
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive multiphase NMR applied to a living organism†
Yalda Liaghati
Mobarhan
a,
Blythe
Fortier-McGill
a,
Ronald
Soong
a,
Werner E.
Maas
b,
Michael
Fey
b,
Martine
Monette
c,
Henry J.
Stronks
c,
Sebastian
Schmidt
d,
Hermann
Heumann
d,
Warren
Norwood
e and
André J.
Simpson
*a
aDepartment of Physical and Environmental Science, University of Toronto, 1265 Military Trail, Toronto, ON, Canada M1C 1A4. E-mail: andre.simpson@utoronto.ca
bBruker BioSpin Corp., 15 Fortune Drive, Billerica, Massachusetts, USA 01821-3991
cBruker BioSpin Canada, 555 Steeles Avenue East, Milton, ON, Canada L9T 1Y6
dSilantes GmbH, Gollierstr. 70 C, 80339 München, Germany
eEnvironment Canada, 867 Lakeshore Rd., Burlington, ON, Canada L7R 4A6
First published on 18th April 2016
Abstract
Comprehensive multiphase (CMP) NMR is a novel technology that integrates all the hardware from solution-, gel- and solid-state into a single NMR probe, permitting all phases to be studied in intact samples. Here comprehensive multiphase (CMP) NMR is used to study all components in a living organism for the first time. This work describes 4 new scientific accomplishments summarized as: (1) CMP-NMR is applied to a living animal, (2) an effective method to deliver oxygen to the organisms is described which permits longer studies essential for in-depth NMR analysis in general, (3) a range of spectral editing approaches are applied to fully differentiate the various phases solutions (metabolites) through to solids (shell) (4) 13C isotopic labelling and multidimensional NMR are combined to provide detailed assignment of metabolites and structural components in vivo. While not explicitly studied here the multiphase capabilities of the technique offer future possibilities to study kinetic transfer between phases (e.g. nutrient assimilation, contaminant sequestration), molecular binding at interfaces (e.g. drug or contaminant binding) and bonding across and between phases (e.g. muscle to bone) in vivo. Future work will need to focus on decreasing the spinning speed to reduce organism stress during analysis.
Introduction
Living organisms are defined as systems where metabolism is identifiable. A wide range of environmental factors including physical (temperature, light), chemical (nutrients, drugs, contaminants) and biological (pathogens) can impact metabolism and lead to subsequent disease or change in physiology.1 Living organisms encompass a range of materials from soluble metabolites at one extreme, through gel-like components (e.g. proteins, muscle, membranes) to true solids (bone, shells) at the other. Changes in shell thickness and structure in the organism can result from environmental and contaminant stress, similarly in human, changes in bone function develop conditions such as osteoporosis2 and Paget's disease.3 The overall integrating theme is that living systems are multifarious and it is the delicate synergism between physical organization, chemical reactivity, and biological processes that give rise to life. As such, to truly understand biological function and response, analytical tools that can comprehensively study all components within living organisms at the molecular level resolution are desperately required.Solution-state NMR and MRI have traditionally been used to study living organisms. To date MRI has been arguably the most powerful tool for the study of living systems. Routine MRI studies map water concentrations giving rise to images from which critical diagnoses can be made.4 Advanced studies even permit localized spectroscopy which can identify dissolved metabolites. However, there are numerous limitations to MRI based methods which include: (1) only true dissolved molecules can be observed; (2) magnetic susceptibilities lead to broad spectra making identification and quantification challenging; (3) the larger sample volume in MRI systems results in less homogeneous magnetic fields; (4) a lack of lock circuity reduces stability overtime (broad lines); (5) limited spectrometer channels and multinuclear capabilities impede the collection of multinuclear correlation spectroscopy critical for providing spectral dispersion and molecular assignment in complex samples. One simple option is therefore to study living organisms with high resolution solution-state NMR spectrometers. Indeed such studies are highly informative, however, as with MRI, information can only be extracted from the truly dissolved components.5 When considering molecular processes (for example, incorporation of dissolved nutrients into solid bone, the crystallization of soluble amyloids to form crystalline fibrils in Alzheimer's, Huntington's and Parkinson's disease) or bioaccumulation of dissolved contaminant or drug within the tissue, the ability to monitor the conversion of one phase into another or the transport across different phase boundaries is essential.
The use of magic angle spinning (MAS) NMR is needed to average chemical shift anisotropy and reduce magnetic susceptibilities in swollen samples permitting both solution-state and swellable (dynamic gels) materials to be investigated. Pioneering studies have been performed showing that organisms can be kept alive under MAS conditions for short periods and high resolution spectral information can be extracted.6,7 However, all studies to date have utilized High Resolution Magic Angle Spinning probes (HR-MAS) which can only handle low power radio frequency (RF) fields and thus cannot be used to study true solids or rigid gels which require high power decoupling and cross polarization for detection.8
In this study comprehensive multiphase NMR (CMP-NMR) spectroscopy, introduced in 2012, is applied for the first time to a living system. CMP-NMR probes contain a lock, pulse field gradients, high power RF circuitry and are fully susceptibility matched.8 The result is that all components in all phases can be studied and differentiated in vivo and the full range of solution-state, gel-state and solid-state NMR experiments applied without compromise. To demonstrate the proof of principle in this study the organism Hyalella azteca (fresh water shrimp) is investigated. One of critically important applications of in vivo NMR is the application to understand environmental toxicity and stress.9 Traditionally toxicity has been routinely assessed using growth, reproduction rates or mortality as endpoints. However, over time it has been recognized that this approach alone is insufficient and more information regarding toxic mechanisms and the biochemical pathways perturbed is critically needed to explain, how and why specific chemicals are toxic. This need is summarized in a report “Toxicity Testing in the 21st Century” by the National Academy of Sciences (commissioned by the Environmental Protection Agency) which concludes “The new paradigm should facilitate evaluating the susceptibility of different life-stages, understanding the mechanisms by which toxicity occurs, and considering the risks of concurrent, cumulative exposure to multiple and diverse chemicals”.10,11
An in vivo approach with the ability to study all phases would be capable of directly evaluating the mode-of-action, bioaccumulation, biotransformation, molecular reactivity, excretion and binding in vivo in response to the organisms surroundings. If specific biological responses can be correlated to certain toxins, then it may eventually be possible to interpret what stressors are truly problematic in a contaminated environment by interpreting the biological fingerprints of the native organisms.
H. azteca is considered one of the most sensitive organisms to its environmental conditions and is frequently used and recommended for aquatic and sediment toxicity testing by environmental organizations.10,11 The organism is used here as a model organism to demonstrate the application of CMP-NMR to study of living organisms in general.
Results and discussion
Conditions for survival
To permit a wide range of NMR experiments required to extract detailed metabolic and structural information, it is imperative that the physical conditions required for in vivo magic angle spinning be optimized. The most obvious source of stress is the spinning itself. Fig. 1A depicts the survival rate at various spinning speeds. Numerous repetitions demonstrate that 2.5 kHz is the highest speed that can be used without affecting survival and to ensure the 1H spinning sidebands are outside the spectral region using a 500 MHz NMR spectrometer. In future, while considerable additional research is required to reduce spinning sidebands and suppress water, it should be possible to combine slow magic angle spinning approaches such as PHORMAT,12 which have been shown effective at spinning speeds as low as 1 Hz and for animals as large as rats to be studied. While successful studies so far have identified a handful of metabolites12 a full multiphase approach could provide information on all components within the organisms and low spinning speed should eliminate spinning stress. In this proof of principle study due to complications of sideband and water suppression at low speeds 2.5 kHz spinning was selected as compromise between NMR spectral information and organism stress.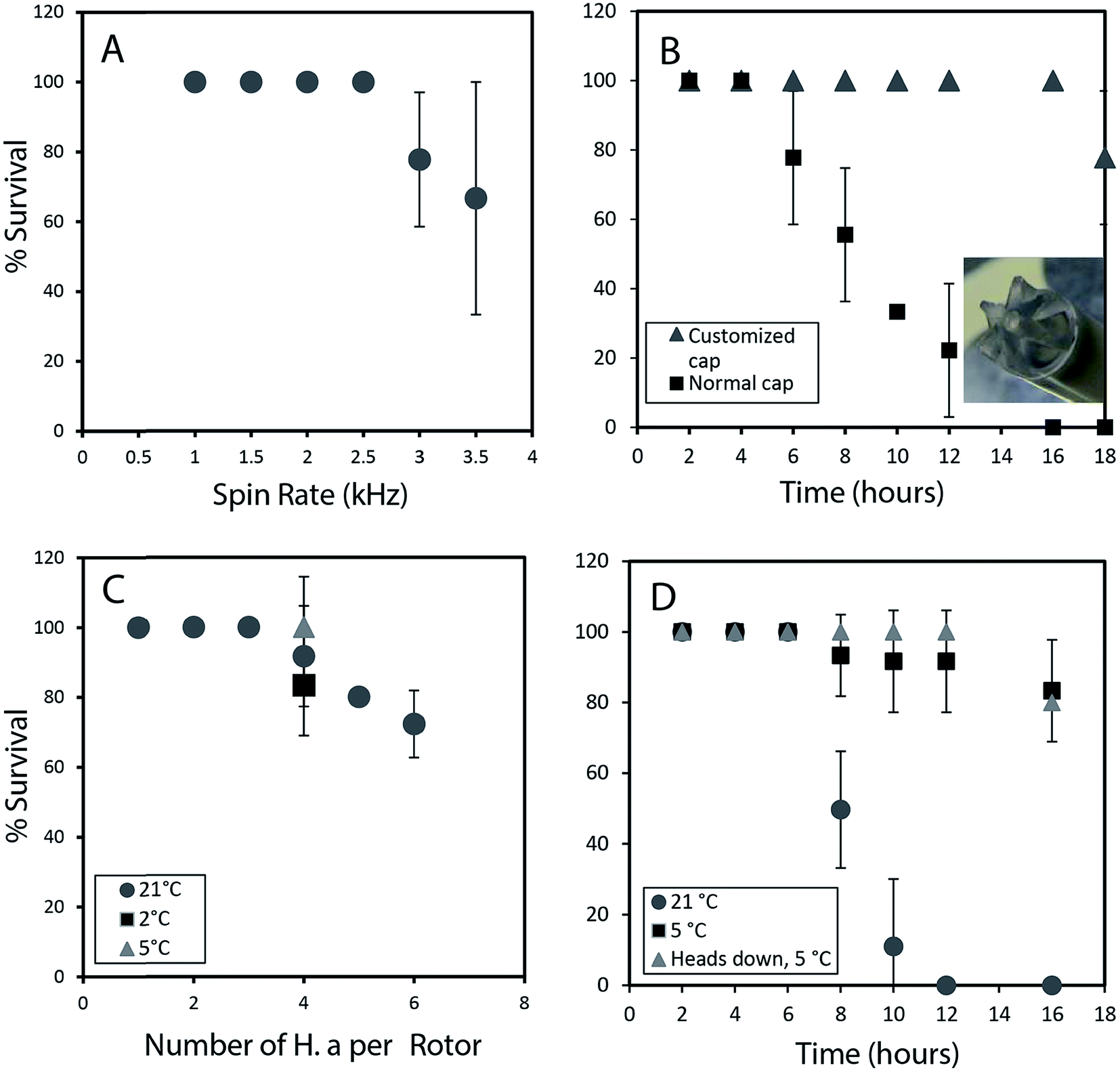 | ||
| Fig. 1 (A) Survival rate in percentage is plotted against several different spin rates (1 h of spinning). (B) The influence of oxygen delivery using a customized cap on improving survival rates is depicted. (C) The effect of loading different number of organisms on the survival is studied. At 5 °C, with lower oxygen consumption survival rates increase to almost 100%. (D) Survival rate up to 16 hours is monitored at different temperatures. When the organism are loaded head first into the rotor there is 100% survival up to 12 h. All experiments were performed in replicates of 3. All studies were performed using the NMR probe external to magnet controlled by a separate variable temperature unit. This permitted use of the NMR spectrometer while these spinning tests were ongoing. | ||
In addition to spinning, oxygen availability was found to be extremely important to the longevity of the organisms. In a conventional NMR rotor the sample is sealed air-tight to prevent the water from leaking. While a wide range of approaches were tested to permit oxygen exchange, the simplest solution was to drill a central hole in the rotor cap and proved to be most effective (inset Fig. 1B). Conventional logic would suggest drilling a hole in the cap would lead to water leakage, however, it was found that while spinning, water pushes against the walls of the seal and a tiny vortex permits air exchange in the center of the rotor. Fig. 1B shows that the use of a modified cap significantly improved the survival rate. Readers should note that Fig. 1B was collected without spinning such that the influence of spinning (Fig. 1A) and oxygen availability (Fig. 1B) can be easily discerned.
The number of organisms per rotor was found to be very important. Tests were carried out with 2–6 week old organisms, and results revealed that H. azteca survive the spinning rate at any age. However, packing more than three organisms result in higher mortality (Fig. 1C). The tests were performed using the modified cap and at 2.5 kHz spinning for 1 hour. In a previous in vivo study under MAS, anaesthetization was used to improve the recovery rate of the organisms. However, the use of anaesthetic is not ideal as it alters metabolisms and introduces strong signals into the NMR spectra masking key information. As an alternative, here the experiments were performed at a lower temperature. Fig. 1C shows that reducing the temperature to 5 °C is beneficial with a 100% survival rate at 2.5 kHz at this temperature observed (Fig. 1C). To further investigate the effects of temperature organisms were spun at three different temperatures (Fig. 1D) and their survival was monitored overtime. Organisms at 5 °C had a much higher survival rate, in part due to their reduced heart rate and oxygen consumption at lower temperature.13 Ultimately, it was found that for the majority of cases spinning at 2.5 kHz for up to 12 hours at 5 °C were good experimental conditions yet often one organism out of 4 would die. Further investigations demonstrated this was caused by evaporation of water through the aeration hole which is accelerated with spinning. The organism closest to the cap would die while organisms lower in the rotor survived. The simplest solution is to use two medium-large organisms or one very large organism and place them with their head down towards the bottom of the rotor.
Throughout this study however, NMR data for single adult shrimp is reported. Fig. S.3† compares the NMR profile for 3 replicates of a single large shrimp (to demonstrate the reproducibility of the approach) with the profile of a single rotor of 3 smaller shrimp for comparison. In general it was found that smaller shrimp would move in an out of the coil while a single larger shrimp always remained within the coil region throughout. In addition, larger numbers of shrimp resulted in a lactate signal appearing in the NMR as a result of anaerobic stress5 as-well as alanine, a general stress indicator.14 The lactate arises likely due to the higher oxygen consumption from a larger number of organisms, also with more organisms packed into the rotor the organisms on top may be forced up towards the water surface. As such we recommend a single large shrimp be used and placed head down such that the gills remain submerged even if some evaporation occurs.
Fig. 1D shows 100% survival at 2.5 kHz for more than 12 hours at 5 °C. Using larger specimens prevents them from turning around within the rotor allowing their gills to always be submerged. The fact the organisms with their gills submerged survive longer demonstrates the limiting factor (at 2.5 kHz spinning, 5 °C) preventing longer experiments is not the NMR experiments or spinning itself but loss of water due to evaporation from the existing hole in the cap. Future work focusing on air permeable, water impermeable membranes may be able to solve this. ESI (Video 1†) shows the H. azteca used to collect the vast majority of data used in the paper. Video 2† shows the H. azteca after nearly 14 hours of spinning at 2.5 kHz and 5 °C using the modified cap, 3 weeks following the end of experiments.
Basic NMR data
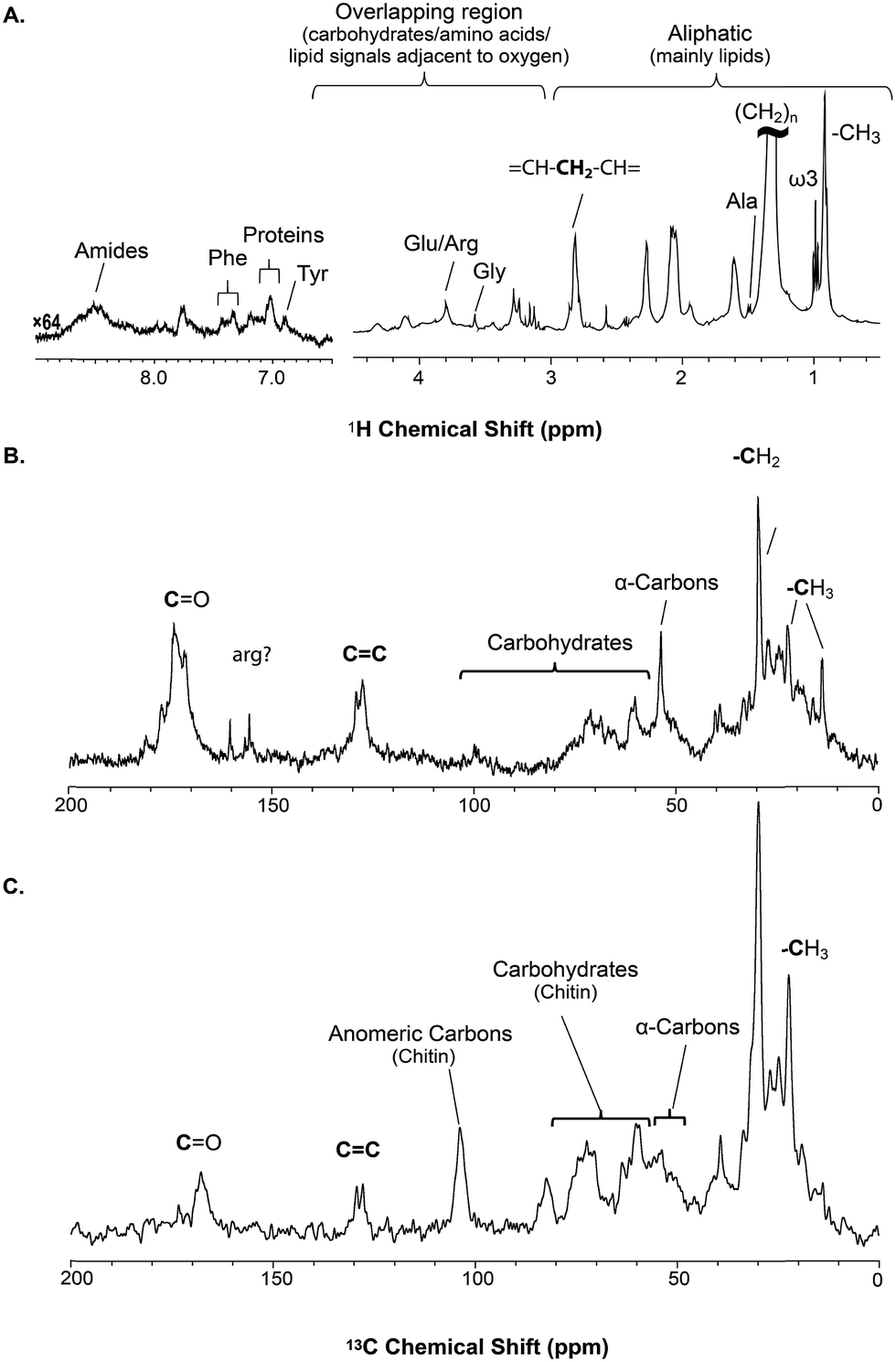 | ||
| Fig. 2 A series of 1D direct NMR experiments are performed, each spectra provides complementary information; (A) 1H NMR represents an overview of dynamic components (solutions and gels) in H. azteca with overlapping resonances dominated by lipids (B) 13C NMR collected with low power 1H and provides an overview as to all the carbon in the organism with the true solids suppressed (C) 1H-13C CP (cross polarization) highlights the solid components in the organisms. | ||
The solid component, more detailed assignments, and full spectral editing approaches (to separate the solution, gel, semi-solids and solids phase) will be discussed later. The spectrum here was collected using 4096 scans taking 2 h 47 min. Fig. S.1† compares the signal obtained in 256 scans (10.5 min). It is clear that in the aliphatic region the vast majority of information is retained with only 256 scans, but unfortunately in the aromatic region the signal-to-noise is too low for in-depth analysis. As such if relatively concentrated and resolved resonances in the 1H spectrum are of interest then collecting real time (every ∼10 min) data should be possible. This however becomes more challenging for metabolites signals buried under the lipid profile which are discussed further in the section “Inverse Diffusion Editing (IDE)”.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and (CH2)n resonances. In addition, contributions from carbohydrates and protein (α-carbons and broad aliphatic distribution 10–40 ppm) are apparent.
C and (CH2)n resonances. In addition, contributions from carbohydrates and protein (α-carbons and broad aliphatic distribution 10–40 ppm) are apparent.
The 1H-13C CP-MAS of the shrimp (Fig. 2C) shows strong contributions from lipids, and the carbohydrate chitin (the major component of shrimps shell). These components are further discussed later. When considered holistically, the 1H, 13C and CP-MAS NMR provide an overview of all the components inside the living H. azteca from soluble metabolites to the most rigid solids. However, due to spectral overlap detailed molecular information is challenging to extract. Hence, a series of spectral editing approaches can be extremely useful, in both reducing overlap and providing complimentary information, by highlighting the dissolved, gel, semi-solid, and solid sub-components within the sample.
Spectral editing
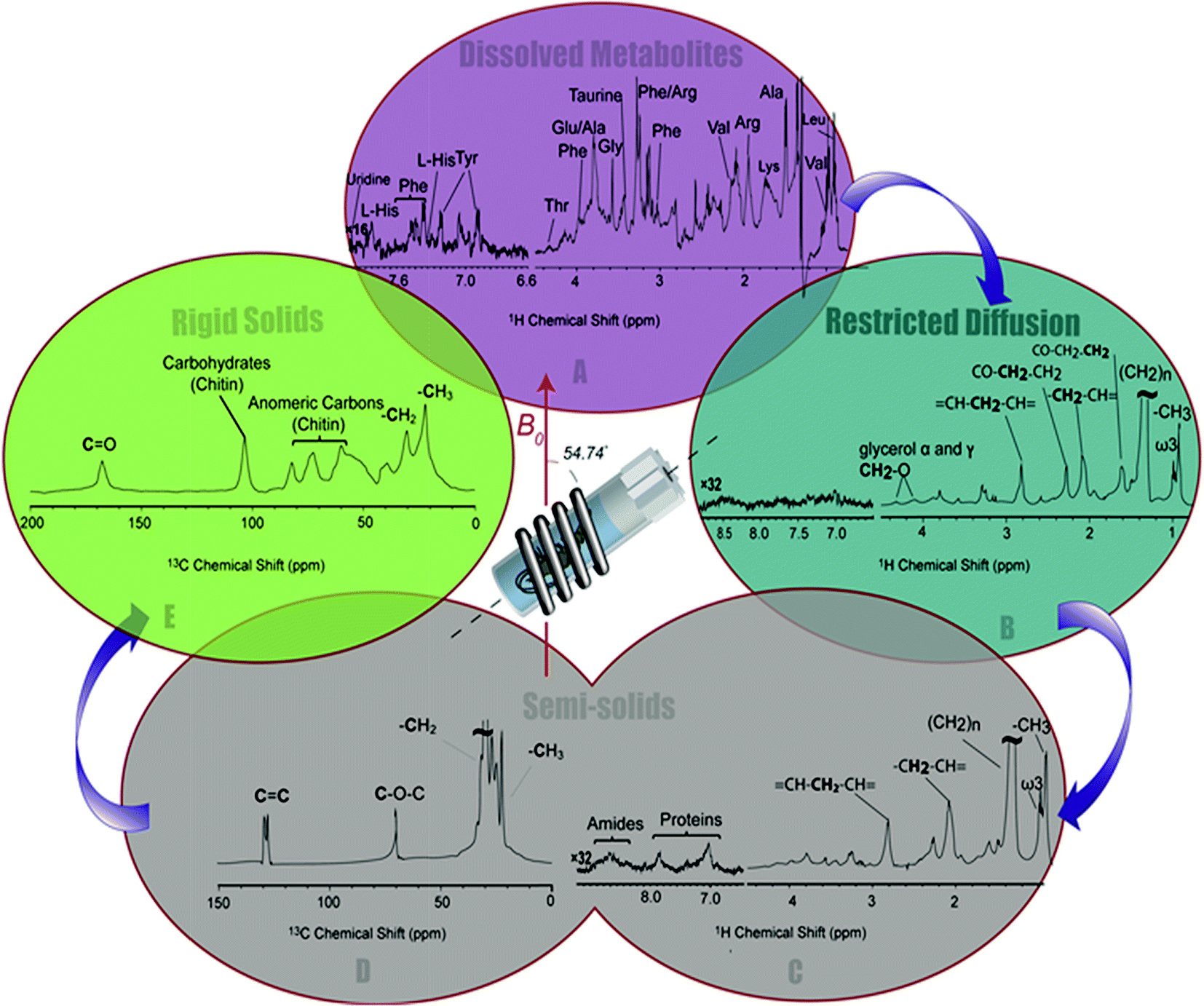 | ||
| Fig. 3 A series of spectral editing approaches applied to differentiate the overlapping resonances are presented. (A) The dissolved metabolites in 1H NMR spectra of H. azteca are observed in Inversed Diffusion Editing (IDE), (B) gel like metabolites are emphasized in the Diffusion Edited experiments (DE), (C) resonances from the semi-solid components are observed in the 1H RADE NMR spectrum, “proteins” refers to aromatic amino acids in macromolecular structures, (D) the semi-solid components are highlighted in 13C (CP-T2) and (E) the most rigid components are selected in the inverse CP-T2 experiment. | ||
2D NMR identification of metabolites
2D NMR provides both additional molecular connectivity information (critical for structural assignment) and affords increased spectral dispersion (reducing spectral overlap). Heteronuclear Single-Quantum Coherence (HSQC) correlates directly bonded 1H and 13C units and can, in simple terms, be thought of as a high resolution (theoretical peak capacity as a measure of resolution per unit is reported as ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 for HSQC23) fingerprint of the H–C framework in a complex mixture. 1H-1H correlation spectroscopy identifies protons on adjacent carbons. When combined, HSQC identifies the H–C fragments and COSY identifies how these units connect to form a molecule.
000000 for HSQC23) fingerprint of the H–C framework in a complex mixture. 1H-1H correlation spectroscopy identifies protons on adjacent carbons. When combined, HSQC identifies the H–C fragments and COSY identifies how these units connect to form a molecule.
Fig. 4A shows a HSQC spectrum of the H. azteca with major regions labelled; aromatics, nucleic acids, carbohydrates, peptides/proteins, lipids and various other aliphatic units. Exact assignments can be made by matching both HSQC and COSY data against bio-reference NMR databases. Approximately 40 metabolites including a range of amino acids, carbohydrates, nucleotides, and other bioactive compounds were identified (Fig. 2B–D and ESI Fig. S.5† for assignment confirmation). The potential role of these compounds as stress biomarkers is addressed bellow. The identification of additional metabolites is challenging, in part due to spectral overlap arising from the complexity of the whole organisms and in part due to limitations of currently available NMR databases. To our knowledge the Bruker Bioreference Databases are one of the most comprehensive metabolites databases commercially available. Still, they only contain 1D and 2D NMR information for ∼650 compounds and no metabolites specific to H. azteca. However, a few years ago, there were no commercially NMR databases available and with the rapid evolution of NMR based metabolomics the development of novel databases and open source repositories such as; http://mmcd.nmrfam.wisc.edu/ are starting to evolve. With this in mind, careful inspection of the HSQC shows a truly in vivo system, this is actually highly encouraging and exemplifies the wealth of information that eventually will be accessible from MAS in vivo spectroscopy as assignment resources improve. Further experiments that should facilitate more detailed assignments are considered in ESI.†
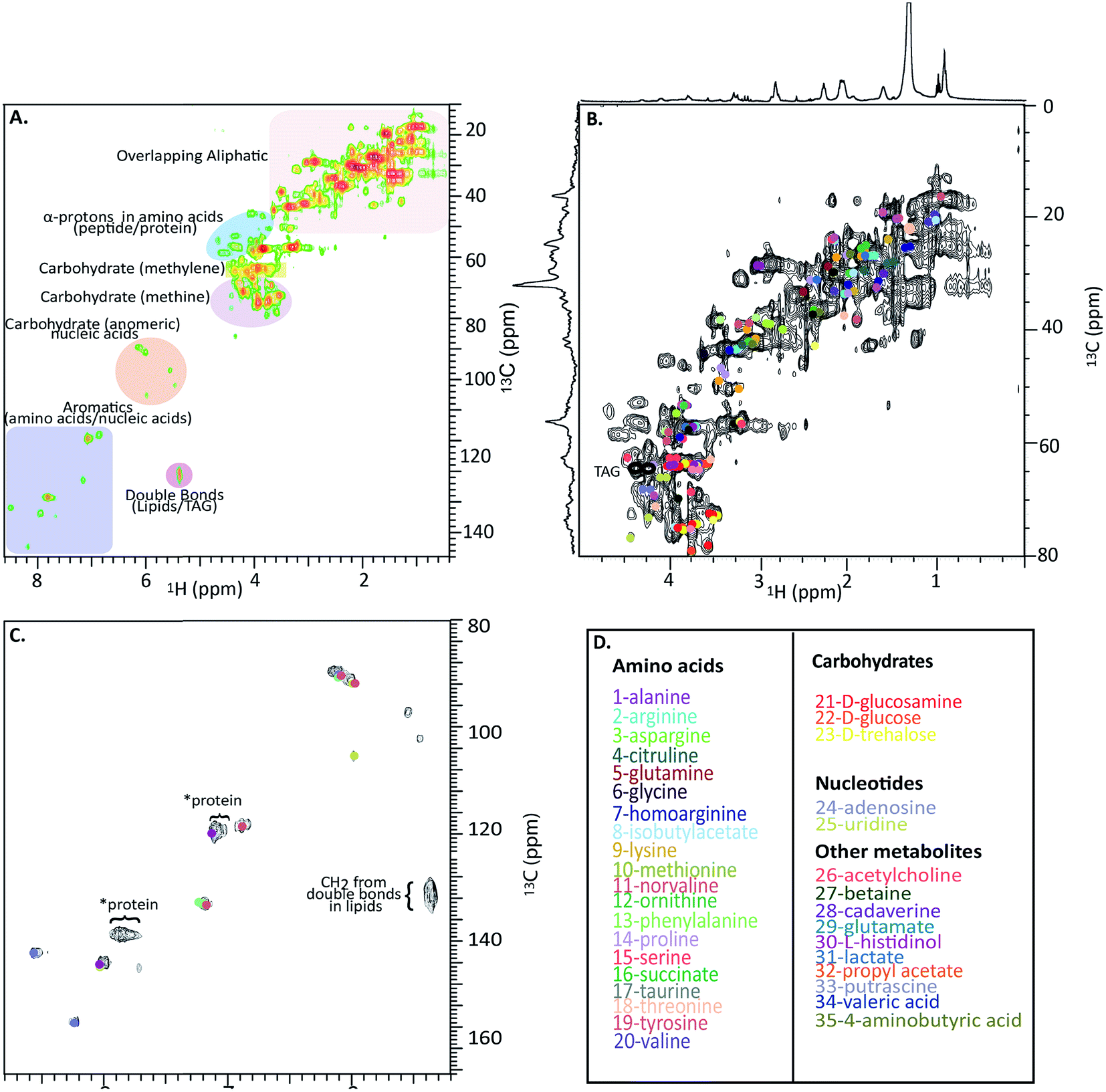 | ||
| Fig. 4 In vivo 1H-13C HSQC of 13C enriched H. azteca. (A) HSQC with ∼40 metabolites assigned using Bruker's Bio-reference databases. (B) Aliphatic region (C) aromatic region (D) color coded assignments corresponding to the dots in B and C. *These signal dominate the semi-solids spectrum (Fig. 3D) and are consistent with aromatic residues in proteins. | ||
Biological significance
Nearly 40 metabolites were identified in the current study, the most noteworthy of which are discussed briefly here. Close to 20 free amino acids, such as leucine, isoleucine, valine, lysine, and other essential amino acids observed in previous metabolomic studies, appear in the IDE and are confirmed by HSQC.24 Other identified metabolites include, lipids, choline, nucleosides, carbohydrates, osmolytes as-well as, structural components such as chitin. For a complete list of the identified metabolites see Fig. 4D where the biological significance of these identified components is discussed next.Although all amino acids are the basic building blocks of proteins, most amino acids also play major roles in multiple bio-molecular events. The three branched amino acids (leucine, isoleucine and valine) for example are crucial in muscular protein and neurotransmitter synthesis through regulating the mRNA translation.25 They can also potentially act as novel biomarkers that may aid in understanding cardiometabolic health.26 Corresponding resonances from free tyrosine and phenylalanine are evident in the expanded aromatic region, these two amino acids act as main precursors in many biological processes such as dopamine biosynthesis, which is an intercellular transmitter in multicellular organisms and nervous systems in larger animals.
Aspartic acid, glutamic acid and alanine are found largely as free amino acids mainly in the axoplasm the nervous systems of marine crustacean and account for 20% of the dry weight of the nerves.27 Free taurine and glutamic acid may serve as receptors in crustacean antennae and aid in locating the prey in the depth of the ocean or lakes in benthic organisms.28 Taurine, glycine, proline, glutamic and alanine may likely be important substances for the regulation of osmotic pressure in the crustaceans muscles.29 Other free amino acids are being targeted as potential biomarkers and differentiators of cancers cells30 and other disease in general.
The diffusion edited spectra identify an abundance of lipid reserves which were dominated by triacylglycerides (TAG) as major energy storage in most plants and animals.31,32 The ability to monitor the lipid composition is very important as they are intimately tied to energy metabolism a process commonly perturbed by a wide range of contaminants and other external stressors.33 Lipids also serve as a source of essential biomolecules such as hormones. Many invertebrates such as D. magna are of particular interest as they cannot synthesize lipids de novo and therefore rely on food sources for these molecules.34 However, molecular understanding of the process is not well understood and being able to monitor the consumption of food (algae) in vivo or examine the transfer of carbon between trophic levels represents a key tool to better understand the ecology and chemistry of food webs. Particularly interesting are the resonances corresponding to ω3 fatty acids in the diffusion edited data, which are an important component of cell membranes as well as precursors to many other substances such as the hormones central to reproduction in many organisms. In addition, the ω3 content of the marine organisms is a “quality indicator” in the food industry, since they are the primary source of ω3 for human consumption and cannot be biosynthesized in mammals.35
The diamines putrescine (1,4-diaminobutane), cadaverine (1,5-diaminopentane)36 produced by the deamination of lysine or ornithine37 are found in high abundance in all major groups of marine organisms such as invertebrates. They are naturally occurring polyamines, which regulate the concentrations of the cations in macromolecular structure of DNA and RNA or their transport through cellular membranes.
The nucleosides adenosine and uridine, represent important biomarkers related to DNA/RNA damage, which have been implicated to be an indication of carcinogenesis. DNA/RNA monitoring can act as a measure of hereditary risk of cancer or many other diseases.38 Choline is another identified metabolite that appears in the head groups of some phospholipids. It is also a precursor needed to form acetylcholine which is a neurotransmitter that controls memory and muscle movements. Therefore, choline plays major part in many nervous system related disorders.39 Also, fluctuation in choline level is a well-established biomarker for different cancer types.30
Chitin is the main structural polysaccharide that forms the protective exoskeleton in all arthropods, which makes up to 60% of their mass and serves as an important structural component in their body. It is periodically shed and causes their growth in several stages of molting.40,41 Several steps take place to convert trehalose to chitin in which mainly glucose, glutamine and uridine triphosphate (UTP) serve as precursors of its synthesis. The presence of most of the precursors and products through these stages are identified in the 2D spectra. The ability to study soluble precursors, through conversion into a gel and finally a solid shell are only possible using a CMP-NMR probe where all phases can be studied and differentiated in situ. The approach opens up exciting possibilities to follow process such as bone formation. Indeed not just the formation but the degradation of the solid materials could be an important complimentary source of information42 itself key to stress induced thinning in bird egg shells and shells of marine animals, as well as the degradation of bone with age.
Towards in vivo organism monitoring using CMP-NMR
The major drawback of in vivo CMP-NMR is the potential stress from the spinning itself. Fig. S.2† compares the 1H NMR over a 20 hour period of continuous spinning at 2.5 kHz. In this case a large single shrimp was used and the organism was still alive after 20 hours of spinning. The main 1H NMR profile remains consistent. The main difference is the appearance of alanine, which has been reported as a general indicator of stress.14 The lack of lactate (an indicator of anaerobic stress) observed when numerous organisms are placed in a rotor (see Fig. S.3D†), suggests that spinning rate (rather than lack of oxygen) likely plays a role in the alanine production. While it can be argued that moving forward, spinning stress could be reduced (e.g. lower spinning rates) eliminating it completely may never be possible. As such it is important to discuss potential ways in which this stress could be accounted and the best way to incorporate CMP-NMR into an in vivo monitoring program. This can be achieved through using CMP-NMR as a stand-alone tool or as part of a larger in vivo NMR framework, the approaches will be discussed briefly and separately.Using CMP-NMR as a part of a larger in vivo NMR framework
Recently our group has demonstrated that flow based in vivo NMR is an excellent low-stress approach to understand metabolic change.5 Organisms are placed in a NMR flow tube (which is kept static) and oxygenated water (with or without food) is continually recirculated. Contaminants, stressors or nutrients can be easily introduced into the flow making it ideal for monitoring metabolic response to stress. The drawback however is the low resolution from the 1H NMR spectra. While this can be overcome via spectral dispersion afforded by 1H-13C HSQC, information is still only obtained for the truly dissolved metabolites. Conversely, in CMP-NMR the NMR profile is sharpened through MAS and information on the gels and solids can also be obtained. CMP-NMR would be best employed at time point when flow-NMR identifies responses and the additional capabilities of CMP-NMR used to provide additional resolution to aid with metabolite assignment as-well as provide information on gels and solids only detectable by CMP-NMR. In this way CMP-NMR plays more of a supporting role in terms of metabolic change, and a primary tool for observing structural change. This approach leverages the low stress attributes of flow NMR to monitor metabolic change while benefitting from CMP-NMR for assignment. In such a combined approach as conclusions regarding stress fluxes are drawn from the low stress technique, additional stress from the spinning in CMP-NMR becomes less critical.CMP-NMR as a stand-alone tool
While the combined approach would be recommended for large studies aimed at truly targeting the metabolic stress, there are situations where the approach may not be feasible or available. In such a scenario spinning stress would have to be differentiated from impacts of exposure (contaminant, drug etc.) via controls. The controls should be identical to the dosed organisms with the exception of the addition of the stressor. In its simplest form differences after chemometrics analysis between the control and dosed organisms, spun for the same amount of time, should arise solely from the stressor as stress arising from spinning should cancel. Such studies should be carefully controlled in terms of spinning time and if multiple NMR experiments are performed they should be run in the same order and started at the same time after spin initiation on both the dosed and control subjects such that spinning effects cancel. As the stress from spinning likely manifests over time (evidenced by the increase in alanine in Fig. S.2†) more advanced studies could use time based chemometric trajectory analysis to further differentiate metabolites co-involved in the separate processes.43 Of particular interest may be to introduce the chemical stressor midway the analysis such that trajectory from spinning remains linear throughout while the influence of the stressor impacts only the later datasets.44 Currently our group is working toward protocols for using CMP-NMR effectively to understand stress processes. Given the unique and comprehensive in vivo information that CMP-NMR can provide over more traditional NMR and other non-NMR based methods stress from spinning is an unavoidable side-effect that must be accounted for.Experimental
Results (Fig. 1) suggest that H. azteca spun at a reasonably slow rate 2.5 kHz are limited by the oxygen content in approximately 80 μL of water in the rotor, but are not limited by the spinning itself. To minimize this oxidative stress a hole is drilled into the cap. Rotor caps with o-ring seals were purchased from Wilmad Glass and drilled with a 0.022′′ inch drill bit to produce a hole allowing for air exchange thereby extending the survival of H. azteca under MAS conditions. Viability studies were performed in triplicate and the percentage survival rate along with the standard deviation were reported (see Fig. 1).The original Hyallela azteca culture was provided by Environment Canada from their main colony. The organisms were then cultured within the University of Toronto laboratory and living conditions for the specimen were controlled according to methodologies by Environment Canada10 and kept similar to their natural habitat in fresh water. They were kept in 20 L tanks with a 2 cm layer of sand in dechlorinated, aged tap water, continuously aerated as their medium. 20% of the overlaying water was changed 3 times a week before feeding time. The tanks were exposed to 16:8 h light to dark photoperiod using a fluorescence commercial lamp and the temperature was kept at constant 24 °C. Carbonate hardness of 124 mg CaCO3 L1 (consistent with local freshwater conditions). 13C isotopically labelled Chlamydomonas reinhardtii were cultivated in a small scale closed loop system. A custom photobioreactor built by Silantes GmbH was used for production of algae biomass. Each fermentation has been conducted autotrophically and exclusively with 13CO2 (98% enriched with stable isotope 13C). The cultivation parameters including media, temperature, light intensity, and pH were adjusted to gain a maximum growth rate. The harvested algal biomass was fed at a rate of 2 mg per H. azteca, 3 times a week over their life span and represents their sole carbon source. For more details regarding the culturing of the isotopically enriched algae please refer to ESI.†
A 4 mm zirconium rotor was filled with water. For offline viability studies (Fig. 1) 1-6 H. azteca were used. The H. azteca were selected based on size with <2 mm representing smaller shrimp, 2–5 mm medium shrimp and >5 mm large shrimp. Size was gauged using a microscope and males were selected for analysis. Different sexes can be recognized by the absence of an enlarged gnathopod or presence of egg case in females. For our experimental purpose adult males were selected due to large body size as well as to avoid the inconsistencies due to additional lipid storage within the eggs in the females. For all the NMR data reported here (with the exception of Fig. S.3D†) a single large shrimp (length ∼ 7 mm) was used and placed in the rotor with its gills towards the bottom. H. azteca were loaded from the droplet at the tip of a plastic pipette and let swim into the rotor (refer to Video S.1 in the ESI†). A trace of D2O (∼3 μL) was added to act as a spectrometer lock. The rotor was sealed using a top Kel-F cap with an o-ring seal customized with a hole as described above. All NMR spectra were acquired using a Bruker Avance 500 MHz Bruker Avance III Spectrometer at a spinning rate of 2.5 kHz fitted with a prototype CMP MAS 4 mm 1H-13C-2H probe with an actively shielded magic angle gradient (Bruker BioSpin). All experiment were performed at 5 °C and locked on D2O, sample temperature was not seen to increase more than 1 °C during any experiments. The lock was maintained for all experiments including solids experiments. After each run the organism was monitored for 1 week before being returned to the main colony. Only NMR data from organisms that survived and fully recovered were used in this paper.
Additional information including total experiment time, T1 values and delays used are provided in Table S.1.†1H NMR SPR-W5 WATERGATE pulse sequence was used for water suppression15,45 with garp-4 decoupling to remove 13C splitting. The shaped pre-saturation prior to W5 was achieved using a train of selective pulses: 1000, 2 ms, calibrated rectangular pulses were used, each separated by a 4 μs delay. 256–4096 scans (see Table S.1†) were collected with 8192 time domain points, 20 ppm sweep width, a 125 μs binomial delay and a recycle delay 5 × T1. Spectra were processed using an exponential function corresponding to a line broadening of 2 Hz in the transformed spectrum and a filling factor of 2. Carbon spectra were obtained using spectral width of 400 ppm, 16384 time domain points, 5000 scans inverse gated decoupling (Waltz-16) and a recycle delay 5 × T1. Spectra were processed using an exponential function corresponding to a line broadening of 5 Hz in the transformed spectrum and a filling factor of 2.
1H diffusion based editing was performed with a bipolar pulse pair longitudinal encode-decode (BPPLED) sequence.19 Scans were collected using an encoding/decoding gradients of 1.8 ms at ∼50 gauss per cm (65 gauss per cm max for current probe) and a diffusion time of 180 ms. Inverse diffusion editing (IDE), relaxation Recovery Arising from Diffusion Editing (RADE) and inverse T2 filtered 13C CP/MAS were done by appropriate spectral subtraction as previously described.8 Briefly, the spectra are scaled until the spectra being subtracted was nulled with the resulting difference spectra containing positive peaks.
CP/MAS was performed using linear ramp defined by 1000 points from 80–100% during a contact time of 2 ms, high power composite pulse decoupling (Spinal 64) and Total Suppression of Sidebands (TOSS).16 Decoupling was applied using an RF field of 50 kHz, improvements were not seen beyond this as such the RF field strength was applied at 50 kHz to prevent additional sample heating. 30720 scans were collected using 1024 points and a recycle delay of ∼5 × T1 (1H macromolecules, see Table S.1†). Spectra were processed using an exponential function corresponding to a line broadening of 25 Hz in the transformed spectrum and a filling factor of 2. For the selection of semi-solids (CP-T2) a train of 2 × 7.5 μs CPMG echoes were applied to the 1H spins prior to contact.8 The inverse CP-T2 (solids) was created by difference. Singular-value decomposition46 was applied to CP-T2 and ΔCP to reduce spectral noise (an example of which is shown in ESI Fig. S.4†).
1H-13C HSQC (Heteronuclear Single-Quantum Coherence) spectra were collected in phase sensitive mode using Echo/Antiecho-TPPI gradient selection, with 600 transient for each of the 128 increments in the F1 dimension. 2048 time domain points were collected in F2 and a 1J 1H-13C of 145 Hz. F2 was processed using an exponential function corresponding to line broadening of 15 Hz and F1 using sine-squared functions with a π/2 phase shift. 2D COSY (correlation spectroscopy) spectrum was acquired to confirm HSQC assignments of metabolites. The COSY experiments were collected using an in-phase approach,47 using gradients for coherence selection and low power 13C garp decoupling throughout. 128 scans and 2048 data points were collected for each of the 196 increments in the F1. Both dimensions were processed using sine-squared functions shifted by 90°, zero filling factor of 2. Compound identification and quantification were performed using AMIX (Analysis of MIXtures software package, version 3.9.15, Bruker BioSpin, in combination with the Bruker Bioreference NMR databases, versions 2-0-0 to 2-0-4) as previously reported.48 Only assignments with an R2 correlation >0.99 between the observed and databases shifts were retained. Where possible correlations were also confirmed with COSY.
Conclusions
In this study the main objective was to demonstrate the feasibility and applicability of CMP-NMR to a whole living organism, while retaining the biological function. The study demonstrates all structural components can be identified, from metabolites to true solids. The ability to monitor potential real-time molecular fluxes and relate this directly to biological response, activity or behavior provides an essential link to better understand biological processes and ultimately the biochemistry and chemistry that define these events. The potential of the current work goes way beyond the freshwater shrimp studied here and could theoretically be applied to a wide range of small organisms, complete tissue sections (including bone), and to follow almost any biological process, including, growth, reproduction, disease, stress, and others. To our knowledge, this approach arguably provides the greatest amount of molecular information in vivo compared to any modern scientific tool, and the versatility to extract information from all components, solutions, gels and solids, is quite profound. Of course the approach does not come without limitations, some of which are discussed and addressed in ESI† section.In summary, this study demonstrates it is possible to study a whole living organism by CMP-NMR. The unique capabilities afford the possibilities to study and differentiate all phases (solutions, gels, semi-solids, true solids) in vivo. Theoretically, this provides the potential to study all organic components within a living organism, providing an unprecedented window into biological processes and stress responses. Considering the huge potential along with the unique and unprecedented molecular information the approach affords, it is clear in vivo CMP-NMR will have an important role to play in many areas of research. The technique acts as the ideal “molecular interpreter” providing the desperately needed connection between the physical (for example environmental stress and disease) to the chemistry that ultimately defines these processes.
Acknowledgements
A. J. S. thanks NSERC, (Strategic and Discovery Programs), the Canada Foundation for Innovation (CFI), and the Ministry of Research and Innovation (MRI) and Krembil Foundation for providing funding. A. J. S. also thanks the Government of Ontario for an Early Researcher Award.References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, fifth edn, 2008 Search PubMed.
- G. Walker, The British Journal of Chiropractic, 1999, 3, 95 CrossRef.
- M. P. Whyte, N. Engl. J. Med., 2006, 355, 593–600 CrossRef CAS PubMed.
- J. S. Lewin, J Magn. Reson. Im., 2003, 17, 383 CrossRef.
- R. Soong, E. Nagato, A. Sutrisno, B. Fortier-McGill, M. Akhter, S. Schmidt, H. Heumann and A. J. Simpson, Magn. Reson. Chem., 2015, 53, 774–779 CrossRef CAS PubMed.
- R. A. de Graaf, in In Vivo NMR Spectroscopy, John Wiley & Sons, Ltd, 2007, pp. 43–110 Search PubMed.
- D. Bon, V. Gilard, S. Massou, G. Pérès, M. Malet-Martino, R. Martino and F. Desmoulin, Biol. Fertil. Soils, 2006, 43, 191–198 CrossRef CAS.
- D. Courtier-Murias, H. Farooq, H. Masoom, A. Botana, R. Soong, J. G. Longstaffe, M. J. Simpson, W. E. Maas, M. Fey, B. Andrew, J. Struppe, H. Hutchins, S. Krishnamurthy, R. Kumar, M. Monette, H. J. Stronks, A. Hume and A. J. Simpson, J. Magn. Reson., 2012, 217, 61–76 CrossRef CAS PubMed.
- A. J. Simpson, Y. Liaghati, B. Fortier-McGill, R. Soong and M. Akhter, Magn. Reson. Chem., 2015, 53, 686–690 CrossRef CAS PubMed.
- Biological Test Method: Test for Survival and Growth in Sediment and Water Using the Freshwater Amphipod Hyalella azteca Environment Canada (EC), 2013.
- D. Krewski, D. Acosta, M. Andersen, H. Anderson, J. C. Bailar, K. Boekelheide, R. Brent, G. Charnley, V. G. Cheung, S. Green, K. T. Kelsey, N. I. Kerkvliet, A. A. Li, L. McCray, O. Meyer, R. D. Patterson, W. Pennie, R. A. Scala, G. M. Solomon, M. Stephens, J. Yager and L. Zeise, Toxicol. Env. Heal. B , 2010, 13, 51–138 CrossRef CAS PubMed.
- R. A. Wind, J. Z. Hu and D. N. Rommereim, Magn. Reson. Med., 2003, 50, 1113–1119 CrossRef PubMed.
- K. Halcrow and C. M. Boyd, Comp. Biochem. Physiol., 1967, 23, 233–242 CAS.
- H. Hummel, C. Amiard-Triquet, G. Bachelet, M. Desprez, J. Marchand, B. Sylvand, J. C. Amiard, H. Rybarczyk, R. H. Bogaards, J. Sinke and L. de Wolf, Sci. Total Environ., 1996, 188, 233–241 CrossRef CAS.
- B. Lam and A. J. Simpson, Analyst, 2008, 133, 263–269 RSC.
- W. T. Dixon, J. Schaefer, M. D. Sefcik, E. O. Stejskal and R. A. McKay, J. Magn. Reson., 1969, 1982(49), 341–345 Search PubMed.
- S. Patel and S. Ahmed, J. Pharm. Biomed. Anal., 2015, 107, 63–74 CrossRef CAS PubMed.
- C. Wu, H. K. Kim, G. P. van Wezel and Y. H. Choi, Drug Discovery Today: Technol., 2015, 13, 11–17 CrossRef PubMed.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- J. D. O'Connor and L. I. Gilbert, Am. Zool., 1968, 8, 529–539 CrossRef.
- A. Bunescu, J. Garric, B. Vollat, E. Canet-Soulas, D. Graveron-Demilly and F. Fauvelle, Mol. BioSyst., 2010, 6, 121–125 RSC.
- F. Desmoulin, D. Bon, R. Martino and M. Malet-Martino, C. R. Chim., 2008, 11, 423–433 CrossRef CAS.
- N. Hertkorn, C. Ruecker, M. Meringer, R. Gugisch, M. Frommberger, E. M. Perdue, M. Witt and P. Schmitt-Kopplin, Anal. Bioanal. Chem., 2007, 389, 1311–1327 CrossRef CAS PubMed.
- B. P. Lankadurai, E. G. Nagato and M. J. Simpson, Environ. Rev., 2013, 21, 180–205 CrossRef CAS.
- A. Tom and K. S. Nair, J. Nutr., 2006, 136, 324S–330S CAS.
- B. C. Batch, S. H. Shah, C. B. Newgard, C. B. Turer, C. Haynes, J. R. Bain, M. Muehlbauer, M. J. Patel, R. D. Stevens, L. J. Appel, L. K. Newby and L. P. Svetkey, Metabolism, 2013, 62, 961–969 CrossRef CAS PubMed.
- P. R. Lewis, Biochem. J., 1952, 52, 330–338 CrossRef CAS PubMed.
- Z. M. Fuzessery and J. J. Childress, Biol. Bull., 1975, 149, 522–538 CrossRef CAS.
- Y. Itoh, A. Oguro, K. Nakamura, Y. Ishizuka and K. Yamane, Biosci., Biotechnol., Biochem., 1992, 56, 867–871 CrossRef CAS PubMed.
- B. Sitter, T. F. Bathen, M.-B. Tessem and I. S. Gribbestad, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 239–254 CrossRef CAS.
- A. C. Giese, Lipids in the economy of marine invertebrates, 1966 Search PubMed.
- J. G. Bell, C. Ghioni and J. R. Sargent, Aquaculture, 1994, 128, 301–313 CrossRef CAS.
- B. K. Dutra, R. B. Santos, A. A. P. Bueno and G. T. Oliveira, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2008, 151, 322–328 CrossRef CAS PubMed.
- D. Martin-Creuzburg, E. von Elert and K. H. Hoffmann, Limnol. Oceanogr., 2008, 53, 456–468 CrossRef.
- M. T. Arts, R. G. Ackman and B. J. Holub, Can. J. Fish. Aquat. Sci., 2001, 58, 122–137 CrossRef CAS.
- E. C. C. Silva, D. C. Masui, R. P. M. Furriel, F. L. M. Mantelatto, J. C. McNamara, H. Barrabin, F. A. Leone, H. M. Scofano and C. F. L. Fontes, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2008, 149, 622–629 CrossRef CAS PubMed.
- M. G. Höfle, Appl. Environ. Microbiol., 1984, 47, 843–849 Search PubMed.
- K. Broedbaek, H. E. Poulsen, A. Weimann, G. D. Kom, E. Schwedhelm, P. Nielsen and R. H. Böger, Free Radicals Biol. Med., 2009, 47, 1230–1233 CrossRef CAS PubMed.
- B. L. Miller, L. Changl, R. Booth, T. Ernst, M. Cornford, D. Nikas, D. McBride and D. J. Jenden, Life Sci., 1996, 58, 1929–1935 CrossRef CAS PubMed.
- P.-Y. Chen, A. Y.-M. Lin, J. McKittrick and M. A. Meyers, Acta Biomater., 2008, 4, 587–596 CrossRef PubMed.
- D. Doucet and A. Retnakaran, Adv. Insect Physiol., 2012, 43, 437 Search PubMed.
- F. Gagné, C. Blaise and J. Pellerin, Environ. Res., 2005, 98, 89–99 CrossRef PubMed.
- J. Teul, A. Garcia, J. Tuñón, J. L. Martin-Ventura, N. Tarín, L. L. Bescós, J. Egido, C. Barbas and F. J. Rupérez, J. Pharm. Biomed. Anal., 2011, 56, 343–351 CrossRef CAS PubMed.
- H. C. Keun, T. M. D. Ebbels, M. E. Bollard, O. Beckonert, H. Antti, E. Holmes, J. C. Lindon and J. K. Nicholson, Chem. Res. Toxicol., 2004, 17, 579–587 CrossRef CAS PubMed.
- B. G. Pautler, A. J. Simpson, M. J. Simpson, L.-H. Tseng, M. Spraul, A. Dubnick, M. J. Sharp and S. J. Fitzsimons, Environ. Sci. Technol., 2011, 45, 4710–4717 CrossRef CAS PubMed.
- P. P. Man, C. Bonhomme and F. Babonneau, Solid State Nucl. Magn. Reson., 2014, 61–62, 28–34 CrossRef CAS PubMed.
- Y. Xia, G. Legge, K.-Y. Jun, Y. Qi, H. Lee and X. Gao, Magn. Reson. Chem., 2005, 43, 372–379 CrossRef CAS PubMed.
- G. C. Woods, M. J. Simpson, P. J. Koerner, A. Napoli and A. J. Simpson, Environ. Sci. Technol., 2011, 45, 3880–3886 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00329j |
| This journal is © The Royal Society of Chemistry 2016 |