrutinize so many candidate features is not practical, we prioritized and examined 250 and 65 most statistically significant candidate features from the LMM and the OPLS-DA approaches, respectively. From these, 114 had database IDs (36% of the most significant candidate features). After Checkpoint 2, 15 preliminary identifications remained (Table S1, Fig. S7c, ESI†). Due to the lack of commercially available standards when this work was completed, preliminary features were not further validated.Discussion
In this study, we used UPLC/MSe to analyze pairs of related samples and implemented a data processing workflow to compare and select preliminary identifications of small molecules that are different between each sample in a pair (Fig. 1). Two checkpoints based on (1) manual observation of XICs of chemometrically selected features and (2) comparison of fragmentation spectra with in silico fragmentation patterns of database IDs were essential to exclude false positives and provide high-confidence preliminary identifications. Comparison of enriched rat liver autophagosomes (versus liver homogenate), enriched L6 myoblast autophagosomes (versus cell homogenate), and secretions from activation of mast cells (versus non-activated or after a second activation) showed 20, 8, and 15 preliminary identifications, respectively (see examples of structures in Fig. S7† and mass error of preliminary IDs in Table S2, ESI†).We used two of three chemometric/statistical approaches (OPLS-DA, t-test, and LMM) to select unique features (Fig. 1). The OPLS-DA was used in the three studies discussed here. The t-test was useful when only one technical replicate was done (e.g. due to rat liver sample limitations). The LMM was useful when three technical replicates were carried out. Because only 17% of the candidate features were present in the two chemometric/statistical analyses used for each specific study (see % CCF in Table S1, ESI†), the combined outputs of the chemometric/statistical approaches were used for each study. This improved the number of final preliminary IDs (43 in total), in which 23 of them were initially selected by only the OPLS-DA or only the LMM/t-test approach (Table S2, ESI†).
Checkpoint 1 was essential to eliminate selected features with inadequate XICs (false positives). Those that passed checkpoint 1 (candidate features) were 67%, 86% and 63% of selected features in the liver, myoblast, and secretions of activated mast cells, respectively (Table S1, ESI†). Thus, the respective rates of false positives were 33%, 14%, and 37%. We cannot attribute this rate of false positives to S/N issues. With signal threshold of 100 counts and a peak-to-peak baseline noise of 8.0 counts, values for S/N were greater or equal to 12. Indeed, once the candidate features are known, one could retroactively optimize MarkerLynx parameters (e.g. mass tolerance and retention time tolerance) to reduce the initial number of data features until the number of false positives is minimized. Indeed, this optimization would be impractical without the knowledge of true positives that passed Checkpoint 1 in a given sample. Future investigation on the optimization of these parameters may reduce the rate of false positives in a sample-specific manner, thereby reducing the manual effort spent on applying Checkpoint 1.
A total of 46% of the candidate features searched in chemical databases (Chemical Entities of Biological Interest, Human Metabolome Database, Lipid MAPS, and ChemSpider) had a matching ID. The other 54% are considered false negatives because they are likely true compounds that are not represented in the databases searched here. This is in agreement with previous GC- and LC/MS-based metabolomic analyses reporting ∼50%46,47 as the yield for searching databases. To eliminate the possibility of inadvertent elimination of true positives, we conducted the database searches with an ‘inflated’ mass error (i.e. 10 mDa or 11 ppm), which indeed may result in incorrect ID assignments. This is not a major concern here because these are subsequently eliminated in Checkpoint 2 described below. Searching other databases (e.g. MyCompoundID, MZmine, Massbank, METLIN, mz Cloud), may reduce the number of false negatives, but will not eliminate the fact that representation of chemical entities of biological interest is currently a major bottleneck in metabolomics and other unbiased analysis of small molecules in biological systems.29–31
Checkpoint 2 (Fig. 1) was critical to discard Database IDs with theoretical fragmentation patterns that were inconsistent with fragmentation patterns observed in the high-collision energy mass spectra (81% in Table S1, ESI†). Because we conducted searches with an ‘inflated mass tolerance’ (11 ppm), it is not surprising to find incorrect ID assignments in the database searches. Given the final mass error of the preliminary identifications (≤2 ppm, Table S2, ESI†) that passed Checkpoint 2, there is a low probability of false preliminary identifications. One concern though is that of incorrect prediction of fragmentation patterns. Better predictive algorithms for in silico fragmentation would reduce the number of false negatives caused by incorrect fragmentation predictions.48,49 In addition, comparison of predicted and observed isotope patterns could provide complementary scrutiny to in silico fragmentation comparisons currently done at Checkpoint 2.
Validating preliminary identifications via comparison of the UPLC/MSe of their respective commercially available standards in one of the biological systems is the gold standard in metabolomics studies. One such validation in our study was that of 1-octadecanoyl-glycero-3-phosphocholine (LMGP01050026), found in the comparative analysis of enriched autophagosomes and homogenate rat liver. UPLC retention times, XICs of the parent and fragment, and fragmentation patterns are remarkably similar between the sample (Fig. 3) and the data obtained with the standard (Fig. 4). Three other commercially available standards were also used to validate their respective preliminary identifications (Table S1 and Fig. S8–S10†). Although validation results could not be extended to other preliminary identifications, due to the lack of commercial standards, 100% of the preliminary identifications tested against standards were successful, giving credence to the use of Checkpoints 1 and 2 to increase the confidence in the preliminary identifications from comparative analysis of small molecules in biological samples.
The biological context is also critical to support the chemical identity of mass spectral features that were selected as preliminary identifications (Checkpoint 2). For instance, the preliminary identification of 1-octadecanoyl-glycero-3-phosphocholine (LMGP01050026) (Fig. 3) is also biologically validated because of the role that glycerophospholipids play in autophagy.38 Indeed, further biological insight could be gained through parallel studies of diseases associated with autophagy50–54 as well as the composition and origin of autophagosome membranes.55–57 Although not explored here, the biological context gives credence to preliminary identifications in the comparative studies of activated mast cells (see ESI†) highlighting the power of biological context to further support scrutiny through application of Checkpoints 1 and 2 (Fig. 1).
Conclusions
In this study, manual evaluation of XICs and comparison of in silico fragmentation of database IDs reduced the number of false positives and resulted in preliminary identifications with error masses within 2 ppm. Each of the validations of preliminary identifications against commercial standards were successful, increasing confidence in the preliminary identifications. When known, the biological context further increases confidence of the preliminary identifications. The workflow described here could be a benchmark for optimization of initial data processing (i.e. reduce false positives currently detected through XICs), and would be further improved with inclusion of more databases in ID searches, better fragmentation algorithms, and inclusion of isotopic data in the annotation of each feature. We acknowledge that such improvements and the incorporation of emerging data processing resources will still require a great deal of validation, which is possible through workflows such as the one described here. Careful consideration and rigorous validation of data processing resources, prior to their application to biological systems, will help prevent ‘omic's pollution, which can easily proliferate when data are not properly scrutinized.Animal considerations
All rat studies for autophagosome enrichment were conducted under an animal study protocol approved by the Albert Einstein College of Medicine Animal Institute Animal Care and Use Committee. Calorically restricted rats were allowed free access to water. All mice for mast cells were raised and euthanized according to an animal study protocol #0806A37663 approved by the University of Minnesota Institutional Animal Care and Use Committee.Acknowledgements
This work was supported by the Center for Analysis of Biomolecular Signaling (University of Minnesota) and NIH grant AG020866. Individual support was provided to C. P. S. (NIH GM8347), J. S. K. (NIH CA077598), J. A. R. N. (NIH AG031782, Spanish Ministerio de Educacion y Ciencia Fellowship, Revson Foundation Fellowship), T. H. T. and M. M. H. (NIH Chemistry Biology Interface Training Grant GM008700), and C. L. H. (NIH DP2 OD004258-01). Ana Maria Cuervo provided scientific insight in the preparation of autophagosomes.References
- I. D. Wilson, R. Plumb, J. Granger, H. Major, R. Williams and E. A. Lenz, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 817, 67–76 CrossRef CAS PubMed.
- J. M. Castro-Perez, J. Kamphorst, J. DeGroot, F. Lafeber, J. Goshawk, K. Yu, J. P. Shockcor, R. J. Vreeken and T. Hankemeier, J. Proteome Res., 2010, 9, 2377–2389 CrossRef CAS PubMed.
- X. J. Wang, H. Sun, A. H. Zhang, P. Wang and Y. Han, J. Sep. Sci., 2011, 34, 3451–3459 CrossRef CAS PubMed.
- V. B. Ivleva, Y. Q. Yu and M. Gilar, Rapid Commun. Mass Spectrom., 2010, 24, 2631–2640 CrossRef CAS PubMed.
- J. J. Ma, L. P. Kang, W. B. Zhou, H. S. Yu, P. Liu and B. P. Ma, J. Med. Plants Res., 2011, 5, 6152–6159 CAS.
- L. P. Kang, K. T. Yu, Y. Zhao, Y. X. Liu, H. S. Yu, X. Pang, C. Q. Xiong, D. W. Tan, Y. Gao, C. Liu and B. P. Ma, J. Pharm. Biomed. Anal., 2012, 62, 235–249 CrossRef CAS PubMed.
- J. Trygg, E. Holmes and T. Lundstedt, J. Proteome Res., 2006, 6, 469–479 CrossRef PubMed.
- G. Theodoridis, H. G. Gika and I. D. Wilson, Mass Spectrom. Rev., 2011, 30, 884–906 CAS.
- J. Trygg and S. Wold, J. Chemom., 2002, 16, 119–128 CrossRef CAS.
- M. Barker and W. Rayens, J. Chemom., 2003, 17, 166–173 CrossRef CAS.
- M. Bylesjö, M. Rantalainen, O. Cloarec, J. K. Nicholson, E. Holmes and J. Trygg, J. Chemom., 2006, 20, 341–351 CrossRef.
- J. A. Westerhuis, H. C. J. Hoefsloot, S. Smit, D. J. Vis, A. K. Smilde, E. J. J. van Velzen, J. P. M. van Duijnhoven and F. A. van Dorsten, Metabolomics, 2008, 4, 81–89 CrossRef CAS.
- J. Westerhuis, E. J. Velzen, H. J. Hoefsloot and A. Smilde, Metabolomics, 2010, 6, 119–128 CrossRef CAS PubMed.
- G. Verbeke and G. Molenberghs, Linear mixed models for longitudinal data, Springer, 2000 Search PubMed.
- M. Daszykowski and B. Walczak, TrAC, Trends Anal. Chem., 2006, 25, 1081–1096 CrossRef CAS.
- J. Boccard, J. L. Veuthey and S. Rudaz, J. Sep. Sci., 2010, 33, 290–304 CrossRef CAS PubMed.
- D. C. Rubinsztein, M. DiFiglia, N. Heintz, R. A. Nixon, Z.-H. Qin, B. Ravikumar, L. Stefanis and A. Tolkovsky, Autophagy, 2005, 1, 11–22 CrossRef CAS PubMed.
- Z. P. Xie and D. J. Klionsky, Nat. Cell Biol., 2007, 9, 1102–1109 CrossRef CAS PubMed.
- D. C. Rubinsztein, Nature, 2006, 443, 780–786 CrossRef CAS PubMed.
- W. H. Yu, A. M. Cuervo, A. Kumar, C. M. Peterhoff, S. D. Schmidt, J.-H. Lee, P. S. Mohan, M. Mercken, M. R. Farmery, L. O. Tjernberg, Y. Jiang, K. Duff, Y. Uchiyama, J. Näslund, P. M. Mathews, A. M. Cataldo and R. A. Nixon, J. Cell Biol., 2005, 171, 87–98 CrossRef CAS PubMed.
- T. C. Theoharides and D. Kalogeromitros, Ann. N. Y. Acad. Sci., 2006, 1088, 78–99 CrossRef CAS PubMed.
- L. Marzella, J. Ahlberg and H. Glaumann, J. Cell Biol., 1982, 93, 144–154 CrossRef CAS PubMed.
- H. Koga, S. Kaushik and A. M. Cuervo, FASEB J., 2010, 24, 3052–3065 CrossRef CAS PubMed.
- P. O. Seglen and M. F. Brinchmann, Autophagy, 2010, 6, 542–547 CrossRef CAS PubMed.
- P. Masson, A. C. Alves, T. M. D. Ebbels, J. K. Nicholson and E. J. Want, Anal. Chem., 2010, 82, 7779–7786 CrossRef CAS PubMed.
- R. D. C. Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2011.
- Y. Benjamini and D. Yekutieli, Annals of Statistics, 2001, 29, 1165–1188 CrossRef.
- K. A. Azizan, S. N. Baharum, H. W. Ressom and N. M. Noor, Am. J. of Appl. Sci.,, 2012, 9, 1124–1136 CrossRef CAS.
- J. F. Xiao, B. Zhou and H. W. Ressom, TrAC, Trends Anal. Chem., 2012, 32, 1–14 CrossRef CAS PubMed.
- T. Pluskal, S. Castillo, A. Villar-Briones and M. Oresic, BMC Bioinf., 2010, 11, 395 CrossRef PubMed.
- T. Huan, C. Tang, R. Li, Y. Shi, G. Lin and L. Li, Anal. Chem., 2015, 87, 10619–10626 CrossRef CAS PubMed.
- J. Sun, L. Schnackenberg, L. Pence, S. Bhattacharyya, D. Doerge, J. Bowyer and R. Beger, Metabolomics, 2010, 6, 550–563 CrossRef CAS.
- M. Hodson, G. Dear, J. Griffin and J. Haselden, Metabolomics, 2009, 5, 166–182 CrossRef CAS.
- K. Chalcraft, J. Kong, S. Waserman, M. Jordana and B. McCarry, Metabolomics, 2013, 1–9, DOI:10.1007/s11306-013-0589-7.
- H. Pereira, J. F. Martin, C. Joly, J. L. Sebedio and E. Pujos-Guillot, Metabolomics, 2010, 6, 207–218 CrossRef CAS.
- O. Fiehn, D. Robertson, J. Griffin, M. Werf, B. Nikolau, N. Morrison, L. Sumner, R. Goodacre, N. Hardy, C. Taylor, J. Fostel, B. Kristal, R. Kaddurah-Daouk, P. Mendes, B. Ommen, J. Lindon and S.-A. Sansone, Metabolomics, 2007, 3, 175–178 CrossRef CAS.
- C. Dall'Armi, A. Hurtado-Lorenzo, H. Tian, E. Morel, A. Nezu, R. B. Chan, W. H. Yu, K. S. Robinson, O. Yeku, S. A. Small, K. Duff, M. A. Frohman, M. R. Wenk, A. Yamamoto and G. Di Paolo, Nat. Commun., 2010, 1, 142 CrossRef PubMed.
- R. F. S. Menna-Barreto, K. Salomao, A. P. Dantas, R. M. Santa-Rita, M. J. Soares, H. S. Barbosa and S. L. de Castro, Micron, 2009, 40, 157–168 CrossRef CAS PubMed.
- Y. Ichimura, T. Kirisako, T. Takao, Y. Satomi, Y. Shimonishi, N. Ishihara, N. Mizushima, I. Tanida, E. Kominami, M. Ohsumi, T. Noda and Y. Ohsumi, Nature, 2000, 408, 488–492 CrossRef CAS PubMed.
- R. Nebauer, S. Rosenberger and G. Daum, J. Biol. Chem., 2007, 282, 16736–16743 CrossRef CAS PubMed.
- F. Scarlatti, C. Bauvy, A. Ventruti, G. Sala, F. Cluzeaud, A. Vandewalle, R. Ghidoni and P. Codogno, J. Biol. Chem., 2004, 279, 18384–18391 CrossRef CAS PubMed.
- W. Zheng, J. Kollmeyer, H. Symolon, A. Momin, E. Munter, E. Wang, S. Kelly, J. C. Allegood, Y. Liu, Q. Peng, H. Ramaraju, M. C. Sullards, M. Cabot and A. H. Merrill Jr, Biochim. Biophys. Acta, 2006, 1758, 1864–1884 CrossRef CAS PubMed.
- C. D. Pacheco and A. P. Lieberman, Expert Rev. Mol. Med., 2008, 10, e26 CrossRef PubMed.
- M. M. Young, M. Kester and H.-G. Wang, J. Lipid Res., 2013, 54, 5–19 CrossRef CAS PubMed.
- M. Høyer-Hansen, S. P. S. Nordbrandt and M. Jäättelä, Trends Mol. Med., 2010, 16, 295–302 CrossRef PubMed.
- S. Krueger, P. Giavalisco, L. Krall, M. C. Steinhauser, D. Bussis, B. Usadel, U. I. Flugge, A. R. Fernie, L. Willmitzer and D. Steinhauser, PLoS One, 2011, 6, 1–16 Search PubMed.
- N. Benkeblia, T. Shinano and M. Osaki, Metabolomics, 2007, 3, 297–305 CrossRef CAS.
- S. Wolf, S. Schmidt, M. Muller-Hannemann and S. Neumann, BMC Bioinf., 2010, 11, 148 CrossRef PubMed.
- M. Sugimoto, M. Kawakami, M. Robert, T. Soga and M. Tomita, Curr. Bioinf., 2012, 7, 96–108 CrossRef CAS PubMed.
- A. M. Cuervo, Trends Cell Biol., 2004, 14, 70–77 CrossRef PubMed.
- A. M. Cuervo, E. Bergamini, U. T. Brunk, W. Dröge, M. Ffrench and A. Terman, Autophagy, 2005, 1, 131–140 CrossRef PubMed.
- M. Martinez-Vicente and A. Cuervo, Lancet Neurol., 2007, 6, 352 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069–1075 CrossRef CAS PubMed.
- A. M. Cuervo, Autophagy, 2009, 5, 904–905 Search PubMed.
- A. Longatti and S. A. Tooze, Cell Death Differ., 2009, 16, 956–965 CrossRef CAS PubMed.
- A. M. Cuervo, Nat. Cell Biol., 2010, 12, 735–737 CrossRef CAS PubMed.
- S. A. Tooze and T. Yoshimori, Nat. Cell Biol., 2010, 12, 831–835 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary table, supplementary data. See DOI: 10.1039/c6ay02500e |
| This journal is © The Royal Society of Chemistry 2017 |