
Understanding undesirable anode lithium plating issues in lithium-ion batteries
Qianqian Liu
,
Chunyu Du
*,
Bin Shen,
Pengjian Zuo,
Xinqun Cheng,
Yulin Ma,
Geping Yin and
Yunzhi Gao
MIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: cydu@hit.edu.cn; Fax: +86-451-86418616; Tel: +86-451-86403961
First published on 1st September 2016
Abstract
Lithium-ion batteries (LIBs) are attractive candidates as power sources for various applications, such as electric vehicles and large-scale energy storage devices. However, safety and life issues are still great challenges for the practical applications of LIBs. Metallic lithium plating on the negative electrode under critical charging conditions accelerates performance degradation and poses safety hazards for LIBs. Therefore, anode lithium plating in LIBs has recently drawn increased attention. This article reviews the recent research and progress regarding anode lithium plating of LIBs. Firstly, the adverse effects of anode lithium plating on the electrochemical performance of LIBs are presented. Various in situ and ex situ techniques for characterizing and detecting anode lithium plating are then summarized. Also, this review discusses the influencing factors that induce anode lithium plating and approaches to mitigating or preventing anode lithium plating. Finally, remaining challenges and future developments related to anode lithium plating are proposed in the conclusion.
 Qianqian Liu | Qianqian Liu received her Bachelor's degree from Harbin Institute of Technology, China in 2012. Currently, she is pursuing her PhD degree at Harbin Institute of Technology. Her main research focus is on capacity fading of graphite anodes in Li ion batteries, especially undesired lithium plating on graphite, and ways to mitigate/suppress anode lithium plating. |
 Chunyu Du | Dr. Chunyu Du is a Professor at the School of Chemistry and Chemical Engineering at Harbin Institute of Technology, China. He has engaged in research in the field of Li ion batteries and fuel cells for more than ten years and has published about 80 research papers in refereed journals. His main research interests include the development of novel electrode materials for high energy density batteries and the degradation mechanisms of lithium ion batteries. |
1. Introduction
Lithium ion batteries (LIBs) have been widely used as energy storage devices for portable applications such as cell phones, laptops and digital electronics; this is due to their unique advantages, such as high energy and power density, low memory effect and environmental friendliness.1–3 Recently, the application of LIBs has also been extended to the fields of large-scale energy storage and electric vehicles (EVs) to replace conventional energy sources such as fossil fuels. Unfortunately, to become commercially viable and competitive, LIBs still face some critical technological challenges, including short cycling life and poor safety, especially for EV and large-scale energy storage applications.4The cycling life and safety of LIBs depend on many complex factors, including their material properties, manufacturing techniques, battery designs, and practical operating conditions.5–8 Among these, metallic lithium plating (or deposition) on the anode, usually in the form of dendrites or mosses, is one of the major causes of aging and safety accidents of LIBs.5,9 Under normal charging conditions, Li+ ions shuttle from the cathode to the anode and intercalate quickly into the anode active material (most commonly graphite), which does not induce anode lithium plating. However, Li plating is kinetically favorable, as the working potential of graphite is very close to that of metallic Li deposition. The propensity towards anode Li plating in LIBs has a close relationship not only with the charging conditions, such as low temperature,10,11 high charging rate and overcharging,12,13 but also with aspects of the battery design, such as low anode/cathode ratio and manufacturing defects.14 These conditions result in high anode polarization and force the anode potential to the threshold of metallic Li plating, thus leading to anode lithium plating. Actually, in addition to graphite, some alternative anode candidates, such as Si or Sn materials, also have working potentials close to that of Li metal and are also likely to encounter the difficulty of anode Li plating.15,16
The deposited Li metal easily reacts with the electrolyte, which, on the one hand, consumes active lithium and electrolyte, and on the other hand, causes the loss of electrical contact of some deposited Li with the anode (referred to as dead lithium), thus accelerating capacity fading.17 Moreover, the reaction between Li metal and electrolyte forms a redundant interfacial film which increases the anode polarization and, in turn, promotes further anode Li plating. More importantly, the continuous growth of dendritic Li may pierce the separator and induce an internal short circuit, which can result in thermal runway and safety accidents of LIBs.18,19
Therefore, in order to improve the cycling life and safety of LIBs, it is very urgent and imperative to prevent anode lithium plating, especially the formation of dendritic Li. The importance of anode Li plating has triggered extensive investigations, including analysis of the mechanism and conditions, developing effective characterization methods, and designing strategies for restraint of anode Li plating. For example, suitable charging protocols, battery designs and modifications of the anode and electrolyte have been suggested to prevent anode lithium plating.20–22 However, anode Li plating is not completely understood or suppressed, due to its complex mechanisms. Thus, it is of significance to summarize recent progress to better understand and suppress anode Li plating.
In this review, recent investigations into anode lithium plating in the literature are systematically reviewed. We firstly describe the electrochemical or chemical reactions associated with anode lithium plating and its adverse effects on the electrochemical performance of LIBs. The in situ and ex situ methods of detecting anode lithium plating are then presented. After that, the causes of anode lithium plating are analyzed and approaches to preventing anode Li plating are discussed. Finally, we conclude with recent developments and suggest further studies of anode lithium plating in LIBs.
2. Reactions associated with anode lithium plating
When anode lithium plating occurs in the charging process, two processes (Li interaction into active materials and anode Li plating) coexist, shown as follows:23| xLi+ + LiδC6 + xe− → Liδ+xC6 | (1) |
| yLi+ + ye− → yLi | (2) |
Under these conditions, the total charging current is divided into intercalation current and Li plating current. With continuing charging, due to the decreasing vacancy sites for Li intercalation and the limited solid-state diffusion in graphite, the charge current for Li intercalation is gradually reduced. Simultaneously, the current for Li plating is increased because the transport rate of Li+ from electrolyte exceeds the Li-intercalation rate, which induces the accumulation of more Li+ on the surface and drives the anode potential to below 0 V (Fig. 1a). As a consequence, anode lithium plating becomes more severe with charging time.
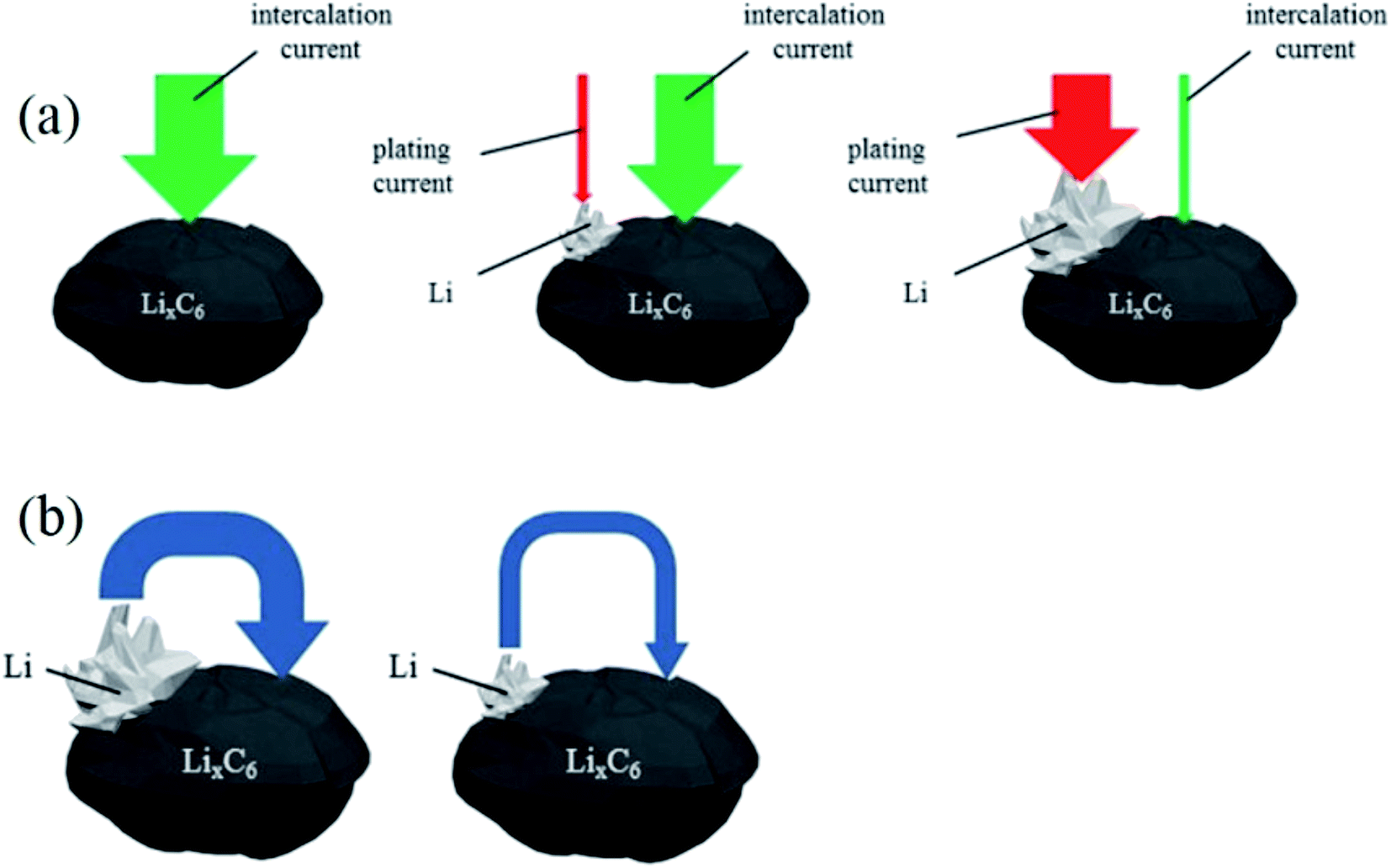 | ||
| Fig. 1 Schematic of (a) the intercalation and plating currents during charging to higher states of charge, and (b) the re-intercalation of deposited lithium during rest (adapted from ref. 23). | ||
Regardless of any external current, when anode lithium plating occurs, a portion of the deposited lithium may undergo two subsequent reactions:23 reinsertion into the graphite anode (shown in Fig. 1b) during the tapering charging or resting time (eqn (3)), and reaction with the electrolyte (eqn (4)):
| εLi + Liδ+xC6 → Liδ+x+εC6 | (3) |
| R + Li → R–Li (R represents electrolyte) | (4) |
During discharge, two reactions can successively proceed at the anode:
| Li → Li+ + e− | (5) |
| Liδ+x+εC6 → 6C + (δ + x + ε)Li+ + (δ + x + ε)e− | (6) |
Fig. 1b clearly shows the reinsertion current of deposited Li during relaxation or tapering charging. When lithium deposits on the graphite surface, a potential difference exists between the deposited Li and the LixC6 in the graphite bulk. The potential difference pushes the deposited Li to reinsert into the graphite, given sufficient time, until the potential difference or deposited active Li disappears. The reinsertion current depends on the amount of deposited Li, which means the greater the amount of deposited Li, the more reinsertion occurs during relaxation. The re-intercalation (eqn (3)) and Li stripping (eqn (5)) reactions of deposited lithium do not cause any capacity loss; this is called reversible deposited lithium. However, the reaction of deposited lithium with the electrolyte forms surface films and results in permanent capacity loss.
3. Adverse effects of anode lithium plating
The adverse effects of anode lithium plating on LIBs mainly involve rapid performance degradation and safety issues. When anode lithium plating occurs during abnormal charging, the deposited metallic lithium reacts spontaneously with the electrolyte until an intact passivation layer forms, i.e., solid electrolyte interphase (SEI) film, because metallic lithium is thermodynamically active in polar aprotic solvents.24 The reaction between metallic lithium and the electrolyte consumes the active lithium and electrolyte, which will induce capacity degradation and poor coulombic efficiency. Simultaneously, the formed SEI film increases the internal resistance of LIBs, causing larger polarization during the intercalation/deintercalation of Li ions.25 After anode lithium plating, partial deposited lithium, especially dendritic lithium, may lose electrical contact with the anode and may even form floating fragments or structures in the electrolyte (known as dead lithium) during the subsequent discharge. Both dead lithium and the formation of SEI film will result in permanent loss of active lithium and are responsible for the fast capacity degradation during anode lithium plating. It should be noted that anode lithium plating is a self-accelerating process, since the reaction of metallic Li with electrolyte leads to an increase in internal resistance due to thickening of the SEI film and drying out of the electrolyte.17,26,27 The increased internal resistance enhances the likelihood of further anode lithium plating during the subsequent charging; this was confirmed by Petzl et al.,28 who demonstrated that the rate of battery aging was accelerated when anode lithium plating occurred. It has already been proved that the dramatically increased overall impedance induced by continuous growth of SEI film will even result in ultimate failure of metal lithium electrodes during high current density cycling;29 this may also be the case for LIBs that are subject to anode lithium plating.The safety issues induced by anode lithium plating include internal short circuits caused by the dendritic lithium30,31 and thermal runaway due to the intense exothermic reaction of deposited lithium with the electrolyte. The deposited lithium is often present in the form of dendrites or needles which are very similar to the dendrites of the metal Li electrode; therefore, the dendrites in LIBs and lithium metal batteries have analogous detrimental effects on cell safety.32,33 The formation of dendritic lithium is closely related to the charge current, SEI film and concentration of Li+ ions on the anode surface. According to Chazalviel's model,34 the formation and growth of Li dendrites occur where the Li+ concentration is close to zero (Li+ depletion zone). High charging current leads to rapid Li+ depletion at the anode and thus induces the formation of Li dendrites.35,36 Also, the local non-uniformity of the electrode/electrolyte interface,37 which results in a large concentration variation of Li+ ions, causes the formation of lithium dendrites. The continuous growth of dendritic or needle-like lithium is prone to penetrate the separator and cause internal short circuits in LIBs and even cell explosion. Pinholes were observed on the separator after dissembling a LIB overcharged to 150% state of charge (SOC); this was believed to be triggered by the growth of lithium dendrites.25
Another safety problem is that the deposited lithium, even when not in dendritic form, accelerates the thermal runaway of LIBs.38,39 The rapid exothermic reaction between deposited lithium with electrolyte will produce significant heat, increasing the cell temperature and further causing fire or explosion of LIBs.40 When 18650-type cells with different SOCs were subjected to thermal runaway testing, the onset exothermic temperature of these cells overcharged to Li plating decreased dramatically from 140 °C to as low as 65 °C due to the exothermic reaction between the deposited lithium and the electrolyte.41 Based on the accelerating rate calorimeter (ARC) and thermal analysis combined with mass spectrometry, Fleischhammer et al. found that the graphite anode with lithium plating generated more heat and strongly reduced the safety of LIBs.42 Yuan et al.43 studied the overcharge failure of a 32 A h battery and found that massive anode lithium plating was the main reason for the increase of internal temperature (more than 200 °C) when the battery was overcharged to 180% SOC.
4. Detection of anode lithium plating
Since anode lithium plating is one of the major reasons for aging and safety issues, which deteriorate the reliability and durability of LIBs, it should be avoided or at least easily detected by effective methods. Tremendous efforts have been devoted to characterizing or detecting anode lithium plating during charging in order to gain deeper understanding and further avoid anode lithium plating. These detection techniques are roughly divided into electrochemical and physical categories.4.1. Electrochemical techniques
Based on the reaction between metal lithium and the electrolyte and the difference in electrochemical behaviors of deposited lithium and intercalated carbon (LixC6), various electrochemical techniques have been employed to detect anode lithium plating in LIBs, which involve the measurement of anode potential, coulombic efficiency, charge/discharge curves and corresponding differential curves, and voltage relaxation during resting. Due to their high sensitivity and non-destructivity to LIBs, electrochemical techniques are regarded as ideal methods for in situ detection of anode lithium plating which are easily applicable to battery management systems (BMS).Typically, metallic lithium foil is used as the reference electrode for a three-electrode cell.44,45 However, the potential of metallic lithium is highly dependent on its surface properties, particularly the SEI layer. Moreover, the lithium foil reference electrode must be prepared under inert atmosphere, leading to inconvenience in operation. Alternative reference electrodes include lithium alloys, such as Li–Sn46 and Li–Al,47 or lithium intercalation compounds with stable equilibrium potentials, such as Li4Ti5O12 and LiFePO4.48 Compared with metallic lithium, these reference electrodes have the advantage of easy assembly into LIBs in open air. Among these, Li4Ti5O12 and LiFePO4 are regarded as the most promising reference electrodes due to their stable working potential and minimal reaction with the electrolyte.48–50
Using an inserted reference electrode, the effects of cell design and charging conditions, such as temperature and charging rate, on anode lithium plating can be determined. Using pouch cells with metallic Li foil as the reference electrode, Wu et al. studied the effect of the capacity ratio of the anode to the cathode on the anode potential. When the ratio was 0.9, the potential of mesocarbon microbeads (MCMB) was reduced to −0.1 V with occurrence of lithium plating during 0.2 C charging, while the potential of MCMB remained above 0 V when the ratio was 1.05, suggesting that the capacity ratio of the anode to the cathode must be controlled within a certain range.51 Meanwhile, using a LiySn micro-reference electrode, Jansen et al. found that the potential of graphite anode dropped below 0 V even when charging to the cutoff voltage of 4.1 V at low temperatures.46
Although reference electrodes have been promisingly used to detect anode lithium plating, which can provide helpful guidance for the design and the charge protocol optimization of practical LIBs, it is noteworthy that the introduction of a reference electrode should not significantly disturb the current distribution between the anode and the cathode in LIBs.52 It was suggested that a large-size reference electrode could interfere with the ionic pathways between the anode and the cathode; therefore, a micro-reference electrode made by depositing metallic lithium or lithium alloys onto inert substrates such as thin Ni or Cu wires is a good choice.53,54 Moreover, the position of the reference electrode may also interrupt the current distribution and influence the accurate measurement of anode potential. Hoshi et al. observed the dissolution of a metallic lithium reference electrode when it was placed between the anode and the cathode; they suggested that the optimum position for a reference electrode may be outside the area between the anode and the cathode.52 However, the reference electrode should not be far away from the electrodes in order to minimize ohmic drop.55 The major drawback of monitoring the anode potential is that the built-in reference electrode requires modifications of the battery design and fabrication process and may interfere with the electrochemical processes in LIBs.
Coulombic efficiency as the detection signal of anode lithium plating is highly feasible and appropriate for different types of LIBs, if an accurate coulombmeter is used. However, current commercial coulombmeters do not provide the required accuracy when measuring coulombic efficiency. Moreover, other parasitic reactions may also result in a decrease of coulombic efficiency during charging and should be taken into consideration, such as oxidation of the electrolyte on the positive electrode and loss of active materials during cycling. If these parasitic reactions are severe, coulombic efficiency may not be used as an efficient indicator for anode lithium plating.
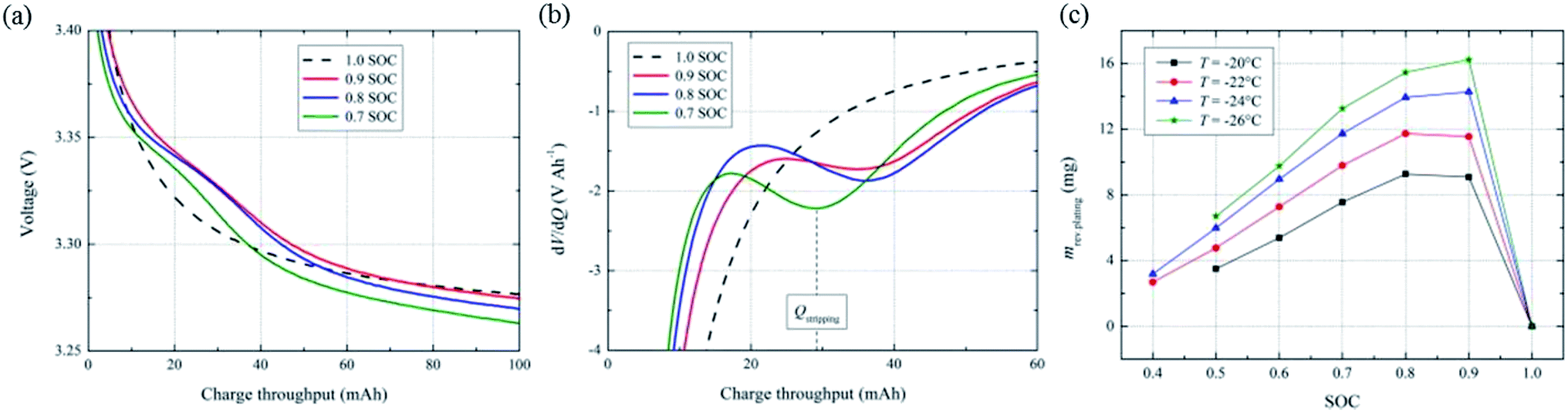 | ||
| Fig. 2 (a) Discharge voltage profiles and (b) differential voltage plots (dV/dQ) of the 26650-type LiFePO4/graphite battery after 1 C charging to different SOC levels at −20 °C, and (c) reversible lithium calculated from the dV/dQ plots (adapted from ref. 10). | ||
The key to the stripping discharge method is that the stripping voltage plateau can be clearly identified and does not overlap with that of the cathode material. As a result, this method only applies to cathode materials without high potential plateaus, such as LiFePO4. Moreover, this method can be used to detect anode lithium plating by characterizing the reversible deposited lithium. Thus, when there is no stripping of reversible metallic lithium in the subsequent discharge, we cannot detect anode lithium plating even if it actually occurs, as in the case discussed above of tapering the charging to 100% SOC. For this reason, this method is only applicable at low temperatures (lower than −20 °C reported to date10) because the deposited metal lithium is more likely to reinsert into graphite and react with the electrolyte at higher temperatures, which reduces the reversible lithium and causes the disappearance of the high voltage plateau during stripping discharge.
In addition to stripping discharge, anode lithium plating has recently been detected in situ by voltage relaxation during the resting of LIBs after charging at low temperatures.59 A mixed potential of the intercalated graphite phase and deposited metallic lithium is formed during resting because the deposited metallic lithium reinserts into graphite even if no external current passes the LIBs. A modified differential voltage method (dV/dt), similar to dV/dQ in Fig. 2b, was developed by Schindler et al.59 The changes in dV/dt during resting were attributed to the decay of the mixed potential because the reintercalation of reversible deposited lithium into the anode led to an increase in the mixed potential. Therefore, this dV/dt plot could indirectly detect anode lithium plating and provide the amount of deposited metallic lithium. However, they did not show any direct/indirect proof to confirm this correlation between anode lithium plating and the dV/dt plot, such as post-mortem analysis of the anode.
4.2. Physical characterization techniques
Anode lithium plating can also be distinguished by various physical properties, including morphology, interface properties, chemical composition and microstructure. Thus, some physical characterization techniques, such as optical microscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM), nuclear magnetic resonance spectroscopy (NMR) and neutron diffraction, were used to detect anode Li plating.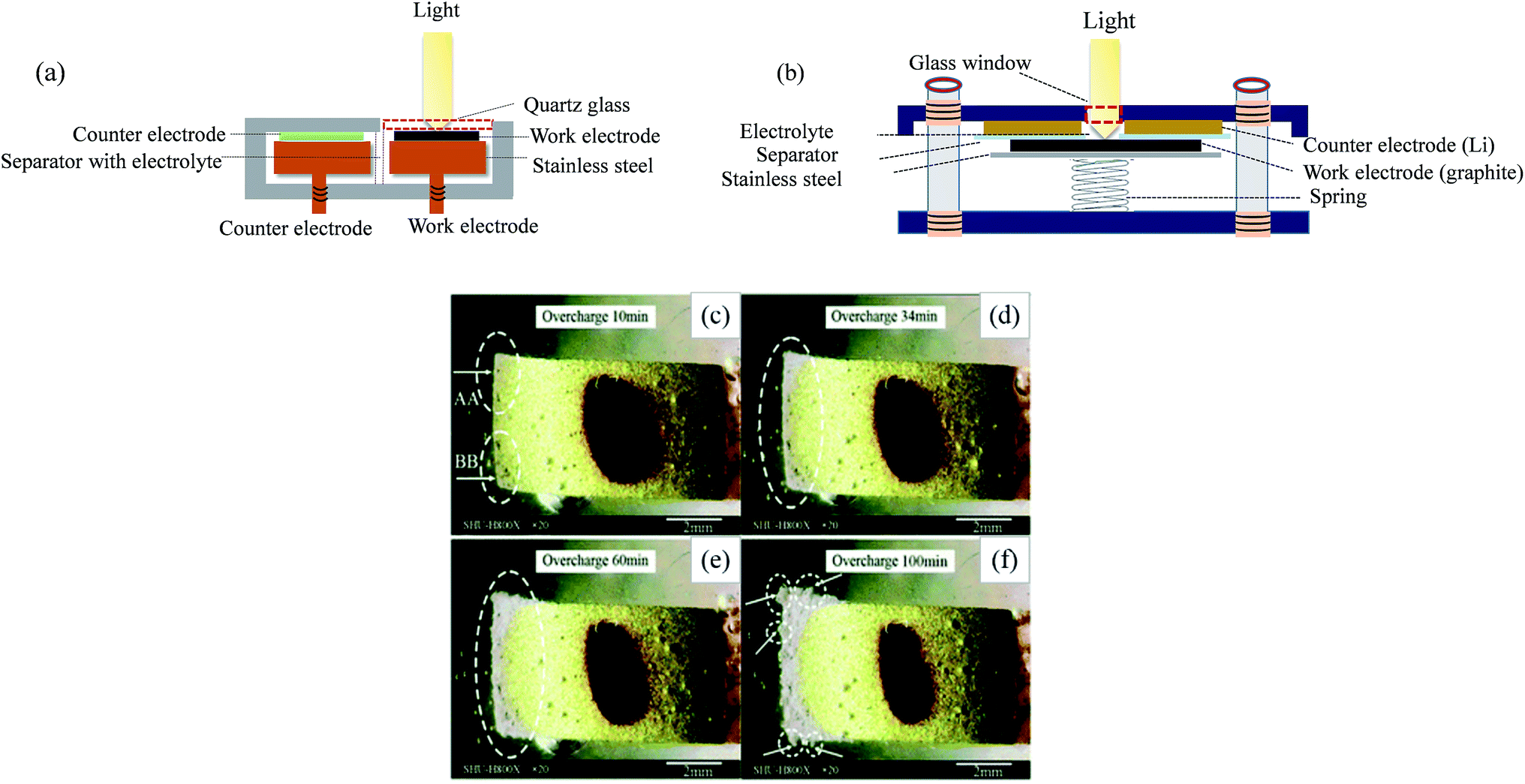 | ||
| Fig. 3 In situ optical testing cells with (a) side by side and (b) face to face arrangements, and in situ observation of graphite during overcharge at 0.2 C for (c) 10 min, (d) 34 min, (e) 60 min and (f) 100 min ((c)–(f) are adapted from ref. 65). | ||
SEM has been widely used to analyze the morphologies of deposited lithium under different experimental conditions, such as charge/discharge current, working temperature and electrolyte composition.66–68 During ex situ SEM investigation, in order to protect the sample from air exposure, special transfer equipment was designed with a movable airlock to transfer the sample from the glove box to the SEM.36,69 In situ SEM is more helpful to understand the dynamics of Li nucleation, growth and dissolution. However, due to the requirement of high vacuum, in situ SEM could only be developed with extremely low vapor electrolytes, such as solid/polymer electrolytes70,71 and ionic liquids.72,73 Sagane et al. observed Li morphology during lithium plating and stripping at a lithium phosphorus oxynitride glass electrolyte and copper current collector (LiPON/Cu) interface by in situ SEM.71 During plating, as shown in Fig. 4a–e, the lithium nucleated first, then grew with time, and finally became needle-like. The dynamic stripping process (Fig. 4f and g) demonstrated that the core region of deposited lithium was mostly stripped, while its surface lost electrochemical activity, resulting in the formation of a large amount of electrically-isolated dead lithium and the reduction of coulombic efficiency during cycling. However, in situ SEM has not yet been reported to study anode Li plating behavior on graphite anode. Moreover, in situ SEM with common liquid organic electrolytes is quite difficult due to the high vacuum operation of SEM. To overcome this restriction, in situ environmental SEM that requires moderate vacuum may enable monitoring of electrode surface changes in less volatile organic electrolytes.74 Also, it is worth mentioning that during SEM observation, the materials should not be completely submerged in the liquid electrolyte because the electrode must be exposed to the electron beam.
 | ||
| Fig. 4 In situ SEM studies of morphology during lithium plating for (a) 0 s, (b) 300 s, (c) 600 s, (d) 1800 s, and (e) 3600 s (also 0 s of stripping); stripping at 50 mA cm−2 for (f) 600 s, (g) 1200 s, and (h) 2160 s (adapted from ref. 71). | ||
Compared with SEM, TEM has higher resolution and is a powerful tool to characterize the morphology and structure of deposited lithium. A detailed discussion of in situ TEM and its application can be seen in a previous review.75 A closed liquid cell has been successfully demonstrated with two electron transparent silicon nitride protection windows.76 Using in situ TEM observation, the dynamic changes in nucleation, growth and stripping of lithium dendrites in ionic and carbonate-based liquids have been studied.77–79 Leenheer et al. imaged lithium electrodeposition/dissolution in organic carbonate electrolyte during the first few cycles at submicron resolution and observed increased dendritic lithium with increasing current density.78 More importantly, in situ TEM could probe the dynamic evolution of interfaces and understand the mechanisms of SEI formation on the nanoscale, which cannot be easily achieved by other techniques. Mehdi et al. distinguished Li deposited from the SEI layer and quantified the thickness of the SEI layer on Pt in LiPF6/PC electrolyte directly from TEM observations.77 However, TEM also has some drawbacks. On the one hand, real-time TEM characterization with liquid cells is limited by low resolution because multiple scattering of electrons in the liquid layer and protection window material will weaken the signal of the sample in the closed liquid cell.80 On the other hand, the sample should be thin enough to be transmitted by electron beam, which may be different from practical electrode systems.
Recently, several groups have studied the growth of Li dendrites on graphite electrodes by in situ NMR. A main benefit of in situ NMR is that it can be used to quantify deposited Li using the intensities of the 7Li signals. Gotoh et al. designed a full cell for in situ 7Li NMR to study Li plating behavior during overcharging.90 They found that the deposited Li could reinsert into carbon within a few hours of resting, which is called the “relaxation effect”. This was the first experimental proof for the relaxation of deposited Li. Arai et al. investigated the effects of graphite and hard carbon as anodes on Li plating using in situ 7Li NMR;91 they concluded that Li deposition was more difficult and Li stripping was much easier on hard carbon than on graphite. Accordingly, in situ NMR is a nondestructive and convenient technique for detecting and quantifying anode Li plating in LIBs.
In addition to the common NMR, magnetic resonance imaging (MRI) can be more helpful in providing multidimensional and spatially resolved information by computing the resonance signals of certain atomic nuclei. Through in situ MRI, the locations of Li deposition and two-dimensional or three-dimensional images of Li dendrites were reported;92 this is helpful to understand the mechanisms of Li microstructure growth and to find ways to suppress dendrite growth.
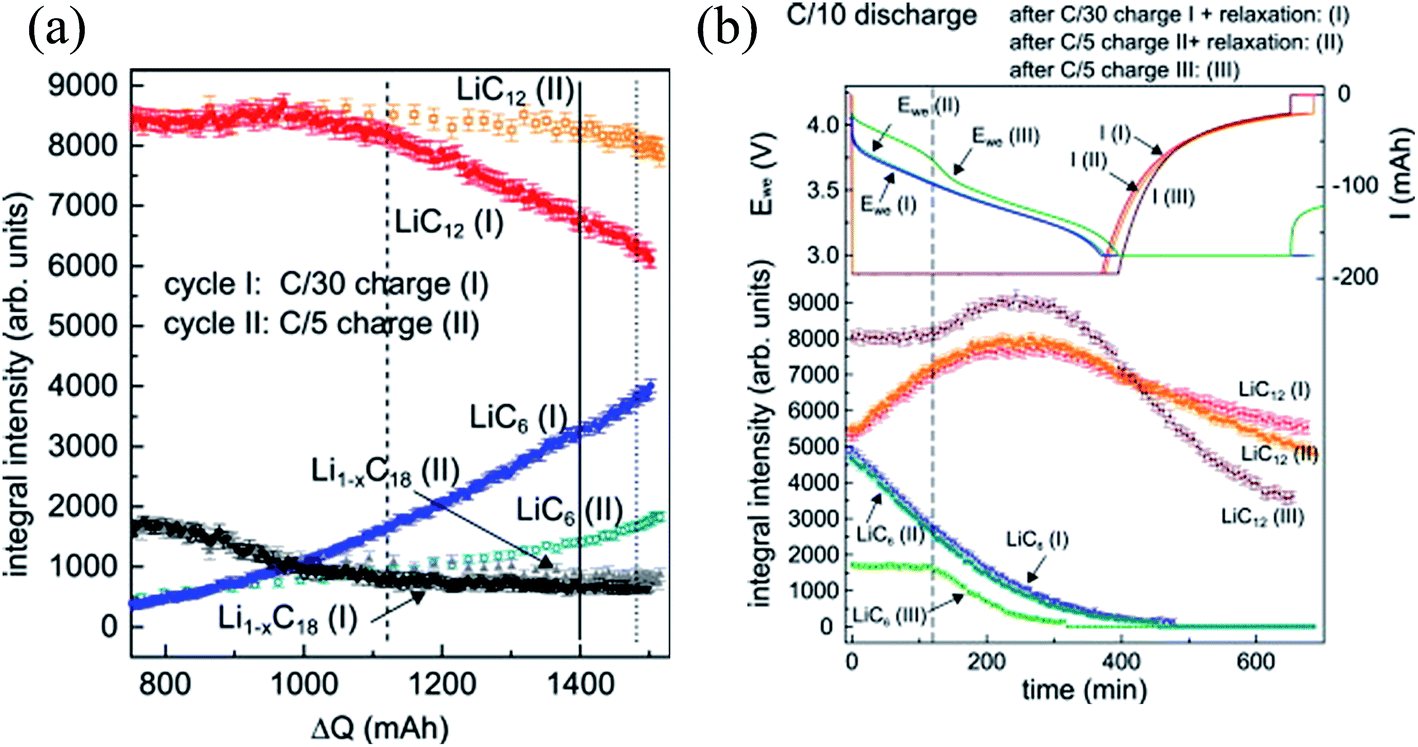 | ||
| Fig. 5 (a) Integral reflection intensities of LiC6, LiC12, and Li1−xC18 during C/30 (I) and C/5 (II) charging; (b) integral reflection intensity of LiC6, LiC12, and Li1−xC18 during C/10 discharging after C/30 charging + 20 h rest (I), C/5 charging + 20 h rest (II) and 5/C charging + no rest (III) (adapted from ref. 95). | ||
Moreover, since lithium atoms and heavy atoms such as Cu and Al have different interactions with neutrons and thus have varied neutron attenuations, neutron imaging is very effective to study the distribution of metallic lithium in electrode materials without the influence of the container and current collector on the internal electrodes. Same et al. visualized metallic lithium formation and its location on a graphite electrode during overcharging of a graphite/Li coin cell with neutron imaging.96 In situ neutron diffraction is a promising method to detect anode Li plating and reveal the associated mechanisms in commercial LIBs without special designs. However, the expensive neutron source is the main limitation for wide application of this technique.
Using isothermal microcalorimetry, Downie et al. detected the occurrence of lithium plating in LiNi0.4Mn0.4Co0.2O2/graphite coin cells during charging based on the thermal behavior of the anode lithium plating.99 When the intercalation sites in graphite were fully occupied, a significant drop of thermal flow, which was caused by the entropy changes of intercalation sites and overpotential, occurred. A small exothermic response would emerge when anode lithium plating occurred during the drop of heat flow; this could be recorded by isothermal microcalorimetry, indicating the full lithiation of graphite and the occurrence of anode lithium plating. This detection method is limited by the requirements of a high-precision isothermal calorimeter and adiabatic conditions. Furthermore, the thermal behavior of deposited metallic Li in commercial LIBs may not be identified by this method because of the complex thermal behaviors in large commercial LIBs.
5. Influencing factors of anode Li plating
Generally, anode lithium plating occurs when the intercalation of Li+ ions into graphite becomes sluggish, causing an increase of anode polarization. The lithium ion intercalation process involves three successive steps: the diffusion of solvated Li+ ions in the electrolyte, the interfacial charge-transfer process and the diffusion of Li in the solid electrode material. The slowing of any step will result in anode lithium plating. Many factors (shown in Table 1) influence the lithium ion intercalation process and, thus, anode lithium plating; these include low-temperature charging, overcharging, high-rate charging, non-uniform current and potential distributions and long-term cycling. In this section, we discuss the main factors that induce anode lithium plating under different working conditions and in different cell designs.| Working conditions | Causes of anode lithium plating |
|---|---|
| Low temperature | Limited lithium solid diffusion100 and sluggish charge transfer process101,102 |
| High rate charging | Lithium solid diffusion and charge transfer limitations103–105 |
| Non-uniform charging | Unreasonable cell design and local defects106 |
| Long-term cycling | Continued growth of SEI film26,107 |
5.1. Low temperature
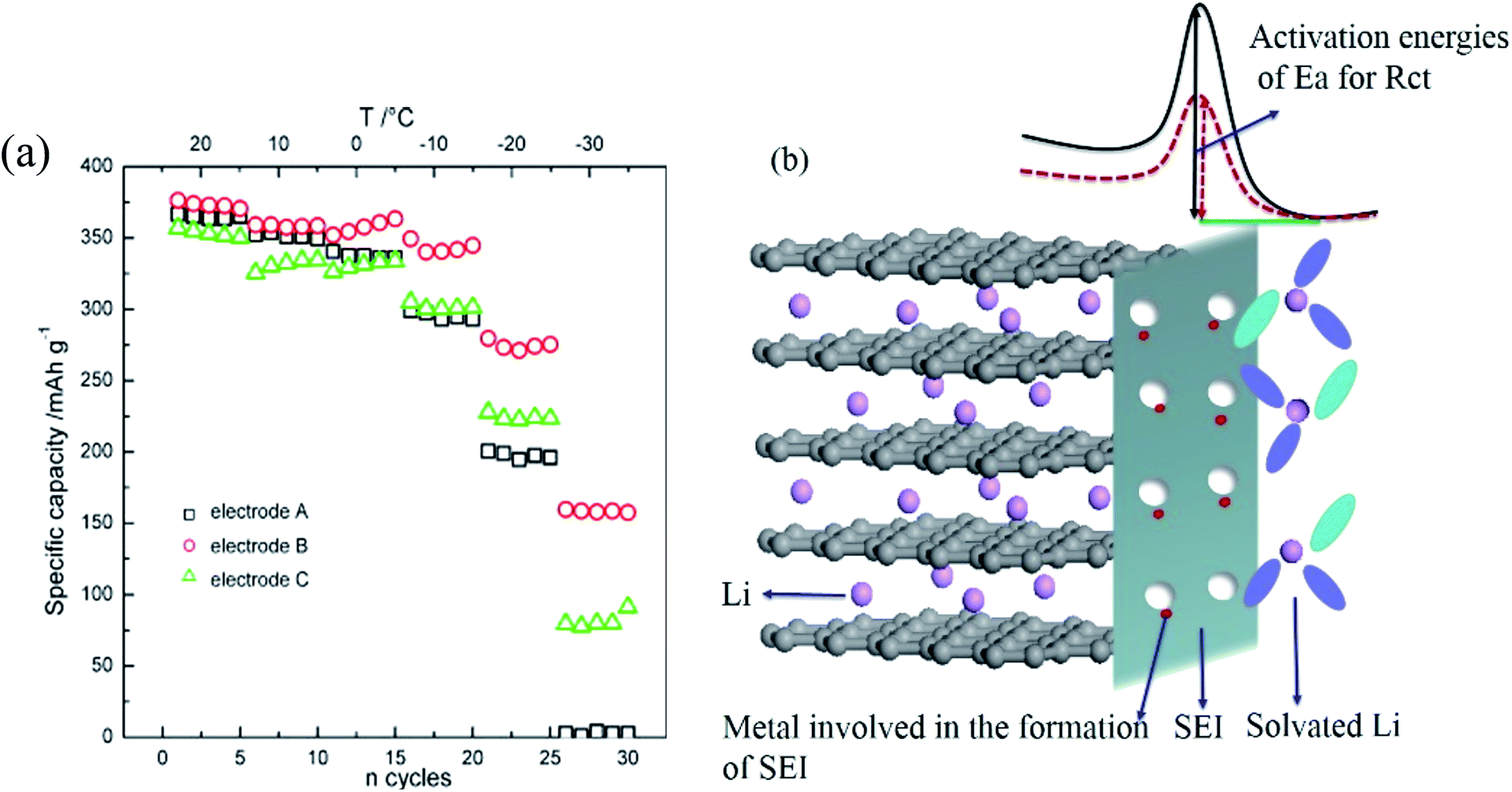
The capacities and power capabilities of LIBs are severely limited at low temperature, especially below −20 °C.108 Many studies have shown that the poor low-temperature performance of LIBs is mainly related to polarization of the anode.109,110 The increased polarization at low temperatures drives the anode potential to below 0 V, leading to anode lithium plating before the anode intercalation sites are fully utilized. The increased polarization is ascribed to reduced ionic conductivity of the electrolyte, high resistance of the solid electrolyte interphase (SEI) film, increased charge transfer resistance and slow lithium diffusion within graphite. Among these, the dominant factors are still controversial. It has been proved that the ionic conductivity of the electrolyte is not the key factor in poor low-temperature performance because lithium ion diffusion in the electrolyte is much faster than in the solid electrode (solid lithium diffusion).109 The main debate regarding the influencing factors of anode lithium plating at low temperatures is charge-transfer resistance, Li+ solid diffusion in graphite or a combination of these.109,111Lithium ion diffusion coefficient in a solid electrode (DLi+) can be determined by some electrochemical techniques, including electrochemical impedance spectroscopy (EIS), slow scan cyclic voltammetry (SCCV), galvanostatic intermittent titration technique (GITT) and potential step chronoamperometry (PSCA). Using the PSCA technique, DLi+ in graphite was obtained during the charge and discharge processes at −20 °C.112 It was found that DLi+ was significantly reduced to only 12% of that at room temperature, consistent with the low capacity of graphite at −20 °C. Moreover, DLi+ in the delithiated graphite was higher than that in the lithiated graphite, which indicates that Li intercalation is more difficult than Li deintercalation at low temperatures.112 Using the GITT technique, DLi+ in graphite was found to decrease from 10−8 cm2 s−1 at room temperature to 10−13 cm2 s−1 at −32 °C, indicating that the limiting factor for low temperature charging is Li+ solid diffusion in graphite.100 Meanwhile, Jiang et al.11 found that 4 h of resting were required for the deposited lithium to diffuse into graphite at −20 °C, while this process was rather rapid at 25 °C, demonstrating that lithium solid diffusion in graphite is limited at low temperatures. Using an electrochemical-thermal coupled model with different electrolyte conductivities, solid state diffusivities and interfacial kinetic resistances, Wang et al.113 concluded that Li+ diffusion in electrolyte and bulk graphite limits the low-temperature discharge behavior.
At the same time, much experimental evidence has also demonstrated that sluggish charge transfer kinetics are the limiting factor of low temperature performance. The charge transfer resistance was obtained by EIS and hybrid pulse power characterization (HPPC) techniques. It was observed that the interface kinetics of electrochemical reactions declined sharply below 0 °C, regardless of the anode materials.101,102 Equivalent circuit simulation of the EIS results confirmed that interfacial charge-transfer resistance was the limiting factor at low temperatures.53 The charge transfer process contains two steps: (1) desolvation of solvated lithium ions and (2) naked Li+ migration through SEI along with electrons originating from the current collector, as shown in Fig. 6. The overall activation energy of the graphite/electrolyte interface was 60 to 70 kJ mol−1 as calculated by the temperature dependent Arrhenius behavior of the charge transfer resistance. Among these, the Li+ desolvation process (about 52 ± 3 kJ mol−1) was the main contributor to the charge transfer process and may be more sluggish at low temperatures.114–116
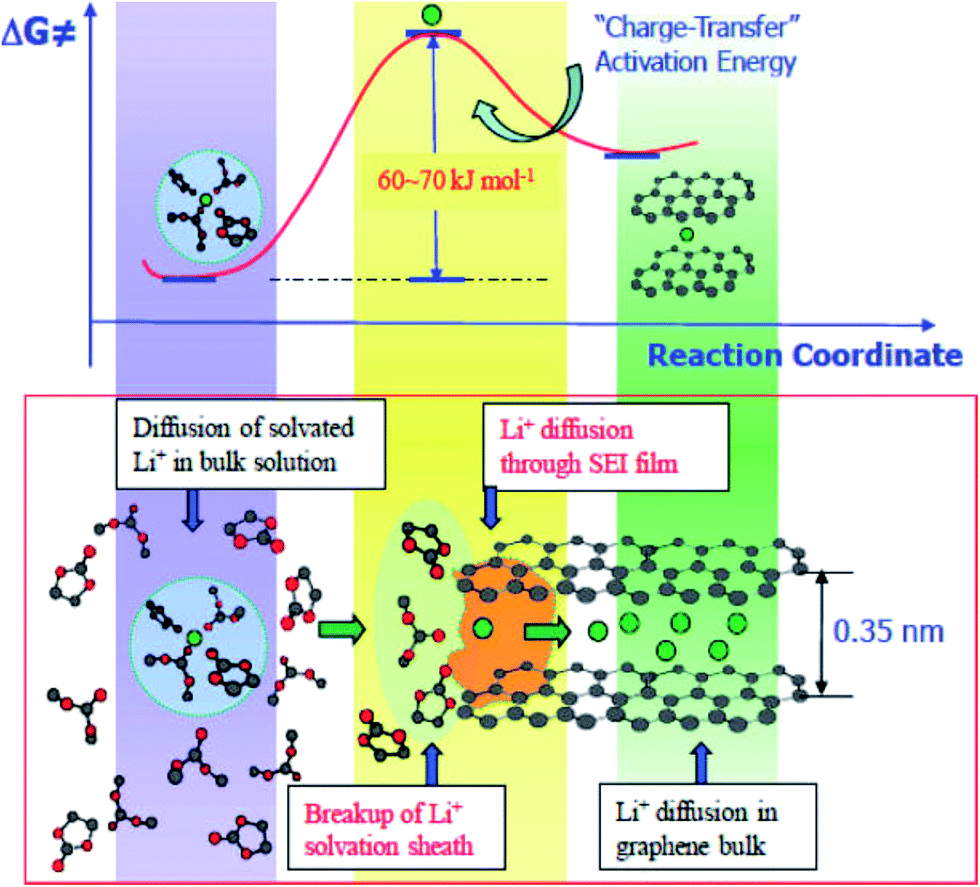 | ||
| Fig. 6 Li ion intercalation process and energy barrier for charge transfer from the electrolyte to bulk graphite (adapted from ref. 116). | ||
To date, although numerous investigations on the low-temperature behavior of LIBs have been conducted, the rate determining step of anode lithium plating at low temperatures has still not been identified. In most cases, the mechanism of low-temperature anode lithium plating depends on the diagnostic technique. However, most of the testing techniques are performed near the equilibrium states and thus cannot reveal real kinetic behaviors at low temperatures. Accordingly, the mechanism of low-temperature anode lithium plating cannot be deeply understood, and it is difficult to avoid anode lithium plating at low temperatures. New experimental and modeling techniques are urgently needed to fully understand the poor low-temperature behaviors of LIBs.
5.2. High charging rate
In LIBs, moderate charge currents are recommended to ensure the favorable insertion of lithium ion into graphite (shown in Fig. 7a and b). Fast charging of LIBs, which is highly required for electric vehicles, is mainly limited by the Li+ insertion process at the graphite anode.117 It is generally believed that the limited solid diffusion of lithium in graphite dominates the lithiation process at high charge currents.103–105 When charging at high charge rates, a large amount of Li+ ions accumulate on the electrode/electrolyte interface because the Li solid diffusion (DLi+ = 10−11 to 10−8 cm2 s−1)118 is significantly lower than Li+ diffusion in the electrolyte. This accumulation results in a high concentration gradient of Li+ ions on the interface of graphite. If the concentration of Li ions on the interface is saturated, anode lithium plating will occur (Fig. 7c). However, the charge transfer kinetics should not be ignored during high rate charging. Charge transfer kinetics would be the rate-limiting factor at sufficiently high charge rates, such as pulse charging.119 Dokko et al. investigated the charge transfer kinetics of a single MCMB particle and found that Li diffusion in the MCMB particle did not limit the Li extraction rate, even at the current rate of 50 C.120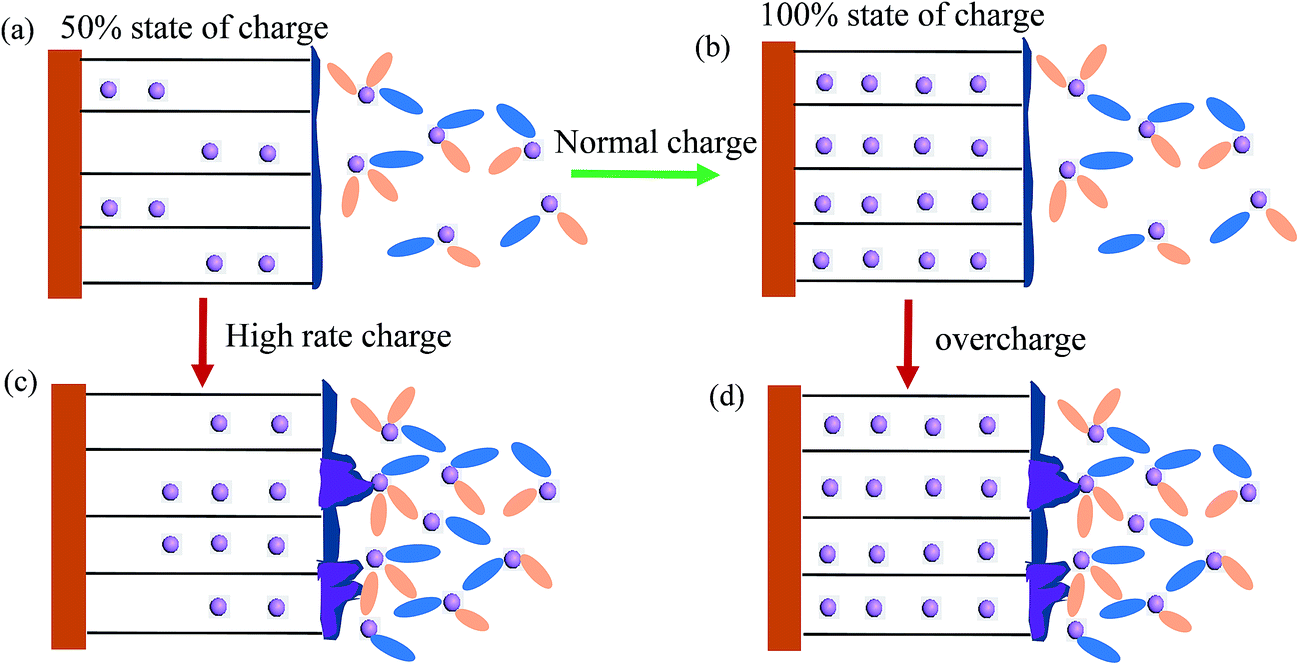 | ||
| Fig. 7 Illustration of different charge protocols: (a) charging to 50% SOC at low rates, (b) charging to 100% SOC, (c) charging at high rates, and (d) overcharging. | ||
5.3. Overcharge
Accidental overcharge of LIBs also facilitates anode lithium plating. Under the normal charging voltage, Li+ ions transferred from the cathode can smoothly insert into graphite (Fig. 7a), and no anode lithium plating occurs until full lithiation (Fig. 7b). When LIBs are charged above the normal upper cutoff voltage (overcharged), the concentration of lithium ions on the anode/electrolyte interface exceeds that which the anode can accommodate, leading to anode lithium plating121 (Fig. 7d). Verbrugge et al.13 studied the effect of overcharging on a single-fiber electrode made from partially graphite carbon. They illustrated that no anode lithium plating was observed during the initial stage when the anode potential was below 0 V (vs. Li+/Li) because the lithium insertion process was more facile compared with lithium plating. However, anode lithium plating occurred and became severe when the negative potentials were maintained below 0 V for a long time. In order to avoid anode lithium plating, the anode/cathode capacity ratio in LIBs is generally designed to be greater than 1. Therefore, anode lithium plating did not occur until charging to 120% SOC at the current rate of 0.2 C.91 However, a large amount of anode Li plating occurred after overcharging the commercial prismatic 0.72 A h Li-ion cell to 4.6 V (150% SOC).122 In addition, another undesired side reaction at high charge voltage is oxidation of the electrolyte on the positive electrode,5 which is beyond the scope of this review.5.4. Non-uniform charging
Non-uniform charging at room temperature is mainly caused by unreasonable cell designs and local defects (see Table 2 for details). Non-uniform charging will result in inhomogeneous distributions of current density and temperature, which are likely to induce local anode lithium plating.123 Cannarella et al. demonstrated well how a locally deformed separator influenced anode lithium plating through assembling coin cells with local closure on the separator. The separator was locally compressed (Fig. 8a)106 to produce closed pores (the transparent part in Fig. 8b). The morphology of graphite after charging is presented in Fig. 8c and d. Compared with the intact separator, apparent anode lithium plating could be observed around the region of pore closure of the separator (Fig. 8d) during normal charging. The reason for the local anode lithium plating was further explained by the axisymmetric finite element model. It was illustrated that the defect separator was lithium ion-insulated, which locally generated high Li+ concentration and overpotential on the open/closed separator interface, thus inducing anode lithium plating around the defect separator.| Non-uniform charging | Specific correlations |
|---|---|
| Unreasonable design | Cell mismatch, e.g., excess cathode active material5,124 |
| Geometric misfits, e.g., large cathodes overlapping anodes at the edges124 | |
| Large scale cells, e.g., the distribution of temperature and current density in large scale cells are more likely to be inhomogeneous during charging125 | |
| Local defects | Locally deformed separators106 |
| Local drying out of electrolyte5 | |
| Partial delamination between the active material and current collector126 | |
| Non-uniform compression/mechanical stress127 |
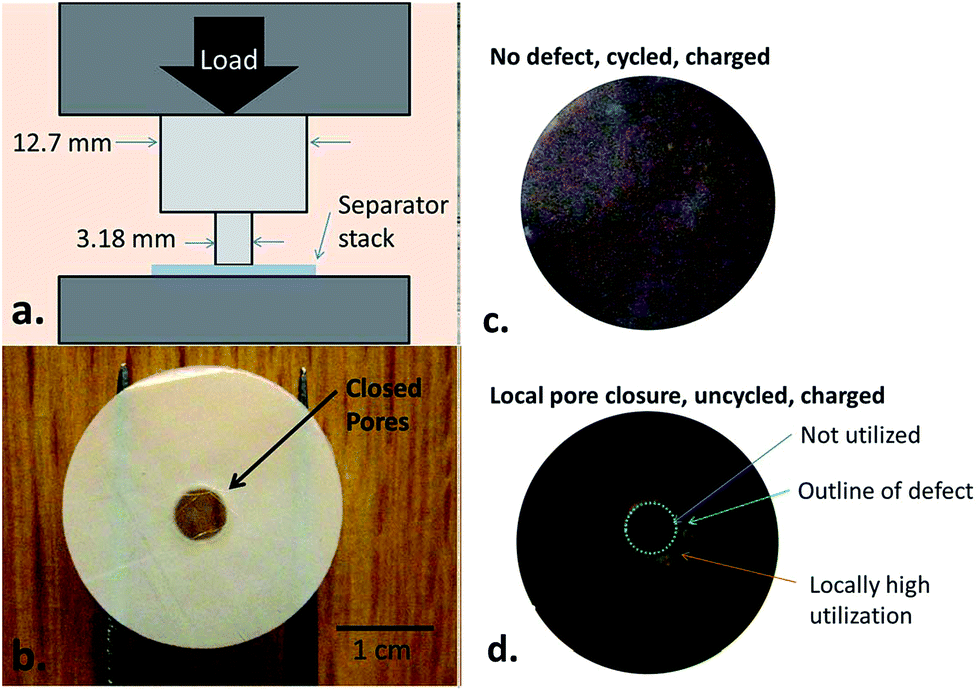 | ||
| Fig. 8 (a) Schematic of a device designed to deliberately create local deformation on the separator, (b) photograph of the local-defect separator showing that the deformed part appears transparent, (c) cell with a defect-free separator in the charged state, showing uniform color, and (d) cell with a defect-containing separator in the lithiation state, showing local plating around the defect (adapted from ref. 106). | ||
6. Restraint of anode lithium plating
It is well known that Li dendrite growth is still a major challenge for the wide application of Li metal secondary batteries. The electrode/electrolyte interface is the most critical factor for the Li metal electrode, which is the same as graphite anode in lithium ion batteries. To prevent dendrite growth for lithium metal, the formation of a stable and flexible interface on the Li metal anode through electrolyte composition,128–131 an ex situ-formed protective layer,132,133 special design of Li electrodes with nanostructures or current collectors with 3D structures134–138 and cell designs (e.g., novel separator)139,140 are essential to the successful application of Li secondary batteries. Like the suppression of Li dendrite growth in Li metal anodes, for lithium ion batteries, improving the electrode/electrolyte interface is key to facilitate lithium insertion kinetics. Therefore, electrolyte components such as solvents, Li salts, and additives used to suppress Li dendrite growth could be applied in lithium ion batteries as well. In addition, as Li deposition on graphite anode is an undesired parasitic reaction, which is different from the case of Li metal batteries, special methods such as graphite modification, cell design (e.g., the capacity ratio of anode/cathode) and appropriate charge protocols (e.g., avoiding high charge rate and low temperature) are required for LIBs.Based on the above influencing factors of anode lithium plating and their mechanisms, extensive investigations, including designing the electrolyte composition, modifying the interfacial properties of the anode, and optimizing cell design and charge protocols, have been carried out in order to prevent anode lithium plating.
6.1. Optimization of electrolyte composition
The electrolyte used in LIBs, which is typically composed of a mixture of organic solvents and lithium salt, not only determines the ionic conductivity of the electrolyte, but also has significant effects on the SEI film. Ethylene carbonate (EC) has the advantages of a high dielectric constant and SEI-formation ability on graphite; lithium hexafluorophosphate (LiPF6) is the most commonly used electrolyte in EC-based organic solvents. However, the high melting point and viscosity of EC solvent reduce the ion conductivity of the electrolyte and limit the low temperature performance of LIBs, especially below −20 °C.141 To improve the low-temperature properties of the electrolyte, several strategies have been suggested, such as employing co-solvents with low melting points and low viscosities, alternative Li salts and electrolyte additives.Ternary and quaternary carbonate-based electrolytes with low EC content have been extensively studied by Jet Propulsion Laboratory and have exhibited improved low-temperature performance.142–144 The employed co-solvents, with low melting points and low viscosities, include linear carbonates such as ethyl methyl carbonate (EMC) and diethyl carbonate (DEC), cyclic carbonates such as propylene carbonate (PC), and aliphatic esters such as methyl acetate (MA), ethyl acetate (EA), ethyl propionate (EP) and ethyl butyrate (EB).145 For example, a ternary solvent system of EC–DMC–EMC with LiPF6 as the lithium salt showed a specific conductivity of >1 mS cm−1 at −40 °C.146 PC is an attractive low-temperature co-solvent due to its low melting point and high boiling point. However, PC will co-intercalate into graphite and destroy the structure of graphite during the first charging process. In order to solve this problem, SEI-forming additives, such as vinylene compounds and sulfites, that can be predominantly reduced on the graphite surface were suggested to protect the graphite structure. For example, the addition of 5% ethylene sulfite (ES) could effectively prevent the co-intercalation of PC into graphite, as the ES could form an effective SEI layer at about 2.1 V (vs. Li+/Li).147,148
In addition to co-solvents, the low-temperature performance of LIBs has been improved by the choice of Li salts, including lithium tetrafluoroborate (LiBF4), lithium bis(oxalate)borate (LiBOB), and lithium difluorooxalatoborate (LiDFOB). It has been found that LiBF4 provides better low-temperature performance by decreasing the charge transfer resistance, probably owing to the less resistive anode SEI film formed in the LiBF4-based electrolyte.149 However, the SEI film formed in LiBF4-based electrolyte does not fully protect the PC-containing electrolyte and is unstable at high temperatures. Therefore, a small amount of LiBOB is added to the LiBF4–PC based electrolyte because BOB anion is effectively involved in the SEI formation and can form a stable SEI film by reduction on the anode.150–152 Unfortunately, LiBOB as the Li salt has low conductivity and limited solubility in organic carbonates; it is generally used as an electrolyte additive. LiDFOB contains the molecular moieties of both LiBOB and LiBF4 and thus combines their advantages, such as SEI-forming ability in organic carbonates and low charge-transfer resistance.153 In addition, LiDFOB has relatively high solubility in organic carbonates and improves the ionic conductivity. LiDFOB has been reported to improve the rate capacity and subzero-temperature performance of graphite.154,155 Therefore, LiDFOB is one of the most promising lithium salts for wide-temperature operation applications. Recently, high concentration lithium salts (>3 mol L−1) in various solvents such as PC, ether, nitrile, sulfoxide and sulfone have been found to improve the capability of Li+ intercalation into graphite because the superconcentrated electrolytes allow for reversible lithium intercalation rather than solvent co-intercalation into graphite.117,156–158 These findings provide new insights for designing fast-charging and low-temperature electrolytes.
The use of electrolyte additives is also a convenient and efficient way to improve the electrochemical performance and safety of LIBs.159 As is well known, vinylene carbonate (VC), fluoroethylene carbonate (FEC), vinyl ethylene carbonate (VEC), ES and propylene sulfite (PS)148 can help form effective SEIs on graphite, particularly in non-carbonate solvents. However, some SEI-forming additives are more prone to anode lithium plating. For example, VC in low EC-based electrolyte increased the likelihood of anode lithium plating during low temperature charging due to the highly resistive surface films formed by VC on the anode, which retarded the anode lithium intercalation kinetics.57 Therefore, additives that can reduce anode interface resistance are needed to prevent anode lithium plating. The use of 2% allyl sulfide (AS) in 1.3 M LiPF6 EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EMC:DEC (3:2:5) can improve the low-temperature (−30 °C) performance and mitigate lithium plating.160 The addition of 0.22 mol L−1 NaClO4 into 1 mol L−1 LiClO4 electrolyte can improve the low-temperature performance of LIBs due to the modification of Na+ on SEI film.161,162 Meanwhile, cesium or rubidium ions formed a positively charged electrostatic shield around the protuberances of Li dendrites, which could prevent the formation of Li dendrites.163 In addition, additives such as polysiloxane-based copolymers could enhance ionic conductivities, thus improving the low-temperature performance.164 Details of these electrolytes and their influences on electrode/electrolyte interfaces can be seen in a review.165
:EMC:DEC (3:2:5) can improve the low-temperature (−30 °C) performance and mitigate lithium plating.160 The addition of 0.22 mol L−1 NaClO4 into 1 mol L−1 LiClO4 electrolyte can improve the low-temperature performance of LIBs due to the modification of Na+ on SEI film.161,162 Meanwhile, cesium or rubidium ions formed a positively charged electrostatic shield around the protuberances of Li dendrites, which could prevent the formation of Li dendrites.163 In addition, additives such as polysiloxane-based copolymers could enhance ionic conductivities, thus improving the low-temperature performance.164 Details of these electrolytes and their influences on electrode/electrolyte interfaces can be seen in a review.165
6.2. Modification of anode materials
Undoubtedly, the surface properties of anode materials also have great impacts on not only the electrochemical performance of the anode but also on anode Li plating; the activation energy of the charge-transfer kinetics and the SEI resistance of the anode can be adjusted by surface modification of the anode materials. As carbonaceous materials are the most commonly used anode materials for LIBs, due to their high reversible capacity and low working potential and cost, the modification of carbonaceous materials is widely studied (see Table 3). These modifications generally improve the overall performance of graphite; for example, they can increase the reversible capacity and rate capacity. Detailed descriptions of the modifications of graphite materials can be seen in a previous review.166 Here, the effects of these modifications on the low temperature performance of LIBs and the main limitations of different modifications are discussed.| Modification | Detailed approaches and references | Functions and effects |
|---|---|---|
| Coating non-graphitic carbon layer | Carbonization of the combination of graphite with carbon precursors such as poly(vinyl alcohol) (PVA) and poly(vinyl chloride) (PVC)167,168 | Excellent electrical conductivity, superior chemical/electrochemical stability and higher Li-ion diffusivity |
| Doping or coating with metals/metal oxides (Au, Cu, In, Pb or Sn, Ni, Ag, SnO, SnO2, Al2O3) | Doping: mixing of nanosized powder with graphite; coating: metal evaporation169–171 | Increased electronic conductivity; modified SEI film; may act as hosts for lithium storage |
| Surface decoration | Edge-selective functionalization of graphite172 and mild expansion173 | Enlarged graphite interlayer spacing |
| Nanostructured carbon | Nanoparticulate graphite,174 nanotubes or pores,175,176 graphene nanosheets (GNSs),177 nanostructured graphene framework178 | Shortened lithium diffusion route |
 | ||
| Fig. 9 (a) Reversible capacity of different graphites at C/5 from 20 °C to −30 °C: pristine oxidized graphite anode (A), coated with a 50 Å Sn layer (B) and 1% (w/o) Sn powder in oxidized graphite (C); (b) illustration of the superior performance of Sn coated graphite ((a) adapted from ref. 186). | ||
6.3. Cell design
The optimization of cell design parameters can improve anode performance (including anode lithium plating) by reducing the polarization. It has been found that increasing the porosity and reducing the thickness of the anode could effectively prevent anode lithium plating.111 Meanwhile, increases in residual capacity and anode width are also instrumental to slow or suppress anode lithium plating.13 However, over-increasing the anode capacity would lead to capacity loss and additional cost, which must be considered in the design of practical LIBs. In addition, the number and location of the current collecting tabs also have impacts on the current distribution, especially for large-format cells. In order to increase the uniform distribution of current density and reduce the risk of local anode lithium plating for large-format LIBs, the configuration of positive and negative tabs located at the same ends, which readily induces inhomogeneous current distribution, should be avoided;191 increasing the tab number has been suggested.1926.4. Charging protocols
Appropriate charging protocols have also been suggested to inhibit anode lithium plating, especially at sub-zero temperatures. First of all, high-rate charging at a high state of charge and overcharging should be avoided in order to prevent anode lithium plating. Moreover, the standard constant current–constant voltage (CC–CV) protocol at high rates and low temperatures is more prone to anode lithium plating, resulting in fast capacity fading. Novel charging strategies have been proposed, mainly based on three-electrode measurements and numerical simulations. With a lithium reference electrode, Waldmann et al. described a stepwise charging strategy:193 high-rate charging to a voltage corresponding to the anode potential of 0 V (vs. Li+/Li), followed by low-rate charging to the end of the charge voltage. A model based on the assumption that lithium solid diffusion is the rate limiting factor of charging showed that the capacity range without saturation of lithium ions at the interface could be extended by proper selection of current waveform parameters, thereby significantly enhancing the low temperature performance.104 Based on an electrochemical model with the lithium deposition criterion of anode potential below 0 V (vs. Li+/Li), Tippmann et al. simulated and predicted the CC–CV charging parameters for LIBs without anode lithium plating at low temperatures.20When charging at low temperatures, strong interplay exists between electrochemical and thermal processes.194 Building a thermal strategy is necessary to prevent or reduce degradation by anode lithium plating at low temperatures. Higher charging rates, particularly pulse charging at lower SOC, were suggested to self-heat and warm up the cells. Hasan et al. proposed a subzero temperature charging protocol which involved a decay pulse charging current followed by CC–CV charging through an electrochemical-thermal coupled model.111 In addition, preheating before cell operation has been suggested to reduce anode lithium plating at low temperatures. The preheating methods include external heating using electric resistance heaters, heat pumps such as external jacket heating or airflow heating, and internal heating methods such as high frequency alternating current heating195 and direct current heating, which have been discussed in detail in a review.196
7. Conclusion and prospects
Anode lithium plating is one of the main factors that result in rapid aging and safety issues of LIBs; addressing this issue is critical to designing safer and more durable LIBs. This paper presents a complete review of anode lithium plating, mainly covering its mechanisms, diagnostic techniques, incentives and suppressing approaches. Various techniques have been applied to detect anode lithium plating. Among these, physical characterizations such as solid-state NMR spectroscopy and neutron diffraction are promising for understanding the mechanisms of lithium plating, while electrochemical methods such as coulombic efficiency and discharge voltage profiles are more applicable to practical LIBs. Nevertheless, it is still challenging to sensitively and quantitatively detect anode lithium plating in practical LIBs, which is significant for battery management systems.The cause of anode lithium plating depends strongly on the cell design and operating conditions, especially charge rate and temperature. During high-rate charging at room temperature, the poor solid phase diffusion of lithium ions is responsible for anode lithium plating. As the temperature decreases, the interface reaction is also hindered in addition to solid phase lithium diffusion. Therefore, the main limiting factor of anode lithium plating at low temperatures is still controversial: sluggish charge transfer kinetics and limited lithium ion diffusion in graphite have both been proposed. At present, in order to suppress anode lithium plating, optimizing the electrolyte composition and modifying the graphite surface structure by methods such as coating and doping are usually employed. In addition, appropriate working conditions and charging protocols can reduce the possibility of anode lithium plating. Nevertheless, anode lithium plating has still not been satisfactorily suppressed because the anode lithium plating process is complicated.
Therefore, in spite of significant progress, the problem of anode lithium plating in practical LIBs is still greatly challenging. Substantial further research efforts to fully understand and suppress anode lithium plating are needed, which mainly include: (1) nondestructive and more reliable detecting methods of anode lithium plating (especially trace lithium plating) for commercial LIBs based on their unique characteristics, such as embedding of a reliable reference electrode and elaborate differential voltage/capacity analysis; (2) clarifying the main mechanism that induces anode lithium plating by developing new experimental approaches, such as in situ morphology characterization techniques coupled with in situ electrochemical impedance measurements to analyze Li dendrite formation and growth, as well as models that are more consistent with the normal operating conditions of batteries; (3) delicate anode/electrolyte interface engineering to reduce SEI resistance by efficiently modifying the anode surface and optimizing the electrolyte compositions and the choice of appropriate additives.
Acknowledgements
This work was financially supported by the National High Technology Research and Development Program of China (863 Program) under Contract No. 2012AA110203.References
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- J. Vetter, P. Novák, M. Wagner, C. Veit, K.-C. Möller, J. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- A. Barré, B. Deguilhem, S. Grolleau, M. Gérard, F. Suard and D. Riu, J. Power Sources, 2013, 241, 680–689 CrossRef.
- L. Ma, M. Nie, J. Xia and J. Dahn, J. Power Sources, 2016, 327, 145–150 CrossRef CAS.
- L. Ma, J. Xia, X. Xia and J. Dahn, J. Electrochem. Soc., 2014, 161, A1495–A1498 CrossRef CAS.
- M. Broussely, P. Biensan, F. Bonhomme, P. Blanchard, S. Herreyre, K. Nechev and R. Staniewicz, J. Power Sources, 2005, 146, 90–96 CrossRef CAS.
- M. Petzl and M. A. Danzer, J. Power Sources, 2014, 254, 80–87 CrossRef CAS.
- J. Fan and S. Tan, J. Electrochem. Soc., 2006, 153, A1081–A1092 CrossRef CAS.
- A. Whitehead, M. Perkins and J. Owen, J. Electrochem. Soc., 1997, 144, L92–L94 CrossRef CAS.
- M. W. Verbrugge and B. J. Koch, J. Electroanal. Chem., 1997, 436, 1–7 CrossRef CAS.
- C.-S. Kim, K. M. Jeong, K. Kim and C.-W. Yi, Electrochim. Acta, 2015, 155, 431–436 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed.
- T. Waldmann, M. Wilka, M. Kasper, M. Fleischhammer and M. Wohlfahrt-Mehrens, J. Power Sources, 2014, 262, 129–135 CrossRef CAS.
- F. Orsini, A. Du Pasquier, B. Beaudouin, J. Tarascon, M. Trentin, N. Langenhuizen, E. De Beer and P. Notten, J. Power Sources, 1999, 81, 918–921 CrossRef.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- S. Tippmann, D. Walper, L. Balboa, B. Spier and W. G. Bessler, J. Power Sources, 2014, 252, 305–316 CrossRef CAS.
- R. Chandrasekaran, J. Power Sources, 2014, 271, 622–632 CrossRef CAS.
- P. Arora, M. Doyle and R. E. White, J. Electrochem. Soc., 1999, 146, 3543–3553 CrossRef CAS.
- C. Uhlmann, J. Illig, M. Ender, R. Schuster and E. Ivers-Tiffée, J. Power Sources, 2015, 279, 428–438 CrossRef CAS.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- D. Aurbach, E. Zinigrad, H. Teller and P. Dan, J. Electrochem. Soc., 2000, 147, 1274–1279 CrossRef CAS.
- L. Yang, X. Cheng, Y. Gao, Y. Ma, P. Zuo, C. Du, Y. Cui, T. Guan, S. Lou and F. Wang, RSC Adv., 2014, 4, 26335–26341 RSC.
- M. Ouyang, D. Ren, L. Lu, J. Li, X. Feng, X. Han and G. Liu, J. Power Sources, 2015, 279, 626–635 CrossRef CAS.
- M. Petzl, M. Kasper and M. A. Danzer, J. Power Sources, 2015, 275, 799–807 CrossRef CAS.
- D. Lv, Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. Zhang, M. H. Engelhard, N. T. Saenz and W. A. Henderson, Adv. Energy Mater., 2015, 5, 1402290 CrossRef.
- W. Lu, C. M. López, N. Liu, J. T. Vaughey and A. Jansen, J. Electrochem. Soc., 2012, 159, A566–A570 CrossRef CAS.
- M. Rosso, C. Brissot, A. Teyssot, M. Dollé, L. Sannier, J.-M. Tarascon, R. Bouchet and S. Lascaud, Electrochim. Acta, 2006, 51, 5334–5340 CrossRef CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2015, 1500213 Search PubMed.
- L. Ma, K. E. Hendrickson, S. Wei and L. A. Archer, Nano Today, 2015, 10, 315–338 CrossRef CAS.
- J.-N. Chazalviel, Phys. Rev. A, 1990, 42, 7355 CrossRef CAS.
- M. Z. Mayers, J. W. Kaminski and T. F. Miller III, J. Phys. Chem. C, 2012, 116, 26214–26221 CAS.
- F. Orsini, A. Du Pasquier, B. Beaudoin, J. Tarascon, M. Trentin, N. Langenhuizen, E. De Beer and P. Notten, J. Power Sources, 1998, 76, 19–29 CrossRef CAS.
- M. Rosso, T. Gobron, C. Brissot, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 2001, 97, 804–806 CrossRef.
- Y. Li, Y. Duh, J. Hsu and C. Kao, J. Therm. Anal. Calorim., 2014, 116, 1219–1226 CrossRef CAS.
- Q. Yuan, F. Zhao, W. Wang, Y. Zhao, Z. Liang and D. Yan, Electrochim. Acta, 2015, 178, 682–688 CrossRef CAS.
- T. Ohsaki, T. Kishi, T. Kuboki, N. Takami, N. Shimura, Y. Sato, M. Sekino and A. Satoh, J. Power Sources, 2005, 146, 97–100 CrossRef CAS.
- A. W. Golubkov, S. Scheikl, R. Planteu, G. Voitic, H. Wiltsche, C. Stangl, G. Fauler, A. Thaler and V. Hacker, RSC Adv., 2015, 5, 57171–57186 RSC.
- M. Fleischhammer, T. Waldmann, G. Bisle, B.-I. Hogg and M. Wohlfahrt-Mehrens, J. Power Sources, 2015, 274, 432–439 CrossRef CAS.
- Q. Yuan, F. Zhao, W. Wang, Y. Zhao, Z. Liang and D. Yan, Electrochim. Acta, 2015, 178, 682–688 CrossRef CAS.
- J. R. Belt, D. M. Bernardi and V. Utgikar, J. Electrochem. Soc., 2014, 161, A1116–A1126 CrossRef CAS.
- G. Nagasubramanian and D. Doughty, J. Power Sources, 2005, 150, 182–186 CrossRef CAS.
- A. N. Jansen, D. W. Dees, D. P. Abraham, K. Amine and G. L. Henriksen, J. Power Sources, 2007, 174, 373–379 CrossRef CAS.
- M. Levi, G. Salitra, B. Markovsky, H. Teller, D. Aurbach, U. Heider and L. Heider, J. Electrochem. Soc., 1999, 146, 1279–1289 CrossRef CAS.
- F. La Mantia, C. Wessells, H. Deshazer and Y. Cui, Electrochem. Commun., 2013, 31, 141–144 CrossRef CAS.
- I. J. Gordon, S. Grugeon, A. Débart, G. Pascaly and S. Laruelle, Solid State Ionics, 2013, 237, 50–55 CrossRef.
- M. Dollé, F. Orsini, A. S. Gozdz and J.-M. Tarascon, J. Electrochem. Soc., 2001, 148, A851–A857 CrossRef.
- M.-S. Wu, P.-C. J. Chiang and J.-C. Lin, J. Electrochem. Soc., 2005, 152, A47–A52 CrossRef CAS.
- Y. Hoshi, Y. Narita, K. Honda, T. Ohtaki, I. Shitanda and M. Itagaki, J. Power Sources, 2015, 288, 168–175 CrossRef CAS.
- H.-M. Cho, W.-S. Choi, J.-Y. Go, S.-E. Bae and H.-C. Shin, J. Power Sources, 2012, 198, 273–280 CrossRef CAS.
- C. Thurston, J. Owen and N. Hargreaves, J. Power Sources, 1992, 39, 215–224 CrossRef CAS.
- T. Waldmann, B.-I. Hogg, M. Kasper, S. Grolleau, C. G. Couceiro, K. Trad, B. P. Matadi and M. Wohlfahrt-Mehrens, J. Electrochem. Soc., 2016, 163, A1232–A1238 CrossRef CAS.
- J. Burns, D. Stevens and J. Dahn, J. Electrochem. Soc., 2015, 162, A959–A964 CrossRef CAS.
- M. Smart and B. Ratnakumar, J. Electrochem. Soc., 2011, 158, A379–A389 CrossRef CAS.
- M. Dubarry, V. Svoboda, R. Hwu and B. Y. Liaw, Electrochem. Solid-State Lett., 2006, 9, A454–A457 CrossRef CAS.
- S. Schindler, M. Bauer, M. Petzl and M. A. Danzer, J. Power Sources, 2016, 304, 170–180 CrossRef CAS.
- S. J. Harris, A. Timmons, D. R. Baker and C. Monroe, Chem. Phys. Lett., 2010, 485, 265–274 CrossRef CAS.
- O. Crowther and A. C. West, J. Electrochem. Soc., 2008, 155, A806–A811 CrossRef CAS.
- K. Nishikawa, T. Mori, T. Nishida, Y. Fukunaka, M. Rosso and T. Homma, J. Electrochem. Soc., 2010, 157, A1212–A1217 CrossRef CAS.
- J. Steiger, D. Kramer and R. Mönig, J. Power Sources, 2014, 261, 112–119 CrossRef CAS.
- J. K. Stark, Y. Ding and P. A. Kohl, J. Electrochem. Soc., 2013, 160, D337–D342 CrossRef CAS.
- Z. Guo, J. Zhu, J. Feng and S. Du, RSC Adv., 2015, 5, 69514–69521 RSC.
- F. Ding, W. Xu, X. Chen, J. Zhang, M. H. Engelhard, Y. Zhang, B. R. Johnson, J. V. Crum, T. A. Blake and X. Liu, J. Electrochem. Soc., 2013, 160, A1894–A1901 CrossRef CAS.
- H. Yang, E. O. Fey, B. D. Trimm, N. Dimitrov and M. S. Whittingham, J. Power Sources, 2014, 272, 900–908 CrossRef CAS.
- L. Gireaud, S. Grugeon, S. Laruelle, B. Yrieix and J.-M. Tarascon, Electrochem. Commun., 2006, 8, 1639–1649 CrossRef CAS.
- W. Li, H. Zheng, G. Chu, F. Luo, J. Zheng, D. Xiao, X. Li, L. Gu, H. Li and X. Wei, Faraday Discuss., 2015, 176, 109–124 RSC.
- M. Nagao, A. Hayashi, M. Tatsumisago, T. Kanetsuku, T. Tsuda and S. Kuwabata, Phys. Chem. Chem. Phys., 2013, 15, 18600–18606 RSC.
- F. Sagane, R. Shimokawa, H. Sano, H. Sakaebe and Y. Iriyama, J. Power Sources, 2013, 225, 245–250 CrossRef CAS.
- S. Arimoto, H. Kageyama, T. Torimoto and S. Kuwabata, Electrochem. Commun., 2008, 10, 1901–1904 CrossRef CAS.
- D. Chen, S. Indris, M. Schulz, B. Gamer and R. Mönig, J. Power Sources, 2011, 196, 6382–6387 CrossRef CAS.
- P. Raimann, N. Hochgatterer, C. Korepp, K. Möller, M. Winter, H. Schröttner, F. Hofer and J. Besenhard, Ionics, 2006, 12, 253–255 CrossRef CAS.
- F. Wu and N. Yao, Nano Energy, 2015, 11, 196–210 CrossRef CAS.
- C.-M. Wang, J. Mater. Res., 2015, 30, 326–339 CrossRef CAS.
- B. L. Mehdi, J. Qian, E. Nasybulin, C. Park, D. A. Welch, R. Faller, H. Mehta, W. A. Henderson, W. Xu and C. Wang, Nano Lett., 2015, 15, 2168–2173 CrossRef CAS PubMed.
- A. J. Leenheer, K. L. Jungjohann, K. R. Zavadil, J. P. Sullivan and C. T. Harris, ACS Nano, 2015, 9, 4379–4389 CrossRef CAS PubMed.
- R. L. Sacci, N. J. Dudney, K. L. More, L. R. Parent, I. Arslan, N. D. Browning and R. R. Unocic, Chem. Commun., 2014, 50, 2104–2107 RSC.
- F. M. Ross, Science, 2015, 350, aaa9886 CrossRef PubMed.
- N. M. Trease, T. K. Köster and C. P. Grey, Electrochem. Soc. Interface, 2011, 20, 69 CAS.
- F. Blanc, M. Leskes and C. P. Grey, Acc. Chem. Res., 2013, 46, 1952–1963 CrossRef CAS PubMed.
- P. Harks, F. Mulder and P. Notten, J. Power Sources, 2015, 288, 92–105 CrossRef CAS.
- R. Gerald, C. Johnson, J. Rathke, R. Klingler, G. Sandí and L. Scanlon, J. Power Sources, 2000, 89, 237–243 CrossRef CAS.
- M. Letellier, F. Chevallier and M. Morcrette, Carbon, 2007, 45, 1025–1034 CrossRef CAS.
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznéc, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239–9249 CrossRef CAS PubMed.
- M. Letellier, F. Chevallier, C. Clinard, E. Frackowiak, J.-N. Rouzaud, F. Béguin, M. Morcrette and J.-M. Tarascon, J. Chem. Phys., 2003, 118, 6038–6045 CrossRef CAS.
- F. Chevallier, F. Poli, B. Montigny and M. Letellier, Carbon, 2013, 61, 140–153 CrossRef CAS.
- H. J. Chang, N. M. Trease, A. J. Ilott, D. Zeng, L.-S. Du, A. Jerschow and C. P. Grey, J. Phys. Chem. C, 2015, 119, 16443–16451 CAS.
- K. Gotoh, M. Izuka, J. Arai, Y. Okada, T. Sugiyama, K. Takeda and H. Ishida, Carbon, 2014, 79, 380–387 CrossRef CAS.
- J. Arai, Y. Okada, T. Sugiyama, M. Izuka, K. Gotoh and K. Takeda, J. Electrochem. Soc., 2015, 162, A952–A958 CrossRef CAS.
- S. Chandrashekar, N. M. Trease, H. J. Chang, L.-S. Du, C. P. Grey and A. Jerschow, Nat. Mater., 2012, 11, 311–315 CrossRef CAS PubMed.
- A. Senyshyn, M. Mühlbauer, O. Dolotko and H. Ehrenberg, J. Power Sources, 2015, 282, 235–240 CrossRef CAS.
- N. Sharma and V. K. Peterson, J. Power Sources, 2013, 244, 695–701 CrossRef CAS.
- V. Zinth, C. von Lüders, M. Hofmann, J. Hattendorff, I. Buchberger, S. Erhard, J. Rebelo-Kornmeier, A. Jossen and R. Gilles, J. Power Sources, 2014, 271, 152–159 CrossRef CAS.
- A. Same, V. Battaglia, H.-Y. Tang and J. W. Park, J. Appl. Electrochem., 2012, 42, 1–9 CrossRef CAS.
- B. Bitzer and A. Gruhle, J. Power Sources, 2014, 262, 297–302 CrossRef CAS.
- C. Birkenmaier, B. Bitzer, M. Harzheim, A. Hintennach and T. Schleid, J. Electrochem. Soc., 2015, 162, A2646–A2650 CrossRef CAS.
- L. Downie, L. Krause, J. Burns, L. Jensen, V. Chevrier and J. Dahn, J. Electrochem. Soc., 2013, 160, A588–A594 CrossRef CAS.
- A. Yaqub, Y.-J. Lee, M. J. Hwang, S. A. Pervez, U. Farooq, J.-H. Choi, D. Kim, H.-Y. Choi, S.-B. Cho and C.-H. Doh, J. Mater. Sci., 2014, 49, 7707–7714 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, J. Power Sources, 2003, 115, 137–140 CrossRef CAS.
- J. Li, C. F. Yuan, Z. H. Guo, Z. A. Zhang, Y. Q. Lai and J. Liu, Electrochim. Acta, 2012, 59, 69–74 CrossRef CAS.
- J. Li, E. Murphy, J. Winnick and P. A. Kohl, J. Power Sources, 2001, 102, 302–309 CrossRef CAS.
- B. Purushothaman and U. Landau, J. Electrochem. Soc., 2006, 153, A533–A542 CrossRef CAS.
- J. Marcicki, A. Conlisk and G. Rizzoni, J. Power Sources, 2014, 251, 157–169 CrossRef CAS.
- J. Cannarella and C. B. Arnold, J. Electrochem. Soc., 2015, 162, A1365–A1373 CrossRef CAS.
- K. Jalkanen, J. Karppinen, L. Skogström, T. Laurila, M. Nisula and K. Vuorilehto, Appl. Energy, 2015, 154, 160–172 CrossRef CAS.
- H.-C. A. Shiao, D. Chua, H.-p. Lin, S. Slane and M. Salomon, J. Power Sources, 2000, 87, 167–173 CrossRef CAS.
- H.-P. Lin, D. Chua, M. Salomon, H. Shiao, M. Hendrickson, E. Plichta and S. Slane, Electrochem. Solid-State Lett., 2001, 4, A71–A73 CrossRef CAS.
- C. K. Huang, J. Sakamoto, J. Wolfenstine and S. Surampudi, J. Electrochem. Soc., 2000, 147, 2893–2896 CrossRef CAS.
- M. F. Hasan, C.-F. Chen, C. E. Shaffer and P. P. Mukherjee, J. Electrochem. Soc., 2015, 162, A1382–A1395 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, Electrochim. Acta, 2002, 48, 241–246 CrossRef CAS.
- Y. Ji, Y. Zhang and C.-Y. Wang, J. Electrochem. Soc., 2013, 160, A636–A649 CrossRef CAS.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120–A1123 CrossRef CAS.
- Y. Yamada, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2009, 25, 12766–12770 CrossRef CAS PubMed.
- K. Xu, A. von Cresce and U. Lee, Langmuir, 2010, 26, 11538–11543 CrossRef CAS PubMed.
- Y. Yamada, M. Yaegashi, T. Abe and A. Yamada, Chem. Commun., 2013, 49, 11194–11196 RSC.
- M. Levi and D. Aurbach, Electrochim. Acta, 1999, 45, 167–185 CrossRef CAS.
- N. Legrand, B. Knosp, P. Desprez, F. Lapicque and S. Raël, J. Power Sources, 2014, 245, 208–216 CrossRef CAS.
- K. Dokko, N. Nakata, Y. Suzuki and K. Kanamura, J. Phys. Chem. C, 2010, 114, 8646–8650 CAS.
- S. Hossain, Y.-K. Kim, Y. Saleh and R. Loutfy, J. Power Sources, 2006, 161, 640–647 CrossRef CAS.
- D. Belov and M.-H. Yang, Solid State Ionics, 2008, 179, 1816–1821 CrossRef CAS.
- M. Klett, R. Eriksson, J. Groot, P. Svens, K. C. Högström, R. W. Lindström, H. Berg, T. Gustafson, G. Lindbergh and K. Edström, J. Power Sources, 2014, 257, 126–137 CrossRef CAS.
- M. Tang, P. Albertus and J. Newman, J. Electrochem. Soc., 2009, 156, A390–A399 CrossRef CAS.
- D. Burow, K. Sergeeva, S. Calles, K. Schorb, A. Börger, C. Roth and P. Heitjans, J. Power Sources, 2016, 307, 806–814 CrossRef CAS.
- N. Zhang and H. Tang, J. Power Sources, 2012, 218, 52–55 CrossRef CAS.
- T. C. Bach, S. F. Schuster, E. Fleder, J. Müller, M. J. Brand, H. Lorrmann, A. Jossen and G. Sextl, J. Energy Storage, 2016, 5, 212–223 CrossRef.
- Y. Lu, M. Tikekar, R. Mohanty, K. Hendrickson, L. Ma and L. A. Archer, Adv. Energy Mater., 2015, 5, 1402073 CrossRef.
- R. Younesi, G. M. Veith, P. Johansson, K. Edström and T. Vegge, Energy Environ. Sci., 2015, 8, 1905–1922 CAS.
- X. B. Cheng, T. Z. Hou, R. Zhang, H. J. Peng, C. Z. Zhao, J. Q. Huang and Q. Zhang, Adv. Mater., 2016, 28, 2845 CrossRef CAS.
- C.-Z. Zhao, X.-B. Cheng, R. Zhang, H.-J. Peng, J.-Q. Huang, R. Ran, Z.-H. Huang, F. Wei and Q. Zhang, Energy Storage Materials, 2016, 3, 77–84 CrossRef.
- G. Zheng, S. W. Lee, Z. Liang, H.-W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- A. C. Kozen, C.-F. Lin, A. J. Pearse, M. A. Schroeder, X. Han, L. Hu, S.-B. Lee, G. W. Rubloff and M. Noked, ACS Nano, 2015, 9, 5884–5892 CrossRef CAS PubMed.
- R. Zhang, X. B. Cheng, C. Z. Zhao, H. J. Peng, J. L. Shi, J. Q. Huang, J. Wang, F. Wei and Q. Zhang, Adv. Mater., 2016, 28, 2155–2162 CrossRef CAS PubMed.
- Q. Yun, Y. B. He, W. Lv, Y. Zhao, B. Li, F. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 6932–6939 CrossRef CAS PubMed.
- Y. Sun, G. Zheng, Z. W. Seh, N. Liu, S. Wang, J. Sun, H. R. Lee and Y. Cui, Chemistry, 2016, 1, 287–297 CrossRef.
- L.-L. Lu, J. Ge, J.-N. Yang, S.-M. Chen, H. Yao, F. Zhou and S.-H. Yu, Nano Lett., 2016, 16, 4431–4437 CrossRef CAS PubMed.
- X. B. Cheng, H. J. Peng, J. Q. Huang, F. Wei and Q. Zhang, Small, 2014, 10, 4257–4263 CAS.
- L. Ma, P. Nath, Z. Tu, M. Tikekar and L. A. Archer, Chem. Mater., 2016, 28, 5147–5154 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- M. Smart, B. Ratnakumar and S. Surampudi, J. Electrochem. Soc., 1999, 146, 486–492 CrossRef CAS.
- M. Smart, B. Ratnakumar, S. Surampudi, Y. Wang, X. Zhang, S. Greenbaum, A. Hightower, C. Ahn and B. Fultz, J. Electrochem. Soc., 1999, 146, 3963–3969 CrossRef CAS.
- M. Smart, B. Ratnakumar, L. Whitcanack, K. Chin, S. Surampudi, H. Croft, D. Tice and R. Staniewicz, J. Power Sources, 2003, 119, 349–358 CrossRef.
- E. Plichta, M. Hendrickson, R. Thompson, G. Au, W. Behl, M. Smart, B. Ratnakumar and S. Surampudi, J. Power Sources, 2001, 94, 160–162 CrossRef CAS.
- M. Smart, B. Ratnakumar and S. Surampudi, J. Electrochem. Soc., 2002, 149, A361–A370 CrossRef CAS.
- O. Yariv, D. Hirshberg, E. Zinigrad, A. Meitav, D. Aurbach, M. Jiang and B. R. Powell, J. Electrochem. Soc., 2014, 161, A1422–A1431 CrossRef CAS.
- G. H. Wrodnigg, J. O. Besenhard and M. Winter, J. Power Sources, 2001, 97, 592–594 CrossRef.
- G. H. Wrodnigg, J. O. Besenhard and M. Winter, J. Electrochem. Soc., 1999, 146, 470–472 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, Electrochem. Commun., 2002, 4, 928–932 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, J. Power Sources, 2006, 156, 629–633 CrossRef CAS.
- N. Takami, T. Ohsaki, H. Hasebe and M. Yamamoto, J. Electrochem. Soc., 2002, 149, A9–A12 CrossRef CAS.
- V. Aravindan, J. Gnanaraj, S. Madhavi and H. K. Liu, Chem.–Eur. J., 2011, 17, 14326–14346 CrossRef CAS PubMed.
- S. S. Zhang, Electrochem. Commun., 2006, 8, 1423–1428 CrossRef CAS.
- Z. Chen, J. Liu and K. Amine, Electrochem. Solid-State Lett., 2007, 10, A45–A47 CrossRef CAS.
- S. Li, W. Zhao, Z. Zhou, X. Cui, Z. Shang, H. Liu and D. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4920–4926 CAS.
- S.-K. Jeong, H.-Y. Seo, D.-H. Kim, H.-K. Han, J.-G. Kim, Y. B. Lee, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Commun., 2008, 10, 635–638 CrossRef CAS.
- Y. Yamada, K. Usui, C. H. Chiang, K. Kikuchi, K. Furukawa and A. Yamada, ACS Appl. Mater. Interfaces, 2014, 6, 10892–10899 CAS.
- X.-R. Liu, L. Wang, L.-J. Wan and D. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 9573–9580 CAS.
- L. Ma, J. Xia and J. Dahn, J. Electrochem. Soc., 2015, 162, A1170–A1174 CrossRef CAS.
- S. Jurng, S. Park, T. Yoon, H.-S. Kim, H. Jeong, J. H. Ryu, J. J. Kim and S. M. Oh, J. Electrochem. Soc., 2016, 163, A1798–A1804 CrossRef CAS.
- S. Komaba, T. Itabashi, B. Kaplan, H. Groult and N. Kumagai, Electrochem. Commun., 2003, 5, 962–966 CrossRef CAS.
- S. Komaba, T. Itabashi, M. Watanabe, H. Groult and N. Kumagai, J. Electrochem. Soc., 2007, 154, A322–A330 CrossRef CAS.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie and J. Xiao, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- K. M. Kim, N. V. Ly, J. H. Won, Y.-G. Lee, W. I. Cho, J. M. Ko and R. B. Kaner, Electrochim. Acta, 2014, 136, 182–188 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- L. Fu, H. Liu, C. Li, Y. P. Wu, E. Rahm, R. Holze and H. Wu, Solid State Sci., 2006, 8, 113–128 CrossRef CAS.
- H.-L. Zhang, F. Li, C. Liu and H.-M. Cheng, J. Phys. Chem. C, 2008, 112, 7767–7772 CAS.
- H.-Y. Lee, J.-K. Baek, S.-W. Jang, S.-M. Lee, S.-T. Hong, K.-Y. Lee and M.-H. Kim, J. Power Sources, 2001, 101, 206–212 CrossRef CAS.
- P. Yu, J. A. Ritter, R. E. White and B. N. Popov, J. Electrochem. Soc., 2000, 147, 1280–1285 CrossRef CAS.
- J. Yun, Y. Wang, T. Gao, H. Zheng, M. Shen, Q. Qu and H. Zheng, Electrochim. Acta, 2015, 155, 396–401 CrossRef CAS.
- J. Suzuki, O. Omae, K. Sekine and T. Takamura, Solid State Ionics, 2002, 152, 111–118 Search PubMed.
- J.-S. Park, M.-H. Lee, I.-Y. Jeon, H.-S. Park, J.-B. Baek and H.-K. Song, ACS Nano, 2012, 6, 10770–10775 CAS.
- L. Zou, F. Kang, Y.-P. Zheng and W. Shen, Electrochim. Acta, 2009, 54, 3930–3934 CrossRef CAS.
- D. T. Welna, L. Qu, B. E. Taylor, L. Dai and M. F. Durstock, J. Power Sources, 2011, 196, 1455–1460 CrossRef CAS.
- C. Cui, X. Sun, X. Li, C. Li and Y. Niu, RSC Adv., 2015, 5, 55348–55352 RSC.
- F. Wang, R. Song, H. Song, X. Chen, J. Zhou, Z. Ma, M. Li and Q. Lei, Carbon, 2015, 81, 314–321 CrossRef CAS.
- P. Guo, H. Song and X. Chen, Electrochem. Commun., 2009, 11, 1320–1324 CrossRef CAS.
- X.-B. Cheng, H.-J. Peng, J.-Q. Huang, R. Zhang, C.-Z. Zhao and Q. Zhang, ACS Nano, 2015, 9, 6373–6382 CrossRef CAS PubMed.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, J. Electrochem. Soc., 2000, 147, 1245–1250 CrossRef CAS.
- H.-L. Zhang, S.-H. Liu, F. Li, S. Bai, C. Liu, J. Tan and H.-M. Cheng, Carbon, 2006, 44, 2212–2218 CrossRef CAS.
- N. Ohta, K. Nagaoka, K. Hoshi, S. Bitoh and M. Inagaki, J. Power Sources, 2009, 194, 985–990 CrossRef CAS.
- N. Gunawardhana, N. Dimov, M. Sasidharan, G.-J. Park, H. Nakamura and M. Yoshio, Electrochem. Commun., 2011, 13, 1116–1118 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, T. Umeno, T. Abe and Z. Ogumi, J. Mater. Chem., 2004, 14, 1754–1758 RSC.
- F. Nobili, S. Dsoke, T. Mecozzi and R. Marassi, Electrochim. Acta, 2005, 51, 536–544 CrossRef CAS.
- M. Mancini, F. Nobili, S. Dsoke, F. D'Amico, R. Tossici, F. Croce and R. Marassi, J. Power Sources, 2009, 190, 141–148 CrossRef CAS.
- F. Nobili, M. Mancini, S. Dsoke, R. Tossici and R. Marassi, J. Power Sources, 2010, 195, 7090–7097 CrossRef CAS.
- Y. Lin, Z.-H. Huang, X. Yu, W. Shen, Y. Zheng and F. Kang, Electrochim. Acta, 2014, 116, 170–174 CrossRef CAS.
- T.-H. Kim, E. K. Jeon, Y. Ko, B. Y. Jang, B.-S. Kim and H.-K. Song, J. Mater. Chem. A, 2014, 2, 7600–7605 CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- G. Zhang, C. E. Shaffer, C.-Y. Wang and C. D. Rahn, J. Electrochem. Soc., 2013, 160, A2299–A2305 CrossRef CAS.
- W. Zhao, G. Luo and C.-Y. Wang, J. Power Sources, 2014, 257, 70–79 CrossRef CAS.
- T. Waldmann, M. Kasper and M. Wohlfahrt-Mehrens, Electrochim. Acta, 2015, 178, 525–532 CrossRef CAS.
- Y. Ji and C. Y. Wang, Electrochim. Acta, 2013, 107, 664–674 CrossRef CAS.
- H. Ge, J. Huang, J. Zhang and Z. Li, J. Electrochem. Soc., 2016, 163, A290–A299 CrossRef CAS.
- Z. Li, J. Huang, B. Y. Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |