
Direct and site-selective Pd(II)-catalyzed C-7 arylation of indolines with arylsilanes†
Haiqing Luo*,
Haidong Liu,
Zhipeng Zhang,
Yifan Xiao,
Sihui Wang,
Xuzhong Luo and
Kejun Wang*
Department of Chemistry & Chemical Engineering, Gannan Normal University, Ganzhou 341000, China. E-mail: luohq@gnnu.edu.cn; wangkejun@sina.com
First published on 13th April 2016
Abstract
The palladium-catalyzed oxidative arylation of indolines with arylsilanes at the C-7 position via C–H bond activation has been reported. This transformation has been applied to a wide range of substrates. It represents a facile access to C-7 arylated indolines, which can be conveniently transformed into C-7 arylated indoles.
The indole and indoline scaffolds are important nitrogen-containing heterocycles which can be widely found in natural alkaloids, commercial drugs and other functional molecules.1,2 With the development of C–H bond functionalization,3 it has become the most straight forward protocol leading to C-7 substituted indoles and indolines. Recently, a great deal of effort have been devoted to the formation of C-2 and C-3 functionalized indoles.4 Nevertheless, the C–H bond functionalization of indoles at the C-7 position remains relatively unexplored.5 As far as we know, many examples of transition-metal-catalyzed C-7 C–H bond functionalizations of indolines with various coupling partners have been disclosed such as olefination,6 alkynylation,7 alkylation,8 and carbonylation.9 Equally importantly, arylation in the C-7 position of indoline was first be reported by Sanford group with the hypervalent iodine compound and assisted by acetyl group.10 In 2007, Shi group developed the palladium catalyzed C–H functionalizations of the Suzuki–Miyaura reaction.11 In the same year, Lipshutz group developed a mild condition cross coupling reaction directed by urea group.12 In addition, the Oestreich group developed a dehydrogenative C–H/C–H arylation of indolines in a mild condition.13 However, the methods for the synthesis of C-7 arylated indolindes and C-7 arylated indoles are very limited.
On the other hand, the Hiyama cross-coupling reaction is one of the most useful and reliable approaches for the formation of C–C bonds.14,15 Compared with many other organometallic coupling-partners, organosilicon reagents have many unique advantages, including nontoxicity, high stability, environmental benignity and ease of introduction into substrates. Up to now, there are still very few examples on C–H bond direct arylation using organosilanes as the coupling reagents.16,17 To the best of our knowledge, the C-7 arylation of indoline with arylsilanes has not been reported. Herein, we reported palladium-catalyzed oxidative arylation of indolines with arylsilanes at the C-7 position via C–H bond activation. The reaction proceeds smoothly under very mild conditions and it shows a much wider substrate scope (Scheme 1).
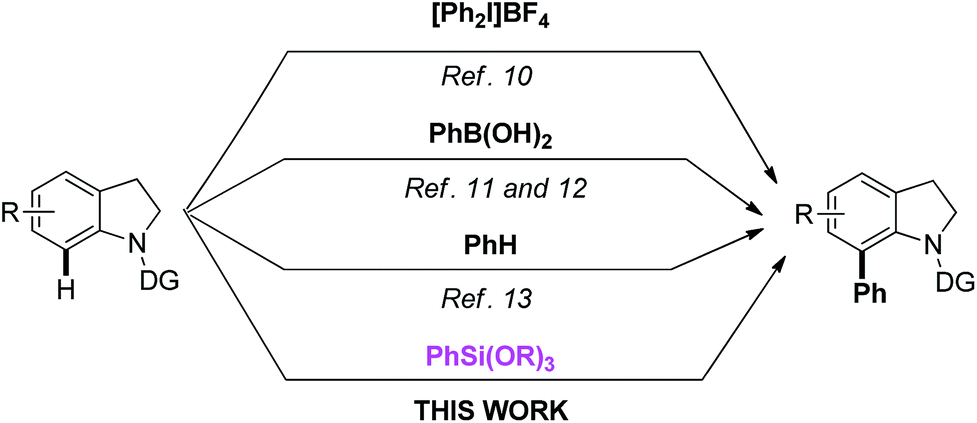 | ||
| Scheme 1 Methods for direct C7 C–H arylation of the indoline. | ||
At the outset of the investigation, we first examined the direct C-7 C–H arylation of N-acetylindoline 1a coupling with phenyltrimethoxysilane 2a in the presence of 10 mol% of Pd(OAc)2 in dioxane at 80 °C, and 2.0 equiv. AgF was used as the fluorine source. After preliminary screening of different copper salts as the oxidants, Cu(OTf)2 turned out to be the suitable additive for the reaction (Table 1, entries 1–4). One of the key points of the Hiyama coupling reaction is to find an appropriate fluorine source to break the C–Si bond. Then, different fluorine sources such as AgF, TBAF, KF, and CsF were examined (Table 1, entries 4–7). All the fluorine sources except TBAF were found to be effective, affording the desired product 3aa. Among these salts, AgF is the best choice for the reaction; affording the desired product in a yield of 79% (Table 1, entries 4). The effect of solvent was also investigated (Table 1, entries 8–11), dioxane and THF were both found to be suitable for the reaction. However, trace amounts of or no desired product could be detected by using DMF, DMSO and toluene as solvents. Furthermore, in order to gain a more mild reaction condition, the reaction temperature was tried to reduce. To our delight, the reaction could proceed smoothly when temperature was reduced to 40 °C or 60 °C, which can afford the better yield in 91% (Table 1, entries 13). It was noted that only 32% yield could be observed when the reaction was performed at room temperature (Table 1, entries 14).
|
||||
|---|---|---|---|---|
| Entry | Oxidant | F source | Solvent | Yieldb (%) |
| a Unless otherwise noted, the reaction conditions are as follows: 1a (0.2 mmol), 2a (0.4 mmol), solvent (4 mL).b Isolated yield after purification by flash column chromatography on silica gel.c Reaction time was 48 h.d T = 60 °C or 40 °C.e Room temperature. | ||||
| 1 | CuCl2 | AgF | Dioxane | 0 |
| 2 | CuBr2 | AgF | Dioxane | 0 |
| 3 | Cu(OAc)2 | AgF | Dioxane | 0 |
| 4 | Cu(OTf)2 | AgF | Dioxane | 79 |
| 5 | Cu(OTf)2 | TBAF | Dioxane | 0 |
| 6 | Cu(OTf)2 | KF | Dioxane | 35 |
| 7 | Cu(OTf)2 | CsF | Dioxane | 41 |
| 8 | Cu(OTf)2 | AgF | DMF | 0 |
| 9 | Cu(OTf)2 | AgF | DMSO | 0 |
| 10 | Cu(OTf)2 | AgF | Toluene | <5% |
| 11 | Cu(OTf)2 | AgF | THF | 85 |
| 12c | Cu(OTf)2 | AgF | THF | 91 |
| 13d | Cu(OTf)2 | AgF | THF | 91 |
| 14e | Cu(OTf)2 | AgF | THF | 32 |
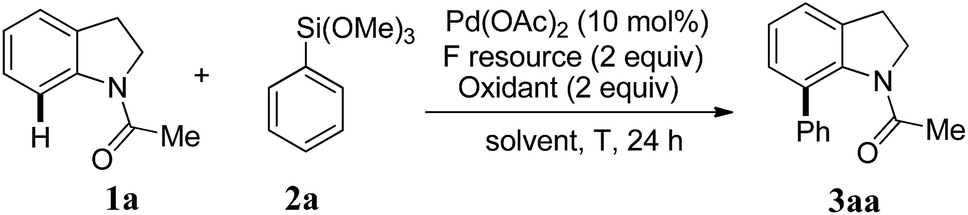
With the optimized reaction condition, we then proceeded to explore the scope of the direct C-7 C–H arylation with various substituted indolines with phenyltrimethoxysilane. The results are summarized in Table 2. Firstly, various indolines bearing carbonyl kind direct groups (3aa–3ad) were investigated, most of them led to moderate to excellent yield. Especially, the indolines with pivaloyl group gave the best yield in 92% (3ab). For the direct group replaced as urea unit, the reaction gave a little diminished yield of the product 3ac in 55%. While the substrate bearing a pyrimidine direct group (3ae) was tested, only trace product was detected by 1H NMR spectra. Using the pivaloyl group as the direct group, various indoline derivatives was used to react with phenyltrimethoxysilane. In general, the indolines bearing electron-donating groups on the aromatic ring such as 4-methyl and 5-methoxyl group react smoothly to afford the corresponding arylated products in very excellent yields (3af–3ag). In addition, the reactions with the indolines bearing electron-drawing group on the aromatic ring also afford moderate to good yield (3ah–3aj). Notably, the substrates with C-2 or C-3 methyl substituted group also show excellent compatibility with the reaction condition (3aj–3ak). But indolines with two substituent groups on both C-2 and C-3 positions were used to carry out the reaction, the diminished yield will obtained in 50% and 31% yield, respectively (3al–3am).
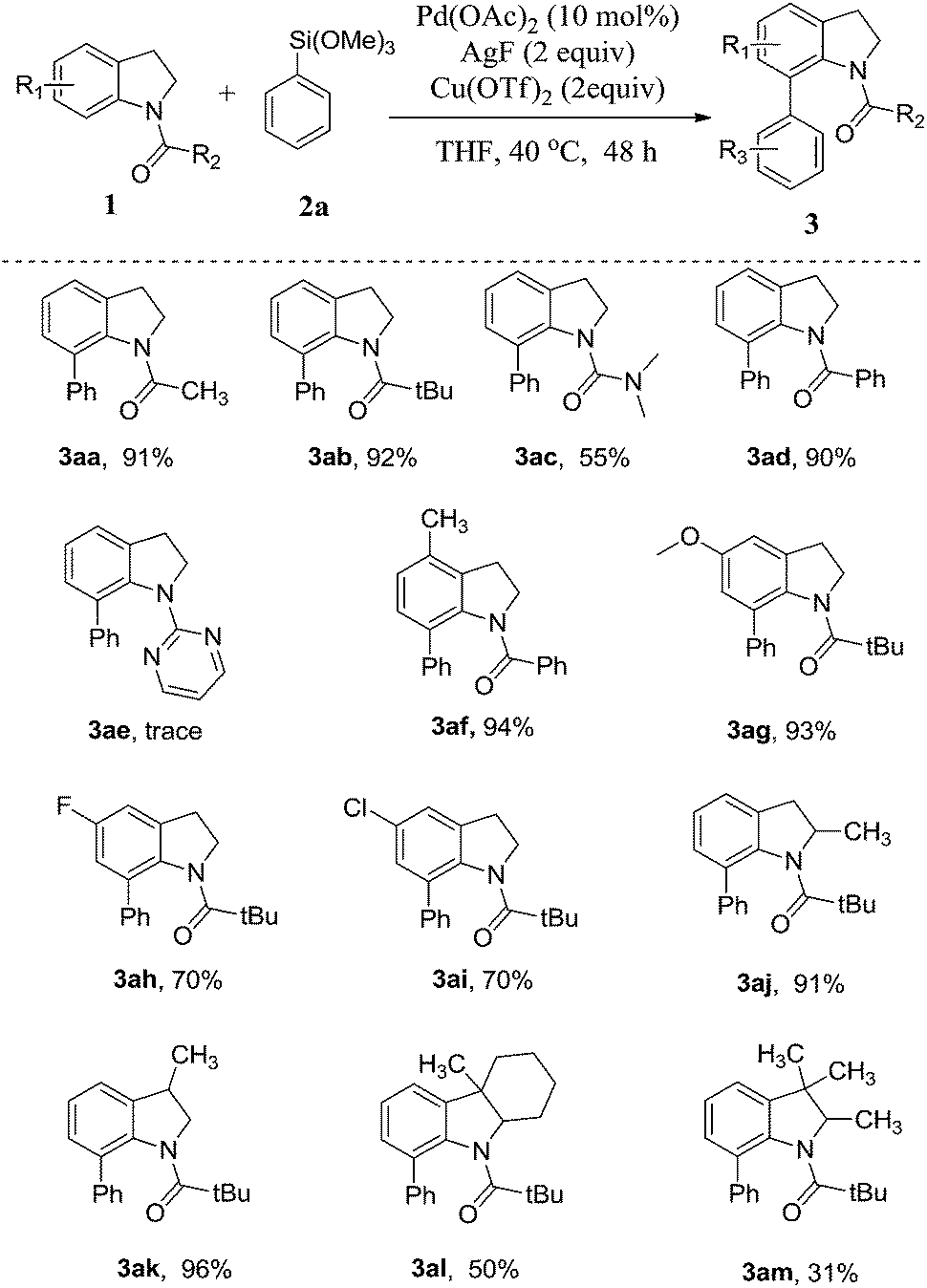
To further evaluate the substrate scope, a series of arylsilanes were used to test the reaction with the N-pivaloylindoline 1b under the optimized reaction conditions. The results are summarized in Table 3. At first, phenyltriethoxysilane was used to test the reaction and it shows a good compatibility in our reaction. For this reaction, all substituted arylsilanes were prepared by following the reported method from the corresponding Grignard reagents.18 To investigate the steric effect, the phenyltriethoxysilanes bearing methyl group on different positions were employed as the substrates. The results indicate that the phenyltriethoxysilane with ortho-methyl substituent was found to afford the desired product for a little diminished yield in 61% (3bd). Essentially no steric effect was observed as shown in the reactions by using the other two meta- and para-methyl phenyltriethoxysilanes as the substrates; affording the same yield in 87% (3bb–bc). The direct C–H arylation of the indolines has shown excellent tolerance to both electron-withdrawing and electron-donating groups of aromatic substituents, including methyl (3bb–bd), methoxy (3be), fluoro (3bf) and chloro groups (3bg). In addition, the heterocyclic triethoxy(thiophen-2-yl)silane was also used to explore the possibility of the C–H arylation, the reaction also afforded the corresponding product, but only low yield (21%) was obtained (3bh).
 | ||
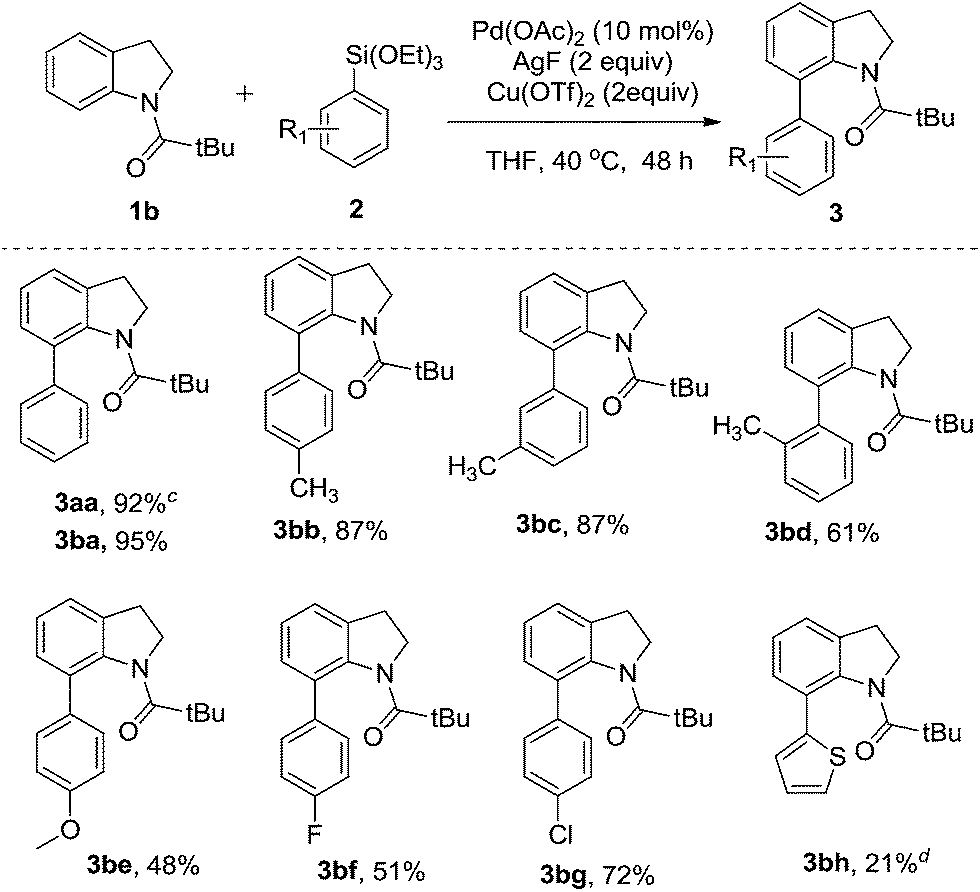
| Scheme 2 Palladium-catalyzed direct C–H arylation of the N-pivindole 3a. | ||
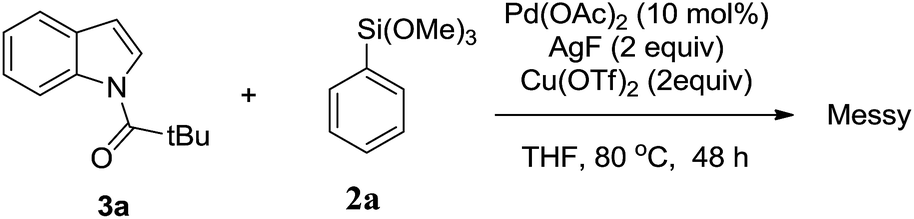
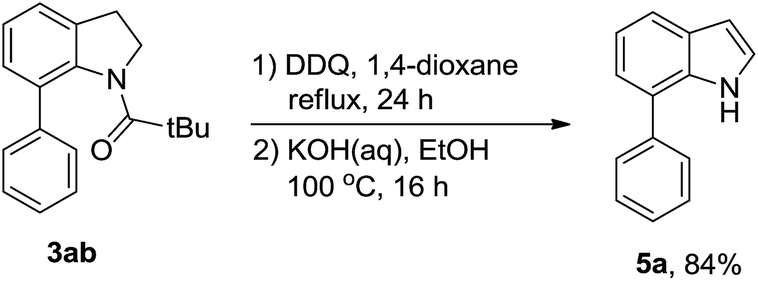
Next, we tried to explore direct C-7 C–H arylation with N-pivindole 3a and phenyltrimethoxysilane 2a under the standard condition. But it was very disappointing that the reaction was totally messy, which was observed by TLC plate (Scheme 2). It means that the reaction cannot afford a single arylated product by using N-pivindole as the substrate under the standard conditions. Then, the transformation of C-7 arylated indoline into C-7 arylated indole was considered to be crucial for this method. As shown in Scheme 3, the transformation was begun with the oxidation of C-7 arylated N-pivindoline by the addition of DDQ (2,3-dicyano-5,6-dichlorobenzoquinone), followed by removing the directing group with the hydrolysis of the amide strategy. Finally, 84% yield of the arylated indole product was successfully obtained.
 | ||
| Scheme 3 Transformation of C7-arylated indoline to the corresponding indole. | ||
Conclusions
In summary, we have demonstrated a direct Pd-catalyzed C-7 arylation of indolines with arylsilanes via C–H activation. In this reaction, carbonyl based directing group is needed on the indoline nitrogen atom. These transformations have been applied to a wide range of substrates. It represents a facile access to 7-arylated indolines, which can be conveniently transformed into 7-arylated indoles. Since this reaction exhibits excellent reactivity and broad substrate scope in very mild conditions, it may be found useful applications in organic synthesis.Acknowledgements
The project is supported by Natural Science Foundation of China (No. 21562003), the Natural Science Foundation of Jiangxi Province (No. 20151BAB203009) and Natural Science Foundation of Jiangxi Provincial Education Department (No. KJLD14080 and 13081).Notes and references
- (a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (b) J. D. Podoll, Y. Liu, L. Chang, S. Walls, W. Wang and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15573 CrossRef CAS PubMed.
- (a) L. Xiang, D. Xing, W. Wang, R. Wang, Y. Ding and L. Du, Phytochemistry, 2005, 66, 2595 CrossRef CAS PubMed; (b) M. E. Kuehne, W. G. Bornmann, I. Markó, Y. Qin, K. L. LeBoulluec, D. A. Frasier, F. Xu, T. Mulamba, C. L. Ensinger and L. S. Borman, Org. Biomol. Chem., 2003, 1, 2120 RSC; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (d) M. G. Bell, D. L. Gernert, T. A. Grese, M. D. Belvo, P. S. Borromeo, S. A. Kelley, J. H. Kennedy, S. P. Kolis, P. A. Lander and R. Richey, J. Med. Chem., 2007, 50, 6443 CrossRef CAS PubMed; (e) T. Owa, A. Yokoi, K. Yamazaki, K. Yoshimatsu, T. Yamori and T. Nagasu, J. Med. Chem., 2002, 45, 4913 CrossRef CAS PubMed.
- For selected reviews on C-H bond functionalization, see: (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (b) J. Wencel-Delord, T. Drçge, F. Kiu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (c) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (d) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., 2012, 124, 10382 (Angew. Chem., Int. Ed., 2012, 51, 10236) CrossRef; (e) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed.
- For selected reviews, see: (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673 CrossRef CAS; (c) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85 CrossRef CAS PubMed; (d) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215 CrossRef PubMed.
- (a) D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068 CrossRef CAS PubMed; (b) Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495 CrossRef CAS PubMed; (c) L. Xu, C. Zhang, Y. He, L. Tan and D. Ma, Angew. Chem., Int. Ed, 2016, 55, 321 CrossRef CAS PubMed.
- (a) X.-F. Yang, X.-H. Hu, C. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 2532 RSC; (b) Z. Song, R. Samanta and A. P. Antonchick, Org. Lett., 2013, 15, 5662 CrossRef CAS PubMed; (c) L.-Y. Jiao and M. Oestreich, Org. Lett., 2013, 15, 5374 CrossRef CAS PubMed; (d) S. Pan, T. Wakaki, N. Ryu and T. Shibata, Chem.–Asian J., 2014, 9, 1257 CrossRef CAS PubMed; (e) B. Urones, R. G. Arrayás and J. C. Carretero, Org. Lett., 2013, 15, 1120 CrossRef CAS PubMed.
- (a) N. Jin, C. Pan, H. Zhang, P. Xu, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2015, 357, 1149 CrossRef CAS; (b) Y. Wu, Y. Yang, B. Zhou and Y. Li, J. Org. Chem., 2015, 80, 1946 CrossRef CAS PubMed.
- (a) C. Premi, A. Dixit and N. Jain, Org. Lett., 2015, 17, 2598 CrossRef CAS PubMed; (b) S. R. Neufeldt, C. K. Seigerman and M. S. Sanford, Org. Lett., 2013, 15, 2302 CrossRef CAS PubMed; (c) S. Pan, N. Ryu and T. Shibata, Adv. Synth. Catal., 2014, 356, 929 CrossRef CAS.
- (a) Y. Shin, S. Sharma, N. K. Mishra, S. Han, J. Park, H. Oh, J. Ha, H. Yoo, Y. H. Jung and I. S. Kim, Adv. Synth. Catal., 2015, 357, 594 CrossRef CAS; (b) M. Kim, N. K. Mishra, J. Park, S. Han, Y. Shin, S. Sharma, Y. Lee, E.-K. Lee, J. H. Kwak and I. S. Kim, Chem. Commun., 2014, 50, 14249 RSC.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed.
- Z. Shi, B. Li, X. Wan, J. Cheng, Z. Fang, B. Cao, C. Qin and Y. Wang, Angew. Chem., Int. Ed., 2007, 46, 5554 CrossRef CAS PubMed.
- T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 4978 CrossRef CAS PubMed.
- (a) L. Y. Jiao and M. Oestreich, Eur. J. Org. Chem., 2013, 19, 10845 CrossRef CAS PubMed; (b) L. Y. Jiao, P. Smirnov and M. Oestreich, Org. Lett., 2014, 16, 6020 CrossRef CAS PubMed.
- For selected reviews on Hiyama coupling reaction, see: (a) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835 CrossRef CAS PubMed; (b) T. Hiyama and E. Shirakawa, Top. Curr. Chem., 2002, 219, 61 CrossRef CAS; (c) S. E. Denmark and M. H. Ober, Aldrichimica Acta, 2003, 36, 75 CAS; (d) S. E. Denmark and J. H.-C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978 CrossRef CAS PubMed; (e) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC.
- (a) S. E. Denmark and S.-M. Yang, J. Am. Chem. Soc., 2004, 126, 12432 CrossRef CAS PubMed; (b) S. E. Denmark, C. S. Regens and T. Kobayashi, J. Am. Chem. Soc., 2007, 129, 2774 CrossRef CAS PubMed; (c) S. E. Denmark, J. H.-C. Liu and J. M. Muhuhi, J. Am. Chem. Soc., 2009, 131, 14188 CrossRef CAS PubMed.
- (a) S. D. Yang, B. J. Li, X. B. Wan and Z. J. Shi, J. Am. Chem. Soc., 2007, 129, 6066 CrossRef CAS PubMed; (b) H. Zhou, Y. H. Xu, W. J. Chung and T. P. Loh, Angew. Chem., Int. Ed., 2009, 48, 5355 CrossRef CAS PubMed; (c) M. Z. Lu, P. Lu, Y. H. Xu and T. P. Luo, Org. Lett., 2014, 16, 2614 CrossRef CAS PubMed; (d) H. Luo, Z. Zhang, H. Liu and H. Liu, Chin. J. Org. Chem., 2015, 35, 802 CrossRef CAS.
- (a) H. Hachiya, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2010, 49, 2202 CrossRef CAS PubMed; (b) Z. J. Liang, B. B. Yao and Y. H. Zhang, Org. Lett., 2010, 12, 3185 CrossRef CAS PubMed; (c) L. Bi and G. I. Georg, Org. Lett., 2011, 13, 5413 CrossRef CAS PubMed; (d) W. Li, Z. W. Yin, X. Q. Jiang and P. P. Sun, J. Org. Chem., 2011, 76, 8543 CrossRef CAS PubMed.
- (a) M. Murata, H. Yamasaki, T. Ueta, M. Nagata, M. Ishikura, S. Watanabe and Y. Masuda, Tetrahedron, 2007, 63, 4087 CrossRef CAS; (b) A. S. Manoso, C. Ahn, A. Soheili, C. J. Handy, R. Correia, W. M. Seganish and P. DeShong, J. Org. Chem., 2004, 69, 8305 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06915k |
| This journal is © The Royal Society of Chemistry 2016 |